

ABSTRACT
Respiratory syncytial virus (RSV) is the most important cause of lower respiratory tract infection in young children and the leading cause of infant hospitalization worldwide. Uncontrolled response to RSV is mediated by a toll-like receptor (TLR)-mediated immune response. Resveratrol possesses anti-RSV activity and is an inhibitor of the TRIF/TBK1/IRF-3 complex. We hypothesize that resveratrol inhibits the TRIF-dependent pathway through upregulation of SARM post-RSV infection. BALB/c mice were infected with RSV and were injected with resveratrol 1 h postinoculation. SARM short interfering RNA was administered to RSV-infected and resveratrol-treated mice. Lung function was measured by whole-body plethysmography, lung histopathology was examined, and lymphocytes in bronchoalveolar lavage fluid were quantified. SARM and TRIF protein expression were detected in the lung by Western blot analyses. The expression of gamma interferon in bronchoalveolar lavage fluid (BALF) was evaluated by enzyme-linked immunosorbent assay (ELISA). SARM expression was reduced and TRIF expression was increased after infection with RSV. Resveratrol increased SARM expression and decreased TRIF expression after RSV infection. SARM knockdown in resveratrol-treated mice enhanced gamma interferon production, RSV-induced airway inflammation, and airway hyperresponsiveness (AHR). Resveratrol decreased TRIF expression and prevented the RSV-mediated reduction of SARM expression. Resveratrol-mediated inhibition of the TRIF-dependent pathway may be dependent on SARM expression.
IMPORTANCE Our study provides insights into the regulation of innate immunity in response to RSV infection. The results suggest that resveratrol-mediated alterations in SARM have therapeutic potential against RSV immunopathology caused by deregulation of the TLR-mediated immune response. Ultimately, improved insight into the complex interplay between TLR adaptor proteins and the occurrence of severe RSV infection might lead to novel therapeutic treatment strategies, such as TLR adjuvants.
INTRODUCTION
Respiratory syncytial virus (RSV) is the most important cause of viral bronchiolitis in infants and young children and the leading cause of infant hospitalization worldwide (1). Nearly all children have been infected with RSV before they reach 2 years of age (2). Surprisingly, infection at a young age does not provide life-long protection. Immunocompromised children and the elderly are also at high risk of developing RSV-associated disease (3, 4). According to WHO, RSV causes 64 million infections annually and approximately 160,000 deaths per year (5). Altogether, RSV causes significant economic burden. Unfortunately there is no RSV vaccine or effective antiviral drug available today.
Toll-like receptors (TLRs) recognize specific structural motifs expressed by microbes that are defined as pathogen-associated molecular patterns (PAMPs). They are type-1 transmembrane proteins composed of an extracellular leucine-rich (LR) domain and a cytoplasmic tail that contains a conserved TIR domain (6). Viral PAMPs bind to TLRs, causing the activation of transcription factors (7). This leads to the induction of cytokines, chemokines, and interferons (IFN) that create an antiviral state and mature the adaptive immune response (8).
To date, five intracellular TLR adaptor proteins containing a TIR domain have been identified: myeloid differential primary response protein (MyD88), MyD88-adaptor-like (Mal or TIRAP), TIR domain-containing adaptor inducing beta interferon (TRIF or TICAM-1), TRIF-related adaptor molecule (TRAM or TICAM-2), and sterile-alpha and Armadillo motif protein (SARM). SARM was the last of the adaptor proteins to be discovered. It contains sterile-α (SAM) and HEAT/armadillo (ARM) motifs (9, 10) and is highly conserved from arthropod to human. SARM is the most conserved TIR domain-containing protein (11). In Caenorhabditis elegans, TIR-1, a SARM homologue, plays a fundamental role in host defense against bacterial and fungal infections. Human SARM functions as a negative regulator of the TRIF-dependent pathways in innate immunity (12, 13). It has been shown to inhibit both TRIF- and MyD88-mediated activation of the transcription factor AP-1 (14). SARM contributes to the initiation, elongation, and maintenance of dendritic arbors and influences axonal death and neuronal polarization (15–17). Recent studies have shown that SARM directly binds to mitochondria (18) and induces apoptosis in T cells (19). However, whether human SARM has an antiviral role has not yet been elucidated.
Resveratrol (trans-3,5,4-trihydroxystilbene), one of the nonflavonoid polyphenolic phytoalexins found in grapes and red wines (20), is a potent inhibitor of TRIF-dependent signaling (21, 22). Resveratrol has been shown to prevent cancer (23), cardiovascular disease (24), and ischemic injuries (25) and to possess anti-RSV activity (26, 27). It also protects against airway remodeling and airway hyperreactivity in asthma (28). Resveratrol can reduce RSV titers in the lung, the number of infiltrating lymphocytes present in bronchoalveolar lavage fluid (BALF), and inflammation (21, 22). It reduces airway inflammation following RSV infection, significantly decreases IFN-γ (27), and downregulates IFN-γ-inducible inflammatory genes in macrophages (29).
Previously, we reported that RSV infection induced TLR3 and activated TRIF-dependent signaling, which was associated with the induction of IFN-γ (27). IFN-γ from Th1 cells is required for the induction of severe airway hyperresponsiveness (30). Furthermore, we demonstrated that resveratrol inhibits TLR3 signaling, M2R expression, and IFN-γ production.
Taken together, these data indicate that RSV functions as a trigger to activate innate immune responses, and that the anti-inflammatory function of resveratrol involves TLR-associated signaling. However, several interesting issues remain to be determined, including whether resveratrol inhibits the TRIF-dependent pathway through the upregulation of SARM after RSV infection.
MATERIALS AND METHODS
Cell lines and cell culture conditions.
The 9HTEo cell line was provided gratis by Hans D. Ochs (University Washington School of Medicine, Seattle, WA, USA). 9HTEo cells and HEp2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum (FBS; GIBCO), 100 U/ml penicillin (Invitrogen, Carlsbad, CA), and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA) at 37°C under 5% CO2.
Virus preparation and titration.
The RSV viral stock (A2 strain) was obtained from the viral laboratory at Beijing Children's Hospital (Capital University of Medical Sciences, Beijing, China) and was grown in HEp2 cells as described elsewhere (31). RSV A2 was treated with UV light irradiation at 9 × 105 μJ/cm2 for 30 min using a UV cross-linker (Thermo Scientific, USA). Titers of viable virus were determined by plaque assay (32). A master stock and working stock of RSV were prepared as described previously (33).
Infection of 9HTEo cells.
An overnight culture of 9HTEo cells in a 6-well plate was infected with RSV at a multiplicity of infection (MOI) of 10 for 2 h. To remove extracellular RSV, the cells were washed twice with 1 ml of PBS. The infection was allowed to continue for 12, 24, 36, 48, or 72 h at 37°C under 5% CO2. Cells were collected for analysis at each time point.
Animals.
Six- to 8-week-old female BALB/c mice, free of specific pathogens, were purchased from Chongqing Medical University Animal Laboratory and housed in individual filtered cages. Cages, bedding, food, and water were sterilized before use. Room temperature was maintained at 23°C, and we provided a 12-h on/12-h off light cycle. All animal handling procedures were performed under clean-bench policy conditions. This study was performed in strict accordance with the recommendations in the guide for the care and use of laboratory animals of Chongqing Medical University. The protocol was approved by the Committee on the Ethics of Animal Experiments of Chongqing Medical University [permit number SYXK-(YU) 2012-0001]. All surgery was performed under sodium pentobarbital anesthesia, and every effort was made to minimize suffering. Experiments were performed three times using three mice per group unless otherwise noted.
Experimental design and sample collection.
Mice were infected intranasally with 4.5 × 107 PFU of RSV in a 100-μl volume. Mock-infected mice were inoculated intranasally with the same amount of HEp-2 cell culture supernatant in parallel. One hour postinoculation, mice were injected intraperitoneally with either resveratrol (Sigma-Aldrich Corp., St. Louis, MO) or placebo (phosphate-buffered saline [PBS]) as previously described (27). The lung function of the mice was measured at serial time points after infection (days 3, 5, and 7) prior to sacrificing the animal. The lungs were removed at each time point for detection of virus, protein extraction, and histopathological analysis. BALF was collected at 5 days after infection to determine the total number of cells, cell phenotypes, and cytokine expression.
Western blot analysis.
Total protein extracts from lung tissues or cells were obtained using a total protein extraction kit (KeyGEN, Nanjing, China). The protein concentration was determined using bicinchoninic acid assay reagent (Bioteke) according to the manufacturer's protocol. Equal amounts of the isolated proteins from lung or cell extracts were separated on an 8% SDS-PAGE gel and then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). The membranes were probed with primary antibodies against SARM (1:500; Santa Cruz Biotechnology, USA), TRIF (1:500; Abcam, Cambridge, MA), or β-actin (1:5,000; cwbiotech, Beijing, China). Alkaline phosphatase-conjugated goat anti-rabbit secondary antibody (1:10,000; MultiSciences, China) and goat anti-mouse antibody (1:10,000; MultiSciences, China) were used to detect the presence of the respective protein bands. Signals were quantified by use of Quantity One software (Bio-Rad, Hercules, CA) and normalized relative to β-actin.
Total differential cell count in BALF.
To obtain the BALF, the trachea of each mouse was cannulated to access the lungs. The lungs were washed three times with 0.5 ml ice-cold PBS. For total cell counts, BALF cells were pelleted by centrifugation at 1,000 × g for 5 min at 4°C. The cells were resuspended in 1 ml of PBS and counted with a hemocytometer. The BALF was centrifuged, and supernatants were collected and stored at −80°C to test for cytokine production. The cells were pelleted to cytospin slides, air dried, and used to quantify lymphocytes in the BALF. Cytospin slides were fixed and stained with DiffQuik (Baxter Healthcare Corp., Miami, FL) for leukocyte differential analysis. The number of monocytes, lymphocytes, neutrophils, and eosinophils in at least 200 cells per slide were counted. These experiments were performed three times with three mice per group.
Lung histopathology.
The lung tissue was fixed in 10% (vol/vol) neutral buffered formalin for 24 h and then embedded in paraffin. The blocks were cut into 5 μm-thick sections and stained with hematoxylin and eosin (H&E) solution (hematoxylin, MHS-16; eosin, HT110-1-32; both from Sigma). Tissue was subsequently mounted and coverslipped using Dako mounting medium (Dakocytomation, Denmark, CA). The degree of airway inflammatory cell infiltration was scored in double-blind screening by two independent investigators as described previously (34).
Pulmonary function tests.
Five days post-RSV infection, airway hyperresponsiveness (AHR) was assessed in conscious and unrestrained mice by means of whole-body plethysmography (Emca instrument; France). Each mouse was placed in a plastic chamber and exposed to aerosolized PBS, which was followed by increasing concentrations of aerosolized methacholine solutions (3.125, 6.25, 12.5, 25, and 50 mg/ml; Sigma, USA) in PBS for 3 min per exposure. Bronchoconstriction was recorded for 5 min after each dose of methacholine. The highest Penh value (airway resistance) obtained during each methacholine challenge was expressed as a proportion of the basal Penh value seen in response to PBS challenge.
Preparation and administration of in vivo siRNA.
SARM short interfering RNA (siRNA) and negative-control siRNA tagged internally with green fluorescent protein (GFP) were purchased from Invitrogen (Shanghai, China). The siRNA sequences were the following: SARM:237868(3-1) sense, TGCTGTGAAGAAGCGGCACAGTTTGTGTTTTGGCCACTGACTGACACAAACTGCCGCTTCTTCA; antisense, CCTGTGAAGAAGCGGCAGTTTGTGTCAGTCAGTGGCCAAAACACAAACTGTGCCGCTTCTTCAC; 237868 (negative control) sense, tgctgAAATGTACTGCGCGTGGAGACGTTTTGGCCACTGACTGACGTCTCCACGCAGTACATTT; antisense, cctgAAATGTACTGCGTGGAGACGTCAGTCAGTGGCCAAAACGTCTCCACGCGCAGTACATTTc.
The siRNA was dissolved in a solution of 5% glucose and in vivo jetPEI (Polyplus Transfection, New York, NY, USA) to an N/P ratio of 7 (number of nitrogen residues of jetPEI per molecule of RNA phosphate) according to the manufacturer's instructions. A total of 80 μl of siRNA-jetPEI complex was administered intranasally to RSV-infected and resveratrol-treated mice as previously described (35). The siRNA knockdown experiments were performed at least three times with five mice.
Confocal analysis.
Lungs from wild-type control or transgenic mice expressing GFP fluorescence were freshly excised and frozen sectioned. Airway sections were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Beyotime, China) for nuclear staining. Confocal images were acquired using a confocal microscope (A1R; Nikon, Japan). These experiments were performed three times with five mice.
Cytokine levels in BALF measured by ELISA.
The level of IFN-γ contained in BALF was measured using a specific mouse IFN-γ ELISA kit (Sizhengbai, Beijing, China). ELISAs were performed per the manufacturer's specifications.
Quantitative PCR (qPCR).
Lungs were harvested 5 days post-RSV infection. The RSVA N gene-specific primers and probe were the following: RSVA-F, 5′-AGATCAACTTCTGTCATCCAGCAA-3′; RSVA-R, 5′-TTCTGCACATCATAATTAGGAGTATCAAT-3′; RSVA-P, 6-carboxyfluorescein-5′-CACCATCCAACGGAGCACAGGAGAT-3′-black hole quencher 1 (36). The plasmid-amplified target fragment was cloned into the pMD19-T vector (TaKaRa Biotechnology, Dalian, China) and verified by sequencing. The real-time PCR instrument (Applied Biosystems) used the following conditions: one cycle at 50°C for 2 min, one cycle at 95°C for 10 min, 40 cycles at 95°C for 15 s, and one cycle at 60°C for 1 min. RSV load values were expressed as log10 copy numbers of RSV RNA/ml. RSV subtype A plasmid was the positive control. Negative controls and serial dilutions of positive controls were included in every PCR assay.
Statistical analysis.
Statistical analyses were performed with a two-way analysis of variance (ANOVA) or a Student t test between all groups using Prism GraphPad software (La Jolla, CA). P < 0.05 was considered significant in the present experiments.
RESULTS
Live RSV but not UV-inactivated RSV reduces SARM expression.
To investigate SARM expression in 9HTEo cells at the protein level, we infected 9HTEo cells with RSV at an MOI of 10. After 12 h, 24 h, 36 h, 48 h, or 72 h of infection, the cells were harvested and the expression of SARM protein was determined by immunoblotting. RSV was able to suppress SARM expression within 36 h of infection. SARM expression levels continued to gradually decrease with time (Fig. 1A1 and A2). In contrast, TRIF expression was increased in a time-dependent manner (Fig. 1A1 and A3).
FIG 1.

RSV reduced SARM expression in vivo and in vitro. (A) Cells were infected with RSV at an MOI of 10. (A1) SARM and TRIF protein levels were detected by immunoblotting at the time points indicated. (A2) RSV reduced the SARM/β-actin ratio compared to the control group. *, P < 0.05 48 h and 72 h after RSV infection versus the control. (A3) RSV infection increased the TRIF/β-actin ratio compared to that of the control group. **, P < 0.01 48 h after RSV infection versus the control. *, P < 0.05 72 h after RSV infection versus the control. (B) BALB/c mice were infected by live RSV at 4.5 × 107 PFU. The lungs of the infected mice were harvested at days 3, 5, and 7 after infection. (B1) SARM and TRIF protein levels were detected by immunoblotting at the time points indicated. (B2) RSV reduced the SARM/β-actin ratio compared to that of the control group. ***, P < 0.001 5 and 7 days after RSV infection versus the control. (B3) RSV infection increased the TRIF/β-actin ratio compared to that of the control group. **, P < 0.01 5 and 7 days after RSV infection versus the control. (C) BALB/c mice were infected by UV-inactivated RSV. (C1 and C2) Days 3, 5, and 7 after infection, the lungs of the infected mice were harvested and the protein levels were detected by immunoblotting. (D) BALB/c mice were infected with live RSV (4.5 × 107 PFU) and then treated intraperitoneally with either resveratrol (RES) or placebo. (D1) The lungs were harvested, and the protein expression of TRIF and SARM was determined by immunoblotting. (D2) RSV reduced the SARM/β-actin ratio compared to that of the control group. ***, P < 0.001 5 days after RSV infection versus the control. The SARM/β-actin ratio was elevated in RES-treated mice compared to untreated mice. **, P < 0.01 for RES-treated mice versus untreated mice infected with RSV. (D3) RSV infection increased the TRIF/β-actin ratio compared to that of the control. **, P < 0.01 5 days after RSV infection versus the control. The TRIF/β-actin ratio was reduced in RES-treated mice compared to that of untreated mice. **, P < 0.01 for RES-treated mice versus untreated mice. All experiments were performed three times with three mice per group.
To determine whether RSV inhibited SARM expression in vivo, BALB/c mice were infected intranasally with live RSV or UV-inactivated RSV. Plaque assay confirmed that the mice were infected by the RSV challenge. Similar to results in the 9HTEo cell line, RSV reduced SARM expression 3 days after infection (Fig. 1B1 and B2). The levels of SARM remained low, but the levels of TRIF increased at day 5 and day 7 postinfection (Fig. 1B1 and B3). Unlike live virus, UV-inactivated RSV was not able to reduce SARM expression in a time-dependent manner in vivo (Fig. 1C1 and C2).
SARM is a functional inhibitor of RSV-mediated activation of TRIF-dependent pathway induced by resveratrol.
Resveratrol and SARM are inhibitors of TRIF- and MyD88-independent pathways (21). To investigate the interplay between resveratrol and SARM expression after RSV infection, BALB/c mice were treated with resveratrol for 1 h post-RSV infection. RSV increased TRIF expression by Western blotting, while at the same time infection reduced SARM expression. In contrast, in mice treated with resveratrol after RSV infection, TRIF expression was inhibited and SARM expression increased. These results indicated that resveratrol-mediated inhibition of the TRIF-dependent pathway relies on SARM expression (Fig. 1D1 to D3).
Resveratrol reduces airway inflammation and AHR by upregulating SARM expression.
We have previously established that IFN-γ causes severe airway inflammation in a model of RSV infection in immunocompromised mice (27). To understand whether the airway inflammation we observed was SARM mediated, mice were treated with cyclophosphamide (CYP) as described previously (37). Briefly, CYP was administered in a single dose of 100 mg/kg of body weight, and 5 days later, mice were intranasally infected with RSV. SARM expression was elevated in the lungs of BALB/c mice treated with resveratrol after RSV infection compared to infection with RSV alone. siRNA was used to knock down the SARM mRNA level and reduce the protein expression level in resveratrol-treated mice. To test whether transgenic SARM siRNA-GFP was able to function in lungs of treated mice, confocal microscopy was used to confirm the proper insertion and function of siRNA-GFP (Fig. 2A). The SARM siRNA construct effectively suppressed the level of SARM protein expression in the lungs of RSV-infected BALB/c mice treated with resveratrol, and the negative-control siRNA vector had no effect. However, following SARM knockdown, TRIF expression was increased and the TRIF-dependent pathway was induced (Fig. 2B1 to B3).
FIG 2.
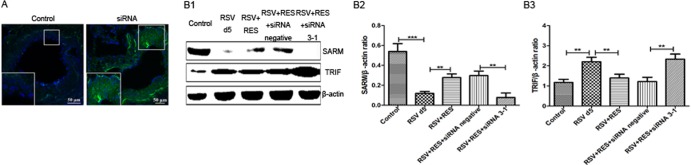
SARM siRNA 3-1-GFP or SARM siRNA-negative-control-GFP organization and protein expression. (A) The SARM siRNA-GFP signal in freshly harvested lungs demonstrates polymer formation by the SARM siRNA transgene, i.e., GFP in lungs of transgenic mice, compared to the wild-type control. Images show cross-sections of mouse lung proximal and distal airway sections from transgenic mice or wild-type control mice stained with DAPI as a nuclear stain. Corresponding differential interference contrast/DAPI images are shown. Wild-type images are shown as a control. (B1) Western blotting was used to evaluate the SARM and TRIF expression in wild-type control mice, RSV-infected mice, and RSV-infected mice treated with resveratrol (RES) (lanes 1 to 3). RSV-infected mice treated with RES were either transfected with SARM siRNA negative control (lane 4) or SARM siRNA 3-1 (lane 5). (B2) SARM siRNA reduced the SARM/β-actin ratio in the lungs of RES-treated mice. **, P < 0.01 for RES-treated, SARM siRNA 3-1-transfected mice versus RES-treated, siRNA negative-control-transfected mice. (B3) SARM siRNA increased the TRIF/β-actin ratio in the lungs of RES-treated mice. **, P < 0.01 for RES-treated, SARM siRNA 3-1-transfected mice versus RES-treated, siRNA negative-control-transfected mice. These experiments were performed three times with three mice per group.
Mice infected with RSV had severe airway inflammation compared to uninfected control mice. However, treatment with resveratrol reduced inflammation in mice infected with RSV. The effect of resveratrol was reduced when SARM expression was knocked down using siRNA. Resveratrol-treated mice with SARM knocked down had histologic findings similar to those for mice inoculated with RSV in the absence of resveratrol (Fig. 3A1 to A2).
FIG 3.

SARM knockdown increased inflammation in RSV-infected lungs. (A1) Histological examination of lung tissues was performed 5 days postresveratrol treatment. The lung tissues were fixed and stained with H&E (magnification, ×100). (A2) Lung tissue inflammatory cell infiltration scores. ***, P < 0.001 for resveratrol (RES)-treated SARM siRNA 3-1-transfected mice versus RES-treated siRNA negative-control-transfected mice. (B) Cells were isolated by cytospin and stained with DiffQuik. Cells were counted using a hemocytometer. Values are expressed as the means; error bars are standard deviations (SD) (n = 3/group). Total numbers of cells were present in the BALF of the respective treatment groups. ***, P < 0.001 for RSV-infected versus RES-treated mice and RES-treated SARM siRNA 3-1-transfected mice versus RES-treated siRNA negative-control-transfected mice. Differences were found for lymphocytes between treatment groups. **, P < 0.01 for untreated versus RES-treated RSV-infected mice and untreated SARM siRNA 3-1-transfected mice versus RES-treated siRNA negative-control-transfected mice. (C) AHR was measured 5 days postresveratrol treatment and 1 day post-siRNA transfection in mice treated with increasing methacholine concentrations (3.125 to 50.0 mg/ml) by plethlysmography. Values are expressed as means ± SD (n = 5/group). ***, P < 0.001 for RES- and SARM siRNA 3-1-treated mice versus RES- and siRNA negative-control-transfected mice. **, P < 0.01 for untreated mice versus RES-treated mice infected with RSV. (D) IFN-γ was measured by ELISA from BALF harvested 5 days post-RSV infection (n = 8/group). IFN-γ levels in BALF are shown. Values are expressed as means ± SD (n = 8/group). *, P < 0.05 for untreated mice versus RES-treated mice. ***, P < 0.001 for RES-treated SARM siRNA 3-1-transfected mice versus RES-treated siRNA negative-control-transfected mice. (E) Viral titer was measured by qPCR. RSV-infected lungs were harvested 5 days post-RES treatment and 1 day post-SARM siRNA transfection. Values are expressed as means ± SD (n = 5/group). There was no significant difference between untreated mice and RES-treated SARM siRNA 3-1-transfected mice.
RSV-infected mice had significantly more cells in the BALF than uninfected mice. However, treatment with resveratrol reduced the cellularity of the BALF (Fig. 3B). SARM knockdown was sufficient to significantly increase the total number of cells present in the BALF of resveratrol-treated mice compared to the negative-control siRNA-treated group. There were qualitative differences in cell types observed between treatment groups. The RSV-infected group treated with resveratrol and siRNA 3-1-treated had significantly increased lymphocyte numbers (P < 0.01) compared to RSV-infected mice that were treated with resveratrol and siRNA negative control (Fig. 3B).
Aerosolized methacholine elicited significantly increased AHR in mice infected with live RSV compared to all other groups. Five days after inoculation, mice infected with live RSV had significantly greater AHR than uninfected controls at methacholine concentrations between 12.5 and 50.0 mg/ml. Treatment with resveratrol significantly reduced AHR caused by RSV. However, in mice with SARM knocked down after transfection, AHR was significantly higher than in mice transfected with the negative-control siRNA vector and mice treated with resveratrol after RSV infection (Fig. 3C).
Depletion of SARM may cause more severe airway inflammation. To investigate a possible mechanism of action, the IFN-γ expression level in the BALF was assessed using ELISA-based assays. RSV infection significantly increased the IFN-γ level in BALF compared to the uninfected control mice (Fig. 3D). Resveratrol treatment showed some protection and significantly reduced the level of IFN-γ compared to the RSV group. However, SARM knockdown abrogated the protective effect of resveratrol treatment (Fig. 3D). The level of IFN-γ was significantly increased in the SARM knockdown group compared to the negative-control siRNA vector group (Fig. 3D). RSV-infected mice and RSV-infected mice treated with RES and transfected with SARM siRNA 3-1 had a similar RSV viral titer (Fig. 3E).
DISCUSSION
In the present study, we discovered that SARM expression was reduced after infection with live, but not inactivated, RSV in vivo and in vitro. Furthermore, we found that resveratrol was able to prevent the RSV-mediated reduction of SARM expression levels. Resveratrol was also able to decrease the airway inflammation and AHR caused by RSV. The anti-inflammatory function of resveratrol involved TLR-TRIF-associated signaling.
TLRs are expected to play an essential role in innate immune activity against RSV infection. The activation of cytokines, chemokines, and IFNs in the host after binding viral PAMPs leads to an antiviral state and activates the adaptive immune response. Upon RSV infection, the signal initiated from TLR3 and TLR4 activates the adaptor protein TRIF (7, 27, 30). Specifically, the RSV fusion protein can bind to TLR4, which induces the production of interleukin-1β (IL-1β), IL-6, IL-8, and tumor necrosis factor alpha (TNF-α) (38). RSV is a negative-strand, nonsegmented RNA pneumovirus. Double-stranded RNA replication intermediates formed during the RSV replication cycle activate TLR3 (39) and the RIG-like helicases RIG-I (40) and MDA-5 (41). Our previous studies showed that RSV replication activates TLR3 and activates a TRIF-dependent signaling pathway (27, 42). Some reports have demonstrated that mice deficient in TLR4 showed an impaired ability to clear the virus (43). In contrast, Douville et al. showed that responses to RSV infection are not dependent upon TLR4-mediated stimulation and are associated with substantial innate immunity and robust Th1 activation (44). Moreover, SARM is an inhibitor of TRIF-dependent signaling (13). Peng et al. showed that SARM inhibits both TRIF- and MyD88-mediated AP-1 activation (14), indicating that SARM is a negative regulator in TLR-mediated innate immunity. The results presented here suggest that RSV suppressed SARM expression in vivo and in vitro. UV-inactivated RSV, which cannot replicate in the host, was not able to reduce the expression of SARM in vivo. This suggests that active viral replication is necessary to reduce SARM expression.
Resveratrol significantly reduces influenza virus replication by inhibiting protein kinase C (PKC) phosphorylation and its dependent pathways, JNK and p38 mitogen-activated protein kinase (MAPK) (45). Resveratrol also reduces the inflammation and AHR caused by enhanced IFN-γ after RSV infection (27). However, the mechanism underlying the link between resveratrol and IFN-γ remains unclear. Resveratrol exerts its broad-spectrum anti-inflammatory effects through inhibition of the TRIF/TBK1/IRF-3 complex (22). Furthermore, SARM acts as a specific inhibitor of TRIF-dependent signaling (13). In this study, resveratrol acted as a promoter of SARM; thus, it inhibited TRIF expression and reduced airway inflammation and AHR after RSV infection. Furthermore, SARM knockdown reversed the protective effects of resveratrol when SARM siRNA was transfected in the lungs of BALB/c mice.
The Caenorhabditis elegans SARM homologue TIR-1 plays a crucial role for efficient immune responses against bacterial infections (12). SARM has been shown to restrict West Nile virus infection and influence TNF-α production, microglia activation, and neuronal cell death (46). RSV infection activates TLRs and induces the production of inflammatory cytokines that direct the differentiation of naive Th0 cells to Th1 or Th2-type CD4+ T-helper cells (47). This gives rise to Th1-type responses (48). Importantly, TRIF-dependent signaling is associated with IFN-γ production (49). Recent reports suggest that IFN-γ produced by Th1 cells is required in the induction of severe AHR (30). During RSV infection, TRIF-dependent signaling was induced and the level of IFN-γ was elevated (27). Our results show that SARM interacts with TRIF and that once SARM expression is induced by resveratrol, the level of IFN-γ is reduced, as is airway inflammation and AHR. On the contrary, SARM knockdown established an uncontrolled immune response against RSV in which TRIF expression was enhanced. It is important that a robust Th1 response was induced and the IFN-γ level was elevated, which subsequently caused an exacerbation of airway inflammation and AHR.
In summary, these results suggest that resveratrol-mediated alterations in SARM have therapeutic potential against RSV immunopathology caused by deregulation of the TLR-mediated immune response.
ACKNOWLEDGMENTS
We thank the Experimental Animal Center at Chongqing Medical University for providing the BALB/c mice.
This work was supported by the second Colleges and Universities Excellent Talents Program in Chongqing (2011.1-2012.12), the National Natural Science Foundation of China (81170010), and the National Natural Science Foundation of China for Young Scholars (31100125).
Footnotes
Published ahead of print 29 January 2014
REFERENCES
- 1.Hall C. 1994. Prospects for a respiratory syncytial virus vaccine. Science 265:1393–1394. 10.1126/science.7915433 [DOI] [PubMed] [Google Scholar]
- 2.Tregoning JS, Schwarze J. 2010. Respiratory viral infections in infants: causes, clinical symptoms, virology, and immunology. Clin. Microbiol. Rev. 23:74–98. 10.1128/CMR.00032-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall CB, Powell KR, MacDonald NE, Gala CL, Menegus ME, Suffin SC, Cohen HJ. 1986. Respiratory syncytial viral infection in children with compromised immune function. N. Engl. J. Med. 315:77–81. 10.1056/NEJM198607103150201 [DOI] [PubMed] [Google Scholar]
- 4.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 352:1749–1759. 10.1056/NEJMoa043951 [DOI] [PubMed] [Google Scholar]
- 5.Littel-van den Hurk SVD, Mapletoft JW, Arsic N, Kovacs-Nolan J. 2007. Immunopathology of RSV infection: prospects for developing vaccines without this complication. Rev. Med. Virol. 17:5–34. 10.1002/rmv.518 [DOI] [PubMed] [Google Scholar]
- 6.Athman R, Philpott D. 2004. Innate immunity via Toll-like receptors and Nod proteins. Curr. Opin. Microbiol. 7:25–32. 10.1016/j.mib.2003.12.013 [DOI] [PubMed] [Google Scholar]
- 7.Haeberle HA, Takizawa R, Casola A, Brasier AR, Dieterich H-J, van Rooijen N, Gatalica Z, Garofalo RP. 2002. Respiratory syncytial virus-induced activation of nuclear factor-κB in the lung involves alveolar macrophages and Toll-like receptor 4-dependent pathways. J. Infect. Dis. 186:1199–1206. 10.1086/344644 [DOI] [PubMed] [Google Scholar]
- 8.Ubol S, Halstead SB. 2010. How innate immune mechanisms contribute to antibody-enhanced viral infections. Clin. Vaccine Immunol. 17:1829–1835. 10.1128/CVI.00316-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Neill LAJ, Fitzgerald KA, Bowie AG. 2003. The Toll–IL-1 receptor adaptor family grows to five members. Trends Immunol. 24:286–289. 10.1016/S1471-4906(03)00115-7 [DOI] [PubMed] [Google Scholar]
- 10.Couillault C, Pujol N, Reboul J, Sabatier L, Guichou J-F, Kohara Y, Ewbank JJ. 2004. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat. Immunol. 5:488–494. 10.1038/ni1060 [DOI] [PubMed] [Google Scholar]
- 11.O'Neill LAJ, Bowie AG. 2007. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7:353–364. 10.1038/nri2079 [DOI] [PubMed] [Google Scholar]
- 12.Belinda LW-C, Wei WX, Hanh BTH, Lei LX, Bow H, Ling DJ. 2008. SARM: a novel Toll-like receptor adaptor, is functionally conserved from arthropod to human. Mol. Immunol. 45:1732–1742. 10.1016/j.molimm.2007.09.030 [DOI] [PubMed] [Google Scholar]
- 13.Carty M, Goodbody R, Schröder M, Stack J, Moynagh PN, Bowie AG. 2006. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol. 7:1074–1081. 10.1038/ni1382 [DOI] [PubMed] [Google Scholar]
- 14.Peng J, Yuan Q, Lin B, Panneerselvam P, Wang X, Luan XL, Lim SK, Leung BP, Ho B, Ding JL. 2010. SARM inhibits both TRIF- and MyD88-mediated AP-1 activation. Eur. J. Immunol. 40:1738–1747. 10.1002/eji.200940034 [DOI] [PubMed] [Google Scholar]
- 15.Yu XM, Luo L. 2012. Neuroscience. dSarm-ing axon degeneration. Science 337:418–419. 10.1126/science.1226150 [DOI] [PubMed] [Google Scholar]
- 16.Chen CY, Lin CW, Chang CY, Jiang ST, Hsueh YP. 2011. Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J. Cell Biol. 193:769–784. 10.1083/jcb.201008050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Conforti L, Coleman M, Tessier-Lavigne M, Zuchner S, Freeman MR. 2012. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337:481–484. 10.1126/science.1223899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panneerselvam P, Singh Laishram P, Ho B, Chen J, Ding Jeak L. 2012. Targeting of pro-apoptotic TLR adaptor SARM to mitochondria: definition of the critical region and residues in the signal sequence. Biochem. J. 442:263–271. 10.1042/BJ20111653 [DOI] [PubMed] [Google Scholar]
- 19.Panneerselvam P, Singh LP, Selvarajan V, Chng WJ, Ng SB, Tan NS, Ho B, Chen J, Ding JL. 2013. T-cell death following immune activation is mediated by mitochondria-localized SARM. Cell Death Differ. 20:478–489. 10.1038/cdd.2012.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bråkenhielm E, Cao R, Cao Y. 2001. Suppression of angiogenesis, tumor growth, and wound healing by resveratrol, a natural compound in red wine and grapes. FASEB J. 15:1798–1800. 10.1096/fj.01-0028fje [DOI] [PubMed] [Google Scholar]
- 21.Youn HS, Lee JY, Fitzgerald KA, Young HA, Akira S, Hwang DH. 2005. Specific inhibition of MyD88-independent signaling pathways of TLR3 and TLR4 by resveratrol molecular targets are TBK1 and RIP1 in TRIF complex. J. Immunol. 175:3339–3346 [DOI] [PubMed] [Google Scholar]
- 22.Kim MH, Yoo DS, Lee SY, Byeon SE, Lee YG, Min T, Rho HS, Rhee MH, Lee J, Cho JY. 2011. The TRIF/TBK1/IRF-3 activation pathway is the primary inhibitory target of resveratrol, contributing to its broad-spectrum anti-inflammatory effects. Pharmazie 66:293–300. 10.1691/ph.2011.0798 [DOI] [PubMed] [Google Scholar]
- 23.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CWW, Fong HHS, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. 1997. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275:218–220. 10.1126/science.275.5297.218 [DOI] [PubMed] [Google Scholar]
- 24.Bradamante S, Barenghi L, Villa A. 2004. Cardiovascular protective effects of resveratrol. Cardiovasc. Drug Rev. 22:169–188. 10.1111/j.1527-3466.2004.tb00139.x [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Xu J, Rottinghaus GE, Simonyi A, Lubahn D, Sun GY, Sun AY. 2002. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 958:439–447. 10.1016/S0006-8993(02)03543-6 [DOI] [PubMed] [Google Scholar]
- 26.Xie X-H, Law HKW, Wang L-J, Li X, Yang X-Q, Liu E-M. 2009. Lipopolysaccharide induces IL-6 production in respiratory syncytial virus-infected airway epithelial cells through the Toll-like receptor 4 signaling pathway. Pediatr. Res. 65:156–162. 10.1203/PDR.0b013e318191f5c6 [DOI] [PubMed] [Google Scholar]
- 27.Zang N, Xie X, Deng Y, Wu S, Wang L, Peng C, Li S, Ni K, Luo Y, Liu E. 2011. Resveratrol-mediated gamma interferon reduction prevents airway inflammation and airway hyperresponsiveness in respiratory syncytial virus-infected immunocompromised mice. J. Virol. 85:13061–13068. 10.1128/JVI.05869-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Royce SG, Dang W, Yuan G, Tran J, El Osta A, Karagiannis TC, Tang MLK. 2011. Resveratrol has protective effects against airway remodeling and airway hyperreactivity in a murine model of allergic airways disease. Pathobiol. Aging Age Relat. Dis. 2011:1. 10.3402/PBA.v1i0.7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung EY, Kim BH, Hong J-T, Lee C-K, Ahn B, Nam S-Y, Han S-B, Kim Y. 2011. Resveratrol down-regulates interferon-γ-inducible inflammatory genes in macrophages: molecular mechanism via decreased STAT-1 activation. J. Nutr. Biochem. 22:902–909. 10.1016/j.jnutbio.2010.07.012 [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi M, Ashino S, Shiohama Y, Wakita D, Kitamura H, Nishimura T. 2012. IFN-γ elevates airway hyper-responsiveness via up-regulation of neurokinin A/neurokinin-2 receptor signaling in a severe asthma model. Eur. J. Immunol. 42:393–402. 10.1002/eji.201141845 [DOI] [PubMed] [Google Scholar]
- 31.Jafri HS, Chávez-Bueno S, Mejías A, Gómez AM, Ríos AM, Nassi SS, Yusuf M, Kapur P, Hardy RD, Hatfield J, Rogers BB, Krisher K, Ramilo O. 2004. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J. Infect. Dis. 189:1856–1865. 10.1086/386372 [DOI] [PubMed] [Google Scholar]
- 32.McKimm-Breschkin JL. 2004. A simplified plaque assay for respiratory syncytial virus–direct visualization of plaques without immunostaining. J. Virol. Methods 120:113–117. 10.1016/j.jviromet.2004.02.020 [DOI] [PubMed] [Google Scholar]
- 33.Graham BS, Bunton LA, Wright PF, Karzon DT. 1991. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J. Clin. Investig. 88:1026–1033. 10.1172/JCI115362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myou S, Leff AR, Myo S, Boetticher E, Tong J, Meliton AY, Liu J, Munoz NM, Zhu X. 2003. Blockade of inflammation and airway hyperresponsiveness in immune-sensitized mice by dominant-negative phosphoinositide 3-kinase–TAT. J. Exp. Med. 198:1573–1582. 10.1084/jem.20030298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogawa H, Azuma M, Uehara H, Takahashi T, Nishioka Y, Sone S, Izumi K. 2012. Nerve growth factor derived from bronchial epithelium after chronic mite antigen exposure contributes to airway hyperresponsiveness by inducing hyperinnervation, and is inhibited by in vivo siRNA. Clin. Exp. Allergy 42:460–470. 10.1111/j.1365-2222.2011.03918.x [DOI] [PubMed] [Google Scholar]
- 36.van Woensel JB, Lutter R, Biezeveld M, Dekker T, Nijhuis M, Van Aalderen WM, Kuijpers TW. 2003. Effect of dexamethasone on tracheal viral load and interleukin-8 tracheal concentration in children with respiratory syncytial virus infection. Pediatr. Infect. Dis. J. 22:721–726. 10.1097/01.inf.0000078165.62923.15 [DOI] [PubMed] [Google Scholar]
- 37.Kong X, Hellermann GR, Patton G, Kumar M, Behera A, Randall TS, Zhang J, Lockey RF, Mohapatra SS. 2005. An immunocompromised BALB/c mouse model for respiratory syncytial virus infection. Virol. J. 2:3. 10.1186/1743-422X-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1:398–401. 10.1038/80833 [DOI] [PubMed] [Google Scholar]
- 39.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413:732–738. 10.1038/35099560 [DOI] [PubMed] [Google Scholar]
- 40.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737. 10.1038/ni1087 [DOI] [PubMed] [Google Scholar]
- 41.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. 10.1038/nature04734 [DOI] [PubMed] [Google Scholar]
- 42.Xie X-H, Zang N, Li S-M, Wang L-J, Deng Y, He Y, Yang X-Q, Liu E-M. 2012. Resveratrol inhibits respiratory syncytial virus-induced IL-6 production, decreases viral replication, and downregulates TRIF expression in airway epithelial cells. Inflammation 35:1392–1401. 10.1007/s10753-012-9452-7 [DOI] [PubMed] [Google Scholar]
- 43.Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. 2001. Involvement of Toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 75:10730–10737. 10.1128/JVI.75.22.10730-10737.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Douville RN, Bastien N, Li Y, Simons FER, HayGlass KT. 2007. Adult asthmatics display exaggerated IFNγ responses to human metapneumovirus and respiratory syncytial virus. Biochem. Cell Biol. 85:252–258. 10.1139/O07-005 [DOI] [PubMed] [Google Scholar]
- 45.Fioravanti R, Celestino I, Costi R, Cuzzucoli Crucitti G, Pescatori L, Mattiello L, Novellino E, Checconi P, Palamara AT, Nencioni L, Santo RD. 2012. Effects of polyphenol compounds on influenza A virus replication and definition of their mechanism of action. Bioorg. Med. Chem. 20:5046–5052. 10.1016/j.bmc.2012.05.062 [DOI] [PubMed] [Google Scholar]
- 46.Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS. 2009. The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile virus pathogenesis. J. Virol. 83:9329–9338. 10.1128/JVI.00836-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Netea MG, Van der Meer JWM, Sutmuller RP, Adema GJ, Kullberg B-J. 2005. From the Th1/Th2 paradigm towards a Toll-like receptor/T-helper bias. Antimicrob. Agents Chemother. 49:3991–3996. 10.1128/AAC.49.10.3991-3996.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker Y. 2006. Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy: a review. Virus Genes 33:235–252. 10.1007/s11262-006-0064-x [DOI] [PubMed] [Google Scholar]
- 49.Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R, Yanai H, Seko Y, Shitara H, Bishop K, Yonekawa H, Tamura T, Kaisho T, Taya C, Taniguchi T, Honda K. 2008. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. U. S. A. 105:20446–20451. 10.1073/pnas.0810372105 [DOI] [PMC free article] [PubMed] [Google Scholar]