

Abstract
Chronic hepatitis B virus (HBV) infection via perinatal transmission is common in the Asia-Pacific region, but related quasispecies (QS) characteristics are not yet defined. To investigate the homologous, full-length HBV QS after perinatal infection and their clinical implications, five pairs of mother-daughter patients with chronic HBV infection (one patient with liver cirrhosis, one with immune tolerance, and eight with chronic hepatitis) were included. Full-length HBV were amplified by PCR from serum samples before antiviral treatment and cloned; an average of 17 clones per sample were sequenced, and the QS complexities, diversities, and evolution patterns were analyzed. Full-length HBV sequence similarities within mother-daughter pairs were 91.3 to 98.3%. The QS complexities of full-length HBV were similar between mothers and daughters (median of 0.9734 compared to 0.9688, P > 0.05), as were the diversities (median of 3.396 × 10−3 compared to 4.617 × 10−3substitutions/site, P > 0.05). However, the distribution patterns of QS complexities and diversities within specific genes were different, and QS genetic distances of the mothers were higher than those of daughters, both more evident in pairs with different antiviral responses and different immune phases or stages. The nucleotide substitution rate of full-length HBV was 14.388 × 10−5 substitutions/site/year, whereas the preC/C gene rate was the highest. Mutations and indels were more common in clones from the mothers, which decreased the affinity of epitopes by 6- to 89-fold. The various genes from homologous HBV genomes evolved in different patterns despite numerically comparable full-length QS complexities and diversities. The different patterns may correlate with the immune stages of chronic HBV infection, which warrants further study.
INTRODUCTION
Chronic hepatitis B virus (HBV) infection is still a serious public health problem, with potential adverse sequelae, such as liver cirrhosis, liver failure, and hepatocellular carcinoma. More than 350 million people are chronically infected with HBV, and 75% reside in the Asia-Pacific region (1, 2), where the infection is usually acquired perinatally or in early childhood. In contrast to a very short immune-tolerant (IT) phase in adult-acquired chronic HBV infection, the perinatally or early-childhood-acquired chronic HBV infection has a long immune-tolerant phase, followed by an immune-reactive phase, inactive carrier state, HBeAg-negative chronic hepatitis B, and HBsAg-negative phase. The clinical and laboratory characteristics of each phase have been described in detail previously (3, 4).
Due to absence of proofreading during reverse transcription and a high replication rate, the HBV population consists of genetically distinct but closely related variants known as quasispecies (QS). QS means a spectrum of mutants that possess different fitness levels in certain environments (5, 6). Mutants with higher fitness levels predominate by competitive replication, and the predominant mutant may differ in a changing environment (7). In chronic HBV infection via perinatal transmission, the different immune phases of chronic HBV infection confer different environments on HBV QS; thus, the characteristics of HBV QS may differ. However, the characteristics of homologous HBV QS via perinatal transmission in different phases are not yet defined, and the differences of full-length HBV QS between mothers and their progeny have not been investigated, either.
The aim of our present study was to investigate the characteristics of homologous full-length HBV QS evolving in different hosts and its potential correlation with different immune phases and antiviral response.
MATERIALS AND METHODS
Patients.
Five pairs of patients (mother-daughter) with chronic HBV infection at our medical center from May 2008 to July 2010 were retrospectively included in our present study. Written informed consent was obtained from all patients, and the study protocol was approved by the Ethics Committee of Ruijin Hospital in accordance with the Declaration of Helsinki. Each pair of patients met with the following criteria: presence of HBsAg for at least 6 months, elevated HBV DNA levels (>2,000 IU/ml) with elevated or normal serum alanine aminotransferase (ALT) levels (upper limit of normal, 40 U/liter), and no signs of decompensated liver disease (e.g., variceal bleeding, ascites, or encephalopathy). Patients were required to be naive to antiviral treatment history (alpha interferon [IFN-α] or nucleoside/nucleotide analogues) before serum samples were collected.
The perinatal infection in each pair was confirmed in our study by the following criteria: presence of HBsAg in mothers before giving birth and in their daughters during childhood and of homologous sequences documented by the phylogenetic analysis of HBV surface (S) genes from the mothers and daughters (8, 9).
Methods. (i) Liver biochemistry, HBV serology, and HBV DNA tests.
Liver biochemical parameters were tested using an automated chemistry analyzer (Beckman Coulter, Fullerton, CA). HBV serological markers were determined by chemiluminescent microparticle immunoassay using the Abbot Architect immunoassay system (Abbott Laboratories, Abbott Park, IL). The HBV DNA levels were measured by PCR using the Cobas Amplicor HBV monitor test (Roche Diagnostics, Basel, Switzerland) with a low limit of quantification, 300 copies/ml.
(ii) Molecular cloning and sequencing.
HBV genomes were extracted from 1,000-μl serum samples at baseline (before antiviral treatment) using the QIAamp UltraSens virus kit (Qiagen, Hilden, Germany). The complete genomes of HBV were amplified using PCR as described by Gunther et al. (10). Pfu Ultra High-Fidelity DNA polymerase (Agilent, Santa Clara, CA) was used to ensure the high-fidelity PCR. For samples with low templates, the PCR systems were doubled to yield enough PCR products. The primers were as follows: 5′-TTT TTC ACC TCT GCC TAA TCA-3′ (forward; nucleotides [nt] 1821 to 1841) and 5′-AAA AAG TTG CAT GGT GCT GG-3′ (reverse; nt 1825 to 1806). PCR products of about 3,200 bp were purified using the QIAquick gel extraction kit (Qiagen), cloned into the Topo XL PCR cloning vector after the addition of adenylate tails, and transformed into TOP10 Escherichia coli competent cells (Invitrogen, Carlsbad, CA) growing on kanamycin plates. An average of 17 (range of 15 to 24) clones per sample were sequenced randomly using an ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA). Totals of 172 clones were sequenced in the mother and daughter groups.
(iii) Sequence analysis.
Sequence segments were assembled to full-length HBV using the CodonCode Aligner software package (CodonCode Corporation, Dedham, MA) and subsequently were divided into four genes, including S, precore/core (preC/C), polymerase (P), and X genes. The reverse transcriptase (RT) region, preS1, and preS2 were also analyzed. Multiple alignments were carried out on all sequences by using Clustal X, version 2.0 (11). RDP4 software was used to detect recombinant sequences, which were excluded (12). Genotypes of each sequence were determined using the HBV STAR program online (13). Viral QS heterogeneity was evaluated with complexity and diversity. QS complexity was measured using normalized Shannon entropy (Sn) as previously described (14, 15). QS diversity was evaluated by three parameters: the mean genetic distance (d; also called Hamming distance), the number of synonymous substitutions per synonymous site (dS), and the number of nonsynonymous substitutions per nonsynonymous site (dN). All parameters were calculated using MEGA 5.0 software under proper nucleotide/amino acid substitution models (16, 17). The sequence identity matrix and entropy plot were performed using BioEdit software (18) after all gaps were removed.
Net changes in QS diversity, which determine population-based genetic diversity within the context of sequence differences between two populations, estimate the extent of “clustered evolution” in terms of phylogenetic representation. This parameter was selected to clarify the evolutionary pattern of HBV QS after perinatal infection and was calculated using MEGA 5.0 software (19).
PHYML version 3.0 was used to construct phylogenetic trees at baseline using the maximum likelihood method under the general time reversible (GTR) plus proportion of invariant sites (I) plus shape parameter of the gamma distribution (Γ) model, which was estimated by ModelTest 3.7 in advance as previously described (20, 21). Relative rates of nucleotide substitution (six categories), the proportion of invariant sites, and the shape parameter of the gamma distribution were also estimated using ModelTest. The viral evolution rate between the mothers and daughters was calculated using PEBBLE 1.0 software and the serial sample unweighted pair group method with arithmetic mean (sUPGMA) algorithm under the appropriate substitution model for each pair, which was also estimated by ModelTest (22).
(iv) Analysis of immune epitopes.
The sequence of each clone was aligned with that of the wild-type strain (GenBank accession number AB014381, genotype C). The hot-spot mutations in immune epitopes were identified based on previous publications (23–25). Then they were submitted to websites to predict the binding affinity (http://www-bimas.cit.nih.gov/molbio/hla_bind/ and http://www.syfpeithi.de/).
(v) Statistical analysis.
Results were expressed as means or medians and ranges. Variables between the mother and daughter groups were compared using the Student t test, the Mann-Whitney test, the Wilcoxon signed-rank test (related samples), and Fisher's exact test if appropriate. Correlations were analyzed using Spearman's rank correlation. Results were considered statistically significant at a P value of <0.05.
RESULTS
Demographic, clinical, and laboratory data.
Five pairs of patients (mother-daughter) were enrolled in the present study. The demographic, clinical, and laboratory data are shown in Table 1. Three of five pairs were diagnosed with chronic hepatitis B (CHB), one mother was diagnosed with liver cirrhosis (LC), and one mother was in the immune-tolerant (IT) phase (liver biopsy indicated a Metavir score of A0F0). All patients were genotype C. The median age of the daughters was 27 years (range, 22 to 33 years) and that of mothers was 56 years (range, 50 to 62 years), and the baseline ALT and HBV DNA levels were comparable between daughters and mothers.
TABLE 1.
The demographic, clinical, and laboratory data of enrolled patientsa
| Pair | Case no. | Group | Age (yr) | Diagnosis | Genotype | HBsAg (IU/ml) | HBeAg (S/CO) | Baseline ALT level (IU/liter) | Baseline HBV DNA level (log10 copies/ml) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | M | 50 | CHB | C | >250 | 1,471.45 | 225 | 8.50 |
| 2 | D | 22 | CHB | C | >250 | 1,202.27 | 165 | 6.15 | |
| 2 | 3 | M | 56 | CHB | C | >250 | 1.16 | 83 | 6.19 |
| 4 | D | 33 | CHB | C | 27.22 | 1.06 | 108 | 5.39 | |
| 3 | 5 | M | 62 | LC | C | 4,009.61 | 14.17 | 28 | 7.04 |
| 6 | D | 32 | CHB | C | >250 | 152.79 | 190 | 6.75 | |
| 4 | 7 | M | 50 | IT | C | >250 | 65.28 | 20 | 8.68 |
| 8 | D | 26 | CHB | C | >250 | 4.63 | 96 | 6.81 | |
| 5 | 9 | M | 58 | CHB | C | 28.30 | 0.29 | 89 | 5.90 |
| 10 | D | 27 | CHB | C | >250 | 12.37 | 271 | 6.18 |
M, mother; D, daughter; CHB, chronic hepatitis B; LC, liver cirrhosis; IT, immune-tolerant phase; S/CO, signal/cutoff ratio.
Except for one mother in the IT phase, all patients received antiviral therapy, either pegylated IFN-α-2a (PEG-IFN-α-2a) or nucleoside/nucleotide analogues. The detailed antiviral treatments and virological responses are listed in Table 2. Different antiviral responses were compared between the mothers and daughters in pairs 1 and 3. Both received PEG-IFN-α-2a in pair 1; the mother had an undetectable HBV DNA level and HBeAg seroconversion at week 24, whereas the daughter didn't acquire a virological response until the treatment was extended to week 72, and no HBeAg seroconversion was observed. In addition, viral relapse occurred after PEG-IFN-α-2a withdrawal. In pair 3, the mother (LC) experienced viral breakthrough, and an rtM204I mutation was detected at week 32 during telbivudine treatment, whereas the daughter (CHB) had undetectable HBV DNA levels at week 12 and HBeAg seroconversion at week 42.
TABLE 2.
Antiviral treatment and virological responses of enrolled patientsb
| Pair | Case no. | Group | Diagnosis | Treatment | Time to undetectable HBV DNA (wks) | Time to HBeAg seroconversion (wks) | Time to follow-up (mo) |
|---|---|---|---|---|---|---|---|
| 1 | 1 | M | CHB | PEG-IFN-α-2a | 24 | 24 | 53 |
| 2 | D | CHB | PEG-IFN-α-2a | 72 | NA | 60 | |
| 2 | 3 | M | CHB | LAM-ADV | 12 | NA | 35 |
| 4 | D | CHB | LAM-ADV | 12 | NA | 34 | |
| 3 | 5 | M | LC | LdT | NAa | NA | 63 |
| 6 | D | CHB | LdT | 12 | 42 | 33 | |
| 4 | 7 | M | IT | None | 59 | ||
| 8 | D | CHB | LdT | 13 | NA | 58 | |
| 5 | 9 | M | CHB | ETV | 12 | NA | 12 |
| 10 | D | CHB | ETV | 12 | NA | 12 |
Viral breakthrough occurred at week 32 in this patient, and HBV DNA levels decreased to undetectable levels after adding adefovir dipivoxil.
PEG-IFN, pegylated interferon; LAM, lamivudine; LdT, telbivudine; ETV, entecavir; NA, not available.
Defining homologous HBV genomes and viral nucleotide substitution rates.
The homology of the HBV genomes was confirmed by a sequence identity matrix and phylogenetic analysis, in addition to the history of perinatal infection. The sequence identity matrix showed the proportion of identical nucleotides between sequences within one pair (18). The matrix indicated that the lower limit of similarities between the mother and the daughter within pairs ranged from 91.3% to 98.3%. If all gaps and insertions were removed, the lower limit reached as high as 98.0% to 99.3%.
A phylogenetic tree was constructed using the maximum likelihood algorithm. A complete genome of HBV genotype D (GenBank accession no. X85254) served as the outgroup sequence. The phylogenetic trees are shown in Fig. 1. Sequences from the same pair clustered together as expected, which suggested that they were homologous. The branch length of patient 5 (mother group, LC) was much longer than that of the other patients, in accordance with the highest QS diversity among all patients as follows.
FIG 1.

Phylogenetic tree of full-length HBV sequences. The tree was constructed using the maximum likelihood method with a bootstrap of 500. The dark-gray lines represent the mother group, whereas the light-gray ones represent the daughter group. The sequences from a single pair clustered together, indicating they were homologous. The branch length of patient 5 (mother group, liver cirrhosis) was much longer than that of other patients, in accordance with the highest QS diversity among all patients. A genotype D genome (GenBank accession number X85254) served as the outgroup sequence.
Viral nucleotide substitution rates between the mothers and daughters were calculated for both complete genome and specific genes (Table 3). Although the QS characteristics of both the mothers and daughters resulted from the virus evolution since they acquired the infection, the virus strains from the mothers and daughters were the same or the most similar when perinatal infections occurred. Therefore, ages of the daughters were regarded as evolution time (about 30 years). The median nucleotide substitution rate of full-length HBV (14.388 × 10−5 substitutions/site/year) was similar to that of the S gene, P gene, and RT domain. However, the preC/C gene had a much higher rate than the other genes, followed by the X gene.
TABLE 3.
Nucleotide substitution rate of HBV QS
| Gene or domain | Substitution rate (range) (10−5 substitutions/site/yr) |
|---|---|
| Complete genome | 14.388 (10.963∼26.794) |
| S gene | 14.265 (1.763∼16.317) |
| P gene | 14.780 (11.383∼30.844) |
| X gene | 19.419 (8.709∼128.710) |
| PreC/C gene | 36.968 (5.948∼192.732) |
| RT domain | 11.103 (5.238∼12.9539) |
Characteristics of HBV QS.
The QS complexities and diversities of the mother group and the daughter group are listed in Table 4. In general, full-length HBV and specific genes in each group had numerically comparable QS complexities and diversities. For example, the QS complexities of full-length HBV of mothers were comparable to those of daughters overall (median of 0.9734 compared to 0.9688, P > 0.05) and so were the diversities of full-length HBV (median of 3.396 × 10−3 compared to 4.618 × 10−3substitutions/site, P > 0.05). A Spearman correlation analysis showed that QS complexity correlated positively with the length of the genes (r = 0.764, P < 0.001).
TABLE 4.
Characteristics of HBV QS by groups
| Characteristica | Value (range) |
P value | |
|---|---|---|---|
| Mothers (n = 5) | Daughters (n = 5) | ||
| HBV complete genome | |||
| Complexity (nt) | 0.9734 (0.7764∼1) | 0.9688 (0.7611∼1) | 0.548 |
| d (nt) | 3.396 (2.5484∼18.4266) | 4.6171 (3.2639∼10.7356) | 0.841 |
| S gene | |||
| Complexity (nt) | 0.6097 (0.4269∼0.8834) | 0.6671 (0.5195∼0.9688) | 0.548 |
| Complexity (aa) | 0.4940 (0.2192∼0.8834) | 0.5964 (0.4764∼0.8945) | 0.690 |
| d (nt) | 3.1677 (2.0918∼16.2799) | 4.8128 (2.7878∼10.9449) | 0.548 |
| d (aa) | 7.5337 (5.4031∼39.1308) | 11.9728 (6.2915∼20.2155) | 0.690 |
| dS | 1.6991 (0.5249∼6.6704) | 2.2938 (0.6793∼15.3697) | 0.690 |
| dN | 3.3153 (2.3565∼17.0512) | 5.2478 (2.8077∼9.5497) | 0.690 |
| P gene | |||
| Complexity (nt) | 0.9734 (0.8181∼1) | 0.9688 (0.7611∼1) | 0.5478 |
| Complexity (aa) | 0.8668 (0.8181∼1) | 0.8763 (0.7611∼0.9688) | 1 |
| d (nt) | 2.9975 (2.1478∼18.1913) | 4.0352 (2.7891∼11.9128) | 0.690 |
| d (aa) | 4.9776 (3.5722∼27.1359) | 5.1571 (4.3797∼17.845) | 0.690 |
| dS | 5.7286 (2.8398∼25.2360) | 8.1217 (2.9046∼19.4580) | 0.841 |
| dN | 2.5348 (1.7023∼14.6623) | 2.7694 (2.0927∼8.9021) | 0.690 |
| X gene | |||
| Complexity (nt) | 0.6169 (0.4269∼0.7764) | 0.6330 (0.4332∼0.7899) | 0.548 |
| Complexity (aa) | 0.5970 (0.3591∼0.6767) | 0.5153 (0.2659∼0.7899) | 1 |
| d (nt) | 3.43234 (2.4379∼10.0758) | 3.6193 (1.4378∼9.6726) | 1 |
| d (aa) | 9.2298 (5.7517∼17.9920) | 10.20190 (2.6804∼24.6046) | 1 |
| dS | 1.6382 (0∼5.1022) | 2.7177 (0.9540∼5.4788) | 0.310 |
| dN | 4.2825 (2.6844∼9.3927) | 4.8264 (1.2503∼11.5975) | 1 |
| PreC/C gene | |||
| Complexity (nt) | 0.7349 (0.6120∼0.9100) | 0.6014 (0.1450∼0.8632) | 0.222 |
| Complexity (aa) | 0.5083 (0.4269∼0.9100) | 0.42695 (0.1450∼0.9100) | 0.421 |
| d (nt) | 7.8161 (2.6308∼73.8982) | 6.4847 (0.8062∼108.4303) | 0.690 |
| d (aa) | 12.3773 (5.3892∼223.8448) | 9.8675 (1.2092∼21.6277) | 0.690 |
| dS | 5.7235 (1.6433∼86.0777) | 6.3419 (1.4221∼11.0276) | 1 |
| dN | 7.0272 (2.5507∼8.2026) | 5.2271 (0.5691∼10.7794 | 0.690 |
Sn, Shannon entropy; nt, nucleotide; aa, amino acid; dS, number of synonymous substitutions per synonymous site; dN, number of nonsynonymous substitutions per nonsynonymous site.
The complexities of HBV QS (including the complete genome and each gene) in each pair are shown in Fig. 2. Of note, the QS complexities of the preC/C regions of the mothers appeared higher than those of daughters in 4 of 5 pairs. On the contrary, the QS complexities of preS1 regions of the daughters appeared higher than those of the mothers in 4 of 5 pairs. A univariate analysis of variance indicated that the QS complexities among various genes were different (P < 0.001), while the difference between the mothers and daughters did not reach statistical significance (P = 0.704).
FIG 2.

The QS complexity (Shannon entropy [Sn]) in each pair at the nucleotide level. The QS complexity distribution of the mothers differed from that of the daughters. Of note, the QS complexities in the preC/C regions of the mothers were higher than those of the daughters in 4 of 5 pairs, especially in pair 1. On the contrary, the QS complexities in the preS1 regions of the daughters were higher than those of the mothers in 4 of 5 pairs.
As for QS diversity (genetic distance at the nucleotide level), the distribution pattern was even more different (Fig. 3). In general, the genetic distances of various genes of the mothers were higher than those of the daughters (paired nonparametric test, P < 0.05 in 4 pairs; see Fig. 3). The QS diversities of various genes were even higher in patient 5 (the mother in pair 3, LC), especially the preC/C gene. However, in pair 4, the QS diversities of the mother (in the IT phase) were lower than those of the daughter (CHB).
FIG 3.

HBV QS diversities (genetic distance at the nucleotide level) in each pair. In general, the QS genetic distances in mothers were greater than those in daughters (P < 0.05 in 4 pairs). Patient 5 (liver cirrhosis) had a greater genetic distance than the others, and the QS genetic distance in the preC/C region was much greater than that of the other genes. The history of each pair indicated that the higher QS diversities may be due to fluctuation of liver inflammation in addition to the age.
Net changes in QS diversity, which determine population-based genetic diversity, were chosen to estimate the extent of “clustered evolution” between mothers and daughters (Table 5). The net changes in QS diversity of the preC/C gene were highest in all genes, especially in pairs 1 to 3 and 5. In pair 4 (the mother in IT phase and the daughter with CHB), the net changes in QS diversity in the preC/C gene were similar to those in other genes.
TABLE 5.
Net changes of QS diversities between mothers and daughters
| Gene | Net change of QS diversity (range) (10−3 substitutions/site)a |
|||
|---|---|---|---|---|
| d (nt) | d (aa) | dS | dN | |
| Full length | 4.8286 (2.7001∼9.7181) | |||
| S gene | 3.7010 (1.5606∼6.4128) | 9.0828 (5.4650∼20.3159) | 3.2000 (1.3187∼3.346172) | 3.9639 (2.3658∼8.6923) |
| P gene | 3.1570 (2.9243∼8.2818) | 2.6616 (1.7021∼14.3444) | 7.7039 (6.0706∼13.2016) | 1.2412 (0.4210∼6.7103) |
| X gene | 3.7980 (2.0170∼7.1992) | 9.9693 (6.2312∼18.2489) | 0.4319 (0.1326∼2.0897) | 5.2868 (2.9321∼10.2528) |
| PreC/C gene | 7.5979 (5.0589∼63.5208) | 16.5535 (4.3924∼76.3588) | 4.0437 (2.680071∼49.1775) | 7.7852 (2.0488∼67.5542) |
nt, nucleotide; aa, amino acid; dS, number of synonymous substitutions per synonymous site; dN number of nonsynonymous substitutions per nonsynonymous site.
Indels and mutations in the complete HBV genome and their impact on epitopes.
Insertions were found in 4 clones from patient 5 (mother, LC), and they were all duplicate fragments of nt 1611 to 1698 in the X gene from the same patients. Deletions were common in the preS1 (47/172 clones [27.3%], 6/10 patients) and preC/C (26/172 clones [15.1%], 4/10 patients) regions. The most frequent deletions in preS1 were located at nt 2849 to 2866 (41/47 clones [87.2%], 6/10 patients), resulting in the loss of the initiation codon of the preS1 protein. The positions of deletions in the preC/C region varied widely. Of all clones harboring deletions, only 7 in the preS1 region (7/47 clones [14.9%], 3 patients) and 5 in the preC/C region (5/26 clones [19.2%], 2 patients) were from the daughters (Table 6). A χ2 test indicated that the deletions were more common in clones from the mothers (P < 0.001).
TABLE 6.
Summary of indels of HBV sequences
| Gene | Pair no. | Patient no. | Deletion |
Insertion |
||
|---|---|---|---|---|---|---|
| Site (nt) | Clone no. (%) | Site (nt) | Clone (%) | |||
| PreC/C | 3 | 5 | 2010–2051 | 14 (77) | ||
| 3 | 6 | 2088–2232 | 4 (25) | |||
| 4 | 7 | 2088–2232 | 7 (39) | |||
| 4 | 8 | 2054–2167 | 1 (6) | |||
| PreS1 | 3 | 5 | 2854–2877 | 6 (33) | ||
| 3 | 5 | 2849–2866 | 12 (66) | |||
| 3 | 6 | 2849–2866 | 4 (25) | |||
| 4 | 7 | 2849–2866 | 8 (44) | |||
| 4 | 8 | 2849–2866 | 1 (6) | |||
| 5 | 9 | 2849–2866 | 14 (82) | |||
| 5 | 10 | 2849–2866 | 2 (13) | |||
| X | 1 | 1 | 1760–1777 | 4 (27) | ||
| 1 | 1 | 1770–1777 | 7 (47) | |||
| 3 | 5 | 1611–1698 | 4 (22) | |||
Mutations in the basal base core promoter (BCP) and preC region, which are associated with aggressive hepatitis and advanced liver diseases, such as severe and fulminant hepatitis B, cirrhosis, and hepatocellular carcinoma, were detected in 8 patients (4 mothers and 4 daughters), and all these patients had low HBeAg titers. The sG145R mutation, which can lower the affinity to surface antibody, was detected in 2 patients with low HBsAg titers (1 mother and 1 daughter).
The indels listed above caused the loss of some B cell and T cell epitopes; furthermore, the mutations detected within epitopes in the dominant mutants could cause the affinity to change. The indels occurred more frequently in mothers than in daughters (χ2 test, P < 0.001). The entropy at each amino acid and the altered B cell and T cell epitopes in the preC/C region are listed in Fig. 4. The mothers had higher complexity (entropy) within the preC/C gene and more indels, and therefore more altered epitopes were found.
FIG 4.
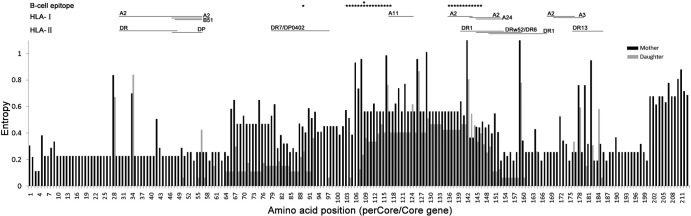
Entropy plot of each amino acid site and related B cell and T cell epitopes in the preC/C genes of mothers and daughters. The dark-gray lines represent the mothers and the light-gray lines represent the daughters. The entropies of most sites in the mothers were higher than those in daughters, which suggested that more mutations in epitopes were found. Mutations and indels were more common in clones from the mothers, which decreased the infinity by 6- to 89-fold or led to a loss of the epitopes. The epitopes were based on references.
All epitopes altered by indels and mutations decreased (by 6- to 89-fold) or maintained their binding affinities, which were predicted by bioinformatics methods; no epitopes with higher affinities altered by indels or mutations were found. Thus, the mutations and indels could lessen or even escape host immunity.
DISCUSSION
In the present study, full-length HBV QS from five pairs of mother-daughter patients with chronic HBV infection were analyzed with bioinformatics methods. The results indicate that the full-length HBV QS complexity and diversity from the mothers and daughters were numerically comparable; however, the distribution patterns of various gene QS differed. The diversity of the preC/C gene was higher in the mothers, in contrast to the higher diversity of the preS1 gene in the daughters. The nucleotide substitution rate of full-length HBV QS between the mothers and daughters was about 1 × 10−4 substitutions/site/year, and the preC/C gene had a higher substitution rate than the P, S, and X genes. Although the HBV QS were homologous, they evolved as different patterns in different hosts, especially in pairs with different antiviral responses and different stages or immune phases of disease. Nucleotide deletions in the preC/C and preS1 regions occurred more frequently in the mothers than in the daughters, which resulted in the absence or lower affinity of epitopes.
The full-length HBV sequence cannot be obtained by splicing the various fragments of PCR amplification due to the existence of QS. The sequence could be an artificial full-length strain sequence that does not actually exist in QS. Ultradeep pyrosequencing was not suitable for determining full-length HBV sequence because of its short read (<400 bp) (26). In this study, the classic PCR amplification of the complete HBV genome by Gunther et al. (10) was adopted to obtain the complete genome of a single strain, and then the PCR products were cloned and sequenced. Currently, this method is still the gold standard in studies of full-length HBV. Meanwhile, the full-length HBV QS may reflect the evolution of HBV more comprehensively than QS of specific genes; furthermore, this method made it possible to explore the relationship between various genes.
Within the same HBV QS, full-length HBV had the highest QS complexity, followed by the P gene, and both were higher than that of the S and X genes, at both the nucleotide level and the amino acid level. One of the major reasons is the positive correlation between QS complexity and the sequence length. However, QS diversities of the specific genes were similarly represented by genetic distance, the average number of synonymous substitutions, and the average number of nonsynonymous substitutions, which were calculated per site with no influence of sequence length.
Despite the numerically comparable full-length HBV QS complexities, the QS complexities of specific genes appeared different, but the difference did not reach statistical significance. As for QS diversities, the distribution patterns were even more different. Three mothers who experienced fluctuating liver inflammation had higher QS diversity than their daughters. Of all the genes, the QS diversity of the preC/C gene from the mothers was much higher than that from the daughters. On the contrary, the preS1 gene from the daughters had a higher diversity than that from the mothers. The different distributions of genes between mothers and daughters were the results of the evolution of homologous HBV QS after perinatal infection, which sheds light on the interaction of specific genes with host immune reactions within the same genome. The preC/C gene is the most important target of host immunity in chronic HBV infection, as many critical T cell and B cell epitopes were clustered in this region (24, 27). Recent data indicated that the increased QS diversity in this region was associated with HBeAg seroconversion in patients receiving IFN-α or nucleotide analogues (28, 29). In our study cohort, the different virological responses acquired by the mother and the daughter in pair 1 were in accordance with those publications. That is, the mother with high preC/C diversity acquired HBeAg seroconversion, while the daughter with low diversity had a delayed virological and poor serological response. The preS1 domain of the L protein is a key determinant for entry of HBV and mediates viral interaction with the cellular receptor(s) on hepatocytes (30, 31). As high QS diversity could confer the RNA virus's higher fitness level or restore pathogenesis as reported by Vignuzzi et al. (32), the higher preS1 diversity might contribute to the virus entry and thus promote perinatal infection. The speculative mechanism or hypothesis requires further experimentation for confirmation.
Net changes in QS diversities, which presented differences both within the HBV QS populations and between the QS populations of the mothers and daughters, reflected the QS evolution better than QS diversity. Of all genes and complete genomes, the preC/C region had the highest net changes in QS diversities. The following reasons may account for this difference. First, the preC/C region is the least overlapping of all HBV genes. The nucleotide mutations in overlapping genes are constrained by more than 1 gene, and thus mutations cannot occur as easily as those in nonoverlapping genes. In fact, mutations in the precore and basal core promoter regions are the most common naturally occurring changes in the HBV genome (29). Second, the preC/C region is abundant with epitopes of both B cells and T cells, which are critical for host immunity to clear virus (24, 27); mutations in this region are associated with advanced liver diseases, including severe and fulminant hepatitis B, cirrhosis, and hepatocellular carcinoma (33–35). HBV could evolve by mutations and indels to increase its adaptation against environmental selection, and these mutations manifest as escaping from host immunity to survival or lowering the binding affinity of epitopes to lymphocytes (36).
Nucleotide substitution rates or evolution rates of full-length HBV were 7.9 × 10−5substitutions/site/year as reported by Osiowy et al. (37), which was slightly higher than previously published estimates (1.5 × 10−5 to 5 × 10−5 substitutions/site/year) (38–40). However, the evolution rates between the mothers and daughters (1.44 × 10−4 to 2.38 × 10−4 substitutions/site/year) are much higher than those figures. The possible reasons are as follows. First, most patients enrolled in our study were CHB patients rather than IT patients. The immune-active phase means a much higher selective pressure from host immunity, which absolutely drives the HBV QS to evolve faster than in the IT phase. Second, the serum samples studied were not serial samples from one patient. Changing hosts means an extraselective pressure for HBV QS; thus, it also acts as a driving force for QS evolution. Finally, the patients included were all genotype C, which has a higher replication level than other genotypes. A higher replication level implies more mutations detected in HBV QS, thus leading to a higher evolution rate.
In addition to higher net changes in QS diversity and evolution rate, more mutations and deletions at epitopes were found in the preC/C region. The affinity of mutated epitopes decreased by order of magnitudes using bioinformatics tools, with some deletions even leading to a loss of epitopes. Lower affinity or loss of epitopes may result in alleviation or absence of host immunity, which absolutely enhances the viral capacity to survive from the immune clearance.
To conclude, although numerically similar QS complexities and diversities of full-length HBV were observed, the specific genes from the homologous HBV genomes are involved in different patterns in different hosts. The different patterns may correlate with the immune phases or stages of chronic HBV infection, and the higher net changes in QS diversity, substitution rates, and more mutations in the preC/C region may confer the virus with high adaptability to escape from or resist host immunity in chronic HBV infection, which warrants further study.
ACKNOWLEDGMENTS
This work was supported by grants for Special Key Projects during the 12th Five-Year Plan of China (no. 2012ZX10002007 and 2013ZX10002001) and by grants from the National Natural Science Foundation of China (no. 81271883 and 81171616).
Footnotes
Published ahead of print 26 February 2014
REFERENCES
- 1.Lavanchy D. 2004. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 11:97–107. 10.1046/j.1365-2893.2003.00487.x [DOI] [PubMed] [Google Scholar]
- 2.Liaw YF, Leung N, Kao JH, Piratvisuth T, Gane E, Han KH, Guan R, Lau GK, Locarnini S. 2008. Asian-Pacific consensus statement on the management of chronic hepatitis B: a 2008 update. Hepatol. Int. 2:263–283. 10.1007/s12072-008-9080-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.European Association for the Study of Liver. 2009. EASL clinical practice guidelines: management of chronic hepatitis B. J. Hepatol. 50:227–242. 10.1016/j.jhep.2008.10.001 [DOI] [PubMed] [Google Scholar]
- 4.Lok AS, McMahon BJ. 2009. Chronic hepatitis B: update 2009. Hepatology 50:661–662. 10.1002/hep.23190 [DOI] [PubMed] [Google Scholar]
- 5.Domingo E, Gomez J. 2007. Quasispecies and its impact on viral hepatitis. Virus Res. 127:131–150. 10.1016/j.virusres.2007.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domingo E, Sheldon J, Perales C. 2012. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76:159–216. 10.1128/MMBR.05023-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domingo E, Martin V, Perales C, Grande-Perez A, Garcia-Arriaza J, Arias A. 2006. Viruses as quasispecies: biological implications. Curr. Top. Microbiol. Immunol. 299:51–82. 10.1007/3-540-26397-7_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin CL, Kao JH, Chen BF, Chen PJ, Lai MY, Chen DS. 2005. Application of hepatitis B virus genotyping and phylogenetic analysis in intrafamilial transmission of hepatitis B virus. Clin. Infect. Dis. 41:1576–1581. 10.1086/497837 [DOI] [PubMed] [Google Scholar]
- 9.Halfon P, Quentin Y, Roquelaure B, Sarles J, Halimi G, Gerolami V, Khiri H, Bourliere M, Cartouzou G. 1999. Mother-to-infant transmission of hepatitis C virus: molecular evidence of superinfection by homologous virus in children. J. Hepatol. 30:970–978. 10.1016/S0168-8278(99)80248-7 [DOI] [PubMed] [Google Scholar]
- 10.Gunther S, Li BC, Miska S, Kruger DH, Meisel H, Will H. 1995. A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J. Virol. 69:5437–5444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882. 10.1093/nar/25.24.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin D, Rybicki E. 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16:562–563. 10.1093/bioinformatics/16.6.562 [DOI] [PubMed] [Google Scholar]
- 13.Myers R, Clark C, Khan A, Kellam P, Tedder R. 2006. Genotyping hepatitis B virus from whole- and sub-genomic fragments using position-specific scoring matrices in HBV STAR. J. Gen. Virol. 87:1459–1464. 10.1099/vir.0.81734-0 [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Zhang Q, Yu DM, Wan MB, Zhang XX. 2009. Early changes of hepatitis B virus quasispecies during lamivudine treatment and the correlation with antiviral efficacy. J. Hepatol. 50:895–905. 10.1016/j.jhep.2008.12.018 [DOI] [PubMed] [Google Scholar]
- 15.Liu F, Chen L, Yu DM, Deng L, Chen R, Jiang Y, Huang SY, Yu JL, Gong QM, Zhang XX. 2011. Evolutionary patterns of hepatitis B virus quasispecies under different selective pressures: correlation with antiviral efficacy. Gut 60:1269–1277. 10.1136/gut.2010.226225 [DOI] [PubMed] [Google Scholar]
- 16.Tamura K. 1992. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 9:678–687 [DOI] [PubMed] [Google Scholar]
- 17.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599. 10.1093/molbev/msm092 [DOI] [PubMed] [Google Scholar]
- 18.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98 [Google Scholar]
- 19.Fan X, Mao Q, Zhou D, Lu Y, Xing J, Xu Y, Ray SC, Di Bisceglie AM. 2009. High diversity of hepatitis C viral quasispecies is associated with early virological response in patients undergoing antiviral therapy. Hepatology 50:1765–1772. 10.1002/hep.23290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guindon S, Lethiec F, Duroux P, Gascuel O. 2005. PHYML online—a Web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33:W557–W559. 10.1093/nar/gki352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Posada D. 2006. ModelTest server: a Web-based tool for the statistical selection of models of nucleotide substitution online. Nucleic Acids Res. 34:W700–W703. 10.1093/nar/gkl042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodrigo AG, Goode M, Forsberg R, Ross HA, Drummond A. 2003. Inferring evolutionary rates using serially sampled sequences from several populations. Mol. Biol. Evol. 20:2010–2018. 10.1093/molbev/msg215 [DOI] [PubMed] [Google Scholar]
- 23.Pumpens P, Grens E. 2001. HBV core particles as a carrier for B cell/T cell epitopes. Intervirology 44:98–114. 10.1159/000050037 [DOI] [PubMed] [Google Scholar]
- 24.Desmond CP, Bartholomeusz A, Gaudieri S, Revill PA, Lewin SR. 2008. A systematic review of T-cell epitopes in hepatitis B virus: identification, genotypic variation and relevance to antiviral therapeutics. Antivir. Ther. 13:161–175 [PubMed] [Google Scholar]
- 25.Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O, Nemazee D, Ponomarenko JV, Sathiamurthy M, Schoenberger S, Stewart S, Surko P, Way S, Wilson S, Sette A. 2005. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol. 3:e91. 10.1371/journal.pbio.0030091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong L, Han Y, Chen L, Liu F, Hao P, Sheng J, Li XH, Yu DM, Gong QM, Tian F, Guo XK, Zhang XX. 2013. Comparison of next-generation sequencing and clone-based sequencing in analysis of hepatitis B virus reverse transcriptase quasispecies heterogeneity. J. Clin. Microbiol. 51:4087–4094. 10.1128/JCM.01723-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torre F, Cramp M, Owsianka A, Dornan E, Marsden H, Carman W, Williams R, Naoumov NV. 2004. Direct evidence that naturally occurring mutations within hepatitis B core epitope alter CD4+ T-cell reactivity. J. Med. Virol. 72:370–376. 10.1002/jmv.20016 [DOI] [PubMed] [Google Scholar]
- 28.Cheng Y, Guindon S, Rodrigo A, Lim SG. 2013. Increased viral quasispecies evolution in HBeAg seroconverter patients treated with oral nucleoside therapy. J. Hepatol. 58:217–224. 10.1016/j.jhep.2012.09.017 [DOI] [PubMed] [Google Scholar]
- 29.Lim SG, Cheng Y, Guindon S, Seet BL, Lee LY, Hu P, Wasser S, Peter FJ, Tan T, Goode M, Rodrigo AG. 2007. Viral quasi-species evolution during hepatitis Be antigen seroconversion. Gastroenterology 133:951–958. 10.1053/j.gastro.2007.06.011 [DOI] [PubMed] [Google Scholar]
- 30.Le Duff Y, Blanchet M, Sureau C. 2009. The pre-S1 and antigenic loop infectivity determinants of the hepatitis B virus envelope proteins are functionally independent. J. Virol. 83:12443–12451. 10.1128/JVI.01594-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1:e00049. 10.7554/eLife.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. 2006. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439:344–348. 10.1038/nature04388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo X, Jin Y, Qian G, Tu H. 2008. Sequential accumulation of the mutations in core promoter of hepatitis B virus is associated with the development of hepatocellular carcinoma in Qidong, China. J. Hepatol. 49:718–725. 10.1016/j.jhep.2008.06.026 [DOI] [PubMed] [Google Scholar]
- 34.Malik A, Singhal DK, Albanyan A, Husain SA, Kar P. 2012. Hepatitis B virus gene mutations in liver diseases: a report from New Delhi. PLoS One 7:e39028. 10.1371/journal.pone.0039028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuen MF, Tanaka Y, Fong DY, Fung J, Wong DK, Yuen JC, But DY, Chan AO, Wong BC, Mizokami M, Lai CL. 2009. Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J. Hepatol. 50:80–88. 10.1016/j.jhep.2008.07.023 [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Ren Y, Wu Y, Zhao B, Qiu L, Li X, Xu D, Liu J, Gao GF, Meng S. 2013. The L60V variation in hepatitis B virus core protein elicits new epitope-specific cytotoxic T lymphocytes and enhances viral replication. J. Virol. 87:8075–8084. 10.1128/JVI.00577-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osiowy C, Giles E, Tanaka Y, Mizokami M, Minuk GY. 2006. Molecular evolution of hepatitis B virus over 25 years. J. Virol. 80:10307–10314. 10.1128/JVI.00996-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fares MA, Holmes EC. 2002. A revised evolutionary history of hepatitis B virus (HBV). J. Mol. Evol. 54:807–814. 10.1007/s00239-001-0084-z [DOI] [PubMed] [Google Scholar]
- 39.Kidd-Ljunggren K, Miyakawa Y, Kidd AH. 2002. Genetic variability in hepatitis B viruses. J. Gen. Virol. 83:1267–1280 [DOI] [PubMed] [Google Scholar]
- 40.Simmonds P, Midgley S. 2005. Recombination in the genesis and evolution of hepatitis B virus genotypes. J. Virol. 79:15467–15476. 10.1128/JVI.79.24.15467-15476.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]