

Abstract
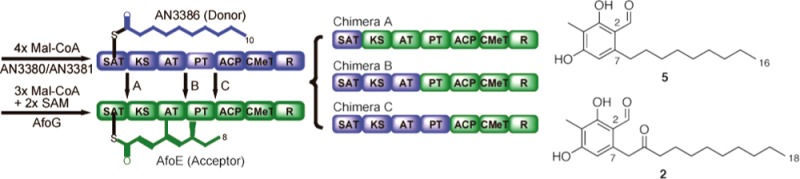
A facile genetic methodology in the filamentous fungus Aspergillus nidulans allowed exchange of the starter unit ACP transacylase (SAT) domain in the nonreduced polyketide synthase (NR-PKS) AfoE of the asperfuranone pathway with the SAT domains from 10 other NR-PKSs. The newly created hybrid with the NR-PKS AN3386 is able to accept a longer starter unit in place of the native substrate to create a novel aromatic polyketide in vivo.
Fungal polyketides are generated by multidomain iterative type I polyketide synthases (PKSs), in which catalytic domains are covalently linked. At a minimum, ketosynthase (KS), acyl transferase (AT), and acyl carrier protein domains (ACP) are featured. Fungal PKSs may be nonreduced, partially reduced, or highly reduced, based on the overall domain composition. Nonreduced PKSs (NR-PKSs) additionally contain starter unit ACP transacylase (SAT) and product template (PT) domains and may also contain a methyltransferase (CMeT) domain as well as domains for product release, including reductase (R).
SAT domains are responsible for the selection of the initial unit, often acetyl-CoA, and its transfer onto the ACP domain. The acetyl unit is relocated onto the KS domain, and the AT domain transfers an extender unit, typically malonyl-CoA, from solution to the ACP domain. The KS domain then catalyzes decarboxylative Claisen condensation between the two units, forming β-ketoacyl-ACP. The polyketide is extended through repeated use of these domains: relocation of the growing chain to KS, transfer of malonyl-CoA by AT to ACP, and condensation of the two units onto ACP. The PT domain in turn is crucial for controlled cyclization of the polyketide. Much research has been performed to more fully understand the function of the domains, including the mechanisms by which the KS domain influences the final polyketide structure.
In previous experiments, we found that a hybrid NR-PKS created by exchanging the AfoE (asperfuranone) SAT domain with the SAT domain from the StcA (sterigmatocystin) PKS generated a new metabolite with the same chain length as the native AfoE product. Because the new starter unit was shorter than the native starter unit, and yet the chain length of the product remained the same, we reasoned that it is the final chain length, not the number of extension rounds, that is fixed during precursor elongation.1 To test whether the preservation of chain length is a general feature of chimeric NR-PKSs, we now have conducted SAT domain swaps between AfoE and nine other characterized NR-PKSs from A. nidulans. AfoE was chosen as the domain acceptor because the native protein, which elongates a medium-length starter unit to a medium length polyketide (C8 to C16),2 was suitable for a challenge with smaller and larger starter units. The nine NR-PKSs selected as SAT domain donors were AN3386/PkiA, which is naturally primed with a C10 starter unit, and eight others associated with acetyl starter units (AN0150/MdpG, AN3230/PkfA, AN6000/AptA, AN6448/PkbA, AN7903/PkeA, AN7909/OrsA, AN8209/WA, AN8383/AusA, using the gene annotations of the Aspergillus Genome Database, http://www.aspgd.org.).3Table S1 lists the products released from these NR-PKSs and the percent protein homologies with AfoE.
To fuse donor SAT sequences to afoE KS-AT-PT-ACP-CMeT-R fragments, we first selected interdomain junction sites using the Udway–Merski algorithm (UMA).4 The chimeric constructs were all generated in nkuAΔ, STΔ (or stcJΔ) parent strains where nkuAΔ improves the frequency of correct integration by homologous recombination and STΔ (or stcJΔ) eliminates the major secondary metabolite sterigmatocystin.2,5,6 AN3386 adopts a C10-starter unit synthesized by the normally silent AN3380 and AN3381.3 To ensure availability of the starter unit, we replaced the promoters of AN3380 and AN3381 with the inducible alcA promoter in the parent strain of the AN3386-AfoE chimera. Next, we deleted the relevant native PKS genes from the donor genomes to prevent possible interference with homologous recombination during transformation. Finally, chimeric PKS promoters and SAT sequences in the acceptor genomes were substituted with alcA and the donor SAT sequences (Figure S1). Extracts from both culture media and mycelia were analyzed.
After induction, only the AN3386-AfoE chimera (Chimera A) produced detectable new products (Figure 1). No products were observed from chimeras bearing SATs that load acetyl units (data not shown). In the mycelium, Chimera A generated a substantial quantity of new compound 5 and lesser amounts of compounds 2 and 3 (Figures 1B, 2 , and 3), and in the media, compound 3 predominated. Compound 2 and a minor amount of 3 had been previously acquired from native AN3386,3 a finding repeated here (Figure 1B, bottom trace). Major product 5 was isolated from a large-scale culture and was characterized by mass spectrometry and NMR (see Supporting Information (SI)).
Figure 1.
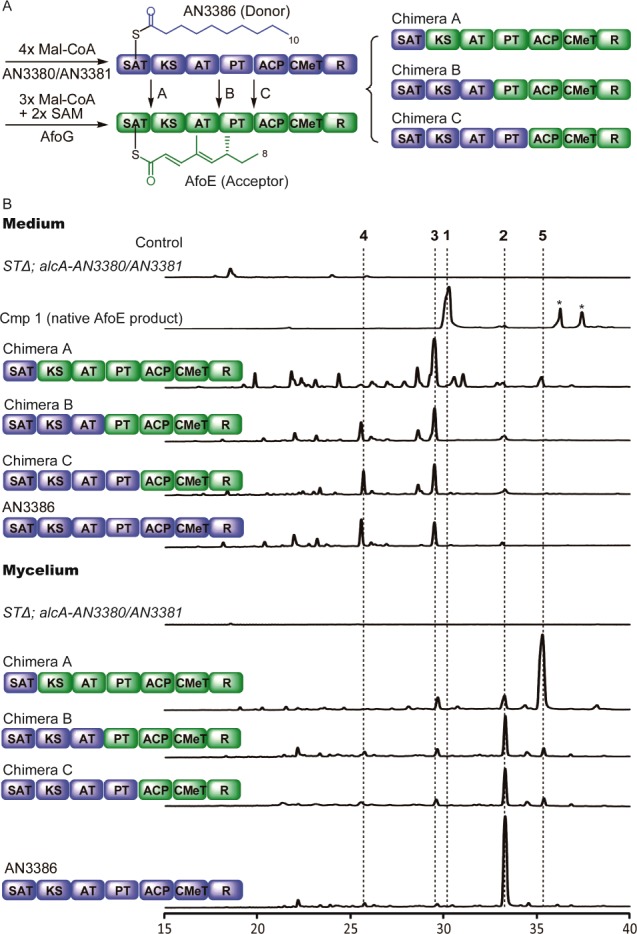
(A) Domain architectures of chimeric AN3386-AfoE PKSs. (B) Total scan HPLC profiles of metabolites extracted from the culture medium and mycelium of mutant strains after induction. Peaks with an asterisk (*) are compounds in equilibrium with 1. (See Supporting Information for a discussion about these compounds.)
Figure 2.
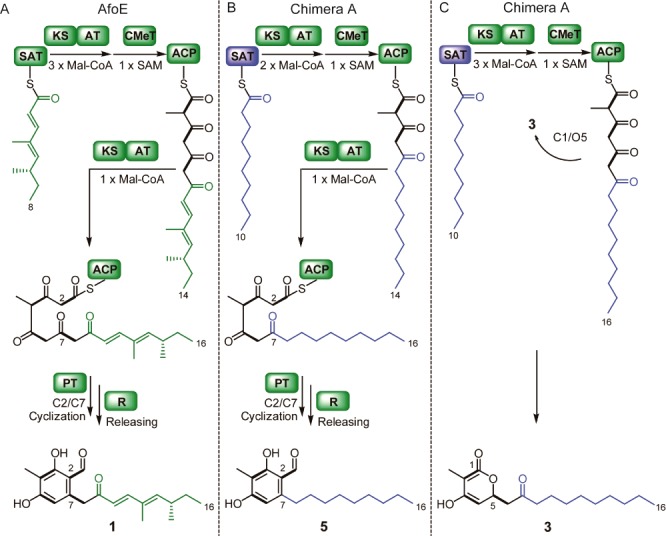
(A) Proposed biosynthetic pathways for native AfoE product 1. (B) Proposed biosynthetic pathway for 5 from Chimera A. (C) Proposed biosynthetic pathway for 3 from Chimera A.
Figure 3.
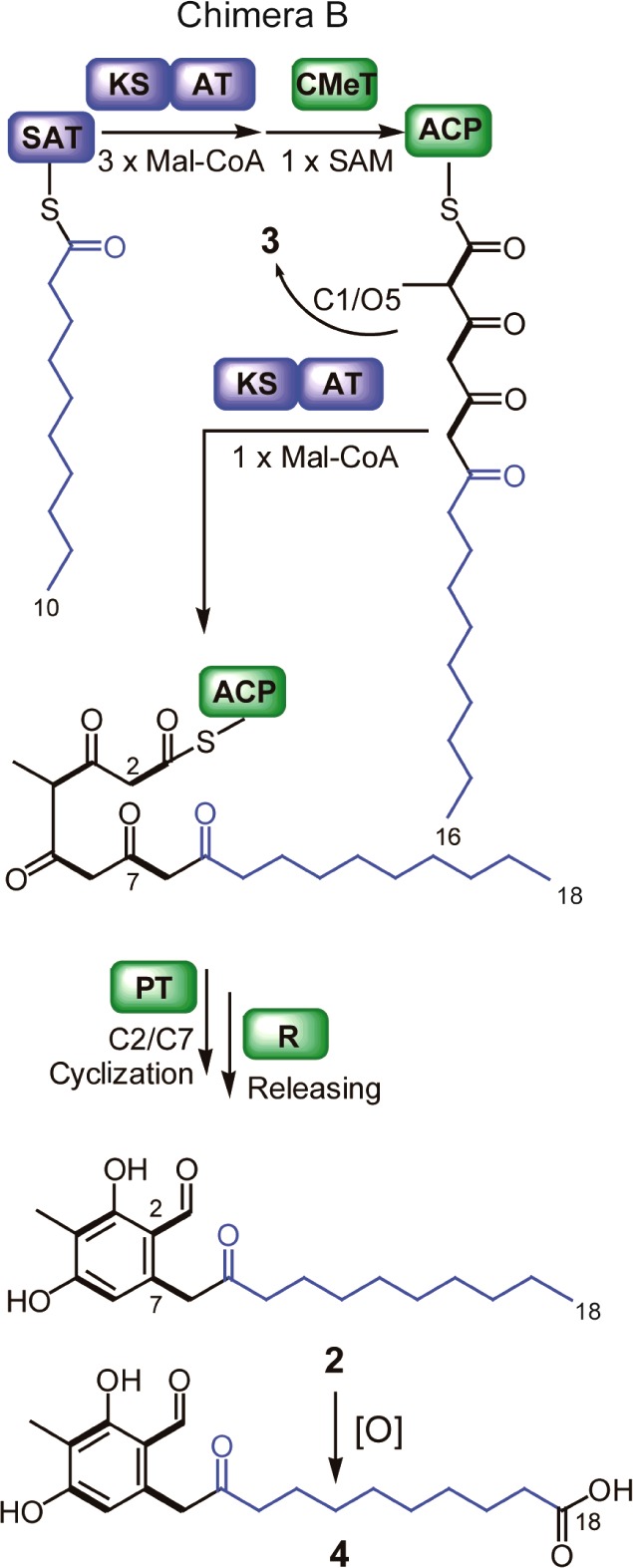
Proposed biosynthetic pathways for 2, 4, and 3 from Chimera B.
Compounds 5 and 3 are both polyketides with a 16-carbon backbone. Compound 1, the native product from AfoE, is also a 16-carbon polyketide, even though the starter unit in Chimera A is longer by two carbon units than in AfoE. The number of rounds of extension with two-carbon malonyl-CoA substrates in Chimera A must have been one fewer than in the native protein in order for the final chain length to be conserved. This finding bookends our previous results, in which a chimera primed with a smaller starting unit extends the chain by one additional round, preserving the same chain length.1 The outcome also recalls work by Nicholson et al.7 in which a minimal actinorhodin type II polyketide synthase from Streptomyces coelicolor was fed starter units of different lengths to yield consistently 16-carbon products.
We deduce that, in the formation of 5, the starting unit is extended by two rounds and methylated at the position that is labeled C4 in the final product. One more round of extension follows, with subsequent C2–C7 cyclization catalyzed by the product template (PT) domain, then reductive release (Figure 2B). For 3, we propose that three rounds of extension occur before methylation at C2, and in lieu of C2–C7 cyclization, nucleophilic attack by the O5 enol liberates the chain to generate a pyrone (Figure 2C). When three rounds of extension have occurred, the methylated C2 carbon cannot act as a nucleophile, and the enzyme active site may not accommodate further extension. Therefore, 3 may represent a stalled intermediate that was spontaneously released.
We continued to probe domain control by adopting the rational domain swap strategy successfully used in PKS-NRPSs by Fisch et al.8 The additional donation of the ketosynthase-acyl transferase (KS-AT) didomain from AN3386 to the chimera (Chimera B) dramatically changed the metabolic profile by shifting the principal product from 5 (C16) to 2 (C18), the major AN3386 product, in the mycelium (Figures 1B, 3). 3 (C16) is still observed in the media. Its generation may be the result of spontaneous cyclization before the last round of extension. 4 (C18), which is an oxidized product of 2 (see Supporting Information for isolation and characterization), is also present in the media (Figure 3). We suspect that one or more endogenous enzymes catalyze the formation of 4 from 2. Because malonyl AT domains demonstrate strong substrate specificity for transferring malonyl-CoA groups,9,10 it can be reasoned that it was the KS domain that was the major determinant of chain length.
The productive SAT swaps between AfoE and StcA,1 and now between AfoE and AN3386, suggest that NR-PKSs may accept starter units that are smaller or greater in length by at least one acetate unit. The lack of detectable metabolites from any of the chimeras with SATs that load acetyl groups raises the possibility that for AfoE these short units fall outside of the range of substrate specificity. Crystal structures of bacterial type II and modular PKS KS domains revealed that a hydrophobic starter unit-binding pocket lies at the end of the KS active site.11−13 Structure comparisons by Pan et al. suggested that the volume of this pocket might determine the substrate specificity of KS for the starter unit.13 However, it should also be considered that metabolites were indeed generated but in low titers or that the products are difficult to detect. Another alternative explanation is that incompatibility between domains rendered the chimeras inactive.
The presence of minor amounts of 2 in Chimera A and 5 in Chimera B (Figure 1B) indicates that chain length control, however, is not absolute. Analogous data in our previous investigation led to the same conclusion.1 Ma et al. similarly reported that donating octanoyl-CoA (C8) to Gibberella fujikuroi PKS4 resulted in compounds generated by five and four rounds of extension. The group suggested the effect was possibly due to the flexibility of the alkyl chain in the KS active site.14
In addition to chain length control, the KS domain may exert an influence on polyketide methylation. In Chimera B, methylation does not occur until a carbon length of 16 is achieved, whether the outcome is 2, 4, or derailment product 3 (Figure 3). With Chimera A, methylation takes place when the polyketide reaches either a length of 14 carbons, ultimately forming 5, or a length of 16 carbons, leading to 3 (Figure 2). Methylation is therefore imprecise in Chimera A, but it should be noted that interdomain effects imperfectly reconstructed in the chimeras may be important in driving metabolic flux toward “intended” products (i.e., compound 5). In fact, there is a lower proportion of 3 in native AN3386 than in any of the chimeras. Nevertheless, the fact that methylation occurs exclusively with a 16-carbon polyketide in Chimera B but not wholly in Chimera A might signal that the KS domain contributes to the methylation pattern.
Further donating the PT domain (Chimera C) led to a metabolite profile highly similar to that of Chimera B (Figure 1B). The PT domains of AN3386 and AfoE are of the same cyclization mode (C2–C7); as a result their exchange does not affect the metabolic profile. Contrasting profiles may have been expected if the two PT domains differed in regiospecificity.15 The profiles of Chimeras B and C are similar to that of native AN3386 (Figure 1B).
Taken as a whole, our studies underscore that SAT domain exchange is emerging as a useful method for generating new polyketides, even in the event that there is some difference in size between starter units. Extended domain swaps help illuminate the range of flexibility in KS domains and their responsibility for fixing chain length (but not extension rounds), an idea proposed by Crawford et al.16 and supported by an increasing body of literature.1,15−18 A role in the control of methylation is also suggested. In turn, these observations provide guidance for further biosynthetic engineering of novel nonreduced polyketides.
Acknowledgments
This work was supported by Grant PO1GM084077 from the National Institute of General Medical Sciences.
Supporting Information Available
A. nidulans strains and primers used in this study, gene deletion and domain swap strategies, a table of NR-PKS products, a discussion on 1 and products in equilibrium, and characterization of compounds 5 and 4 are provided in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Liu T.; Chiang Y. M.; Somoza A. D.; Oakley B. R.; Wang C. C. J. Am. Chem. Soc. 2011, 133, 13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang Y. M.; Szewczyk E.; Davidson A. D.; Keller N.; Oakley B. R.; Wang C. C. J. Am. Chem. Soc. 2009, 131, 2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja M.; Chiang Y. M.; Chang S. L.; Praseuth M. B.; Entwistle R.; Sanchez J. F.; Lo H. C.; Yeh H. H.; Oakley B. R.; Wang C. C. J. Am. Chem. Soc. 2012, 134, 8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udwary D. W.; Merski M.; Townsend C. A. J. Mol. Biol. 2002, 323, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang Y. M.; Oakley C. E.; Ahuja M.; Entwistle R.; Schultz A.; Chang S. L.; Sung C. T.; Wang C. C.; Oakley B. R. J. Am. Chem. Soc. 2013, 135, 7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak T.; Szewczyk E.; Oakley C. E.; Osmani A.; Ukil L.; Murray S. L.; Hynes M. J.; Osmani S. A.; Oakley B. R. Genetics 2006, 172, 1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson T. P.; Winfield C.; Westcott J.; Crosby J.; Simpson T. J.; Cox R. J. Chem Commun 2003, 686–687. [DOI] [PubMed] [Google Scholar]
- Fisch K. M.; Bakeer W.; Yakasai A. A.; Song Z.; Pedrick J.; Wasil Z.; Bailey A. M.; Lazarus C. M.; Simpson T. J.; Cox R. J. J. Am. Chem. Soc. 2011, 133, 16635. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Smith L. H.; Cox R. J.; Beltran-Alvarez P.; Arthur C. J.; Simpson T. J. ChemBioChem 2006, 7, 1951. [DOI] [PubMed] [Google Scholar]
- Crawford J. M.; Vagstad A. L.; Whitworth K. P.; Ehrlich K.; Townsend C. A. ChemBioChem 2008, 9, 1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Kim C. Y.; Mathews I. I.; Cane D. E.; Khosla C. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Chen A. Y.; Kim C. Y.; Cane D. E.; Khosla C. Chem. Biol. 2007, 14, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H.; Tsai S.; Meadows E. S.; Miercke L. J.; Keatinge-Clay A. T.; O’Connell J.; Khosla C.; Stroud R. M. Structure 2002, 10, 1559. [DOI] [PubMed] [Google Scholar]
- Ma S. M.; Zhan J.; Watanabe K.; Xie X.; Zhang W.; Wang C. C.; Tang Y. J. Am. Chem. Soc. 2007, 129, 10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagstad A. L.; Newman A. G.; Storm P. A.; Belecki K.; Crawford J. M.; Townsend C. A. Angew. Chem., Int. Ed. Engl. 2013, 52, 1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford J. M.; Thomas P. M.; Scheerer J. R.; Vagstad A. L.; Kelleher N. L.; Townsend C. A. Science 2008, 320, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A.; Ebizuka Y. Tetrahedron Lett. 2002, 43, 843. [Google Scholar]
- Vagstad A. L.; Bumpus S. B.; Belecki K.; Kelleher N. L.; Townsend C. A. J. Am. Chem. Soc. 2012, 134, 6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.