

ABSTRACT
Human papillomavirus (HPV) can successfully evade the host immune response to establish a persistent infection. We show here that expression of the E7 oncoprotein in primary human keratinocytes results in increased production of interleukin-18 (IL-18) binding protein (IL-18BP). This anti-inflammatory cytokine binding protein is a natural antagonist of IL-18 and is necessary for skin homeostasis. We map increased IL-18BP production to the CR3 region of E7 and demonstrate that this ability is shared among E7 proteins from different HPV types. Furthermore, mutagenesis shows that increased IL-18BP production is mediated by a gamma-activated sequence (GAS) in the IL-18BP promoter. Importantly, the increased IL-18BP levels seen in E7-expressing keratinocytes are capable of diminishing IL-18-mediated CD4 lymphocyte activation. This study provides the first evidence for a virus protein that targets IL-18BP and further validates E7 as a key component of the HPV immune evasion armor.
IMPORTANCE Infection with human papillomavirus is a leading cause of morbidity and mortality worldwide. This study demonstrates that the E7 protein increases production of the anti-inflammatory IL-18BP, a major regulator of epithelial homeostasis. A number of E7 proteins can increase IL-18BP production, and a region within the CR3 of E7 is necessary for mediating the increase. A consequence of increased IL-18BP production is a reduction in CD4-positive lymphocyte activation in response to IL-18 costimulation. These findings may shed light on the immune evasion abilities of HPV.
INTRODUCTION
Human papillomaviruses (HPVs) are small double-stranded DNA (dsDNA) viruses that infect squamous epithelial cells and produce a range of clinical lesions ranging from common warts to cancers of the anogenital tract and oropharynx. More than 100 types have been identified (1) and are classified as either low- or high-risk types, depending on the associated risk of cancer development. High-risk HPVs are responsible for the majority of cervical cancers and a subset of head and neck squamous cell carcinomas (2), and among these, HPV16 is detected in approximately 60% of all cervical cancers worldwide (3). Carcinogenesis is linked to persistent infection with a high-risk HPV type that may last several years. This suggests that HPVs have evolved mechanisms to evade the immune system.
Three virus-encoded proteins, E5, E6, and E7, have been shown to impact the host's ability to mount an effective immune response. Although little is known about the E5 protein, which is a minor oncoprotein, it has been correlated with decreased cell surface expression of major histocompatibility complex (MHC) molecules and reduced activation of the adaptive immune response (4, 5). A significant number of studies have demonstrated the myriad of interactions between the E6 and E7 oncoproteins and the host immune system. It has been shown that they cooperate to downregulate expression of a number of inflammatory cytokines, including interleukin-18 (IL-18) and IL-8, and the chemoattractants monocyte chemoattractant protein 1 (MCP-1) and MIP3α (6–9). Furthermore, they reduce the production of antiviral interferons by subverting the actions of interferon (IFN) regulatory factors (IRFs) (10, 11) and nuclear factor kappa B (NF-κB) (12, 13). More recently, they have been implicated in the reduced production of type III IFNs (14).
Keratinocytes are the targets for infection by HPVs, and they are capable of responding to a wide array of pathogens by secreting cytokines to attract and activate the innate and adaptive immune responses (15), in particular interleukin-18 (IL-18), which is a proinflammatory cytokine that plays a protective role against virus infection by regulating the secretion of gamma interferon (IFN-γ) from TH1 lymphocytes, enhancing the cytotoxicity of natural killer (NK) cells (16), and bolstering the antiviral capacity of keratinocytes (17, 18). The proinflammatory effects of IL-18 are counterbalanced by a natural antagonist, IL-18 binding protein (IL-18BP), which acts to suppress the deleterious effects of excessive IL-18 secretion in vivo (19). Given the critical nature of the IL-18–IL-18BP axis for regulating inflammatory responses of the skin, it is likely to be a critical factor governing the persistence of skin-tropic viruses. Indeed, poxviruses are known to encode functional IL-18BP homologues, which contribute to virus virulence by downregulating IL-18-mediated inflammatory responses (20). Despite the body of evidence regarding the importance of IL-18BP for regulating the epithelial inflammatory response, there is currently no published observation of direct virus modulation of endogenous IL-18BP.
This study shows that following stimulation with IFN-γ, primary human keratinocytes expressing E7 produce significantly higher levels of IL-18BP than control cells. Using a panel of established mutants, the CR3 region of E7 was found to be necessary for increased IL-18BP production. The result of the increased IL-18BP production was decreased CD4-positive lymphocyte activation. These results suggest that increased IL-18BP production may be a novel mechanism by which HPV could evade the immune response and persist in the epithelium.
MATERIALS AND METHODS
DNA manipulations.
The plasmid pBabe-Puro was used to transiently or stably express E6, E7, or E6/E7 products. Constructs containing E6, E7, or E6/E7 from HPV6, -11, or -16 were provided by Dennis McCance (Queen's University Belfast). The HPV18 E7 sequence was amplified from the full-length HPV18 genome (GenBank accession number NC_001357), provided by Sally Roberts (University of Birmingham), and ligated into pBabe-Puro using BamHI and EcoRI sites. HPV16 E7 point and truncation mutants were obtained from Lawrence Banks (ICGEB, Trieste, Italy). The IL-18BP luciferase reporter plasmids have been described previously (21) and were provided by Heiko Muhl (University of Frankfurt, Germany). DNA sequences of all inserts were verified (GATC).
Cell culture.
Primary human foreskin keratinocytes were obtained from Sally Roberts (University of Birmingham) and cultured as described previously (22). Cells were cultured in SFM keratinocyte growth medium (Invitrogen) containing 25 μg/ml bovine pituitary extract and 0.1 ng/ml epidermal growth factor (EGF). Cells were passaged for a maximum of 6 passages. NIH J2 3T3 fibroblasts (a gift from Sally Roberts) were cultured in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) and 2 mM l-glutamine.
Generation of stable keratinocyte lines containing the HPV18 genome.
The HPV18-pGEMII plasmid (Sally Roberts, University of Birmingham), containing the total HPV18 genome, was digested with EcoRI to release the genome, which was then recircularized with T4 ligase (NEB). The genome was cotransfected with a plasmid containing neomycin resistance into SFM keratinocyte growth medium. The cells were selected with 100 μg/ml G418 (Roche) E medium containing 5% (vol/vol) serum, 2 mM l-glutamine, and 5 nM EGF for 8 days. Cells were selected on a layer of J2 3T3 fibroblast feeders (NIH) treated with 8 μg/ml mitomycin C (Roche).
Retrovirus transduction.
Stocks of retrovirus for transduction of primary cells were produced by transfection of the Phoenix packaging cell line. Supernatants from transfected cells were added to target cells with 5 μg/ml Polybrene at 37°C. Virus was removed from cells after 6 h of incubation, and cells were fed fresh medium. After 48 h of recovery at 37°C, cells were placed under selection in 5 μg/ml puromycin.
Isolation of primary CD4 lymphocytes.
Primary cells were extracted and analyzed in accordance with the ethical rules of the University of Hannover Medical School (3362) and the University of Leeds, Faculty of Biological Sciences (BIOSCI09-001). To isolate peripheral blood mononuclear cells (PBMCs), 30 to 60 ml of peripheral blood was collected from healthy donors in sterile Vacutainer lithium heparin (BD Biosciences, Oxford, United Kingdom). Blood was kept at room temperature after donation and used immediately. PBMCs were isolated from peripheral whole blood using density gradients. Peripheral blood was diluted 1:1 with sterile phosphate-buffered saline (PBS) (Gibco, Life Technologies, United Kingdom). An equal amount of diluted blood was then carefully layered onto Ficoll-Paque Plus (GE Healthcare, Buckinghamshire, United Kingdom). This was then centrifuged for 40 min at 400 × g with no brake. PBMCs were then removed and diluted 1:1 with sterile PBS. Cells were then centrifuged at 350 × g for 4 min and supernatant removed. PBMCs were either used immediately or cultured in RPMI 1640 supplemented with 5% fetal calf serum (FCS) (PromoCell, Heidelberg, Germany), 0.5% l-glutamine (Sigma-Aldrich, United Kingdom), and 0.05 mg/ml streptomycin and 50 U/ml penicillin (Sigma-Aldrich, United Kingdom) at 37°C with 5% CO2.
PBMCs were centrifuged at 350 × g for 4 min. The supernatant was removed before washing with sterile PBS. Using the CD4+ T cell isolation kit II (Miltenyi Biotec, Surrey, United Kingdom), cells were then stained with magnetic labels as per the manufacturer's instructions. After magnetic labeling, the cells were then put through the LS columns (Miltenyi Biotec, Surrey, United Kingdom) as per the manufacturer's instructions.
CD4-positive lymphocyte activation assay.
Stable keratinocyte cell lines were stimulated with IFN-γ (10 ng/ml) and incubated for 8 h before the cells were washed thoroughly to remove input IFN-γ and replaced with fresh medium. Cells were incubated for a further 16 h before harvesting. Isolated CD4-positive lymphocytes were stimulated with activating anti-CD3/CD28 (150/200 ng/ml) antibodies and costimulated with IL-18 (20 ng/ml) in the presence of medium taken from keratinocyte cultures. CD4-positive lymphocytes were incubated for 24 h before medium was harvested and analyzed by enzyme-linked immunosorbent assay (ELISA) for secreted IFN-γ production. Keratinocyte medium was analyzed in parallel to confirm the absence of input IFN-γ.
RNA extraction and qRT-PCR.
RNA was extracted from keratinocytes using the Quick-RNA MiniPrep (Zymo Research, Cambridge Bioscience, Cambridge, United Kingdom) and cDNA generated with a cDNA synthesis kit (Fermentas). Reverse transcription-quantitative PCR (qRT-PCR) was performed on a RotorGen (Qiagen, Hilden, Germany) using a ΔΔCT-analysis based on the generation of standard curves for both the housekeeping gene (U6snRNA) and the target gene (IL-18BP) (QuantiTect primer assay; Qiagen) using QuantiFast SYBR green PCR (Qiagen).
Lestaurtinib inhibitor assay.
HPV18-containing keratinocytes grown on mitomycin C-treated J2 3T3 and primary keratinocytes were treated with 100 nM lestaurtinib (Tocris Biosciences) for 1 h at 37°C prior to stimulation with 10 ng/ml IFN-γ (BioLegend) for 24 h. Supernatants were collected for ELISA, and cells were lysed for protein analysis.
Determination of IL-18BP and IFN-γ levels by ELISA.
Quantities of cytokines in cell-free supernatants were determined using the R&D Systems ELISA kit according to manufacturer's instructions.
SDS-PAGE and immunoblotting.
Cells were lysed in Leeds lysis buffer supplemented with protease inhibitor cocktail (Roche) (23). Proteins were separated by SDS-PAGE before transfer onto nitrocellulose membrane (HyBond C Extra; Amersham Biosciences). Membranes were probed with primary antibodies against 16E7 (Santa Cruz) and 18E7, GAPDH (glyceraldehyde-3-phosphate dehydrogenase), and pRb (Abcam) and horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma). Proteins were detected using enhanced chemiluminescence reagent.
Reporter gene assays.
Dual-luciferase reporter assays were performed as previously described (24). Keratinocytes were seeded at 3 × 105 cells/ml in 24-well dishes prior to transfection. Cells were transfected with wild-type and mutant IL-18BP promoter-driven firefly luciferase reporters and a Renilla luciferase (RLTK)-expressing plasmid as a transfection control using FuGene reagent. At 24 h posttransfection, cells were stimulated with IFN-γ (BioLegend) for 12 h prior to lysis in passive lysis buffer (Promega). Firefly luciferase levels were normalized to Renilla levels, and fold induction values were calculated relative to the normalized activity of the mock control.
RESULTS
HPV16 E7 augments IL-18BP expression in primary keratinocytes.
To gain insight into a possible effect of HPV oncoproteins on endogenous IL-18BP secretion, primary human foreskin keratinocytes were transduced with retroviruses expressing HPV16 E6 and E7 in isolation or E6/E7 from a bicistronic transcript. Viral gene expression was confirmed by RT-PCR (data not shown). Cells were stimulated with IFN-γ, a potent inducer of IL-18BP (21), for 16 h and culture supernatants analyzed by ELISA. In control keratinocytes, expressing empty pBabe-Puro plasmid, IFN-γ induced the secretion of IL-18BP (Fig. 1A). Expression of 16E6 induced a modest, but statistically significant, increase in the levels of IL-18BP secretion, whereas 16E7 had the most pronounced effect on IL-18BP secretion, inducing more than a doubling in concentrations of cytokine compared with the control. This was not significantly further increased in the presence of E6. These data demonstrate that HPV oncoproteins, in particular E7, enhance the levels of IL-18BP secreted from primary keratinocytes.
FIG 1.
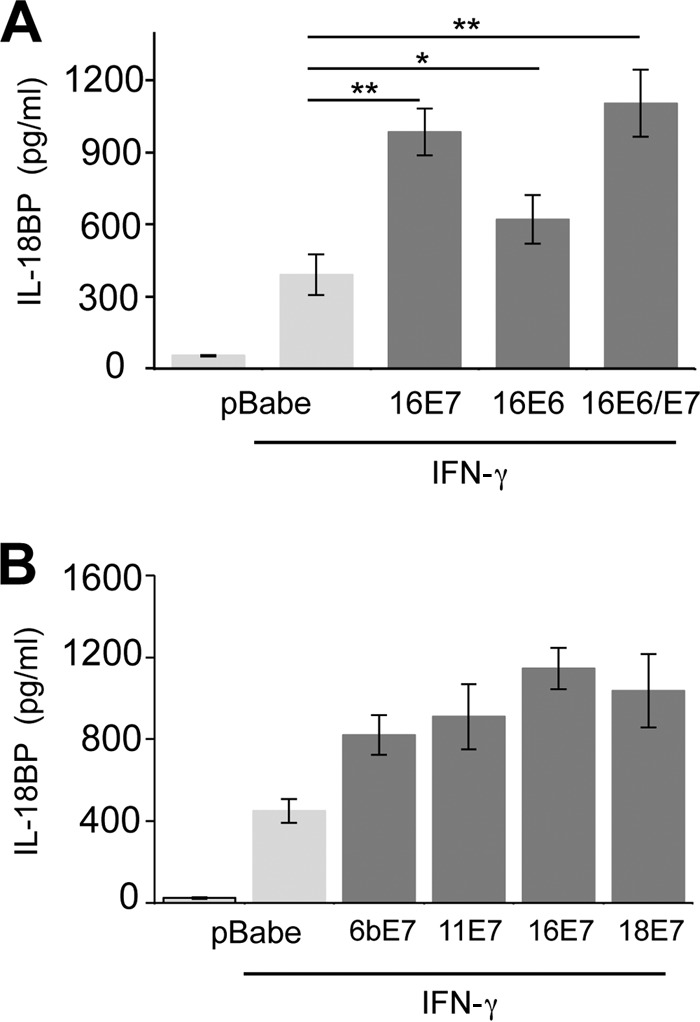
E7 proteins augment IL-18BP expression in primary keratinocytes. (A) Primary human foreskin keratinocytes stably expressing vector alone (pBabe-Puro), HPV16 E7, HPV16 E6, or HPV16 E6sE7 were treated with IFN-γ (10 ng/ml) for 16 h. Following treatment, supernatants were harvested and levels of secreted IL-18BP measured by ELISA. (B) Primary human foreskin keratinocytes expressing E7 from HPV6b, HPV11, HPV16, and HPV18 were treated with IFN-γ (10 ng/ml) for 16 h. Following treatment, supernatants were harvested and levels of secreted IL-18BP measured by ELISA. Results are expressed as the means ± standard deviations (SD) from three independent experiments. *, P < 0.05; **, P < 0.01.
E7 proteins from other HPV types enhance IL-18BP expression.
This is the first report to demonstrate that HPV16 E7 enhances IL-18BP expression. To establish whether enhanced IL-18BP production was a general feature of E7 proteins, keratinocytes stably expressing E7 proteins from several mucosal types were treated with IFN-γ and levels of IL-18BP measured by ELISA (Fig. 1B). E7 from both low-risk (HPV6b and HPV11) and high-risk (HPV16 and HPV18) types increased expression of IL-18BP to comparable levels. These experiments illustrate that IL-18BP is a conserved target for E7 proteins of multiple HPV types, suggesting that increased IL-18BP levels may be necessary during the HPV life cycle rather than associated directly with transformation.
The CR3 region of E7 is necessary for enhanced IL-18BP expression.
Next, the regions of E7 important for enhancing IL-18BP production were identified. Primary keratinocytes were transduced with retroviruses expressing a panel of E7 proteins containing mutations in the amino-terminal disordered conserved region 1 (CR1) and CR2 or in the more structured carboxyl-terminal CR3 (25–29). Figure 2A shows a schematic diagram of E7 and the mutations examined in this study. E7 proteins containing single-amino-acid mutations in CR1 or CR2 (CR1-H2P or CR2-C24G) retained the ability to increase IL-18BP production upon IFN-γ treatment (Fig. 2B). Several mutants containing small deletions in CR3 of E7 were also tested for their ability to augment IL-18BP production. Keratinocytes stably expressing Δ1 (with deletion of amino acids 52 to 56) and Δ4 (with deletion of amino acids 79 to 83) showed IL-18BP levels similar to those in cells expressing wild-type E7 (Fig. 2B). In contrast, cells expressing Δ2 (deletion of amino acids 65 to 67) and Δ3 (deletion of amino acids 75 to 77) produced lower levels of IL-18BP, similar to those with vector (pBabe-Puro) alone. While these data imply that amino acids within Δ2 and Δ3 are necessary for the increased IL-18BP production, it is plausible that the mutations may have had additional impacts on E7 structure, since CR3 has been shown to be highly structured (30, 31). To explore whether the panel of mutants used in this study were broadly functional, we tested their expression level and ability to downregulate the cellular Rb protein, a key target for E7 proteins (28). Stable expression of the mutant proteins was observed (Fig. 2C), although levels of C24G were reduced compared with those of the wild type. Interestingly, this mutant was still capable of increasing IL-18BP levels, suggesting that only low levels of E7 are necessary for this function. Although E7 has been reported to be secreted into the cell medium and to bind directly to the IL-18 receptor (32), we could not detect E7 in the media of our stable keratinocyte cell lines. As such, it is unlikely that any secreted form of E7 was necessary for the observed effects on IL-18BP (Fig. 2C).
FIG 2.
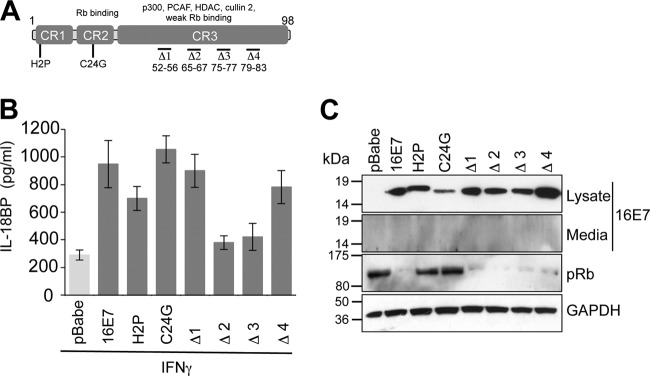
Enhanced IL-18BP expression requires amino acids in the carboxyl terminus of E7. (A) Schematic of E7 and the mutations used in this study. Functional regions of E7 are highlighted. Substitutions are shown, and small deletions are indicated by “Δ”. (B) Amino acids in CR3 are necessary for increased IL-18BP production. Primary human foreskin keratinocytes stably expressing empty plasmid (pBabe), 16E7, 16E7 H2P, 16E7 C24G, and four carboxyl-terminal truncations designated Δ1 (with deletion of amino acids 52 to 56), Δ2 (with deletion of amino acids 65 to 67), Δ3 (with deletion of amino acids 75 to 77), and Δ4 (with deletion of amino acids 79 to 83) were treated with IFN-γ (10 ng/ml) for 16 h. Following treatment, supernatants were harvested and levels of secreted IL-18BP measured by ELISA. Results are expressed as the means ± SD from four independent experiments. (C) Media and cell lysates from representative experiments were subjected to SDS-PAGE and probed with the indicated antibodies to confirm E7 expression and test functionality of the mutations. GAPDH was used to demonstrate equal loading of lysates.
As previously reported, expression of wild-type E7 reduced Rb levels, while mutations in CR1 (H2P) or CR2 (C24G) abrogated this ability and resulted in Rb levels similar to those for the vector alone (Fig. 2C). Expression of the CR3 deletions reduced Rb levels equivalently to the wild type, despite any small differences in E7 expression between samples. These data suggest that amino acids within CR3 are required for increased IL-18BP and that the loss of IL-18BP expression seen in the Δ2 and Δ3 mutant cell lines is not due to gross changes in E7 structure.
Enhanced IL-18BP expression requires a proximal gamma-activated sequence (GAS) element in the IL-18BP promoter.
To address whether the increased levels of IL-18BP protein were due to increased mRNA abundance, time course experiments were performed and levels of mRNA expression determined over a 24-h time period of IFN-γ treatment using quantitative RT-PCR. IL-18BP transcript was detectable at the 6-h time point in both control and E7-expressing keratinocytes, although transcript levels were significantly greater in those expressing E7 (Fig. 3A). Transcript levels continued to increase until the 24-h time point and were greater in all samples containing E7. This trend was also observed with secreted IL-18BP (Fig. 3B). Despite a delay in protein production until 24 h in the control cells, secreted IL-18BP was detectable at as early as 12 h in cells expressing E7 (Fig. 3B).
FIG 3.

E7-dependent IL-18BP transcription is mediated by a proximal GAS element in the IL-18BP promoter. (A) Primary human foreskin keratinocytes stably expressing empty plasmid (pBabe) or 16E7 were treated with IFN-γ (10 ng/ml). RNA was extracted over a 24-h time course and analyzed by qRT-PCR for IL-18BP and U6snRNA expression. Expression levels are shown as the fold IL-18BP mRNA increase compared to untreated controls. (B) As for panel A except that supernatants were taken for measurement of secreted IL-18BP protein by ELISA. (C) Primary foreskin keratinocytes were transfected with the indicated IL-18BP promoter reporter plasmids. After 24 h, cells were untreated or stimulated with IFN-γ (10 ng/ml) for 12 h and activity measured using a dual-luciferase system. Results are expressed as the means ± SD from four independent experiments. (D) Primary normal human foreskin keratinocytes (NHK) and keratinocytes stably harboring the HPV18 genome were treated with IFN-γ (10 ng/ml) in the presence of DMSO (mock) or lestaurtinib (Lest) (100 nM), a small-molecule inhibitor of the Jak-STAT signaling pathway, for 24 h and supernatants taken for measurement of secreted IL-18BP protein by ELISA. Results are expressed as the means ± SD from three independent experiments. **, P < 0.01. Western blot analysis from representative samples of lysates taken from these cells demonstrated that treatment with lestaurtinib did not reduce E7 expression. GAPDH was used as a protein loading control.
IL-18BP transcription is thought to be dependent on the gamma-activated sequence (GAS) within the IL-18BP promoter element, which can be bound by members of the signal transducer and activator of transcription (STAT) family of transcription factors (21). The IL-18BP promoter element contains two conserved GASs, one directly adjacent to the transcriptional start site (bp −25 to −33) and a second, more distal site, located at bp −625 to −633 (21). To begin to decipher the signaling pathways leading to IL-18BP production in E7-expressing cells, luciferase reporter assays were performed. Control and E7-expressing keratinocytes were transfected with a reporter plasmid driving luciferase expression from the IL-18BP promoter (pGL3-BP WT) (Fig. 3C). In addition, three further reporter plasmids were used. These constructs contained mutations that inactivated either the proximal (pGL3-BP prox), distal (pGL3-BP dist), or both (pGL3-BP dist/prox) (21) GASs. Treatment with IFN-γ induced luciferase expression from the wild-type IL-18BP promoter reporter (Fig. 3C). Mutation of the proximal, but not the distal, GAS in the promoter significantly reduced luciferase production in response to IFN-γ treatment in both control and E7-expressing cells. Importantly, the double mutation at the proximal and distal GASs was not associated with any further suppression of luciferase expression compared to that with the single proximal mutant. These results indicate that E7 expression modulates IL-18BP production through the canonical proximal GAS.
It was important to examine the effects of E7 on IL-18BP production in the context of the complete HPV genome in which all viral factors are present. For this, primary human keratinocytes harboring the HPV18 genome were generated using published protocols (22). Control or HPV18-harboring keratinocytes were incubated with dimethyl sulfoxide (DMSO) (mock) or the small-molecule Jak kinase inhibitor lestaurtinib (100 nM) (33) for 1 h prior to stimulation with IFN-γ. As seen in Fig. 3D, levels of IL-18BP were significantly reduced in the media of both control and HPV18-containing keratinocytes. Treatment did not significantly alter levels of the E7 protein, suggesting that abrogation of Jak signaling prevents E7-mediated production of IL-18BP. Overall, these data strongly suggest that IL-18BP production requires a functional Jak-STAT signaling pathway in E7-expressing cells.
The enhanced IL-18BP expression by E7 reduces activation of primary CD4-positive lymphocytes.
The cellular immune response is essential for destruction of HPV-infected keratinocytes. IL-18 is known to augment this by increasing IFN-γ secretion from activated T lymphocytes (16). As IL-18BP antagonizes the effects of IL-18, we wanted to assess the biological consequences of increased IL-18BP production by E7-expressing keratinocytes. Primary human CD4-positive T lymphocytes were isolated from the PBMCs of healthy volunteers and stimulated with anti-CD3/CD28 antibodies. Activated lymphocytes were subsequently incubated with IL-18 in the presence of medium taken from keratinocytes stably expressing vector alone, wild-type E7, or Δ2 E7 and analyzed after 24 h for IFN-γ secretion by ELISA. As shown in Fig. 4, exposure to media from all keratinocytes caused a reduction in IFN-γ release from the CD4-positive lymphocytes. This was to be expected since the media contained various levels of IL-18BP. Importantly, exposure to medium from the wild-type-E7-expressing keratinocytes resulted in the most pronounced inhibition of IFN-γ production. To confirm that increased IL-18BP production was necessary for the reduced IFN-γ levels produced by the CD4-positive lymphocytes, we assessed the effects of the Δ2 E7 cell line on IFN-γ production. CD4-positive lymphocytes incubated in medium from the Δ2 cell line produced more IFN-γ than those from the wild-type E7 cell line, with levels resembling those with the vector alone. To preclude the possibility that IFN-γ was present in the input samples, we tested both the mock samples and keratinocyte medium. These samples contained undetectable levels of IFN-γ. Overall, these data suggest that the increased levels of IL-18BP produced by E7-expressing keratinocytes have an inhibitory effect on CD4-positive lymphocytes.
FIG 4.
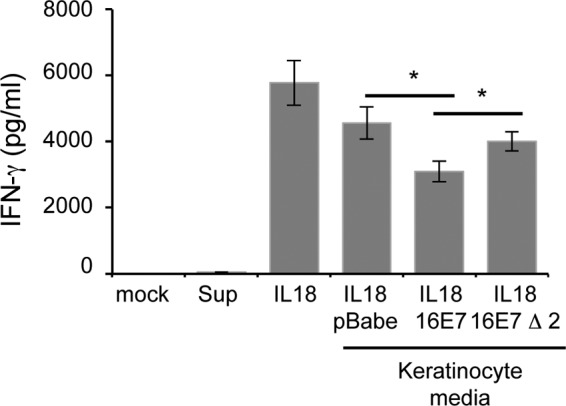
Effect of increased IL-18BP expression on IL-18-induced IFN-γ production in primary human CD4 lymphocytes. Primary human foreskin keratinocytes stably expressing empty plasmid (pBabe), HPV16 E7, or HPV16 Δ2 E7 were treated with IFN-γ (10 ng/ml) for 8 h. Cells were washed to remove input IFN-γ and treated with fresh medium for a further 16 h. Keratinocyte supernatants were collected and added to cultures of anti-CD3/CD28-activated human CD4 lymphocytes, which were costimulated with IL-18 (20 ng/ml) for 24 h. Supernatants were harvested and levels of secreted IFN-γ measured by ELISA. Mock-treated cells and keratinocyte supernatant alone were used as controls to demonstrate absence of IFN-γ from input samples. The data represent the average from five independent experiments, and error bars indicate SD. *, P < 0.05.
DISCUSSION
The pathogenesis of HPV infection is directly linked to the immune responses of the host, which depend on the secretion of specific cytokines to attract components of the adaptive immune system to the site of virus infection. However, HPV infections appear to be immunologically deficient and suffer from reduced inflammatory infiltrate (34). Several mechanisms have been proposed to explain the observed immunodeficiency, and these focus mainly on the E6 and E7 oncoproteins. Both E6 and E7 have been shown to target the IL-18 receptor directly, to prevent activation of target cells (32). E6 and E7 also prevent the transcription of inflammatory cytokines by targeting antiviral signaling pathways (9, 14, 35).
Our data provide the first evidence for upregulation of IL-18BP by a virus protein in the context of both overexpression of the individual protein and the stable expression of the entire HPV genome. Given the critical role of IL-18 in the skin inflammatory response to virus infection, which includes increasing the expression of antiviral molecules such as MHC-II (17), it is likely that the suppressive effects associated with increased IL-18BP present in the vicinity of HPV infection would lead to a diminished cellular immune response associated with HPV infection. The results of this study demonstrate that medium produced from E7-expressing keratinocytes is able to inhibit primary CD4-positive lymphocytes costimulated with IL-18. Importantly, the E7 Δ2 mutant, which was unable to increase the expression of IL-18BP from keratinocytes, was also not able to inhibit IFN-γ production from the CD4-positive lymphocytes, suggesting that IL-18BP is necessary for the effects seen in this assay. Despite this, the use of neutralization assays with IL-18BP-specific antibodies would greatly strengthen these findings.
Interestingly, E7 proteins from low-risk HPV6b and -11 were also capable of increasing IL-18BP expression, suggesting that IL-18BP is an important target for E7 and may contribute to HPV persistence in infected individuals. Mapping studies identified residues in the carboxyl CR3 region of E7 necessary for the increased IL-18BP production. This region contributes to a number of E7 functions, including dimerization (36). It may also mediate interactions with a number of cellular proteins, including cullin 2, HDAC, pCAF, p300, TATA binding protein, and Rb (25, 27, 29, 37, 38). The interaction of E7 with one or several of these proteins may be important for regulating IL-18BP production.
IL-18BP expression is mediated by a number of response elements found within the IL-18BP promoter, and a proximal GAS element is necessary for IFN-γ-mediated expression (21). Members of the STAT family of transcription factors bind GAS elements, and HPV proteins have been shown to differentially regulate these proteins during the virus life cycle (39–42), which may help to coordinate the pleiotropic effects observed on the immune system. The time course experiments demonstrated earlier kinetics of IL-18BP expression in E7-expressing cells than in controls. This is unlikely due to different signaling pathways and so may indicate that E7 also functions through an as-yet-unidentified pathway or acts on a novel negative regulatory protein that functions to suppress IL-18BP expression.
In conclusion, our results add HPV to the short list of persistent viruses that are currently known to modulate the IL-18-IL-18BP axis. Given the critical nature of IL-18 for mounting the defensive response to virus infection, we propose that IL-18BP is likely to be targeted by a growing number of viruses.
ACKNOWLEDGMENTS
We thank Dennis McCance (Queen's University Belfast) for supplying E6 and E7 retrovirus expression plasmids, Lawrence Banks (ICGEB, Trieste, Italy) for E7 mutant plasmids, and Heiko Muhl (Frankfurt, Germany) for IL-18BP reporter plasmids.
This work was supported by grants to M.W. from the Leeds Foundation for Dermatological Research (LFDR) and to A.M. from Yorkshire Cancer Research (L339, LPP041, and PP015), CRUK (C43832/A14246 and C37059/A11941), and the MRC (MR/K012665/1).
Footnotes
Published ahead of print 29 January 2014
REFERENCES
- 1.Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. 2010. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401:70–79. 10.1016/j.virol.2010.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clifford GM, Smith JS, Aguado T, Franceschi S. 2003. Comparison of HPV type distribution in high-grade cervical lesions and cervical cancer: a meta-analysis. Br. J. Cancer. 89:101–105. 10.1038/sj.bjc.6601024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clifford GM, Smith JS, Plummer M, Munoz N, Franceschi S. 2003. Human papillomavirus types in invasive cervical cancer worldwide: a meta-analysis. Br. J. Cancer 88:63–73. 10.1038/sj.bjc.6600688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashrafi GH, Brown DR, Fife KH, Campo MS. 2006. Down-regulation of MHC class I is a property common to papillomavirus E5 proteins. Virus Res. 120:208–211. 10.1016/j.virusres.2006.02.005 [DOI] [PubMed] [Google Scholar]
- 5.Wetherill LF, Ross R, Macdonald 2012. HPV E5: an enigmatic oncoprotein, p 55–71 In Gaston K. (ed), Small DNA tumour viruses. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 6.Cho YS, Kang JW, Cho M, Cho CW, Lee S, Choe YK, Kim Y, Choi I, Park SN, Kim S, Dinarello CA, Yoon DY. 2001. Down modulation of IL-18 expression by human papillomavirus type 16 E6 oncogene via binding to IL-18. FEBS Lett. 501:139–145. 10.1016/S0014-5793(01)02652-7 [DOI] [PubMed] [Google Scholar]
- 7.Huang SM, McCance DJ. 2002. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J. Virol. 76:8710–8721. 10.1128/JVI.76.17.8710-8721.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleine-Lowinski K, Rheinwald JG, Fichorova RN, Anderson DJ, Basile J, Munger K, Daly CM, Rosl F, Rollins BJ. 2003. Selective suppression of monocyte chemoattractant protein-1 expression by human papillomavirus E6 and E7 oncoproteins in human cervical epithelial and epidermal cells. Int. J. Cancer 107:407–415. 10.1002/ijc.11411 [DOI] [PubMed] [Google Scholar]
- 9.Guess JC, McCance DJ. 2005. Decreased migration of Langerhans precursor-like cells in response to human keratinocytes expressing human papillomavirus type 16 E6/E7 is related to reduced macrophage inflammatory protein-3alpha production. J. Virol. 79:14852–14862. 10.1128/JVI.79.23.14852-14862.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonsson A, Payne E, Hengst K, McMillan NA. 2006. The human papillomavirus type 16 E7 protein binds human interferon regulatory factor-9 via a novel PEST domain required for transformation. J. Interferon Cytokine Res. 26:455–461. 10.1089/jir.2006.26.455 [DOI] [PubMed] [Google Scholar]
- 11.Cordano P, Gillan V, Bratlie S, Bouvard V, Banks L, Tommasino M, Campo MS. 2008. The E6E7 oncoproteins of cutaneous human papillomavirus type 38 interfere with the interferon pathway. Virology 377:408–418. 10.1016/j.virol.2008.04.036 [DOI] [PubMed] [Google Scholar]
- 12.Byg LM, Vidlund J, Vasiljevic N, Clausen D, Forslund O, Norrild B. 2012. NF-kappaB signalling is attenuated by the E7 protein from cutaneous human papillomaviruses. Virus Res. 169:48–53. 10.1016/j.virusres.2012.06.028 [DOI] [PubMed] [Google Scholar]
- 13.Vandermark ER, Deluca KA, Gardner CR, Marker DF, Schreiner CN, Strickland DA, Wilton KM, Mondal S, Woodworth CD. 2012. Human papillomavirus type 16 E6 and E 7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology 425:53–60. 10.1016/j.virol.2011.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reiser J, Hurst J, Voges M, Krauss P, Munch P, Iftner T, Stubenrauch F. 2011. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 85:11372–11380. 10.1128/JVI.05279-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Meglio P, Perera GK, Nestle FO. 2011. The multitasking organ: recent insights into skin immune function. Immunity 35:857–869. 10.1016/j.immuni.2011.12.003 [DOI] [PubMed] [Google Scholar]
- 16.Dinarello CA, Fantuzzi G. 2003. Interleukin-18 and host defense against infection. J. Infect. Dis. 187(Suppl 2):S370–S384. 10.1086/374751 [DOI] [PubMed] [Google Scholar]
- 17.Wittmann M, Purwar R, Hartmann C, Gutzmer R, Werfel T. 2005. Human keratinocytes respond to interleukin-18: implication for the course of chronic inflammatory skin diseases. J. Invest. Dermatol. 124:1225–1233. 10.1111/j.0022-202X.2005.23715.x [DOI] [PubMed] [Google Scholar]
- 18.Wittmann M, Macdonald A, Renne J. 2009. IL-18 and skin inflammation. Autoimmun. Rev. 9:45–48. 10.1016/j.autrev.2009.03.003 [DOI] [PubMed] [Google Scholar]
- 19.Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. 1999. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity 10:127–136. 10.1016/S1074-7613(00)80013-8 [DOI] [PubMed] [Google Scholar]
- 20.Krumm B, Meng X, Li Y, Xiang Y, Deng J. 2008. Structural basis for antagonism of human interleukin 18 by poxvirus interleukin 18-binding protein. Proc. Natl. Acad. Sci. U. S. A. 105:20711–20715. 10.1073/pnas.0809086106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachmann M, Paulukat J, Pfeilschifter J, Muhl H. 2009. Molecular mechanisms of IL-18BP regulation in DLD-1 cells: pivotal direct action of the STAT1/GAS axis on the promoter level. J. Cell. Mol. Med. 13:1987–1994. 10.1111/j.1582-4934.2008.00604.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knight GL, Pugh AG, Yates E, Bell I, Wilson R, Moody CA, Laimins LA, Roberts S. 2011. A cyclin-binding motif in human papillomavirus type 18 (HPV18) E1Ê4 is necessary for association with CDK-cyclin complexes and G2/M cell cycle arrest of keratinocytes, but is not required for differentiation-dependent viral genome amplification or L1 capsid protein expression. Virology 412:196–210. 10.1016/j.virol.2011.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gamlen T, Richards KH, Mankouri J, Hudson L, McCauley J, Harris M, Macdonald A. 2010. Expression of the NS3 protease of cytopathogenic bovine viral diarrhea virus results in the induction of apoptosis but does not block activation of the beta interferon promoter. J. Gen. Virol. 91:133–144. 10.1099/vir.0.016170-0 [DOI] [PubMed] [Google Scholar]
- 24.Griffiths DA, Abdul-Sada H, Knight LM, Jackson BR, Richards K, Prescott EL, Peach AH, Blair GE, Macdonald A, Whitehouse A. 2013. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J. Virol. 87:13853–13867. 10.1128/JVI.02159-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernat A, Avvakumov N, Mymryk JS, Banks L. 2003. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene 22:7871–7881. 10.1038/sj.onc.1206896 [DOI] [PubMed] [Google Scholar]
- 26.Bodily JM, Mehta KP, Cruz L, Meyers C, Laimins LA. 2011. The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J. Virol. 85:8852–8862. 10.1128/JVI.00664-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Todorovic B, Hung K, Massimi P, Avvakumov N, Dick FA, Shaw GS, Banks L, Mymryk JS. 2012. Conserved region 3 of human papillomavirus 16 E7 contributes to deregulation of the retinoblastoma tumor suppressor. J. Virol. 86:13313–13323. 10.1128/JVI.01637-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helt AM, Galloway DA. 2001. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 75:6737–6747. 10.1128/JVI.75.15.6737-6747.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Massimi P, Pim D, Banks L. 1997. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J. Gen. Virol. 78:2607–2613 [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Clements A, Zhao K, Marmorstein R. 2006. Structure of the human papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J. Biol. Chem. 281:578–586. 10.1074/jbc.M508455200 [DOI] [PubMed] [Google Scholar]
- 31.Ohlenschlager O, Seiboth T, Zengerling H, Briese L, Marchanka A, Ramachandran R, Baum M, Korbas M, Meyer-Klaucke W, Durst M, Gorlach M. 2006. Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene 25:5953–5959. 10.1038/sj.onc.1209584 [DOI] [PubMed] [Google Scholar]
- 32.Lee SJ, Cho YS, Cho MC, Shim JH, Lee KA, Ko KK, Choe YK, Park SN, Hoshino T, Kim S, Dinarello CA, Yoon DY. 2001. Both E6 and E7 oncoproteins of human papillomavirus 16 inhibit IL-18-induced IFN-gamma production in human peripheral blood mononuclear and NK cells. J. Immunol. 167:497–504 [DOI] [PubMed] [Google Scholar]
- 33.Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, Yang S, Angeles T, Emerson SG, Carroll M, Ruggeri B, Dobrzanski P. 2008. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood 111:5663–5671. 10.1182/blood-2007-04-083402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.al-Saleh W, Delvenne P, Arrese JE, Nikkels AF, Pierard GE, Boniver J. 1995. Inverse modulation of intraepithelial Langerhans' cells and stromal macrophage/dendrocyte populations in human papillomavirus-associated squamous intraepithelial lesions of the cervix. Virchows Arch. 427:41–48 [DOI] [PubMed] [Google Scholar]
- 35.Karim R, Meyers C, Backendorf C, Ludigs K, Offringa R, van Ommen GJ, Melief CJ, van der Burg SH, Boer JM. 2011. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS One 6:e17848. 10.1371/journal.pone.0017848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Todorovic B, Massimi P, Hung K, Shaw GS, Banks L, Mymryk JS. 2011. Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J. Virol. 85:10048–10057. 10.1128/JVI.00643-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. 1999. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 18:2449–2458. 10.1093/emboj/18.9.2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avvakumov N, Torchia J, Mymryk JS. 2003. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene 22:3833–3841. 10.1038/sj.onc.1206562 [DOI] [PubMed] [Google Scholar]
- 39.Hong S, Mehta KP, Laimins LA. 2011. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 85:9486–9494. 10.1128/JVI.05007-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sobti RC, Singh N, Hussain S, Suri V, Bharadwaj M, Das BC. 2010. Deregulation of STAT-5 isoforms in the development of HPV-mediated cervical carcinogenesis. J. Recept. Signal. Transduct. Res. 30:178–188. 10.3109/10799891003786218 [DOI] [PubMed] [Google Scholar]
- 41.Chang YE, Laimins LA. 2000. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 74:4174–4182. 10.1128/JVI.74.9.4174-4182.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sobti RC, Singh N, Hussain S, Suri V, Bharti AC, Das BC. 2009. Overexpression of STAT3 in HPV-mediated cervical cancer in a north Indian population. Mol. Cell. Biochem. 330:193–199. 10.1007/s11010-009-0133-2 [DOI] [PubMed] [Google Scholar]