

ABSTRACT
Paramyxoviruses are a large family of membrane-enveloped negative-stranded RNA viruses causing important diseases in humans and animals. Two viral integral membrane glycoproteins (fusion [F] and attachment [HN, H, or G]) mediate a concerted process of host receptor recognition, followed by the fusion of viral and cellular membranes, resulting in viral nucleocapsid entry into the cytoplasm. However, the sequence of events that closely links the timing of receptor recognition by HN, H, or G and the “triggering” interaction of the attachment protein with F is unclear. F activation results in F undergoing a series of irreversible conformational rearrangements to bring about membrane merger and virus entry. By extensive study of properties of multiple paramyxovirus HN proteins, we show that key features of F activation, including the F-activating regions of HN proteins, flexibility within this F-activating region, and changes in globular head-stalk interactions are highly conserved. These results, together with functionally active “headless” mumps and Newcastle disease virus HN proteins, provide insights into the F-triggering process. Based on these data and very recently published data for morbillivirus H and henipavirus G proteins, we extend our recently proposed “stalk exposure model” to other paramyxoviruses and propose an “induced fit” hypothesis for F-HN/H/G interactions as conserved core mechanisms of paramyxovirus-mediated membrane fusion.
IMPORTANCE Paramyxoviruses are a large family of membrane-enveloped negative-stranded RNA viruses causing important diseases in humans and animals. Two viral integral membrane glycoproteins (fusion [F] and attachment [HN, H, or G]) mediate a concerted process of host receptor recognition, followed by the fusion of viral and cellular membranes. We describe here the molecular mechanism by which HN activates the F protein such that virus-cell fusion is controlled and occurs at the right time and the right place. We extend our recently proposed “stalk exposure model” first proposed for parainfluenza virus 5 to other paramyxoviruses and propose an “induced fit” hypothesis for F-HN/H/G interactions as conserved core mechanisms of paramyxovirus-mediated membrane fusion.
INTRODUCTION
Paramyxoviruses are enveloped, nonsegmented, negative-stranded RNA viruses that infect their hosts by fusing the viral membrane with a host cell membrane at neutral pH (1). The family Paramyxoviridae includes many major clinically and economically important pathogens of humans and animals, including parainfluenza viruses 1 to 5 (PIV1 to PIV5), mumps virus (MuV), Newcastle disease virus (NDV), Nipah virus (NiV), Hendra virus (HeV), measles virus (MeV), canine distemper virus (CDV), respiratory syncytial virus (RSV), and human metapneumovirus (hMPV). Paramyxoviruses are classified into two subfamilies: Paramyxovirinae (including the genera Rubulavirus, Avulavirus, Respirovirus, Morbillivirus, and Henipavirus) and Pneumovirinae. In light of a large number of recently discovered novel, potentially zoonotic paramyxoviruses (2, 3), studies highlighting core mechanisms of paramyxovirus entry have become critical to facilitate the rational design of antiviral drugs and effective vaccine development.
Paramyxoviruses mediate membrane fusion and cell entry by the concerted action of two interacting viral glycoproteins: the attachment protein (HN, H, or G) and the fusion protein (F). The attachment protein binds cellular surface receptors, initiating a sequence of events, involving interactions of the attachment protein with F, followed by activation of the fusion protein (1, 4, 5). Upon activation, the cleaved, metastable, prefusion form of the F protein (6, 7) rearranges to insert a hydrophobic fusion peptide into the target cell membrane and through a pre-hairpin intermediate (8) ultimately refolds irreversibly into a stable, postfusion form (9–12), resulting in viral and cellular membrane merger.
Atomic structures of the attachment proteins (HN, H, or G) reveal a globular head, harboring a typical sialidase domain created by a six-bladed β-propeller fold (13–22). PIV1 to PIV5, MuV, and NDV have HN-type receptor-binding proteins possessing both hemagglutinating and neuraminidase activities, and HN binds sialic acid as a receptor through a central binding site within the β-propeller fold. In contrast, H proteins of MeV and CDV and G proteins of HeV and NiV bind cell surface-expressed protein receptors through specific sites on the globular head. For HN proteins, the neuraminidase activity cleaves sialic acid from complex carbohydrate chains of HN and F and cellular proteins, acting as a receptor-destroying enzyme and preventing virus reattachment to the same cell. The majority of evidence obtained to date suggests that the attachment proteins exist as a dimer-of-dimers, with dimerization occurring through covalent and noncovalent interactions primarily within a stalk domain connecting the globular heads to the transmembrane domain (19, 23–28). Recently obtained atomic structures of HN stalk domains from NDV HN (23) and PIV5 HN (29) showed the stalks to be four-helix bundles (4HB), possessing a C-terminal linear coiled-coil region with an 11-mer hydrophobic central core repeat, adjacent to an N-terminal left-handed supercoiled region with 7-mer repeats. A large body of data suggests that F interacts with the attachment protein through the HN, H, or G stalk domains (30–41); however, the exact nature of the interaction and nature of the F activation process are not clear.
HN, H, or G receptor binding is coupled to F activation to ensure the correct timing of the F fusion peptide insertion into the target membrane. The attachment protein globular heads upon binding receptor are thought to initiate a conformational change or rearrangement within the HN, H, or G proteins that is believed to be responsible for F activation during or after F-HN/H/G interaction (1, 42–45). However, the exact nature of these rearrangements and that of the F-HN/H/G interaction are not well understood. Multiple studies have shown that retargeted MeV H or NDV HN heads binding noncognate receptors are able to trigger F (45–49) and chimeric attachment proteins bearing globular heads or stalks from different viruses in various combinations are functional when paired with the cognate F protein corresponding to the stalk domain portion of the chimeric attachment protein (31, 42, 49–53). Recent data have also shown that “headless” PIV5 HN, MeV H, and NiV G proteins, lacking the globular head domains in their entirety, are sufficient to trigger their cognate F proteins (42, 54, 55). Thus, counterintuitively, it appears that across the paramyxovirus family in general, receptor specificity or even receptor binding by the HN, H, or G heads is not a major requirement for F triggering. However, the critical requirement of initiating F activation at the correct time and place suggests that the globular heads mediate an important regulatory role in F activation through receptor binding, whereas the stalks act as activation domains (42).
Recently solved atomic structures of PIV5 HN and NDV HN show distinct arrangements of the globular head domains (19, 23, 41), with the globular head dimers of HN either attaining a “down” position, where one monomer of each dimer interacts with the stalk 4HB (23), or an “up” position, where this interaction is absent (19). A third hybrid arrangement of PIV5 HN globular heads “2 heads up, 2 heads down” was also observed recently. When HN in the “2 heads up, 2 heads down” form was locked together through disulfide bridges, its ability to trigger PIV5 F was maintained (41), suggesting that only one dimer face of the stalk needs to be exposed.
Based on these data we recently suggested a model (the stalk exposure model) for the activation of paramyxovirus F proteins in which binding of receptor by the attachment protein globular heads results in exposure of an F activation domain in the attachment protein stalk. F, on interaction with this region, is triggered (destabilized), ultimately resulting in membrane fusion (41, 42). Current modeling and mutagenesis data suggest that F-activating regions are at or around the junction of the linear and superhelical regions of 4HB stalks (29, 41, 43, 44). Very recently, Brindley et al. (54) and Liu et al. (55), for MeV and NiV, respectively, provided strong evidence that the “stalk exposure” model could be extrapolated to MeV and NiV. Thus, the “stalk exposure” model links receptor binding to F activation, whereby correct timing of the irreversible F refolding process is achieved by rearrangement of the attachment protein globular head domains, with respect to the attachment protein 4HB stalks.
Here we identified the F-activating regions of PIV5 HN and MuV HN stalk domains and highlighted the roles of individual residues, including that of a single charged amino acid, which determines specificity of F activation between PIV5 and MuV. A comparison of the critical residues identified in the F-activating domains of PIV5 and MuV stalks, and residues identified in previous mutagenesis studies of the NDV HN stalk (33, 36), mapped onto available HN stalk atomic structures revealed a largely hydrophobic F activation interface for PIV5, MuV, and NDV that is sterically protected from F interaction by the globular heads. The similarity of NDV and PIV5 HN stalk F-activating regions are such that maintaining the relative positions of critical hydrophobic residues in the context of the 4HB is sufficient to activate NDV F by a chimeric NDV HN protein containing part of the PIV5 HN F-activating region. In addition, we show a requirement for stalk flexibility at the F activation region of NDV HN similar to that observed for MeV and CDV H proteins (43, 44, 56). Lastly, we show that like PIV5 HN, MeV H, and NiV G, “headless” stalk domains of MuV HN and NDV HN, can trigger F and cause cell-cell fusion. Overall, these results strongly support a “stalk exposure” model, certain features of which are likely to be at the core of a general mechanism of paramyxovirus fusion activation.
MATERIALS AND METHODS
Cells and antibodies.
293T, BHK-21, BSR-T7/5, and Vero cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). BHK-21F cells were additionally supplemented with 10% tryptose phosphate broth, and BSR-T7/5 cells were grown with G418 added every third passage. A PIV5 HN specific polyclonal antibody (R471) (42) was used to detect PIV5 HN wild-type (wt) and PIV5 HN mutant proteins. NDV HN full-length protein and other NDV HN-based chimeric constructs and mutant proteins were detected by using a NDV HN polyclonal antibody (R9722) serum raised in rabbits against the soluble, purified NDV HN ectodomain (23) expressed by a recombinant baculovirus in Sf9 insect cells. MuV full-length HN protein and MuV HN mutant proteins were detected by using a mumps virus neutralizing antiserum raised in rabbits (Denka Seiken Co., Ltd., Niigata-ken, Japan). For the detection of proteins above by flow cytometry, a fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA) was used as a secondary antibody.
Cloning and mutagenesis.
pCAGGS-PIV5 HN and pCAGGS-PIV5 F expression constructs harboring the PIV5 (W3A) F and HN genes were used as described previously (57). Single point mutations in pCAGGS-PIV5 HN were constructed by using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) as described previously (29). The NDV-PIV5-Fact HN chimeras, NDV HN “headless” stalk constructs and NDV HN mutant proteins were constructed by using four-primer PCR, with NDV-HN (Australia-Victoria strain) as the template. The MuV HN “headless” stalk domains and MuV HN constructs bearing single point mutants were generated from a pCAGGS MuV HN (Miyahara strain) construct using four-primer PCR. The resultant PCR products bearing the above modified NDV HN or MuV HN genes were cloned into the pCAGGS vector using KpnI and SacI restriction enzyme sites. The nucleotide sequence of the entire HN open reading frames for each of the generated mutants was verified by using an Applied Biosystems 3100-Avant automated DNA sequencer (Life Technologies Corp., Carlsbad, CA). pT7-luciferase was obtained from Promega Corp. (Madison, WI).
Expression of PIV5, NDV, and MuV F and HN glycoproteins in mammalian cells.
PIV5, NDV, or MuV-F and -HN (wt and mutant) proteins were expressed from pCAGGS constructs in Vero, BHK-21F, and 293T cells by transient transfection using the Lipofectamine Plus transfection reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. For transfection, the cells were incubated with DNA and transfection reagents for 5 h at 37°C, after which protein synthesis and cell surface transport was allowed for 18 h at 37°C in DMEM containing 2% FBS.
Immunoprecipitation and SDS-PAGE.
293T cells were seeded onto BD PureCoat Amine six-well dishes (Becton Dickinson, Franklin Lakes, NJ) overnight, after which the cells were transfected with PIV5, NDV, or MuV HN wt or mutant proteins, as described above. At 18 h posttransfection, the cells were starved in DMEM lacking cysteine and methionine for 30 min, followed by labeling with 50 μCi of Tran35S-label in the same medium for 30 min (pulse). Tunicamycin, if required, was added at a final concentration of 5 μg/ml to this 35S labeling medium. The cells were then incubated for 90 min with complete DMEM containing 10% FBS (chase). After the pulse-chase protocol, the cells were lysed in cold immunoprecipitation assay (RIPA) buffer (58) containing protease inhibitors, 50 mM iodoacetamide, and 2 mM phenylmethylsulfonyl fluoride (PMSF). The lysates were then clarified in a Beckman TLX ultracentrifuge in a Beckman TLA 120.2 rotor at 55,000 rpm for 10 min at 4°C. Proteins in the clarified lysates were allowed to bind with the PIV5 HN polyclonal antibody R471 or the NDV HN polyclonal antibody R4722 for 2 h or the MuV HN neutralizing antiserum overnight at 4°C with rocking, after which protein A-Sepharose beads were added, and the immunocomplexes incubated with the beads at 4°C for 30 min. The antibody-antigen complexes were washed three times with RIPA buffer (58) containing 0.3 M NaCl, twice with RIPA buffer containing 0.15 M NaCl, and once with 50 mM Tris-HCl (pH 7.4)–0.25 mM EDTA–0.15 M NaCl. The proteins were eluted from the beads by boiling for 2 min in protein lysis buffer with or without 15% dithiothreitol and separated on 10, 15, or 17.5% polyacrylamide gels (containing 4 M urea). Radioactivity was detected using a Fuji FLA-5100 image reader with Multi Gauge v3.0 software (Fuji Medical Systems, Stamford, CT).
Flow cytometry.
The amounts of PIV5 HN, NDV HN, or MuV HN wt and mutant proteins present on the surfaces of cells were quantified using flow cytometry. 293T cells were transfected with equal amounts of DNA (1 μg/well of a six-well dish). At 18 h posttransfection, the monolayers were washed with phosphate-buffered saline (PBS) containing 0.02% sodium azide to prevent new protein production and HN internalization. Nonspecific antibody binding was blocked by incubating the cell monolayers in PBS containing 1% bovine serum albumin (BSA) and 0.02% sodium azide for 1 h at 4°C. The cells were then incubated for 1 h at 4°C with the PIV5 HN polyclonal antibody (PAb) R471 or NDV HN PAb R4722 at a dilution of 1:200 or MuV neutralization antiserum at 1:100 dilution in PBS containing 1% BSA. The monolayers were washed thoroughly with PBS to remove unbound antibody and subsequently incubated for 1 h at 4°C with FITC-conjugated goat anti-rabbit IgG (1:200 dilution) (Jackson ImmunoResearch). Cells were washed extensively with PBS to remove nonspecifically bound secondary antibody and resuspended in PBS containing 0.5% formaldehyde to fix the cells. The mean fluorescence intensity (MFI) of 10,000 cells was recorded for each sample using a FACSCanto II flow cytometer (Becton Dickinson). Flow cytometry data were collected using FACSDiva software (Becton Dickinson) and analyzed using BD CellQuest Pro software (Becton Dickinson).
Syncytium formation.
BHK-21F cells were transfected as described above using 1 μg each of pCAGGS containing PIV5, NDV, or MuV HN (wt or mutant constructs) and pCAGGS F from the above viruses. At 18 h posttransfection, unless otherwise specified, the cells were fixed and stained using a Hema3 staining protocol (Fisher Scientific, Pittsburgh, PA) according to the manufacturer's instructions. The monolayers were photographed using an AMG EVOS ×1 inverted microscope (Fisher Scientific, Pittsburgh, PA).
Luciferase reporter assay.
To quantify the fusion observed in the syncytia assay, Vero cell monolayers were transfected with 1 μg each of the pCAGGS-F and pCAGGS-HN wt or pCAGGS constructs containing mutations in PIV5, NDV, or MuV HN and pT7-luciferase, a plasmid that expresses firefly luciferase under T7 polymerase control. At 15 h posttransfection, BSR-T7/5 cells, expressing T7 RNA polymerase, were overlaid on the Vero cell monolayer (at a BSR-T7 cells to Vero cells ratio of 1:2), followed by further incubation for 7 h at 37°C. Subsequently, Glo lysis buffer (Promega, Madison, WI) was used to lyse the cells, and cell debris was removed by centrifugation (13,000 rpm for 3 min). Then, 150 μl of the cleared lysates was added to a 96-well dish, along with 150 μl of the luciferase assay substrate (Promega, Madison, WI), and the luciferase activity in relative light units (RLU) was determined using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
Hemadsorption assay.
Monolayers of 293T cells were transfected with pCAGGS PIV5, NDV, or MuV HN and HN mutants as described above. At 18 h posttransfection, the cells were washed gently with cold PBS. This was followed by incubation for 2 h with 1% chicken red blood cells (RBCs) in PBS at 4°C to allow receptor binding but not fusion. Subsequently, the monolayers were washed thoroughly to remove unbound RBCs. The remaining bound RBCs were lysed in 0.5 ml of cold distilled water by rocking for 2 h at 4°C. The lysate was cleared to remove debris by centrifugation at 5,000 rpm for 3 min at 4°C, and the absorbance of the lysed RBCs in the cleared supernatant was read at 410 or 540 nm on a 730 DU UV/Vis spectrophotometer (Beckman Coulter, Brea, CA).
Neuraminidase activity assay.
293T cells were transfected with PIV5, NDV, or MuV wt HN and HN mutants (1 μg of plasmid each) as described above. Cells were detached from the plates using 50 mM EDTA in PBS. The cells were then centrifuged for 5 min at 3,000 rpm at 4°C, after which they were washed once with PBS and centrifuged as described above to remove the EDTA. The cell pellets were resuspended in 100 μl of 125 mM sodium acetate buffer (pH 4.75) containing 6.25 mM CaCl2. Then, 4-methylumbelliferyl-N-acetyl-α-d-neuraminic acid (Sigma-Aldrich, St. Louis, MO) was added at a final concentration of 5 mM, as the neuraminidase substrate. The reaction was allowed to proceed for 30 min at 37°C with occasional mixing to prevent the cells from settling. Next, 75 μl of 20 mM sodium carbonate buffer (pH 10.4) was added to stop the reaction (59). The cells were pelleted by centrifugation at 14,000 rpm, and 180 μl of the cell-free supernatant was transferred to a black-walled, clear-bottom 96-well plate. The fluorescence of the cleaved substrate was measured at excitation and emission wavelengths of 356 and 450 nm, respectively, using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
RESULTS
Hydrophobic residues of the PIV5 HN stalk domain are crucial for F activation.
For most members of the Paramyxoviridae family the attachment protein stalk domain is believed to interact with and trigger the metastable F protein to initiate fusion. Recent studies on PIV5 HN, NDV-HN, MeV H, and CDV H have implicated the F-interacting and F-activating regions to be in the central part of the HN or H stalk (29, 33, 41, 43, 44, 56, 60). Recently, we identified residues located within and near the PIV5-F “Ig-like” domain as putative HN-interacting residues (61). Given that most of these identified residues were hydrophobic, the nature of corresponding F-interacting residues on the HN stalk and their roles in fusion activation were of great interest and have been investigated here.
We have shown previously that mutation of residues Y77 and T89 to alanine in the PIV5 HN stalk did not significantly alter the F-triggering capabilities of the mutant proteins, whereas mutants PIV5 HN V81T and PIV5 HN L85Q completely ablated fusion (41). These mutations also had similar phenotypes in the context of a “headless” PIV5 HN 1-117 stalk (41). Thus, to understand the role of specific residues in the central part of the PIV5 HN stalk, PIV5 HN mutant proteins harboring single point mutations of residues from positions 81 to 88 on the PIV5 HN stalk (amino acid sequence VALPLQLD) were created. Each amino acid was mutated to a residue similar in nature to the parent amino acid or to a residue that was quite dissimilar in terms of charge or hydrophobicity. The proline at position 84 was mutated to an alanine or a threonine. Previously characterized PIV5 HN mutants V81T and L85Q were also included in this set.
The mutant proteins were expressed in mammalian cells, metabolically labeled and immunoprecipitated (Fig. 1A). A majority of the PIV5 HN mutant proteins were expressed in cells at levels comparable to that of the PIV5 HN wt control, except for the P84A mutant protein and the D88L mutant, both of which showed less than ca. 40 to 50% of the wt PIV5 HN levels (Fig. 1A). When the cell surface expression of the PIV5 HN mutant proteins was measured using flow cytometry, most of the mutant proteins, including D88L, showed surface expression levels between 70 and 120% of the wt PIV5 HN protein (Fig. 1B). However, for mutants P84A and P84T the cell surface expression (ca. 40 to 50% of wt PIV5 HN protein), like its total protein expression in cells (Fig. 1A), was reduced (Fig. 1B), suggesting a possible role for P84 in PIV5 HN protein folding and surface transport.
FIG 1.
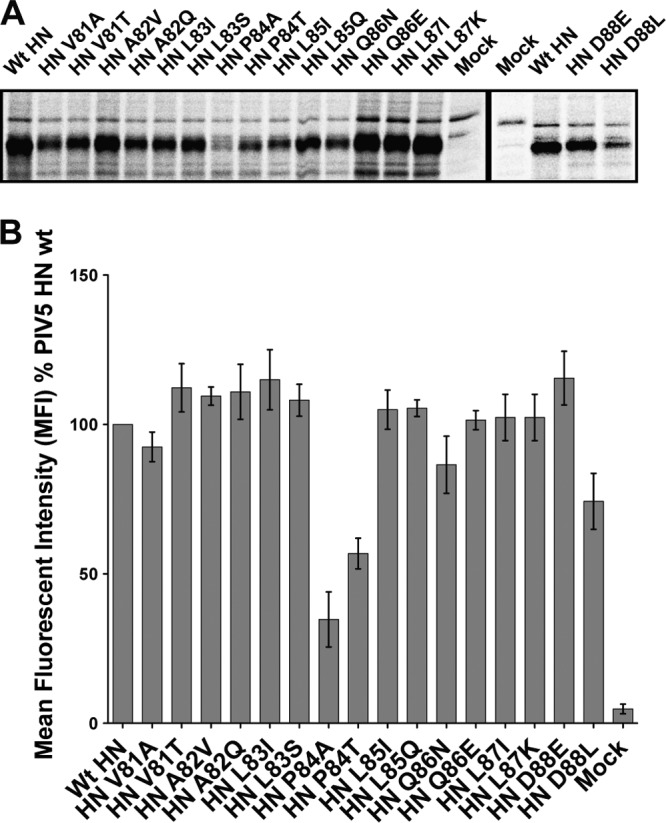
Expression and surface transport of PIV5 HN stalk point mutant proteins. (A) Radioimmunoprecipitation of Tran35S-labeled 293T cell lysates, transfected with PIV5 HN single point mutants in the HN stalk domain. Polypeptides were immunoprecipitated with a PIV5 HN polyclonal antibody (R471) and analyzed on a 10% reducing SDS-PAGE gel. (B) Detection of PIV5 HN point mutant proteins at the surface of 293T cells at 18 h posttransfection. Proteins were detected using the PIV5 HN PAb R471 and a goat α-rabbit fluorescein isothiocyanate (FITC)-conjugated secondary antibody by flow cytometry. The mean fluorescence intensity (MFI) is shown as a percentage of wt PIV5 HN protein expression levels (n = 3).
To test the ability of the above PIV5 HN point mutant proteins to promote fusion, a luciferase reporter assay was used (Fig. 2A). Interestingly, the F-triggering abilities of these HN mutants varied broadly from 3 to 300% compared to fusion measured on coexpression of wt PIV5 F and PIV5 HN.
FIG 2.
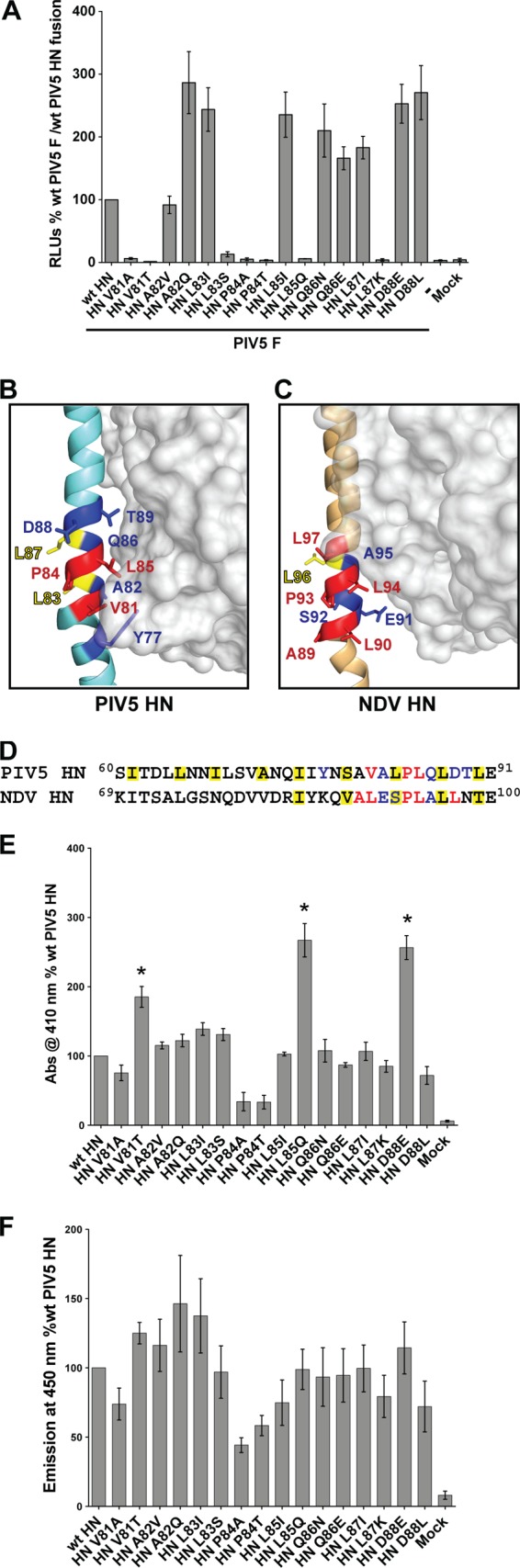
Hydrophobic residues of the PIV5 HN stalk domain form the basis of F activation. (A) Luciferase reporter assay for cell-cell fusion in Vero cells cotransfected with wt PIV5 F and wt PIV5 HN or PIV5 HN stalk point mutants. The extent of cell-cell fusion was measured as relative luciferase units (RLU), expressed as a percentage of wt PIV5 F and PIV5 HN fusion, with data from three independent experiments. (B) Atomic structure of PIV5 HN in the “2 heads up, 2 heads down” form (41) showing where the globular head protomer (surface representation) makes contacts with a single helix from the 4HB stalk domain (diagram representation). Side chains of residues that were mutated are shown as sticks on the stalk helix, and the mutated residues are colored according to their ability to activate PIV5 F, as observed in panel A and as previously described (41). Colors: red, complete block of F activation; blue, no effect on F activation; yellow, core hydrophobic residue within the 4HB that requires a hydrophobic substitution to be functionally active. (C) Atomic structure of NDV HN in the “4 heads down” form (23) showing one globular head protomer (surface representation) forming contacts with one helix of the stalk (diagram representation). NDV HN residues functionally characterized in previous studies (33, 36) are highlighted with their side chains shown as sticks. Color coding is the same as in panel B. (D) Gapless alignment of PIV5 HN (W3A strain) and NDV HN (Australia-Victoria strain) protein sequences showing the part of the HN stalk domains harboring putative F-activating domains. Color coding of residues is the same as in panels B and C, with all of the residues present within the hydrophobic core of the PIV5 HN and NDV HN stalk 4HBs highlighted with a yellow box. (E) Receptor-binding activity of PIV5 HN stalk point mutants, as measured by the ability of these mutant proteins to bind chicken RBCs. Specifically bound RBCs were lysed, and the hemoglobin absorbance (Abs) at 410 nm was expressed as a percentage of the wt PIV5 HN protein hemadsorption activity. The results are from three independent experiments. Asterisks indicate a significant variation in hemadsorption activity compared to that of wt PIV5 HN (P < 0.05). (F) Neuraminidase (receptor-destroying) activity of PIV5 HN stalk point mutants was measured using a fluorogenic substrate (MU-NANA). Emission of the cleaved fluorescent product was measured at 450 nm and is expressed as a percentage of wt PIV5 HN neuraminidase activity (n = 3).
These mutated HN stalk residues were mapped onto the atomic structure of the PIV5 HN “2 heads up, 2 heads down” model (41) (Fig. 2B showing the globular head protomer that is associated with the 4HB stalk). The fusion assay data (Fig. 2A) indicate residues L83 and L87 (yellow), which extend their hydrophobic side chains into the central core of the PIV5 HN stalk 4HB (29), exhibit F activation levels comparable to wt levels or higher than wt levels when the hydrophobic nature of these residues were maintained (L83I or L87I). On the other hand, charged or polar residues (L83S or L87K) at these positions completely ablated fusion, suggesting a role of these residues in maintaining the central core of the PIV5 HN stalk 4HB. Of the solvent-exposed residues on the PIV5 HN stalk, mutating residues A82, Q86, and D88 to either a similar or a dissimilar residue did not disrupt the F-triggering function of PIV5 HN (Fig. 2B, blue). On the other hand, F activation was strongly dependent on the nature of solvent-exposed residues V81 and L85 (Fig. 2B red). Polar or charged residues at these positions completely block fusion promotion (V81T and L85Q). Although a similar hydrophobic residue is tolerated at position 85 (L85I), the valine at position 81 cannot be replaced by alanine (V81A), a small hydrophobic residue (Fig. 2A). Indeed, the L85I mutant, together with mutants in the 4HB central hydrophobic core (L83I and L87I), and surface-exposed A82Q, Q86 mutants, and D88 mutants appear to be more effective than wt HN in activating F for fusion. The proline mutants (P84A and P84T) at position 84 (Fig. 2B, red) also completely inhibit F triggering, despite having ca. 40 to 50% proteins present on the surface. Thus, overall, hydrophobic residues at positions V81 and L85 seem to be critical for F activation, whereas the P84 residue also appears to have an effect on the integrity of the F activation region in PIV5 HN.
To compare F activation regions between HN proteins within the paramyxovirus family, single point mutations in the NDV-HN stalk with known phenotypes (33, 36) were mapped onto the NDV HN “4 heads down” structure (23) (Fig. 2C). For this analysis, mutations that introduce N-linked carbohydrate chains on the stalk were excluded since potentially they could inhibit F-HN interactions through extensive steric effects. Of the mutated NDV HN residues, L96 was found to be in the 4HB core (23) and as expected, similar to the corresponding L83 and L87 residues of PIV5 HN, NDV HN L96 requires a hydrophobic amino acid in order to maintain function (36). Of the solvent-exposed residues that are mapped onto the NDV-HN stalk domains (23), A89, L90, P93, L94, and L97 appear to be critically involved in F interactions and activation (33, 36).
A comparison of the PIV5 HN residues important for F activation to those of NDV HN through a gap-less sequence alignment revealed interesting features (Fig. 2D). Most of the critical residues identified as primary F-activating residues for the two HNs are hydrophobic in nature. The two hydrophobic PIV5 HN stalk residues that are solvent exposed and critical for F activation (V81 and L85) correspond by sequence alignment to NDV HN residues (L90 and L94), which are also in similar positions on the 4HB stalk (Fig. 2B and C) and display similar phenotypes (33). Additionally, the conserved prolines for both viruses (P84 for PIV5 HN and P93 for NDV HN) appeared to have an effect on F activation (Fig. 2A) (33). However, on the other hand, since the negatively charged D88 residue of PIV5 HN (Fig. 2A) did not affect F activation like the corresponding hydrophobic residue L97 of NDV HN (36, 62), the hydrophobic F activation area of NDV HN may be more extensive than that of PIV5 HN. Nonetheless, many of the critical core residues are conserved between the two HN proteins.
To ensure the integrity of the HN “head” domain in the PIV5 HN stalk mutants the receptor-binding ability of the PIV5 HN mutant proteins was tested by measuring hemadsorption. Overall, most of the mutants had close to wt PIV5 HN levels of receptor-binding activity (Fig. 2E). Mutant proteins P84A and P84T had a somewhat lowered ability to bind RBCs, which is expected given their lowered cell surface transport (Fig. 1B). However, there were some unexpected variations. Mutants V81T, L85Q, and D88E had significantly increased receptor-binding ability compared to that of wt PIV5 HN, despite having normal levels of protein expression and surface transport (Fig. 1A and B). Notably, all three are surface-exposed residues on the PIV5 HN stalk, and V81 and L85 are residues critical for F triggering (Fig. 2A). When these PIV5 HN mutant proteins were analyzed for their ability to cleave receptor in a neuraminidase assay, there were a few minor variations compared to the neuraminidase activity of PIV5 HN wt. Mutants V81T, A82Q, and L83I showed slightly higher neuraminidase activity, whereas mutants V81A, L85I, and D88L had slightly lower neuraminidase activity. As expected, the proline 84 mutants (P84A and P84T) had lowered neuraminidase activity (Fig. 2F), possibly due to their reduced cell surface expression levels. Thus, overall, the PIV5 HN mutant proteins that reached the cell surface at levels comparable to wt PIV5 HN did not have any significant defects in neuraminidase activity. Mutants V81T and L85Q were unique in that they possess normal cell surface expression and neuraminidase activity, while displaying increased receptor binding and an inability to activate F.
The PIV5 HN F activation region is able to substitute for the NDV HN F activation domain.
The major residues (V81, L85, and P84) identified to be important for PIV5 F activation are located in the central part of the PIV5 HN stalk structure (residues 81 to 87) (Fig. 3A); hence, we investigated whether replacing the corresponding region of NDV HN (residues 90 to 96) with the putative PIV5 HN F-activating region (residues 81 to 87) would transfer the PIV5 F-activating capability to the NDV HN stalk. A chimeric NDV HN construct (NDV-PIV5-Fact-HN) was created that contained a sequence of seven amino acid residues (VALPLQL) from the PIV5 HN F-activating region, replacing NDV HN residues 90 to 96 (LESPLAL) (Fig. 3B). The relative expression levels of NDV-PIV5-Fact-HN protein were compared to that of wt NDV HN protein by metabolic labeling and immunoprecipitation (Fig. 3C), and it was found that the NDV-PIV5-Fact-HN chimera protein was expressed in abundance similar to that of wt NDV HN. The cell surface expression of wt NDV HN and NDV-PIV5-Fact-HN proteins was analyzed by flow cytometry and found to be very similar (Fig. 3D).
FIG 3.

The PIV5 HN F activation region is able to substitute for the NDV HN F activation domain. (A) Gapless protein sequence alignment of portions of HN, H, or G stalk domains from various paramyxoviruses. Predicted transmembrane domains (TM), identified as a stretch of hydrophobic residues flanked by charged residues, are underlined in red. The sequence of seven residues of the putative F-activating domain of PIV5 HN and those replaced in the NDV HN stalk domain to create the NDV-PIV5-Fact-HN chimera, are shown enclosed within black boxes. Color coding of boxes highlighting individual residues correspond to that described in Fig. 2B. Negatively charged aspartic acid or glutamic acid residues of rubulaviruses, including D88 of PIV5 HN and E105 of MuV HN, are colored red. (B) Schematic representation of the NDV HN wt and the NDV-PIV5-Fact-HN chimera proteins. The PIV5 HN region (residues 81 to 87) that replaced the corresponding NDV HN region (residues 90 to 96) is highlighted in black on the NDV-PIV5-Fact-HN stalk. CT, cytoplasmic tail. (C) NDV HN wt and NDV-PIV5-Fact-HN expression in 293T cells labeled with Tran35S-label. The proteins were immunoprecipitated from transfected cell lysates using a NDV HN polyclonal antibody (R4722). The proteins were analyzed on a 10% reducing gel. Numbers on the right are molecular masses in kilodaltons. (D) Surface expression of the NDV-PIV5-Fact-HN chimera protein compared to wt NDV HN surface expression determined by flow cytometry. Proteins were detected on the surfaces of transfected cells using the NDV HN R4722 antibody and labeled with a goat α-rabbit FITC-conjugated secondary antibody. The results from three independent experiments. (E) Cell-cell fusion mediated by the NDV-PIV5-Fact-HN chimeric protein when cotransfected with PIV5 F or NDV F, as measured using a luciferase reporter assay for fusion. Fusion activity was measured as RLU and is expressed as a percentage of wt NDV F and wt NDV HN fusion activity. Results from three independent experiments are shown. (F) Representative micrographs showing cell-cell fusion in BHK-21 cells transfected with NDV F or PIV5 F alone or in combination with wt NDV HN, PIV5 HN, or the NDV-PIV5-Fact-HN chimera. Cells were fixed, stained, and photographed at 18 h posttransfection. (G) Receptor-binding ability of the NDV-PIV5-Fact-HN chimera protein as measured using a hemadsorption assay. Absorbance of hemoglobin from RBCs specifically bound by the expressed proteins on 293T cells is measured at 410 nm and shown as a percentage of wt NDV HN hemadsorption activity. Asterisk indicates a P value of <0.05 from three independent experiments. (H) Receptor-destroying activity of the NDV-PIV5-Fact-HN chimera protein as measured by a neuraminidase assay using a fluorogenic substrate. The fluorescent emission is measured at 450 nm and expressed as a percentage of wt NDV HN neuraminidase activity (n = 3).
To examine the fusion promotion activity, the NDV-PIV5-Fact-HN chimeric protein was coexpressed with NDV F or PIV5 F proteins in a luciferase reporter assay (Fig. 3E). Interestingly, the NDV-PIV5-Fact-HN chimera could still activate NDV F, despite having most of the NDV F-activating region (LESPLAL) replaced by the PIV5 F-activating region (VALPLQL). On the other hand, this chimeric HN protein was unable to trigger PIV5 F (Fig. 3E). A comparison of the sequence of the F activation region of the NDV-PIV5-Fact-HN chimeric protein to that of the parent NDV HN showed that an alanine at position 89, the highly conserved proline at position 93, and leucines at positions 94 and 97 in the NDV-PIV5-Fact-HN chimera are maintained in similar positions, whereas the leucine at position 90 in NDV HN is replaced by a valine in the NDV-PIV5-Fact-HN chimera (Fig. 3A). Thus, despite extensive mutagenesis of many residues in the NDV HN F-activating region, the presence of a few critical residues in the context of the 4HB of the NDV-PIV5-Fact-HN chimera stalk is sufficient to maintain the ability of this chimeric HN to activate NDV F. However, overall, the F-activating regions of NDV HN and PIV5 HN are sufficiently different to prevent significant noncognate F activation (Fig. 3E). These results observed in the luciferase reporter assay data were recapitulated in the syncytium assay in BHK-21 cells (Fig. 3F).
To investigate the receptor-binding capability of the NDV-PIV5-Fact-HN chimera, a hemadsorption assay was performed (Fig. 3G). Interestingly, the NDV-PIV5-Fact-HN chimera showed higher receptor binding compared to wt NDV HN despite having wt levels of protein expression (Fig. 3C) and cell surface transport (Fig. 3D). Neuraminidase activity was also measured. It was found that despite having an increased receptor-binding activity, the NDV-PIV5-Fact-HN chimera possessed wt levels of neuraminidase activity (Fig. 3H), suggesting that the increase in receptor-binding activity (Fig. 3G) is not due to a failure of the globular head domain active sites to cleave its receptor.
A single charged amino acid in the HN stalk domain determines F activation promiscuity across two rubulaviruses.
Since the F-interacting regions between NDV (genus Avulavirus) HN and PIV5 (genus Rubulavirus) HN are similar in terms of the hydrophobic F-activating residues, yet divergent overall to prevent heterotypic F-HN interactions, we investigated whether certain F-HN heterotypic interactions observed in more closely related paramyxoviruses could be explained through point mutations within their individual F-activating domains. Ito et al. had demonstrated that MuV HN could activate PIV5 F protein (W3A strain) to mediate cell-cell fusion (63), whereas heterotypic combinations between many other rubulavirus F and HN proteins were functionally inactive. Even the reverse combination of MuV F coexpressed with PIV5 HN failed to initiate cell-cell fusion (63). Thus, it seemed likely that there would be significant similarity in the F-activating region between MuV HN and PIV5 HN to enable both HN proteins to activate PIV5 F.
Examination of HN stalk sequences of rubulaviruses indicated that the PIV5 HN F-activating region (VALPLQLD) appeared very similar to the corresponding region of the MuV HN stalk (VSLPLQIE), whereas corresponding regions for other rubulaviruses such as hPIV2 or hPIV4 were quite different in sequence (Fig. 3A). Interestingly, one of the major differences between the two sequences above, A82 in PIV5 HN (corresponding to S99 in MuV HN), did not appear to play a major role in F activation for PIV5 HN (Fig. 2A). Thus, we reasoned that the similarity between the MuV HN and PIV5 HN stalk sequence in the portion of the PIV5 HN stalk containing the F-activating region is an explanation for how MuV HN can trigger PIV5 F. Furthermore, the predicted transmembrane domains of PIV5 HN and MuV HN are the same length (stretches of 19 residues each flanked by two charged residues, underlined in red in Fig. 3A), and the alignment suggests that the potential F activation regions of these two HN proteins would be located at very similar distances from the membrane.
To understand the roles of MuV HN stalk domain residues that correspond to the PIV5 HN F-activating residues, single point mutants in MuV HN were created, i.e., V98T (corresponding to PIV5 HN residue V81), L102T (corresponding to PIV5 HN residue L85), and E105L (corresponding to PIV5 HN residue D88) (Fig. 4A), and the mutants were expressed and analyzed. All of the MuV HN mutant V98T, L102T, and E105L proteins were expressed at levels comparable to wt MuV HN levels (Fig. 4B). When cell surface expression was analyzed by flow cytometry, the mutant proteins MuV HN V98T, L102T, and E105L all showed surface expression levels on 293T cells comparable to that of wt MuV HN (Fig. 4C). As observed in a hemadsorption assay, the receptor-binding abilities of these proteins were also relatively unaffected (Fig. 4D). When the receptor-destroying ability of the MuV HN single point mutants was tested using a neuraminidase assay, mutants V98T and L102T had neuraminidase activities ∼85% of that of wt MuV HN, whereas E105L had ∼60% of the wt activity (Fig. 4E).
FIG 4.

A single charged amino acid in the HN stalk domain determines F activation promiscuity of two rubulaviruses. (A) Sequence alignment of the PIV5 HN F activation domain and the corresponding region of MuV HN. PIV5 HN residues critical for F activation are shown in red. Arrowheads mark the MuV HN residues that were mutagenized. (B) Protein expression levels of MuV HN single point mutants, as observed after radioimmunoprecipitation of Tran35S-labeled proteins from transfected 293T cell lysates using whole MuV polyclonal antisera. Samples were analyzed on a SDS–10% PAGE gel. Numbers on the left indicate molecular masses markers in kilodaltons. (C) Protein expression and transport to the surface of 293T cells were quantified in cells transfected with MuV HN point mutants. MuV HN proteins were detected on the surfaces of cells using the MuV antisera and fluorescently labeled with a goat α-rabbit FITC-conjugated secondary antibody for detection by flow cytometry. MFI values were expressed as a percentage of wt MuV HN surface expression, and the results are representative of three independent experiments (n = 3). (D) Hemadsorption assay of MuV HN stalk point mutants. Absorbance of hemoglobin from lysed RBCs that were specifically bound to HN proteins expressed on 293T cells was measured at 410 nm and expressed as a percentage of wt MuV HN hemadsorption. (n = 3). (E) Neuraminidase assay of MuV HN stalk point mutants. Emission at 450 nm on cleavage of MU-NANA was expressed as a percentage of the wt MuV HN neuraminidase activity (n = 3). (F) Luciferase reporter assay for fusion showing quantitative cell-cell fusion for MuV HN point mutant proteins coexpressed in Vero cells with MuV F (gray bars) or PIV5 F (white bars). The results are expressed in RLU as a percentage of the wt MuV F and MuV HN fusion activity (n = 3). (G) Representative micrographs showing cell-cell fusion observed when MuV HN point mutants are coexpressed with either MuV F or PIV5 F. Cells were fixed, stained, and photographed at 18 h posttransfection.
The ability of the MuV HN mutant proteins bearing mutations V98T, L102T, or E105L to promote fusion when cotransfected with MuV F (gray bars) was tested using a luciferase reporter assay for fusion (Fig. 4F). It was found that replacing the MuV HN hydrophobic residues V98 and L102 with polar residues completely blocked the fusion promoting ability of MuV HN. On the other hand, replacing the negatively charged E105 with a hydrophobic residue (E105L) had minimal effect on fusion promotion. Interestingly, the corresponding residues of PIV5 HN F activation region have the exact same phenotypes (Fig. 2A): PIV5 HN V81T and PIV5 HN L85Q mutant proteins completely lack the capability to activate PIV5 F, whereas the PIV5 HN D88L mutant does not negatively affect F activation capabilities of PIV5 HN.
Given the close functional conservation between critical residues in the F activation region of PIV5 HN and MuV HN, we reasoned that PIV5 F would be activated by MuV HN E105L but not by MuV HN V98T and MuV HN L102T. Surprisingly, none of the MuV HN stalk point mutants were able to activate PIV5 F (white bars) (Fig. 4F), despite having good surface expression, receptor-binding capability, and neuraminidase activity. The results obtained from the luciferase reporter fusion assay were reflected in a syncytium assay (Fig. 4G). Thus, even though the corresponding negatively charged residues, D88 in PIV5 HN and E105 in MuV HN are not important for triggering their cognate F proteins, the MuV HN E105 residue determines the ability of MuV HN to effectively trigger the heterotypic PIV5 F and hence governs the promiscuity of the MuV HN F-activating region.
Disulfide bonds introduced within the NDV HN 4-helix bundle stalk F activation region abrogate fusion promotion.
For CDV and MeV the introduction of disulfide bonds in the central portion of the H stalk have been shown to be critical for F activation (43, 44). Based on the atomic structure of the NDV HN stalk 4HB (23), we investigated whether the introduction of a targeted disulfide bond within the F activation region could disrupt fusion promotion. It was already known that a cysteine substitution at position 92 (S92C) on the NDV HN stalk results in the formation of dimers and tetramers of NDV HN protein on a gel filtration column (23) and that NDV HN residue S92 lies within the F-activating region of the NDV HN stalk, in close proximity to important F-interacting residues A89, L90, P93, and L94 (Fig. 5A). Importantly, Melanson and Iorio (33) showed that mutation of S92 to an alanine (A), a leucine (L), or even a charged residue, arginine (R), did not significantly alter the ability of these mutant NDV HN proteins to activate NDV F (33), suggesting that S92 itself is not directly involved in the F activation process. To test the effects of introducing a disulfide in the central core of the NDV HN 4HB, the following constructs were created: NDV HN S92C (engineered disulfide bond in the stalk, in addition to a natural disulfide at C123), NDV HN C123S (no disulfide bonds in the stalk), and NDV HN C123S/S92C (engineered disulfide bond in the stalk).
FIG 5.

Disulfide bonds introduced within the NDV HN four-helix bundle stalk F activation region abrogate fusion promotion. (A) Top and side views of the NDV HN “4 heads down” structure mapping the positions of surface-exposed residues that are critical for F activation (red) and the S92 residue (green) in the core of the 4HB. (B) Immunoprecipitation of NDV HN point mutants from transfected 293T cell lysates with the NDV HN R4722 polyclonal antibody after labeling with Tran35S-label. The proteins were analyzed on a 10% reducing SDS-PAGE gel. M, monomer. (C) Same as in panel B, but proteins were analyzed on a 10% nonreducing gel. The numbers on the right indicate molecular mass markers in kilodaltons. D, dimers. (D) Surface expression of NDV HN mutants as detected using the above antibody and analyzed by flow cytometry. Mean fluorescent intensities (MFI) of the NDV HN mutant proteins expressed on the surface of 293T cells are shown as a percentage of wt NDV HN (n = 3). (E) Syncytium fusion assay showing cell-cell fusion of the NDV HN mutants cotransfected with NDV F or the NDV F L289A mutant in BHK-21 cells. Samples were fixed, stained, and imaged 18 h posttransfection. (F) Luciferase reporter assay for fusion showing cell-cell fusion activity for NDV HN point mutants cotransfected with the NDV F L289A mutant. The luciferase reporter activity generated in fused cells (RLU) is expressed as a percentage of NDV F L289A and wt NDV HN fusion (n = 3). (G) Hemadsorption assay of NDV HN mutants. RBCs bound specifically to HN proteins on transfected cells were lysed, and the hemoglobin absorbance was measured at 410 nm. The absorbance values are represented as a percentage of the wt NDV HN hemadsorption activity (n = 3). (H) Neuraminidase activity of NDV HN mutant proteins. Emission at 450 nm upon cleavage of MU-NANA by the NDV HN mutant proteins is represented as a percentage of the wt NDV HN neuraminidase activity (n = 3).
The NDV HN cysteine mutants were expressed in 293T cells. Mutant proteins NDV HN-C123S, NDV HN-C123S/S92C, and NDV HN-S92C exhibited protein expression levels comparable to that of wt NDV HN protein and the mutants migrated on an SDS-PAGE gel similar to wt NDV HN (Fig. 5B). To examine disulfide bond formation, wt NDV HN and mutant proteins were analyzed by SDS-PAGE under nonreducing conditions (Fig. 5C). As expected, NDV HN-C123S, lacking any disulfide bonds in the stalk, migrated as a monomer, whereas wt NDV HN migrated as a dimer due to the presence of the natural disulfide bond at C123. NDV HN-S92C migrated as a dimer similar to that of wt NDV HN, whereas NDV HN-C123S/S92C protein migrated as a dimer with a faster mobility on the gel. A possible explanation for the aberrant mobility of the dimer species is that disulfide bond formation results in an altered Stokes' radius on SDS-PAGE. When cell surface expression of the NDV HN cysteine mutants was measured by flow cytometry the mutants had nearly equivalent cell surface expression levels comparable to that of wt NDV HN protein (Fig. 5D).
The fusion promoting activity of the NDV HN mutants was tested in a syncytium assay in BHK-21 cells (Fig. 5E). NDV HN-C123S, which has no disulfide bonds within the stalk 4HB, could promote fusion at levels compared to wt NDV HN. However, mutant proteins NDV HN-C123S/S92C and NDV HN S92C harboring the engineered disulfide bond in the stalk (S92C) had significantly reduced fusion activation abilities (Fig. 5E, top row). This suggests that introduction of a disulfide bond (S92C) within the core of the 4HB of the NDV HN stalk F activation region reduces fusion triggering, despite the likelihood that S92 does not directly participate in interaction and activation of F.
The crystal structures of the wt and S92C NDV HN proteins indicates that there are no major structural alterations of the F activation area of HN stalk (23). We investigated whether the complete loss of F activation caused by introduction of the disulfide bond (S92C) could be compensated by a destabilizing F mutant. To test this, we used a previously characterized hyperfusogenic mutant of NDV F (L289A), which shows an overall reduced stability and as a result increased fusogenicity (64, 65). Fusion mediated on cotransfection of mutant NDV HN-C123S/S92C or NDV HN-S92C with the NDV F-L289A hyperfusogenic mutant showed some degree of fusion in cells cotransfected with NDV F L289A (Fig. 5E, bottom row). These results are also reflected in the luciferase reporter assay for fusion carried out with NDV F L289A cotransfected with NDV HN-C123S, NDV HN-C123S/S92C, or NDV HN-S92C (Fig. 5F). Thus, introduction of the S92C mutation in the F-activating region of the NDV HN 4HB stalk does not completely alter the F-activating surface of NDV HN but possibly results in a loss of flexibility within the 4HB that prevents F activation. Attempts to restore full fusion of NDV HN-C123S/S92C or NDV HN-S92C by reduction using dithiothreitol or tris(2-carboxyethyl)phosphine were unsuccessful.
The receptor-binding abilities (Fig. 5G) and neuraminidase activities (Fig. 5H) of the globular heads of these mutants were tested in order to make sure that a loss of receptor-binding activity or a defect in receptor cleavage due to the mutations in the NDV HN-C123S/S92C and NDV HN-S92C mutant proteins were not the primary reason behind the reduced fusion activity observed in Fig. 5E and F. No major receptor-binding defect or neuraminidase activity defect was detected for the any of the NDV HN mutant proteins (Fig. 5G and H).
A destabilized NDV F protein mediates cell-cell fusion, when triggered by an NDV HN “headless” stalk.
The PIV5 HN “headless” stalk can activate PIV5 F (42), and similar data have been obtained for the MeV H headless stalk (54) and NiV G headless stalk (55). We were interested in investigating whether the stalk of other paramyxoviruses that use sialic acid as a receptor could also activate F. Therefore, we investigated whether the stalk of NDV HN could function as an F-activating protein without its globular head and in the absence of receptor binding.
Two headless NDV HN molecules, NDV HN 1-123 and NDV HN 1-123 S92C were constructed (Fig. 6A). Both of these HN stalk constructs contain the F-activating region as well as the natural cysteine in the NDV HN stalk (C123) that forms a disulfide-linked dimer and NDV HN 1-123 S92C contains a second cysteine in the stalk activating region that in full-length HN inhibited fusion (Fig. 5). On expression both HN stalk constructs could be detected by a polyclonal antibody specific for NDV HN (R4722) (Fig. 6B), and both constructs were expressed as doublets by SDS-PAGE due to glycosylation at residue N119, as shown by the shift of the slower-mobility species to the faster-gel-mobility species of the NDV HN 1-123 protein after tunicamycin treatment of cells (Fig. 6C).
FIG 6.

A destabilized NDV F protein causes significant cell-cell fusion, when triggered by an NDV HN “headless” stalk. (A) Schematic representation of NDV HN full-length and “headless” stalk mutant proteins NDV HN 1-123 and NDV 1-123 S92C. CT, cytoplasmic tail; TM, transmembrane domain. (B) NDV HN “headless” stalk mutants were expressed in 293T cells. The cells were labeled with Tran35S-label and lysed, and proteins were immunoprecipitated with the NDV HN polyclonal antibody R9722. Polypeptides were analyzed by SDS-PAGE on a 15% gel. Numbers indicate molecular masses in kilodaltons; M, HN monomer. (C) 293T cells expressing the NDV HN 1-123 “headless” stalk protein grown in the presence of 5 μg of tunicamycin/ml. Radiolabeled polypeptides were immunoprecipitated and analyzed as in panel B; M, HN monomer; D, HN dimer. (D) Surface expression of NDV HN mutants as detected by the above NDV HN polyclonal antibody on the surface of transfected 293T cells. MFI of the NDV HN mutants are represented as a percentage of wt NDV HN surface expression (n = 3). (E) Receptor binding of the NDV HN full-length and “headless” stalk mutants analyzed using a hemadsorption assay. The ability of the mutant proteins to bind chicken RBCs was quantified and is expressed as a percentage of the wt NDV HN absorbance at 410 nm. (F) Cell-cell fusion as observed in a syncytium assay of NDV F cotransfected with the NDV HN 1-123 headless stalk. Samples were fixed, stained, and photographed at various times posttransfection as indicated. (G) Syncytium assay showing the ability of the NDV HN headless stalk constructs, NDV HN 1-123 and NDV HN 1-123 S92C, to trigger NDV F or the hyperfusogenic NDV F L289A mutant. Cells were fixed, stained, and photographed 20 h posttransfection.
Both NDV HN stalk constructs were detected at the surface of cells by flow cytometry using the NDV HN R4722 antibody (Fig. 6D). The stalk constructs were detected at relatively lower levels compared to the full-length proteins, but this is likely to be a result of differences in the number of antigenic epitopes recognized by the polyclonal antibody on the full-length proteins and the “headless” stalk versions. When the receptor-binding abilities of the NDV HN “headless” stalks were quantified by using a hemadsorption assay, as expected, the NDV HN stalk constructs in the absence of the globular heads failed to show any receptor binding (Fig. 6E).
To test for fusion activation, the NDV HN 1-123 “headless” stalk construct was coexpressed on BHK-21F cells with wt NDV F in a syncytium assay. NDV HN 1-123 caused fusion from 24 or 28 h posttransfection (Fig. 6F). We hypothesized that the reduced efficiency of NDV HN “headless” stalks in triggering NDV F in comparison to PIV5 HN headless stalk triggering PIV5 F (fusion by 18 h posttransfection) (61) could be due to the NDV “headless” stalks adopting a conformation that is not optimal for fusion triggering. To improve fusion promotion detection, the destabilized NDV F-L289A mutant (64) was cotransfected with the NDV HN “headless” stalk constructs and analyzed in a syncytium assay (Fig. 6G). It was observed that the NDV HN 1-123 stalk caused significant cell-cell fusion on BHK-21 cells even at 20 h posttransfection when cotransfected with NDV F-L289A, suggesting that the “headless” NDV HN 1-123 stalk was in a conformation capable of efficient F activation. The destabilized F protein (F L289A) appears to reduce the overall energy barrier for the F trigger, leading to increased F activation by the NDV HN 1-123 headless stalk and an overall increase in cell-cell fusion. Coexpression of NDV HN 1-123 S92C with wt NDV F or NDV F L 289A did not cause detectable fusion consistent with the data obtained with full-length HN S92C.
A mumps virus HN “headless” stalk is able to trigger both cognate and noncognate F proteins.
We examined whether a “headless” MuV HN stalk could trigger MuV F. We also tested whether such a “headless” MuV HN stalk could trigger PIV5 F. Five different MuV HN headless stalks of different lengths were created (Fig. 7A), all of which contained the critical F-activating residues (V98 and L102), as well as residue E105, which affects the promiscuity of the mumps stalk in activating PIV5 F. Whereas the MuV HN 1-119, MuV HN 1-122, and MuV HN 1-125 constructs contained only one of the two disulfide bonds in the MuV HN stalk (at C119), the other two MuV HN “headless” stalk constructs, MuV HN 1-128 and MuV HN 1-132, harbored both the disulfide bonds (at C119 and C128). Expression of the MuV HN constructs was detected by immunoprecipitation and SDS-PAGE (Fig. 7B), and the anticipated difference in size of the species could be observed. Cell surface expression on 293T cells was examined by flow cytometry using a MuV polyclonal sera (Fig. 7C).
FIG 7.

A mumps virus HN “headless” stalk is able to trigger both cognate and noncognate F proteins. (A) Schematic representation of MuV wt HN protein and MuV HN “headless” stalks of various lengths. CT, cytoplasmic tail; TM, transmembrane domain. (B) Migration pattern of MuV HN “headless” stalk proteins on a SDS–15% PAGE gel. Polypeptides were immunoprecipitated from radiolabeled lysates of transfected 293T cells using the MuV polyclonal sera. (C) Expression of MuV HN “headless” stalk domains detected on the surface of transfected 293T cells using the MuV polyclonal sera. A goat α-rabbit FITC-conjugated secondary antibody was used for flow cytometry. The mean fluorescence intensities of the MuV HN “headless” stalks were expressed as a percentage of wt MuV HN surface expression (n = 3). (D) Luciferase reporter assay for fusion showing fusion promotion by the MuV HN “headless” stalk mutants cotransfected with MuV-F, expressed as a percentage of wt MuV-F and wt MuV HN fusion (n = 3). (E) In the first row, representative micrographs of syncytia show cell-cell fusion in BHK-21 cells transfected with MuV F and MuV HN 1-132 “headless” stalk at 18, 20, 22, 24, and 26 h posttransfection. For the second row, a syncytium assay was performed to show the ability of the MuV HN 1-132 “headless” stalk to cause cell-cell fusion when cotransfected with PIV5 F. Cells were fixed, stained, and photographed 18 to 26 h posttransfection. For the third row, a syncytium assay was performed to assess the expression of PIV5 F alone, so that spontaneous cell-cell fusion could be compared to fusion (26 h posttransfection) caused by cotransfection of MuV HN 1-132 and PIV5 F. (F) Receptor-binding ability of the MuV HN 1-132 mutant, as measured with a hemadsorption assay. The hemadsorption activity of the MuV HN 1-132 mutant was expressed as a percentage of the wt MuV HN activity (n = 3).
When the MuV HN stalks were tested for their ability to promote MuV F triggering using a luciferase reporter assay for fusion, only one of the MuV HN headless stalks (MuV HN 1-132) was able to produce readily detectable fusion with coexpression of MuV F, suggesting that the length of the stalk 4HB and/or the conformation attained by the MuV HN 1-132 “headless” stalk is critical for triggering MuV F (Fig. 7D). When the MuV HN 1-132 construct was cotransfected with MuV F in BHK-21 cells, increasing amounts of cell-cell fusion were observed from 20 to 26 h posttransfection (Fig. 7E).
We tested whether the MuV HN 1-132 stalk protein retained the ability to trigger PIV5 F like the capability of full-length MuV HN protein. As shown in Fig. 7E (second row), on coexpression of the MuV HN 1-132 headless stalk with PIV5 F at 24 to 26 h posttransfection a small amount of cell-cell fusion was observed. Although the amount of cell-cell fusion observed was low, it was reproducibly distinguishable from spontaneous fusion caused by PIV5 F expression alone (Fig. 7, third row). Unfortunately, introduction of single point mutants V98T, L102T, and E105L into the MuV HN 1-132 headless stalk background resulted in defects in protein trafficking (data not shown), and thus we could not analyze whether these mutations in the headless MuV HN stalk had similar effects as that in the full-length mutant MuV HN proteins.
In a hemadsorption assay, as expected, the MuV HN 1-132 headless stalk was unable to bind receptor (Fig. 7F). Thus, taken together, these data suggest that the MuV HN 1-132 “headless” stalk maintains the F-interacting region in a “triggering conformation” that allows the activation of both MuV F and PIV5 F proteins to cause cell-cell fusion.
DISCUSSION
Recently, we proposed a model to explain paramyxovirus membrane fusion by which receptor binding can be closely coordinated with F activation (41, 42). In this “stalk exposure” model, the HN, H, or G globular heads, upon binding receptor, undergo rearrangements that expose an F-activating domain on the attachment protein stalk 4HB by disruption of head-stalk contacts. This rearrangement of the heads allows for the HN, H, or G protein stalk domain to interact with F in a manner that subsequently triggers F to undergo refolding from a prefusion state to a postfusion state to mediate membrane merger. In the present study, we highlight critical residues in the PIV5 HN and MuV HN stalk domains that are responsible for F activation and identify a region containing charged or hydrophobic amino acids that determine specificity of the F-HN interaction. When mutants in the stalk of NDV HN with known phenotypes (33, 36) were mapped onto the NDV HN 4HB stalk structure (23), they showed great similarity to the F-activating region of PIV5 HN (and by inference MuV HN). For all three HN proteins, hydrophobic residues appear to be key activity determinants within the F activation domains. For NDV HN, the F-activating region includes the hydrophobic residue L97. However, for PIV5 HN and MuV HN the corresponding residues are D88 and E105, respectively, and even though they are charged residues they have limited effects on F activation. Interestingly, we have recently identified on PIV5 F a putative region of HN interaction that is composed mostly of hydrophobic residues (61), a finding that provides further support for the idea that F-HN interactions probably are primarily hydrophobic in nature.
An important observation from the mutational (33, 36) and structural analyses of the NDV HN and PIV5 HN stalks is that all of the critical F-activating residues known are located in a region that has extensive contacts with the globular heads, when the globular heads are in the “heads down” conformation (23, 41). This suggests that the HN globular heads in this conformation would prevent these residues from interacting with F proteins. It was observed that some point mutants in the PIV5 HN stalk or NDV HN stalk (33, 36), as well as the NDV-PIV5-Fact-HN chimera, showed increased receptor binding. These increases in receptor-binding activity may be a result of potential changes in the head-stalk contacts (23, 41).
We reasoned that a comparison of homotypic and the few known heterotypic F-HN interactions in paramyxoviruses could provide insight into residues that determine F specificity of the HN stalk domain. To test whether the F-activating specificity of the PIV5 HN stalk could be transferred to the NDV HN stalk domain, a chimeric NDV HN protein that contained a stretch of seven residues from the PIV5 HN F-activating region (NDV-PIV5-Fact-HN), was created. Unexpectedly, given that a functional heterotypic interaction between NDV F and PIV5 HN does not occur, maintenance of the NDV HN critical F-activating hydrophobic residues in the context of the 4HB of a chimeric molecule (NDV-PIV5-Fact-HN) allowed this chimeric protein with a transplanted PIV5 HN “F-activating domain” to activate NDV F to wt levels. On the other hand, the NDV-PIV5-Fact-HN chimeric protein failed to activate PIV5 F. A gapless sequence alignment of paramyxovirus HN, H, or G proteins (Fig. 3A) shows some variability in the putative distances at which F-interacting/activating residues are found within these different attachment protein stalks. From biochemical studies, F-activating regions in henipavirus G proteins are considered to be located higher up in the stalk (30, 40, 66). It was also shown using a series of insertion and deletion mutants of MeV H (35) that altering the distances of F-activating residues from the membrane has major effects on functionality. The highly similar PIV5 HN (residues 81 to 87) and NDV HN (residues 90 to 96) F-activating regions differed in heights by three residues. However, raising the height of the PIV5 HN F-activating region by +3 residues within the NDV-PIV5-Fact-HN chimera by inserting 3 residues near the transmembrane domain resulted in defects in protein expression and transport to the cell surface (data not shown). Thus, it is possible that the NDV-PIV5-Fact-HN chimera is unable to activate PIV5 F due to a mismatch of heights with respect to the HN interaction region of PIV5 F. Alternatively, the seven amino acid region swapped may not represent the physical F interaction region in its entirety, and the above observations could be a result of long-range effects. However, this cannot be confirmed from the current data.
Nonetheless, despite differences in heights of F-activating regions in various paramyxovirus attachment protein stalks from the membrane, recent evidence from biochemical, structural, and modeling data, suggests that the F-activating regions of NiV G (55), PIV5 HN (41, 42), NDV HN (23), and MuV H (44) could potentially be shielded by the attachment protein globular heads, presumably in the absence of receptor binding. However, this F-activating region could be exposed by a movement of only two of the globular heads (41, 67).
To further understand the residues of the F-HN stalk that determine the specificity of interaction, we investigated a known heterotypic interaction unusual among paramyxoviruses, where MuV HN is able to activate PIV5 F (W3A strain) and cause cell-cell fusion (63). Interestingly, a single negatively charged residue (E105) was found to determine the promiscuity of the PIV5 F interaction with the MuV HN stalk. The MuV HN mutant E105L was not affected in its ability to activate MuV F, but the mutant lost its ability to activate PIV5 F. On the other hand, the mutation of the corresponding residue of PIV5 HN (D88) had no effect on activation of PIV5 F. Notably, a negatively charged residue is present across all rubulavirus HN proteins that are known be involved in heterotypic paramyxovirus F-HN interactions (Fig. 3A), such as hPIV2 HN being able to substitute for hPIV4a HN or SV41 HN and MuV HN being able to substitute for hPIV2 HN (68, 69). However, for NDV HN (an avulavirus), the corresponding residue (L97) was found to be important for F activation, and the NDV HN L97A mutant maintained its ability to interact with NDV F (62). Overall, these data suggest that in rubulaviruses a negatively charged residue located just above the F activation region can modulate F specificity. Although individual residues critical for F interaction and F activation have been determined for HN, H, or G stalk domains, it is becoming increasingly clear that F interaction with HN, H, or G stalk domain alone does not lead to F activation. Although these two aspects of the F triggering process are functionally linked, they can be separated by targeted mutagenesis (62, 70, 71; reviewed in reference 72).
A number of recent studies have identified conformational changes in attachment protein stalks that are linked to fusion promotion. A requirement of some degree of conformational flexibility in the central part of the 4HB stalk of morbilliviruses (MeV and CDV) (43, 44, 56), as well as energy-dependent conformational changes possibly occurring between globular head dimers (42, 55), has been implicated as critical for the correct timing of F activation on receptor binding. We demonstrate here that a residue within the NDV HN F activation domain (S92) that does not directly affect fusion activation when mutated singly (33) is able to form disulfide-linked dimers and in the process blocks the activation of both NDV F and a hyperfusogenic mutant of NDV F (L289A). Since the NDV HN wt and the NDV HN S92C stalk differ only by the addition of a cysteine cross-link and structural studies have shown little change in the 4HB conformation, these observations suggest that productive F-HN interactions require some additional flexibility within the F activation region of HN. Interestingly, the requirement for stalk flexibility was observed even when the globular heads of NDV HN were absent in a “headless” stalk protein (NDV HN 1-123) that can normally trigger NDV F. Together, these data suggest that such flexibility within the F-activating regions of the attachment protein stalk 4HB is a more general phenomenon across paramyxoviruses.
It is possible that the F activation region allows for an “induced fit” with the F protein, rather than undergoing specific active conformational changes, upon receptor binding being propagated down the highly flexible head-stalk linkers from the head domains of HN, H, or G. Through mixed oligomer experiments, a high degree of flexibility of the F-activating region and higher up in the PIV5 HN stalk has been illustrated by the ability of these regions to tolerate large carbohydrates chain additions within the hydrophobic core of the 4HB (29). Furthermore, introduction of disulfide bonds in the upper region of the MeV H stalk did not cause a major functional disruption (56). Thus, neither the introduction of rigid disulfide bridges nor the addition of bulky carbohydrate moieties in the 4HB of the upper stalk appeared to disrupt any putative conformational changes that may propagate down through this upper stalk region. Chimeric HN, H, or G proteins bearing different stalks and globular heads, as well as engineered noncognate receptor-binding attachment proteins, can trigger F, leading to the argument that specific conformational changes between the globular head domains on receptor binding are unlikely to lead to corresponding specific changes within the 4HB after translocation through a highly flexible stalk. These data suggest an “induced fit” interaction with the F protein, with hydrophobic and charged residues of a flexible central region of the attachment protein 4HB stalk acting as the F activation trigger. In morbilliviruses, F-H glycoprotein complexes assemble during protein trafficking but do not prematurely trigger F (73). Also, certain attachment protein F activation region point mutants such as NDV HN L97A, as well as others in morbillivirus H proteins (39, 43), associate with their respective F proteins but fail to trigger fusion. These observations could potentially be explained by a distinction between nontriggering associations between F-H during intracellular transport versus F triggering “induced fit” interactions at the cell surface. The latter interaction, however, can only be productive when coupled with multiple spatiotemporal conformational changes in the globular heads that exposes an F activation region in the stalk (stalk exposure model), as proposed for PIV5 HN (42) and NiV G (55).
We found that in addition to the PIV5 HN “headless stalk” that could activate PIV5 F (42), a headless stalk of NDV HN and MuV HN could also trigger NDV F and MuV F, respectively. Interestingly, only one of five different headless-stalk constructs of MuV HN could successfully activate MuV F to cause fusion. Similarly, it was found for NiV G that only one of seven different stalk lengths was active (55). Also, a “headless” MeV H protein required tags to provide stabilization in order to achieve a “triggering” conformation (54). In the present study, since the F-triggering capabilities for “headless” MeV H proteins were lowered significantly upon lowering temperature, this result provides further support to the notion that stalk flexibility depends on “breathing” or spontaneous movements within the F activation region (54). Thus, it appears for HN, H, or G “headless” stalks, a degree of flexibility at the F-activating domain, rather than active rearrangements of the stalk domain, could explain the mechanism for an F interaction and F activation. In such HN constructs lacking the globular heads, maintenance of a stalk 4HB conformation that can undergo such F-induced rearrangements is probably dependent on factors such as the length and nature of the stalk 4HB C-terminal residues, which could be important for preserving the structural integrity of the stalk 4HB. In light of the “stalk exposure” model, further support for an “induced fit” F-activating interaction can be provided by the above observations. Headless attachment protein stalks do not require the “stalk exposure” step, but many headless stalks, despite potentially having their F-activating regions exposed, fail to trigger F. Since an “F-triggering” conformation can be attained in the absence of any receptor binding, the “stalk exposure” model coupled with a very specific “induced fit” interaction of F with the HN, H, or G can explain the productive triggering of paramyxovirus F proteins. Further investigation into the F-triggering process is necessary, however, in order to confirm this hypothesis.
Overall, we provide evidence here in support of the “stalk activation” model, extending our previous findings to other paramyxoviruses. Taking together the available biochemical, biophysical, and atomic structure data, we propose that paramyxoviruses from various different subfamilies share core features of the “stalk activation” model. Also analogous to the “headless” influenza virus construct (74), the antigenicity of the headless paramyxovirus stalk domains and the ability of headless stalks to prematurely trigger F proteins are important characteristics that could aid rational drug design. In addition, similarities of F-activating regions across paramyxoviruses provide greater opportunities for development of broadly neutralizing antibodies to HN, H, and G proteins from known and emerging paramyxoviruses, parallel to those recently developed for RSV and hMPV F proteins (75).
ACKNOWLEDGMENTS
We thank Priya A. Shah for help with some of the experiments.
This research was supported in part by National Institutes of Health Research grants AI-23173 (to R.A.L.) and GM-61050 (to T.S.J.). R.A.L. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Published ahead of print 22 January 2014
REFERENCES
- 1.Lamb RA, Parks GD. 2013. Paramyxoviridae: the viruses and their replication, p 957–995 In Knipe DM, Howley PM. (ed), Fields virology, 6th ed, vol 1 Lippincott/The Williams & Wilkins Co, Philadelphia, PA [Google Scholar]
- 2.Drexler JF, Corman VM, Muller MA, Maganga GD, Vallo P, Binger T, Gloza-Rausch F, Rasche A, Yordanov S, Seebens A, Oppong S, Adu Sarkodie Y, Pongombo C, Lukashev AN, Schmidt-Chanasit J, Stocker A, Carneiro AJ, Erbar S, Maisner A, Fronhoffs F, Buettner R, Kalko EK, Kruppa T, Franke CR, Kallies R, Yandoko ER, Herrler G, Reusken C, Hassanin A, Kruger DH, Matthee S, Ulrich RG, Leroy EM, Drosten C. 2012. Bats host major mammalian paramyxoviruses. Nat. Commun. 3:796. 10.1038/ncomms1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marsh GA, de Jong C, Barr JA, Tachedjian M, Smith C, Middleton D, Yu M, Todd S, Foord AJ, Haring V, Payne J, Robinson R, Broz I, Crameri G, Field HE, Wang LF. 2012. Cedar virus: a novel henipavirus isolated from Australian bats. PLoS Pathog. 8:e1002836. 10.1371/journal.ppat.1002836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamb RA, Paterson RG, Jardetzky TS. 2006. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 344:30–37. 10.1016/j.virol.2005.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–636. 10.3390/v4040613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44. 10.1038/nature04322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, Graham BS. 2013. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340:1113–1117. 10.1126/science.1234914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim YH, Donald JE, Grigoryan G, Leser GP, Fadeev AY, Lamb RA, DeGrado WF. 2011. Capture and imaging of a prehairpin fusion intermediate of the paramyxovirus PIV5. Proc. Natl. Acad. Sci. U. S. A. 108:20992–20997. 10.1073/pnas.1116034108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin H-S, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293. 10.1073/pnas.0503989102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLellan JS, Yang Y, Graham BS, Kwong PD. 2011. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 85:7788–7796. 10.1128/JVI.00555-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swanson K, Wen X, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2010. Structure of the Newcastle disease virus F protein in the post-fusion conformation. Virology 402:372–379. 10.1016/j.virol.2010.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swanson KA, Settembre EC, Shaw CA, Dey AK, Rappuoli R, Mandl CW, Dormitzer PR, Carfi A. 2011. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc. Natl. Acad. Sci. U. S. A. 108:9619–9624. 10.1073/pnas.1106536108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowden TA, Aricescu AR, Gilbert RJ, Grimes JM, Jones EY, Stuart DI. 2008. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat. Struct. Biol. 15:567–572. 10.1038/nsmb.1435 [DOI] [PubMed] [Google Scholar]
- 14.Colf LA, Juo ZS, Garcia KC. 2007. Structure of the measles virus hemagglutinin. Nat. Struct. Mol. Biol. 14:1227–1228. 10.1038/nsmb1342 [DOI] [PubMed] [Google Scholar]
- 15.Crennell S, Takimoto T, Portner A, Taylor G. 2000. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat. Struct. Biol. 7:1068–1074. 10.1038/81002 [DOI] [PubMed] [Google Scholar]
- 16.Hashiguchi T, Kajikawa M, Maita N, Takeda M, Kuroki K, Sasaki K, Kohda D, Yanagi Y, Maenaka K. 2007. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. U. S. A. 104:19535–19540. 10.1073/pnas.0707830104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence MC, Borg NA, Streltsov VA, Pilling PA, Epa VC, Varghese JN, McKimm-Breschkin JL, Colman PM. 2004. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J. Mol. Biol. 335:1343–1357. 10.1016/j.jmb.2003.11.032 [DOI] [PubMed] [Google Scholar]
- 18.Xu K, Rajashankar KR, Chan YP, Himanen JP, Broder CC, Nikolov DB. 2008. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. U. S. A. 105:9953–9958. 10.1073/pnas.0804797105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan P, Thompson T, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS. 2005. Structural studies of the parainfluenza virius 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13:1–13. 10.1016/j.str.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 20.Bowden TA, Crispin M, Harvey DJ, Jones EY, Stuart DI. 2010. Dimeric architecture of the Hendra virus attachment glycoprotein: evidence for a conserved mode of assembly. J. Virol. 84:6208–6217. 10.1128/JVI.00317-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan P, Paterson RG, Leser GP, Lamb RA, Jardetzky TS. 2012. Structure of the Ulster strain Newcastle disease virus hemagglutinin-neuraminidase reveals auto-inhibitory interactions associated with low virulence. PLoS Pathog. 8:e1002855. 10.1371/journal.ppat.1002855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santiago C, Celma ML, Stehle T, Casasnovas JM. 2010. Structure of the measles virus hemagglutinin bound to the CD46 receptor. Nat. Struct. Mol. Biol. 17:124–129. 10.1038/nsmb.1726 [DOI] [PubMed] [Google Scholar]
- 23.Yuan P, Swanson KA, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc. Natl. Acad. Sci. U. S. A. 108:14920–14925. 10.1073/pnas.1111691108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowden TA, Crispin M, Harvey DJ, Aricescu AR, Grimes JM, Jones EY, Stuart DI. 2008. Crystal structure and carbohydrate analysis of Nipah virus attachment glycoprotein: a template for antiviral and vaccine design. J. Virol. 82:11628–11636. 10.1128/JVI.01344-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan P, Leser GP, Demeler B, Lamb RA, Jardetzky TS. 2008. Domain architecture and oligomerization properties of the paramyxovirus PIV 5 hemagglutinin-neuraminidase (HN) protein. Virology 378:282–291. 10.1016/j.virol.2008.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng DT, Hiebert SW, Lamb RA. 1990. Different roles of individual N-linked oligosaccharide chains in folding, assembly, and transport of the simian virus 5 hemagglutinin-neuraminidase. Mol. Cell. Biol. 10:1989–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bossart KN, Crameri G, Dimitrov AS, Mungall BA, Feng YR, Patch JR, Choudhary A, Wang LF, Eaton BT, Broder CC. 2005. Receptor binding, fusion inhibition, and induction of cross-reactive neutralizing antibodies by a soluble G glycoprotein of Hendra virus. J. Virol. 79:6690–6702. 10.1128/JVI.79.11.6690-6702.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brindley MA, Plemper RK. 2010. Blue native PAGE and biomolecular complementation reveal a tetrameric or higher-order oligomer organization of the physiological measles virus attachment protein H. J. Virol. 84:12174–12184. 10.1128/JVI.01222-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bose S, Welch BD, Kors CA, Yuan P, Jardetzky TS, Lamb RA. 2011. Structure and mutagenesis of the parainfluenza virus 5 hemagglutinin-neuraminidase stalk domain reveals a four-helix bundle and the role of the stalk in fusion promotion. J. Virol. 85:12855–12866. 10.1128/JVI.06350-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bishop KA, Hickey AC, Khetawat D, Patch JR, Bossart KN, Zhu Z, Wang LF, Dimitrov DS, Broder CC. 2008. Residues in the stalk domain of the Hendra virus G glycoprotein modulate conformational changes associated with receptor binding. J. Virol. 82:11398–11409. 10.1128/JVI.02654-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng R, Wang Z, Mirza AM, Iorio RM. 1995. Localization of a domain on the paramyxovirus attachment protein required for the promotion of cellular fusion by its homologous fusion protein spike. Virology 209:457–469. 10.1006/viro.1995.1278 [DOI] [PubMed] [Google Scholar]
- 32.Deng R, Wang Z, Mahon PJ, Marinello M, Mirza A, Iorio RM. 1999. Mutations in the Newcastle disease virus hemagglutinin-neuraminidase protein that interfere with its ability to interact with the homologous F protein in the promotion of fusion. Virology 253:43–54. 10.1006/viro.1998.9501 [DOI] [PubMed] [Google Scholar]
- 33.Melanson VR, Iorio RM. 2004. Amino acid substitutions in the F-specific domain in the stalk of the Newcastle disease virus HN protein modulate fusion and interfere with its interaction with the F protein. J. Virol. 78:13053–13061. 10.1128/JVI.78.23.13053-13061.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JK, Prussia A, Paal T, White LK, Snyder JP, Plemper RK. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J. Biol. Chem. 283:16561–16572. 10.1074/jbc.M801018200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paal T, Brindley MA, St. Clair C, Prussia A, Gaus D, Krumm SA, Snyder JP, Plemper RK. 2009. Probing the spatial organization of measles virus fusion complexes. J. Virol. 83:10480–10493. 10.1128/JVI.01195-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stone-Hulslander J, Morrison TG. 1999. Mutational analysis of heptad repeats in the membrane-proximal region of Newcastle disease virus HN protein. J. Virol. 73:3630–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takimoto T, Taylor GL, Connaris HC, Crennell SJ, Portner A. 2002. Role of the hemagglutinin-neuraminidase protein in the mechanism of paramyxovirus-cell membrane fusion. J. Virol. 76:13028–13033. 10.1128/JVI.76.24.13028-13033.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao Q, Hu X, Compans RW. 1997. Association of the parainfluenza virus fusion and hemagglutinin-neuraminidase glycoproteins on cell surfaces. J. Virol. 71:650–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plemper RK, Hammond AL, Gerlier D, Fielding AK, Cattaneo R. 2002. Strength of envelope protein interaction modulates cytopathicity of measles virus. J. Virol. 76:5051–5061. 10.1128/JVI.76.10.5051-5061.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aguilar HC, Ataman ZA, Aspericueta V, Fang AQ, Stroud M, Negrete OA, Kammerer RA, Lee B. 2009. A novel receptor-induced activation site in the Nipah virus attachment glycoprotein (G) involved in triggering the fusion glycoprotein (F). J. Biol. Chem. 284:1628–1635. 10.1074/jbc.M807469200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welch BD, Yuan P, Bose S, Kors CA, Lamb RA, Jardetzky TS. 2013. Structure of the parainfluenza virus 5 (PIV5) hemagglutinin-neuraminidase (HN) ectodomain. PLoS Pathog. 9:e1003534. 10.1371/journal.ppat.1003534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bose S, Zokarkar A, Welch BD, Leser GP, Jardetzky TS, Lamb RA. 2012. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc. Natl. Acad. Sci. U. S. A. 109:E2625–E2634. 10.1073/pnas.1213813109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ader N, Brindley MA, Avila M, Origgi FC, Langedijk J, Orvell C, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger F protein refolding for membrane fusion. J. Biol. Chem. 287:16324–16334. 10.1074/jbc.M112.342493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Navaratnarajah CK, Negi S, Braun W, Cattaneo R. 2012. Membrane fusion triggering: three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J. Biol. Chem. 287:38543–38551. 10.1074/jbc.M112.410563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navaratnarajah CK, Oezguen N, Rupp L, Kay L, Leonard VH, Braun W, Cattaneo R. 2011. The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat. Struct. Mol. Biol. 18:128–134. 10.1038/nsmb.1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rasbach A, Abel T, Munch RC, Boller K, Schneider-Schaulies J, Buchholz CJ. 2013. The receptor attachment function of measles virus hemagglutinin can be replaced with an autonomous protein that binds Her2/neu while maintaining its fusion-helper function. J. Virol. 87:6246–6256. 10.1128/JVI.03298-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paraskevakou G, Allen C, Nakamura T, Zollman P, James CD, Peng KW, Schroeder M, Russell SJ, Galanis E. 2007. Epidermal growth factor receptor (EGFR)-retargeted measles virus strains effectively target EGFR- or EGFRvIII-expressing gliomas. Mol. Ther. 15:677–686 [DOI] [PubMed] [Google Scholar]
- 48.Allen C, Vongpunsawad S, Nakamura T, James CD, Schroeder M, Cattaneo R, Giannini C, Krempski J, Peng KW, Goble JM, Uhm JH, Russell SJ, Galanis E. 2006. Retargeted oncolytic measles strains entering via the EGFRvIII receptor maintain significant antitumor activity against gliomas with increased tumor specificity. Cancer Res. 66:11840–11850. 10.1158/0008-5472.CAN-06-1200 [DOI] [PubMed] [Google Scholar]
- 49.Talekar A, Moscona A, Porotto M. 2013. Measles virus fusion machinery activated by sialic acid binding globular domain. J. Virol. 87:13619–13627. 10.1128/JVI.02256-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Porotto M, Salah Z, DeVito I, Talekar A, Palmer SG, Xu R, Wilson IA, Moscona A. 2012. The second receptor binding site of the globular head of the Newcastle disease virus hemagglutinin-neuraminidase activates the stalk of multiple paramyxovirus receptor binding proteins to trigger fusion. J. Virol. 86:5730–5741. 10.1128/JVI.06793-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z, Mirza AM, Li J, Mahon PJ, Iorio RM. 2004. An oligosaccharide at the C terminus of the F-specific domain in the stalk of the human parainfluenza virus 3 hemagglutinin-neuraminidase modulates fusion. Virus Res. 99:177–185. 10.1016/j.virusres.2003.11.010 [DOI] [PubMed] [Google Scholar]
- 52.Farzan SF, Palermo LM, Yokoyama CC, Orefice G, Fornabaio M, Sarkar A, Kellogg GE, Greengard O, Porotto M, Moscona A. 2011. Premature activation of the paramyxovirus fusion protein before target cell attachment with corruption of the viral fusion machinery. J. Biol. Chem. 286:37945–37954. 10.1074/jbc.M111.256248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porotto M, Devito I, Palmer SG, Jurgens EM, Yee JL, Yokoyama CC, Pessi A, Moscona A. 2011. Spring-loaded model revisited: paramyxovirus fusion requires engagement of a receptor binding protein beyond initial triggering of the fusion protein. J. Virol. 85:12867–12880. 10.1128/JVI.05873-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brindley MA, Suter R, Schestak I, Kiss G, Wright ER, Plemper RK. 2013. A stabilized headless measles virus attachment protein stalk efficiently triggers membrane fusion. J. Virol. 87:11693–11703. 10.1128/JVI.01945-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Q, Stone JA, Bradel-Tretheway B, Dabundo J, Benavides-Montano JA, Santos-Montanez J, Biering SB, Nicola AV, Iorio RM, Lu X, Aguilar HC. 2013. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced Nipah virus fusion and cell entry. PLoS Pathog. 9:e1003770. 10.1371/journal.ppat.1003770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Apte-Sengupta S, Navaratnarajah CK, Cattaneo R. 2013. Hydrophobic and charged residues in the central segment of the measles virus hemagglutinin stalk mediate transmission of the fusion-triggering signal. J. Virol. 87:10401–10404. 10.1128/JVI.01547-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paterson RG, Russell CJ, Lamb RA. 2000. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology 270:17–30. 10.1006/viro.2000.0267 [DOI] [PubMed] [Google Scholar]
- 58.Paterson RG, Lamb RA. 1993. The molecular biology of influenza viruses and paramyxoviruses, p 35–73 In Davidson A, Elliott RM. (ed), Molecular virology: a practical approach. IRL Oxford University Press, Oxford, England [Google Scholar]
- 59.Connolly SA, Leser GP, Jardetzky TS, Lamb RA. 2009. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J. Virol. 83:10857–10868. 10.1128/JVI.01191-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Melanson VR, Iorio RM. 2006. Addition of N-glycans in the stalk of the Newcastle disease virus HN protein blocks its interaction with the F protein and prevents fusion. J. Virol. 80:623–633. 10.1128/JVI.80.2.623-633.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bose S, Heath CM, Shah PA, Alayyoubi M, Jardetzky TS, Lamb RA. 2013. Mutations in the parainfluenza virus 5 fusion (F) protein reveal domains important for fusion triggering and metastability. J. Virol. 87:13520–13531. 10.1128/JVI.02123-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirza AM, Iorio RM. 2013. A mutation in the stalk of the NDV HN protein prevents triggering of the F protein despite allowing efficient HN-F complex formation. J. Virol. 87:8813–8815. 10.1128/JVI.01066-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ito M, Nishio M, Kawano M, Kusagawa S, Komada H, Ito Y, Tsurudome M. 1997. Role of a single amino acid at the amino terminus of the simian virus 5 F2 subunit in syncytium formation. J. Virol. 71:9855–9858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sergel TA, McGinnes LW, Morrison TG. 2000. A single amino acid change in the Newcastle disease virus fusion protein alters the requirement for HN protein in fusion. J. Virol. 74:5101–5107. 10.1128/JVI.74.11.5101-5107.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li V, Melanson VR, Mirza AM, Iorio RM. 2005. Decreased dependence on receptor recognition for the fusion promotion activity of L289A-mutated Newcastle disease virus fusion protein correlates with a monoclonal antibody-detected conformational change. J. Virol. 79:1180–1190. 10.1128/JVI.79.2.1180-1190.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maar D, Harmon B, Chu D, Schulz B, Aguilar HC, Lee B, Negrete OA. 2012. Cysteines in the stalk of the Nipah virus G glycoprotein are located in a distinct subdomain critical for fusion activation. J. Virol. 86:6632–6642. 10.1128/JVI.00076-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brindley MA, Takeda M, Plattet P, Plemper RK. 2012. Triggering the measles virus membrane fusion machinery. Proc. Natl. Acad. Sci. U. S. A. 109:E3018–E3027. 10.1073/pnas.1210925109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsurudome M, Kawano M, Yuasa T, Tabata N, Nishimo M, Komada H, Ito Y. 1995. Identification of regions on the hemagglutinin-neuraminidase protein of human parainfluenza virus type 2 important for promoting cell fusion. Virology 213:190–203. 10.1006/viro.1995.1559 [DOI] [PubMed] [Google Scholar]
- 69.Tsurudome M, Ito M, Nishio M, Kawano M, Okamoto K, Kusagawa S, Komada H, Ito Y. 1998. Identification of regions on the fusion protein of human parainfluenza virus type 2 which are required for haemagglutinin-neuraminidase proteins to promote cell fusion. J. Gen. Virol. 79:279–289 [DOI] [PubMed] [Google Scholar]
- 70.Zhu Q, Biering SB, Mirza AM, Grasseschi BA, Mahon PJ, Lee B, Aguilar HC, Iorio RM. 2013. Individual N-glycans added at intervals along the stalk of the Nipah virus G protein prevent fusion but do not block the interaction with the homologous F protein. J. Virol. 87:3119–3129. 10.1128/JVI.03084-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corey EA, Iorio RM. 2007. Mutations in the stalk of the measles virus hemagglutinin protein decrease fusion but do not interfere with virus-specific interaction with the homologous fusion protein. J. Virol. 81:9900–9910. 10.1128/JVI.00909-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iorio RM, Melanson VR, Mahon PJ. 2009. Glycoprotein interactions in paramyxovirus fusion. Future Virol. 4:335–351. 10.2217/fvl.09.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plemper RK, Hammond AL, Cattaneo R. 2001. Measles virus envelope glycoproteins hetero-oligomerize in the endoplasmic reticulum. J. Biol. Chem. 276:44239–44246. 10.1074/jbc.M105967200 [DOI] [PubMed] [Google Scholar]
- 74.Steel J, Lowen AC, Wang TT, Yondola M, Gao Q, Haye K, Garcia-Sastre A, Palese P. 2010. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. mBio 1:e00018–10. 10.1128/mBio.00018-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Corti D, Bianchi S, Vanzetta F, Minola A, Perez L, Agatic G, Guarino B, Silacci C, Marcandalli J, Marsland BJ, Piralla A, Percivalle E, Sallusto F, Baldanti F, Lanzavecchia A. 2013. Cross-neutralization of four paramyxoviruses by a human monoclonal antibody. Nature 501:439–443. 10.1038/nature12442 [DOI] [PubMed] [Google Scholar]