

ABSTRACT
New human norovirus strains emerge every 2 to 3 years, partly due to mutations in the viral capsid that allow escape from antibody neutralization and herd immunity. To understand how noroviruses evolve antibody resistance, we investigated the structural basis for the escape of murine norovirus (MNV) from antibody neutralization. To identify specific residues in the MNV-1 protruding (P) domain of the capsid that play a role in escape from the neutralizing monoclonal antibody (MAb) A6.2, 22 recombinant MNVs were generated with amino acid substitutions in the A′B′ and E′F′ loops. Six mutations in the E′F′ loop (V378F, A382K, A382P, A382R, D385G, and L386F) mediated escape from MAb A6.2 neutralization. To elucidate underlying structural mechanisms for these results, the atomic structure of the A6.2 Fab was determined and fitted into the previously generated pseudoatomic model of the A6.2 Fab/MNV-1 virion complex. Previously, two distinct conformations, A and B, of the atomic structures of the MNV-1 P domain were identified due to flexibility in the two P domain loops. A superior stereochemical fit of the A6.2 Fab to the A conformation of the MNV P domain was observed. Structural analysis of our observed escape mutants indicates changes toward the less-preferred B conformation of the P domain. The shift in the structural equilibrium of the P domain toward the conformation with poor structural complementarity to the antibody strongly supports a unique mechanism for antibody escape that occurs via antigen flexibility instead of direct antibody-antigen binding.
IMPORTANCE Human noroviruses cause the majority of all nonbacterial gastroenteritis worldwide. New epidemic strains arise in part by mutations in the viral capsid leading to escape from antibody neutralization. Herein, we identify a series of point mutations in a norovirus capsid that mediate escape from antibody neutralization and determine the structure of a neutralizing antibody. Fitting of the antibody structure into the virion/antibody complex identifies two conformations of the antibody binding domain of the viral capsid: one with a superior fit and the other with an inferior fit to the antibody. These data suggest a unique mode of antibody neutralization. In contrast to other viruses that largely escape antibody neutralization through direct disruption of the antibody-virus interface, we identify mutations that acted indirectly by limiting the conformation of the antibody binding loop in the viral capsid and drive the antibody binding domain into the conformation unable to be bound by the antibody.
INTRODUCTION
RNA viruses undergo error-prone replication to generate large, highly diverse but genetically related virus populations called quasispecies (1–3). This capacity to generate and maintain mutations allows viruses to rapidly adapt to changing selection pressures in their environment. Human noroviruses (HuNoV) are positive-stranded RNA viruses in the family Caliciviridae and are the major cause of acute viral gastroenteritis, resulting in worldwide epidemics every 2 to 3 years (4, 5). The abundance of norovirus outbreaks (6) and the continuous emergence of new genogroups and variants (4, 7–10) are driven in part by mutations in the major capsid protein of HuNoV that may mediate escape from antibody neutralization (5, 11, 12). However, the lack of a tissue culture system and, until recently, a small animal model (13) has made it difficult to understand the mechanisms of HuNoV antibody neutralization escape and to develop an effective vaccine for HuNoV (5). Murine norovirus (MNV) shares many molecular and biological properties with HuNoV, it grows well in tissue culture and mice, and it has reverse-genetics systems available (14–18). Therefore, MNV is often used to study general mechanisms in norovirus biology (18).
Norovirus particles contain 180 copies of the major capsid protein (VP1; ∼58 kDa), which is divided into the N-terminal (N), shell (S), and C-terminal protruding (P) domains (19–22). The S domain forms a shell around the viral RNA genome, while the P domain dimerizes to form arch-like structures on the capsid surface. The P domain is subdivided into P1 and P2 subdomains, with the latter containing the binding sites for cellular receptors (23, 24) and neutralizing antibodies (25–27). For MNV, the neutralizing monoclonal antibody (MAb) A6.2 binds to the P2 domain and blocks capsid-cell interactions (22, 27). Notably, the crystal structure of the MNV-1 P domain revealed two conformations of the two loops (A′B′ and E′F′) that are thought to bind to MAb A6.2 (22, 27). However, due to the absence of an A6.2/MNV costructure, the preferred epitope conformation was unclear.
Using the previous A6.2/MNV-1 docking model (27) as a guide for mutagenesis, we have identified six single point mutations in the E′F′ loop of the MNV-1 P domain that completely abrogated MAb A6.2 binding to MNV-1 and allowed MAb A6.2 neutralization escape in culture and in mice. Furthermore, the atomic structure of the A6.2 Fab fragment was determined and used to refine the pseudoatomic electron microscopy model (22). This new structure, combined with the mutagenesis results, suggested that a number of escape mutants block antibody binding distal to the epitope by limiting the conformational repertoire of the MAb epitope in the E′F′ loop. These studies are the first to suggest that escape mutations may act by limiting the flexibility of an epitope or by driving the conformation toward a weak binding structure.
MATERIALS AND METHODS
Cell culture and virus stocks.
RAW 264.7 (murine macrophages) and 293T cells were purchased from ATCC (Manassas, VA) and maintained as described previously (17, 27). The plaque-purified MNV-1 clone (GV/MNV1/2002/USA) MNV-1.CW3 (28) (referred to herein as MNV-1) was used at passage 6 (P6) for in vitro passaging experiments.
Plaque assay and plaque neutralization assay.
MNV-1 (5 × 104 PFU) was diluted with Dulbecco's modified Eagle's medium (DMEM) and 0, 20, 60, 200, or 600 ng of purified anti MNV-1 MAb A6.2 or purified isotype control MAb CV (coxsackievirus B4) in a total volume of 150 μl. The mixture was then incubated for 30 min at 37°C. Virus titers were determined by plaque assay as described previously (29).
Generation of MAb A6.2 neutralization escape mutants.
Neutralization escape mutants were generated as previously described (30) with some modifications. RAW 264.7 cells (2 × 106 per well) were plated in 6-well plates and incubated overnight. MNV-1 was preincubated for 30 min with 20 ng MAb A6.2 or the isotype control at 37°C before infecting cells at a multiplicity of infection (MOI) of 0.025 for 1 h at room temperature. The inoculum was removed, and 2 ml of medium containing 1/10 the original amount of antibody was added before the infection was allowed to proceed at 37°C for 48 h. The resulting passage 1 (P1) cell lysates were centrifuged for 5 min at 5,000 × g following two freeze-thaw cycles. The process was repeated for 5 passages, using the previous passage as the inoculum for the subsequent round and increasing the MAb concentration to 60 ng for P2 and 100 ng for P3 to P5. Viral titers were determined by plaque assay after each passage. Plaque neutralization assays were performed for virus samples from indicated passages to monitor the ability of MAb A6.2 to neutralize the infection.
PCR amplicon sequencing analysis of MNV-1 P0-to-P5 samples.
Aliquots of lysates collected from P1 to P5 supernatants and the starting virus stock (P0) were prepared for 454 sequencing at Bode Technology (Lorton, VA). The sequencing and denoising analysis pipeline was performed at Johns Hopkins APL. Specifically, each sample contained amplicons from among three overlapping open reading frame 2 (ORF2) regions spanning positions 5473 to 6681 of the MNV-1 genome. Raw reads were initially filtered to isolate sequences containing only DNA characters and lengths ranging from 410 to 455 bp for each amplicon. Isolated reads were then aligned to the reference sequence with the sequence alignment program Exonerate (http://www.ebi.ac.uk/~guy/exonerate/) using the Smith-Waterman algorithm to produce a gapped local alignment using the affine gap penalty function. Since indels in viral sequences introduce frameshifts that are lethal with high probability and the 454 system produces indel errors at a high rate (∼0.004 per base), we focused only on substitutions for this analysis. Thus, any postalignment insertions with respect to the reference sequence were removed, and any deletions were replaced with the corresponding base from the reference sequence. Forward- and reverse-direction reads for each amplicon were then clustered into unique candidate haplotypes, and the relative proportions of each unique sequence in the corresponding direction were computed. To remove systematic errors, we required each candidate haplotype to be confirmed in the forward and reverse directions and required that the ratio of the higher-to-lower population proportion did not exceed 2.5 for the two read directions. Haplotypes surviving these criteria were then tested for statistical significance using a hypothesis test. The null hypothesis presumed that a haplotype candidate that is one substitution away from another haplotype in the population with greater proportion is generated by sequencing errors in the reading of the higher-proportion sequences in the sample. We computed the expected proportion and its sampling distribution with a simple generative model using a base-calling substitution rate of 0.0005 per base and then calculated the probability that the observed proportion was derived from this distribution. Sequences with a P value of <0.001 were considered to be statistically significant and included in the set of local haplotypes for each amplicon, representing the viral quasispecies.
Determination of significance.
First, the Hamming distance between all pairs of haplotypes was calculated. For each haplotype, the total number of reads was counted from other haplotypes that both had a Hamming distance of 1 away from this haplotype (“one-away haplotypes”) and also had more reads than this haplotype. If there were no other one-away haplotypes that had more reads, or if there were no other one-away haplotypes at all, the haplotype was deemed significant automatically and given a probability of 10 to 100. Probabilities were determined for the remaining reads using the complementary cumulative density function (CCDF) of a binomial function with a significance threshold of 0.001 used to identify significant haplotypes. Analysis of statistically significant viral haplotypes was carried out with the BioEdit Sequence Editor software version 7.2.0, Lasergene 10 (DNASTAR, Inc.), and MEGA 5 (31).
Mutagenesis of MNV-1.
P domain residues were mutated in the vector pT7MNV3′RZ using the QuikChange II XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. The list of primers used is shown in Table 1. Recombinant MNV-1 was generated as described previously (32). Briefly, plasmids encoding mutant genomes were transcribed with T7 polymerase (NEB), and the resulting RNA was capped in vitro using ScriptCap enzyme (Epicentre). RNA was transfected into BSR-T7 cells using Lipofectamine 2000 (Invitrogen). After 24 h, cells and media were subjected to freeze-thaw and titers of lysates were determined by 50% tissue culture infective dose (TCID50) on RAW 264.7 cells. Successfully recovered mutant viruses were amplified in RAW 264.7 cells, and viral titers were determined by TCID50. In case viruses were not recovered, the mutated region of the MNV-1 plasmid was excised via PsiI (NEB) and cloned into pMNV* (16), containing the cDNA of the entire genome of MNV-1.CW1. The sequence of each MNV-1 mutant was confirmed after Sanger sequencing by at least two independent sequence determinations. Restored plasmids were transformed into TOP10 competent cells (Invitrogen) for amplification and purification. New plasmid was transfected into 293T cells as described for BSR-T7 cells. MNV-1 mutants were plaque purified as described previously (17), and the P domain mutation was confirmed by Sanger sequencing. Viruses containing the desired mutation were amplified in RAW 264.7 cells and used at passage 3 for all neutralization studies.
TABLE 1.
Primers for MNV-1 virus site-directed mutagenesis
| Mutation | Direction | Primer sequence (5′→3′) |
|---|---|---|
| E296K | Forward | GAGGCTGCCTATGAATTCCAATCGG |
| Reverse | CCGATTGGAATTCATAGGCAGCCTC | |
| Q298E | Forward | CTGCCTATGAGTTCGAATCGGGCACC |
| Reverse | GGTGCCCGATTCGAACTCATAGGCAG | |
| Q298S | Forward | GCCTATGAGTTCTCATCGGGCACCG |
| Reverse | CGGTGCCCGATGAGAACTCATAGGC | |
| S299A | Forward | GCCTATGAGTTCCAAGCGGGCACC |
| Reverse | GGTGCCCGCTTGGAACTCATAGGC | |
| S299R | Forward | CTATGAGTTCCAACGGGGCACCGG |
| Reverse | CCGGTGCCCCGTTGGAACTCATAG | |
| G300F | Forward | GAGTTCCAATCGTTCACCGGTGAGG |
| Reverse | CCTCACCGGTGAACGATTGGAACTC | |
| G300K | Forward | GAGTTCCAATCGAAGACCGGTGAGG |
| Reverse | CCTCACCGGTCTTCGATTGGAACTC | |
| G300R | Forward | GAGTTCCAATCGAGGACCGGTGAGG |
| Reverse | CCTCACCGGTCCTCGATTGGAACTC | |
| T301I | Nonea | |
| V378A | Forward | GTTCGCCAGCCTCACTGCTGC |
| Reverse | GCAGCAGTGAGGCTGGCGAAC | |
| V378F | Forward | CAGGGTGTTCGCCAGCTTCACTGCTGCGGCCTC |
| Reverse | GAGGCCGCAGCAGTGAAGCTGGCGAACACCCTG | |
| V378L | Forward | GTGTTCGCCAGCGCCACTGCTGCGG |
| Reverse | CCGCAGCAGTGGCGCTGGCGAACAC | |
| A381F | Forward | CAGCGTCACTGCTTTTGCCTCTCTTGAC |
| Reverse | GTCAAGAGAGGCAAAAGCAGTGACGCTG | |
| A381G | Forward | GTCACTGCTGGGGCCTCTCTTGACTTG |
| Reverse | CAAGTCAAGAGAGGCCCCAGCAGTGAC | |
| A382G | Forward | GTCACTGCTGCGGGCTCTCTTGACTTG |
| Reverse | CAAGTCAAGAGAGCCCGCAGCAGTGAC | |
| A382K | Forward | GTCACTGCTGCGAAATCTCTTGACTTGG |
| Reverse | CCAAGTCAAGAGATTTCGCAGCAGTGAC | |
| A382P | Forward | GCGTCACTGCTGCGCCCTCTCTTGACTTGG |
| Reverse | CCAAGTCAAGAGAGGGCGCAGCAGTGACGC | |
| A382R | Forward | GTCACTGCTGCGCGCTCTCTTGAC |
| Reverse | GTCAAGAGAGCGCGCAGCAGTGAC | |
| D385E | Forward | GGCTCTCTTGAGTTGGTGGATGGCAG |
| Reverse | CTGCCATCCACCAACTCAAGAGAGCC | |
| L386F | Forward | CTCTCTTGACTTCGTGGATGGCAGGG |
| Reverse | CCCTGCCATCCACGAAGTCAAGAGAG |
Mutation arose naturally.
Sequencing of MNV-1 mutants.
Total genomic RNA was extracted from 0.14 ml lysate from virus-infected RAW cells using the QIAamp viral RNA minikit (Qiagen) according to the manufacturer's instruction. The MNV P domain (residues 225 to 541) (27) was amplified with ATGAGGATGAGTGATGGCGCAG and TTATTGTTTGAGCATTCGGCCTG primers specific to the viral sequence that harbors the A′B′ and E′F′ loops using 5 μl RNA extract in 50 μl OneStep reverse transcription-PCR (RT-PCR) mixture (Qiagen). The PCR product was confirmed using 1% agarose gel and then purified using the QIAquick PCR purification kit (Qiagen). Sanger sequencing of the PCR product was performed at the DNA Sequencing Core at the University of Michigan.
Virus growth curves.
Overnight cultures of RAW 264.7 cells (2 × 105 per well) in 12-well plates were infected with the respective virus on ice at an MOI of 2. After 1 h, the inoculum was removed and cells were washed twice with ice-cold phosphate-buffered saline (PBS). Medium was added, and cells were incubated at 37°C for the indicated times. Virus titers were determined by plaque assay after 2 freeze-thaw cycles.
Mice.
Mouse studies were performed in accordance with local and federal guidelines as outlined in the Guide for the Care and Use of Laboratory Animals from the National Research Council (33). Protocols were approved by the University of Michigan Committee on Use and Care of Animals (UCUCA no. 09710).
Four- to 8-week-old STAT1−/− mice (strain 2045) were purchased from Taconic Farms, Inc., and housed at the University of Michigan animal care facility, where all experiments were conducted. Mice were simultaneously infected orally with 106 PFU of virus and intraperitoneally with the indicated concentration of purified MAb A6.2 or the isotype control Ab. Two days later, mice were humanely euthanized according to approved protocol. Tissues of the gastrointestinal tract (stomach, duodenum/jejunum, proximal ileum, distal ileum, cecum, and colon), the mesenteric lymph nodes, spleen, and liver were harvested. Samples were homogenized in 1.0 ml PBS with 500 mg of 1.0-mm-diameter silica beads (BioSpec) using a MagNA Lyser (Roche) for 1 min at 6,000 rpm, centrifuged at 15,000 rpm using an Eppendorf 5424 tabletop centrifuge for 2 min, and stored at −80°C. Virus titers were determined by plaque assay.
Statistical analyses.
The software program GraphPad Prism V6 (GraphPad, La Jolla, CA) was used to perform statistical analyses.
MAbs.
The isotype control IgG directed against coxsackievirus B4 (CV) (clone 204-4) was purchased from ATCC (HB 185). It and anti-MNV-1 MAb A6.2 (IgG2a) were grown in Bioreactor CELLine CL 1000 flasks (Sigma-Aldrich) at the Hybridoma Core, University of Michigan, using standard protocols (34). Antibodies were purified over a HiTrap protein A column (GE Healthcare) according to the manufacturer's instruction, dialyzed against PBS, and stored at −20°C.
Generation and purification of Fab fragments.
The hybridoma clone that was specifically used for crystallization was MAb A6.1 and arose from the same fusion as MAb A6.2 (17). At the start, the heavy and light chains of MAbs A6.1 and A6.2 were sequenced by Syd Labs, Inc. (Boston, MA), and found to be identical. Therefore, it is very likely that both clones came from the same mother cell. Therefore, for simplicity, the MAb A6.1 clone was renamed MAb A6.2. Hybridoma cells were grown in Bioreactor CELLine CL 1000 flasks (Sigma-Aldrich). Monoclonal antibodies were purified from cell culture supernatant with a 5-ml HiTrap protein G HP column (GE Healthcare). The column was first equilibrated with 20 mM sodium phosphate buffer (pH 7.0), and the supernatant was passed over the column and was washed with several volumes of 20 mM sodium phosphate buffer (pH 7.0). The bound antibody was eluted with 50 mM sodium citrate buffer (pH 2.0), and the pH was quickly raised to neutrality.
To generate Fab fragments, purified monoclonal antibodies were dialyzed against 0.1 M sodium phosphate buffer (pH 7.0). The antibody was then digested by papain agarose (1:100 [wt/wt]) in the presence of 25 mM β-mercaptoethanol for 17 h at 37°C. Digestion was stopped by removal of the papain beads via centrifugation at 40,000 × g for 30 min. The supernatant was dialyzed extensively against 50 mM Tris (pH 7.6) and then applied to the Mono-Q column equilibrated with the same buffer. At this pH, only the Fc fragments and intact antibodies bound to the column, and the Fab fragments were collected from flowthrough.
Crystallization of the MNV Fab and data collection.
The purified Fab fragments were concentrated to 9.7 mg/ml in 50 mM Tris (pH 7.6) and crystallized by vapor diffusion and the sitting drop method. The reservoir contained 18% polyethylene glycol 8000 (PEG 8000) and 90 mM Tris (pH 8.5). The drop was composed of 5 μl of the Fab solution and 5 μl of the reservoir solution.
Diffraction data were collected from a single crystal frozen at ∼100°K using an Oxford cryosystem and a Proteum R Smart 6000 charge-coupled device (CCD) detector connected to a Bruker-Nonius FR591 rotating anode generator. Prior to data collection, crystals were transferred stepwise in increasing PEG 400 solutions up to 20% (vol/vol) in the reservoir liquid solutions over a 30-min period. An oscillation angle of 0.3° and an exposure time of 10 min per frame were used due to the small size of the crystals. The data were integrated using SAINT and scaled using XPREP in the Bruker analysis package. Crystals belong to the space group P21, with unit cell dimensions a = 42.59 Å, b = 135.8 Å, and c = 83.3 Å, and β = 93.93°. For the final data set, scaled to 2.5 Å, the completeness was 61% with a redundancy of 1.5 and Rsym of 18.0% (Table 2). The low completeness is due to the fact that the crystals were slightly nonisomorphous, and therefore we were only able to combine a limited number of crystals for the final data set. However, this was partially compensated for by the fact that there were two Fab molecules in the asymmetric unit.
TABLE 2.
Refinement statistics for the Fab MAb A6.2 structure
| Parameter | Result for Fab A6.2 structurea |
|---|---|
| PDB accession no. | 4NCC |
| Wavelength (Å) | 1.5418 |
| Data resolution (Å) | 42.5–2.5 (2.55–2.5) |
| No. of reflections | |
| Total | 32,189 |
| Independent | 20,127 |
| Avg I/σ〈I〉b | 11.5 (5.5) |
| Completenessc | 61.1 (48) |
| Redundancy | 1.5 |
| Rsym (%)d | 18.0 (22.2) |
| Refinement resolution (Å) | 42.5–2.5 (2.55–2.5) |
| Total atoms | |
| Nonsolvent | 6,560 |
| Solvent | 370 |
| Rworke | 20.4 (20.4) |
| Rfree (%)f | 27.3 (29.8) |
| Total reflections | 24,470 (930) |
| Avg B (Å2) | |
| Nonsolvent atoms | 19.1 |
| Solvent atoms | 15.7 |
| RMSDg | |
| Bond length (Å) | 0.011 |
| Bond angle (°) | 1.47 |
| Ramachandran analysis (%) | |
| Most favored region | 95.5 |
| Additionally allowed | 3.4 |
| Disallowed | 1.1 |
Statistics for the highest-resolution bin of reflections are in parentheses.
Intensity signal/noise ratio.
Completeness of the unique diffraction data.
Rsym = ΣhΣj|Ihj − 〈Ih〉|/ΣhΣjIhj, where Ihj is the intensity of observation j of reflection h and 〈Ih〉 is the mean intensity for multiply recorded reflections.
Rwork = Σh‖Fo| − |Fc‖/Σh|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes for reflection h.
Rfree was calculated against a 10% random sampling of the reflections that were removed before structure refinement.
RMSD, root mean square deviation.
Structure determination.
Two Fab molecules were observed in the crystallographic asymmetric unit since that organization yielded a Vm (Vm = V/M, where V is the volume of the unit cell and M is the total mass of protein in the unit cell) of ∼2.5. The structure was determined by molecular replacement using the program PHASER in the CCP4 package (35). Coordinates from the heavy chain of PDB 1OTS and the light of PDB 2ZKH were chosen as search models from several Fab structures because of slightly superior cross-rotation function correlations. These two models were broken into two pieces at the hinge region to yield the four individual immunoglobulin domains for searching. The model was refined using PHENIX (36) and rebuilt using the program COOT (37). During each refinement cycle, the noncrystallographic symmetry was highly restrained, the individual B values were restrained, and omit maps (Fo − Fc) were used to direct building efforts. The final model had an Rmodel of 20.4% and an Rfree of 27.3%. Refinement and data statistics are shown in Table 2. These refinement statistics are better than the average PDB structure to this resolution as per analysis using PROCHECK in the CCP4 package (35).
Generation of mutant MNV-1 P domain.
Site-directed mutagenesis was performed on the MNV-1 P domain expression plasmid (27) with QuikChange (Stratagene) following the manufacturer's recommendation. For each mutant plasmid, two complementary PCR primers (Table 3) containing mutations were used to introduce amino acid changes. All plasmids were screened and confirmed by DNA sequencing. All recombinant mutant proteins were expressed and purified as previously reported (27).
TABLE 3.
Primers for MNV-1 P domain site-directed mutagenesis
| Mutation | Direction | Primer sequence (5′→3′) |
|---|---|---|
| Y2965K | Forward | GGCTGCCAAAGAGTTCCAATCGGGCACCGGTG |
| Reverse | GGAACTCTTTGGCAGCCTCCGCCGCAAAGCAGG | |
| F297A | Forward | GAGGCTGCCTATGAGGCCCAATCGGGCACCGG |
| Reverse | CCGGTGCCCGATTGGGCCTCATAGGCAGCCTC | |
| F297I | Forward | CTATGAGATCCAATCGGGCACCGGTGAGGTG |
| Reverse | CGATTGGATCTCATAGGCAGCCTCCGCCGC | |
| F297S | Forward | GCGGAGGCTGCCTATGAGTCCCAATCGGGC |
| Reverse | GCCCGATTGGGACTCATAGGCAGCCTCCGC | |
| Q298E | Forward | CTGCCTATGAGTTCGAATCGGGCACCGGT |
| Reverse | ACCGGTGCCCGATTCGAACTCATAGGCAG | |
| Q298S | Forward | GGCTGCCTATGAGTTCTCATCGGGCACCGGTGAG |
| Reverse | CTCACCGGTGCCCGATGAGAACTCATAGGCAGCC | |
| S299A | Forward | GGCTGCCTATGAGTTCCAAGCGGGCACCGG |
| Reverse | CCGGTGCCCGCTTGGAACTCATAGGCAGCC | |
| S299R | Forward | GGCTGCCTATGAGTTCCAAAGGGGCACCGGT |
| Reverse | ACCGGTGCCCCTTTGGAACTCATAGGCAGCC | |
| G300F | Forward | CTATGAGTTCCAATCGTTCACCGGTGAGGTGGCG |
| Reverse | CGCCACCTCACCGGTGAACGATTGGAACTCATAG | |
| G300K | Forward | CCTATGAGTTCCAATCGAAGACCGGTGAGGTGGCGAC |
| Reverse | GTCGCCACCTCACCGGTCTTCGATTGGAACTCATAGG | |
| G300R | Forward | ATGAGTTCCAATCGCGCACCGGTGAGGTG |
| Reverse | CACCTCACCGGTGCGCGATTGGAACTCAT | |
| V378A | Forward | TGTTCGCCAGCGCCACTGCTGCGGC |
| Reverse | GCCGCAGCAGTGGCGCTGGCGAACA | |
| V378L | Forward | GTGTTCGCCAGCCTCACTGCTGCGG |
| Reverse | CCGCAGCAGTGAGGCTGGCGAACAC | |
| A381F | Forward | GTTCGCCAGCGTCACTGCTTTCGCCTCTCTTGACTTGG |
| Reverse | CCAAGTCAAGAGAGGCGAAAGCAGTGACGCTGGCGAAC | |
| A381G | Forward | AGCGTCACTGCTGGGGCCTCTCTTGAC |
| Reverse | GTCAAGAGAGGCCCCAGCAGTGACGCT | |
| A382G | Forward | GCTGCGGGGTCTCTTGACTTGGTGGATGGCAGG |
| Reverse | CAAGAGACCCCGCAGCAGTGACGCTGGCGAACACC | |
| A382K | Forward | CCAGCGTCACTGCTGCGAAGTCTCTTGACTTGGTGGA |
| Reverse | TCCACCAAGTCAAGAGACTTCGCAGCAGTGACGCTGG | |
| A382P | Forward | GCGTCACTGCTGCGCCCTCTCTTGACTTG |
| Reverse | CAAGTCAAGAGAGGGCGCAGCAGTGACGC | |
| A382R | Forward | GCGTCACTGCTGCGCGCTCTCTTGACTTGG |
| Reverse | CCAAGTCAAGAGAGCGCGCAGCAGTGACGC | |
| D385E | Forward | GCGGCCTCTCTTGAATTGGTGGATGGCAG |
| Reverse | CTGCCATCCACCAATTCAAGAGAGGCCGC |
Peptide ELISA.
For enzyme-linked immunosorbent assays (ELISAs), 96-well enzyme immunoassay/radioimmunoassay (EIA/RIA) Costar plates were first coated with 100 μl of 50 μg/ml P domain protein in 50 mM sodium carbonate buffer (pH 9.6) overnight at 4°C. The coated plates were blocked with a 3% (wt/vol) solution of dry milk dissolved in 50 mM sodium carbonate buffer (pH 9.6) at room temperature for 1.5 h. The plates were rinsed with wash buffer (50 mM sodium chloride, 0.05% Tween 20). To each well, 100 μl of MAb A6.2 diluted in ELISA buffer (0.15 M sodium chloride, 1 mM EDTA, 0.1% BSA, 0.05% Tween 20, in 50 Tris buffer [pH 7.4]) was added, and the mixture was incubated at room temperature for 1.5 h. The plates were then washed and 100 μl of anti-mouse antibody was conjugated with horseradish peroxidase for 1.5 h. The plates were washed, tetramethylbenzidine substrate was added, and the reaction was stopped by addition of 100 μl of 1 N phosphoric acid. Absorption was measured at 450 nm by ELISA plate reader.
Western blot analysis.
Western analysis was performed on all purified P domains in case a mutation aggregated the protein that might affect ELISAs. Approximately 25 μg of purified P domain proteins was separated by reduced 15% SDS-PAGE and blotted to membranes using a Hoefer Scientific Semi-Phor blotter. The membrane was blocked in 3% milk in PBS buffer overnight, and diluted A6.2 was added for 1.5 h. The membranes were washed with PBS buffer and incubated with rabbit anti-mouse IgG peroxidase-conjugated secondary antibody for 1.5 h. The membranes were washed and then developed in tetramethylbenzidine substrate solution.
Western blots and Coomassie-stained gels were scanned, and bands were quantified using ImageJ (http://rsbweb.nih.gov/ij/). The ratio of the peak area of the mutant protein from the Western blot was divided by the Coomassie-stained bands, and this number was divided by the Western blot/Coomassie ratio of the wild-type (WT) protein and expressed as a percentage.
SPR analysis.
Real time biomolecular interaction analysis was performed by surface plasmon resonance (SPR) using a BIAcore 2000 instrument equipped with nitrilotriacetic acid (NTA) sensor chip. Purified polyhistidine-tagged proteins, wild-type MNV-1 P domain proteins, or various mutants were immobilized on flow cell 2. Meanwhile, His-tagged CR3 with a similar RU level was placed on flow cell 1 as in-line negative control. To dampen the tight binding of MAb A6.2, Fab fragments at a range of concentration (25 to 800 nM) with duplicates were used as analytes along with several blanks of running buffer (100 mM sodium phosphate [pH 7.4], 400 mM NaCl, 40 μM EDTA, 0.005% [vol/vol] Tween 20). All experiments were carried out at 25°C at a constant flow rate of 30 μl/min. Briefly, each cycle of running started with charging the NTA chip with 40 μl 0.5 mM NiCl2, followed by injection of 20 μl His-tagged proteins, control (40 nM), and ligands (100 nM) into flow cells 1 and 2, respectively. After a stabilizing wash, the analyte Fab solution was injected with KinInj model (40 μl for association, 3-min dissociation), and the binding signals were monitored. Finally, the NTA surface was regenerated by a pulse of 40 μl EDTA (350 mM). Data analysis was conducted with BIAevaluation package. Curve fittings were done with the 1:1 Langmuir binding model. All fitting quality critiques met requirements.
Fitting of the atomic structures into the cryo-TEM image reconstruction density.
For the fitting process, the inner core (radius, 150 Å) of the electron density was removed from the previous cryo-transmission electron microscopy (cryo-TEM) image reconstruction (22) using the BEDITIMG program in the BSOFT package (38). The crystal structure of the MNV-1 P domain (27) and two copies of the structure of Fab A6.2 were fitted by eye into the molecular envelope. Unlike our previous fitting study when the structure of the Fab was unknown (22), this time a Fab was placed over both the P domain A and B conformations. It should be noted that the elbow angle in the Fab structure was noticeable enough to position the Fab into a unique orientation with regard to the location of the heavy and light chains. The program COLLAGE in the SITUS package was then used to refine the position of the Fab and P domain structures in an unbiased manner.
Protein structure accession numbers.
Coordinates and structure factors for the A6.2 Fab crystal structures were deposited in the Protein Data Bank with the code 4NCC.
RESULTS AND DISCUSSION
Monoclonal antibody A6.2 neutralization escape viruses are generated by serial passaging in cell culture.
A previous study has described that the mutation L386F in the MNV-1 capsid protein arises in vitro after 3 passages of virus in the presence of the neutralizing MAb A6.2 (30). To determine whether other neutralization escape mutants could be generated, MNV-1 was passaged five times through RAW 264.7 cells in the presence of increasing concentrations of MAb A6.2 or a nonneutralizing isotype control antibody. Passage 0 (P0) virus (i.e., starting virus stock) was neutralized by 60 ng and 200 ng MAb A6.2 (P < 0.01). However, P3 and P5 viruses grown under MAb A6.2 selection, but not the isotype control MAb, were resistant to 60 ng and 200 ng of MAb A6.2, respectively (P < 0.05) (Fig. 1A). These data suggest that MAb A6.2 drives the evolution of an MNV-1 quasispecies able to escape MAb neutralization.
FIG 1.
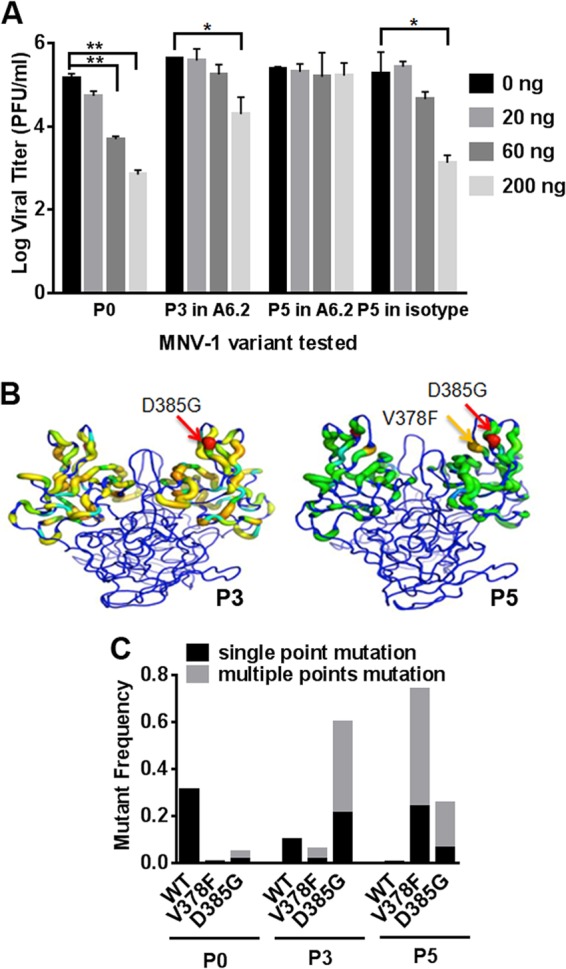
Generation of monoclonal antibody (MAb) A6.2 escape viruses by serial passaging in cell culture. (A) MNV-1 was passaged 5 times through RAW 264.7 cells in the presence of increasing concentrations of the neutralizing MAb A6.2 or a nonneutralizing isotype control antibody. Passage 0 (P0) virus, P3 virus grown with MAb A6.2, and P5 viruses from both conditions were then analyzed in a neutralization assay with 0, 20, 60, and 200 ng MAb A6.2. Data are from two independent experiments. The error bars indicate standard errors. Statistical analysis was performed using the t test. *, P < 0.05; **, P < 0.01. (B) Viruses from RAW 264.7 cell lysates obtained after passage in MAb A6.2 or the isotype control were analyzed by 454 DNA sequencing. Mutations were graphed onto the MNV-1 P domain crystal structure. Ribbon thickness and color (blue to red) are approximately equal to the log frequency of each mutation. The dominant MNV-1 P domain mutants in passages 3 (P3) and 5 (P5) are shown in red (D385G) and orange (V378F), respectively. (C) Clonal sweep trajectory of MNV-1 mutants under MAb A6.2 pressure. Wild-type MNV-1 was the dominant phenotype at P0. MNV-1 containing the mutation 385G became the dominant genotype by P3, with a shift to 378F-containing MNV-1 mutants by P5. The frequency of indicated single point mutations is shown in dark boxes, while gray boxes show the same mutation combined with other mutations.
RNA viruses generate about one mutation per genome per replication cycle (3, 39). Therefore, every point mutation and several double mutations could theoretically be present in the starting virus stock that was used in the passaging experiment. To identify and monitor the effect of MAb treatment on the diversity and frequency of mutations in the virus populations, P0-to-P5 MAb A6.2-treated viruses were analyzed by Roche 454 DNA deep sequencing using PCR amplicons that spanned part of the S domain and the entire P domain (nucleotides [nt] 5473 to 6681). The resulting sequences were compared to the MNV-1 genome, and the total mutations were graphed onto the MNV-1 P domain crystal structure (Fig. 1B). The frequency of each mutation changed per passage. D385G was dominant in P3, while both D385G and V378F were the most abundant mutations by P5 (Fig. 1B).
Among the several point mutations observed in the deep sequencing data, V378F, D385G, and L386F were the only single point mutations with frequencies above 1% in at least one passage. The total diversity of the quasispecies was reduced from 270 different species at P0 to 237 at P5. Wild-type MNV-1 was predominant at P0 (31%), D385G by P3 (21%), and V378F (24%) by P5 (Fig. 1C). The double mutation D385G V378F was observed in P4 and P5, while other multiple mutations involved at least one of these two mutations. In addition, MNV-1 mutants under MAb A6.2 pressure shifted from the predominant mutation D385G at P3 to V378F by P5, suggesting that V378F may be more resistant to MAb A6.2 neutralization than D385G. The dominant D385G mutation at P3 may be a result of its high frequency of 4.9% in P0. Interestingly, the previously identified escape mutation L386F (30) was not the dominant phenotype during these particular passaging experiments. Taken together, these data indicate that multiple MAb A6.2 neutralization escape mutants are generated by serial passaging.
Our findings highlight the advantage of maintaining a diverse and flexible quasispecies, which enables the population as a whole to quickly adapt to new selection pressures. In this instance, the D385G mutant was likely more fit than the wild-type virus in the presence of MAb A6.2. MNV-1 was therefore able to survive MAb A6.2 neutralization and adapt to its new environment after just three rounds of passaging. Similarly, the V378F mutant was already at 5.9% of the quasispecies by P3. Therefore, when MAb A6.2 pressure increased, the V378F mutant became dominant by P5, probably due to its higher fitness in the presence of the MAb.
Mutations in the E′F′ loop of the P domain mediate MNV-1 escape from MAb A6.2 neutralization.
The three identified neutralization escape mutations, V378F, D385G, and L386F, all lie in the E′F′ loop of the MNV-1 P domain. We recently constructed a pseudoatomic model of the MNV-1/A6.2 Fab complex using cryoelectron microscopy image reconstruction and found that the E′F′ loop is part of the MAb A6.2 epitope (22, 27). This was experimentally confirmed because exchanging residues in the E′F′ loop with corresponding amino acids from the human norovirus Norwalk virus led to the loss of neutralization (27). However, the model from electron microscopy suggested that MAb A6.2 epitopes are located on both the A′B′ and E′F′ loops (22, 27). To clarify the relative importance of these two loops, nine A′B′ loop and 12 E′F′ loop MNV-1 mutants were created by site-directed mutagenesis. Amino acids chosen for the mutations were designed to block antibody binding due to drastic changes in size and hydrophobicity, while others were conservative and predicted not to affect MAb A6.2 binding (Table 4). Also included in the analysis were the published attenuation mutation E296K (40), the published MAb A6.2 escape mutation L386F (30), mutation T301I, which spontaneously arose in passage 2 of the molecular clone of MNV-1, and the escape mutations V378F, D385G, and V378F D385G identified during serial passaging.
TABLE 4.
List of mutations in MNV-1 P domains
| Amino acid mutation(s) | Descriptiona |
|---|---|
| A′B′ loop | |
| E296Kb | Same size, added charge |
| Q298E | Same size, reduced charge |
| Q298S | Smaller, still polar |
| S299A | Smaller, no clash |
| S299R | Larger, added charge |
| G300F | Larger, hydrophobic |
| G300K | Larger, added charge |
| G300R | Larger, added charge |
| T301I | Same size, same charge |
| E′F′ loop | |
| V378A | Smaller, less hydrophobic |
| V378Fc | Larger, more hydrophobic |
| V378L | Same size, same charge |
| A381F | Larger, more hydrophobic |
| A381G | Smaller, flexible |
| A382G | Smaller, no clash |
| A382K | Larger, added charge |
| A382P | Larger, structural disruptor |
| A382R | Larger, added charge |
| D385E | Same size, same charge |
| D385Gd | Smaller, added charge |
| L386Fe | Larger, more hydrophobic |
| L378F D385Gc | Double mutant |
Mutant viruses were generated, and the effect of these mutations on viral growth was analyzed (Fig. 2). In all cases, the growth of the mutant viruses was similar to that of wild-type (WT) virus, demonstrating that the point mutations did not affect viral growth kinetics. Each mutant was then assayed for MAb A6.2 neutralization in vitro (Fig. 3). WT MNV-1 and all A′B′ loop mutants were neutralized by 60, 200, and 600 ng of MAb A6.2 (Fig. 3A). Differential susceptibility to neutralization was observed at the lowest concentration of 20 ng of antibody, with almost complete neutralization of S299A, S299R, and G300F viruses, partial neutralization of E296K, Q298S, and G300R viruses, similar to WT, and no neutralization of G300K and T301R viruses. In contrast, all E′F′ loop mutants were resistant to neutralization with 20 ng of MAb A6.2. At 10-fold-higher antibody concentrations, A382K, A382P, and A382R mutants were able to escape neutralization, while the V378A, V378L, A381F, A381G, A382G, and D385E mutants were neutralized (Fig. 3B). In addition, the naturally occurring V378F, L386F, and V378F D385G MAb escape mutants were resistant to all tested concentrations of MAb A6.2 in vitro, while the D385G mutant was neutralized at 600 ng of A6.2 (Fig. 3C). Thus, the D385G mutant has a lower resistance to MAb A6.2 neutralization than the V378F mutant, which may have accounted for the relative decrease of amino acid G385 and increasing dominance of amino acid F378 at P5 during passaging of virus in the presence of MAb A6.2 (Fig. 1).
FIG 2.
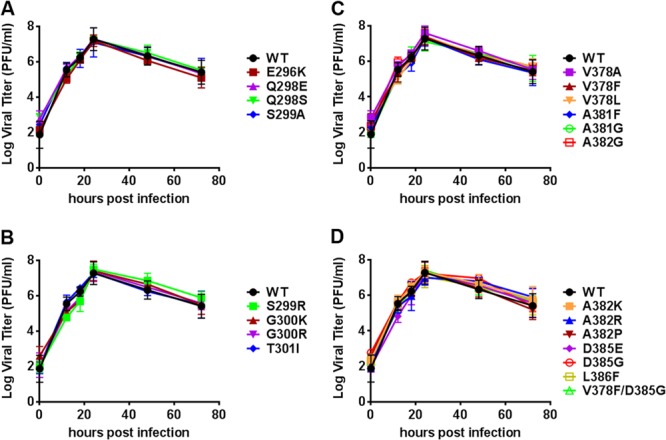
Single amino acid substitutions did not affect MNV-1 growth. RAW 264.7 cells were infected with recombinant viruses having mutations in the A′B′ loop (A and B) or E′F′ loop (C and D) at an MOI of 2 on ice for 1 h. The inoculum was removed, and cells were infected at the indicated time points. Virus titers were determined by plaque assay. The titer for each mutant virus was compared to that for wild-type (WT) MNV. Data are presented as means ± standard errors (SE) from duplicate samples in three independent experiments.
FIG 3.
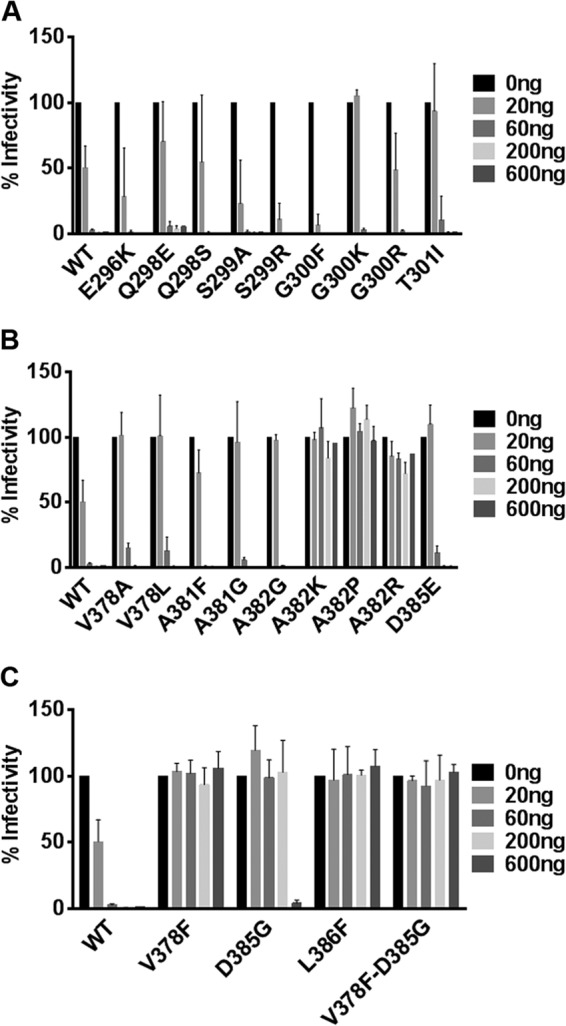
Mutations in the E′F′ loop of the P domain mediate MNV-1 escape from MAb A6.2 neutralization in culture. WT MNV and nine A′B′ loop single point mutants (A), nine E′F′ loop single point mutants (B), and three naturally occurring mutants as well as one double mutant (C) were subjected to in vitro MAb A6.2 neutralization. Virus was incubated with indicated concentrations of MAb A6.2 for 30 min at 37°C before infection of RAW 264.7 cells. Viral titers were measured by plaque assay. Percent infectivity of virus titers at different concentrations of MAb A6.2 was calculated relative to the control without MAb A6.2 set to 100%. Data are presented as means ± SE from at least three independent experiments.
To determine whether escape from in vitro neutralization also occurs in vivo, two nonescape viruses with mutations in the A′B′ loop (Q298E and S299A), three full escape E′F′ loop mutant viruses (A382K, A382R, and L386F), and WT MNV-1 were selected for analysis. To enhance our ability to see MAb neutralization effects, STAT1−/− mice, which support higher viral loads than wild-type mice (15), were chosen. Mice were simultaneously infected with 6 × 105 PFU of each virus orally and injected with 0.5 mg of MAb A6.2 or isotype control MAb intraperitoneally. Tissues were harvested 2 days postinfection, and viral titers were measured by plaque assay (Fig. 4). Viral titers in the gastrointestinal tract and at systemic sites (liver, lung, and spleen) of mice infected with WT or Q298E and S299A mutant viruses were significantly reduced by MAb A6.2 administration but not by the isotype control (Fig. 4A to C). The effect of MAb A6.2 neutralization was greater in liver, lung, and spleen than in the intestine. This may be due to the route of MAb administration, because intraperitoneal injection first delivers the MAb to systemic sites, while virus replication initiates in the intestine. Mice infected with the A382K or A382R mutant did not show significant changes in viral titers between MAb A6.2 and isotype control-treated mice (Fig. 4D and E). These data demonstrated a good correlation between in vitro and in vivo sensitivity or resistance to MAb A6.2 neutralization. In contrast, viral titers in mice infected with the L386F mutant and treated with 0.5 mg MAb A6.2 were significantly different in the liver (P = 0.000), spleen (P = 0.026), colon (P = 0.010), and proximal ileum (P < 0.018) from those in isotype control-treated animals (Fig. 4F). However, viral titers in L386F virus-infected mice were similar between MAb A6.2- and isotype-treated animals when the antibody concentration was reduced to 0.1 mg or 0.015 mg (Fig. 4I and J). Both concentrations were still sufficient to neutralize WT MNV-1 in vivo (Fig. 4G and H). This suggested that the L386F mutant is susceptible to high MAb A6.2 pressure in vivo but not in vitro. Of note, viral titers in feces did not differ between MAb A6.2 and isotype treatment for any virus, suggesting that MAb neutralization does not affect virus shedding.
FIG 4.
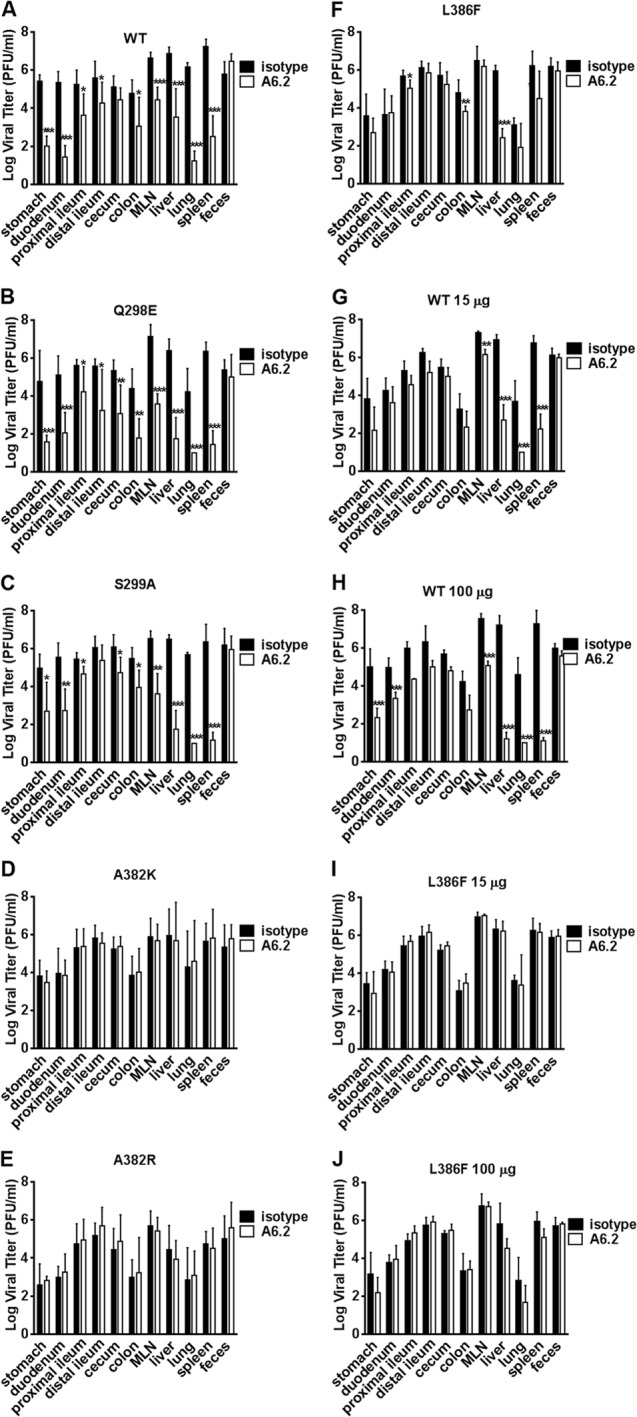
Mutations in the E′F′ loop of the MNV-1 P domain mediate escape from MAb A6.2 neutralization in mice. STAT1−/− mice were simultaneously infected with 106 PFU of the indicated recombinant MNV-1 orally and injected with MAb A6.2 (open box) or isotype control MAb (black box) intraperitoneally. Virus titers were measured 48 h postinfection in indicated tissues from animals infected with the wild type (A) or one of the Q298E (B), S299A (C), A382K (D), A382R (E), or L386F (F) mutants with 500 μg MAb, WT-infected animals injected with 15 (G) and 100 (H) μg MAb, and L386F mutant-infected animals injected with 15 (I) and 100 (J) μg MAb, respectively. Data are presented as means ± standard deviations (SD) from at least five mice per condition from at least two independent experiments. Statistical analysis was performed using the t test to compare the virus titer for each tissue between MAb A6.2-injected mice and the isotype control. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Taken together, these data suggest that none of the single point mutations affected viral growth and that in vitro neutralization escape is generally recapitulated in vivo. More importantly, amino acid mutations in the E′F′ loop of the MNV-1 P domain are critical for escape from MAb A6.2 neutralization in cell culture and in mice.
Viral escape from neutralization correlates with lack of P domain binding to MAb A6.2.
Since MAb A6.2 binds to MNV-1 virions (22), we tested how well neutralization capacity correlated with MAb A6.2 binding. MNV-1 P domains containing the same single amino acid mutations analyzed previously in the context of virions were bacterially expressed and purified. In addition, P domains for the Y295K, F297A, F297I, and F297S A′B′ loop mutants were also generated, as these mutations tested additional changes in the size and/or hydrophobicity of the side chains for their ability to create escape mutations (Table 4).
The binding of MAb A6.2 to the various P domain mutants was analyzed by Western blotting (Fig. 5) and ELISA (Fig. 6). For surface plasmon resonance binding studies (Table 5), Fab fragments were used since, due to avidity, the intact antibodies bound too tightly and eluted too slowly to measure the off-rate constant (koff). In Western blot analysis, MAb A6.2 reacted well with purified WT P domain. Similar or better reactivity compared to the WT P domain was seen with the P domain F297I, S299A, S299R, G300K, V387L, and A381G mutants, reactivity was weaker with the F297S, Q298E, Q298S, G300F, G300R, V378A, A381F, and A382P mutants, and no binding was detected to the P domain Y295K, F297A, A382G, A382K, A382R, and D385E mutants (Table 5 and Fig. 5). Antibody detection of these P domains suggested that MAb A6.2 likely recognizes a linear epitope in the P domains. This is markedly different from other viruses, such as human rhinovirus (HRV), where none of the monoclonal antibodies raised against intact virions reacted to denatured and blotted viral proteins (41). One potential explanation might be that MAb A6.2 appears to mainly bind to the flexible loops on the P domain, while the antibodies to human rhinovirus recognize a larger portion of the three-dimensional surface.
FIG 5.
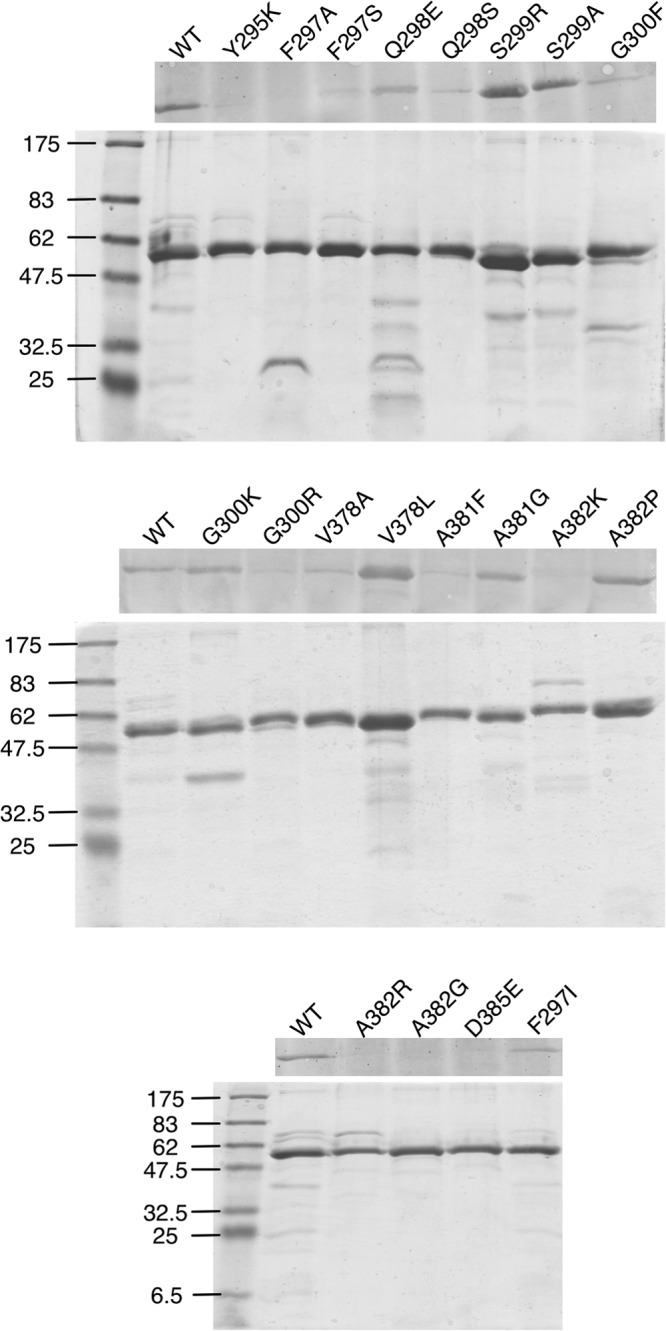
Analysis of recombinant MNV-1 P domains. Shown here are the SDS-PAGE and Western analyses of the various purified P domain mutants. The upper panel shows the Western blots. The lower panel shows the Coomassie-stained SDS-PAGE gels to demonstrate the purity of each and that approximately the same amount of P domain was loaded in each well.
FIG 6.
Binding of recombinant MNV-1 P-domains to MAb A6.2. ELISA plates were coated overnight at 4°C with bacterially expressed, recombinant MNV-1 P domain A′B′ loop (A) and E′F′ loop (B) mutants at 50 μg/ml in each well. Diluted purified MAb A6.2 followed by secondary antibodies was incubated for 60 min at 37°C. MNV.CR3 P domain was used as a negative control. Data are presented as means ± SE from three independent experiments. The three bars in each set of bars represent three dilutions of purified MAb A6.2: 1, 0.1, and 0.01 μg/ml.
TABLE 5.
Summary of MAb A6.2 binding results to the MNV-1 P domain
| Virus | Result by: |
||
|---|---|---|---|
| ELISAa | Western blot (%) | Biacore | |
| WT | + | 100 | 4.6 × 10−8 |
| CR3 (negative control) | − | 0 | 0 |
| Mutants | |||
| Y295K | − | 0 | 0 |
| F297A | + | 0 | 0 |
| F297S | − | 9 | 0 |
| F297I | +/− | 52 | 0 |
| Q298E | + | 67 | 0 |
| Q298S | − | 12 | 1.5 × 10−8 |
| S299A | + | 114 | 5.0 × 10−8 |
| S299R | + | 139 | 2.5 × 10−8 |
| G300F | − | 29 | 0 |
| G300K | + | 116 | 8.0 × 10−7 |
| G300R | +/− | 19 | 0 |
| V378A | +/− | 48 | 1.3 × 10−6 |
| V378L | + | 216 | 1.3 × 10−6 |
| A381F | +/− | 32 | 0 |
| A381G | + | 155 | 7.1 × 10−8 |
| A382G | − | 0 | 0 |
| A382K | − | 0 | 0 |
| A382P | + | 60 | 0 |
| A382R | − | 0 | 0 |
| D385E | − | 0 | 0 |
+, signal within the error of wild-type protein; −, signal within error of controls; +/−, intermediate result.
In general, binding analysis data by ELISA correlated well with those seen by Western blotting, except for the F297A virus (Table 5). Furthermore, Fab binding to the P domain by surface plasmon resonance also generally matched well with either ELISA or Western blotting, except in the case of the Q298E and A382P mutants. A mixed correlation was observed when comparing the data on MAb A6.2 binding to the recombinant P domain with the data on neutralization using intact virions. A good correlation was observed for the A382K and A382R mutants, which did not react to MAb A6.2 in all three assays and were strong escape mutants in vitro and in vivo. Conversely, MAb A6.2 bound to WT virus and the S299A, S299R, G300K, and A381G mutants very well in all three assays and was neutralized in vitro and additionally in vivo for the S299A mutant. No correlation between binding and neutralization was observed with the D385E and A382G mutants, which did not react to MAb A6.2 in all three assays, yet were neutralized in vitro. The reasons for this are not clear but may be due to different sensitivities of the various assays. For example, A382G virus infectivity was not affected at the two lowest dilutions of antibody, perhaps suggesting an intermediate effect not reflected in the in vitro binding assays. There were also a number of mutations that suggested intermediate effects on antibody binding. The V378A mutant had weak signals in ELISAs and Western blots, and the V378L mutant bound well in ELISAs and Western blots, but both P domains bound MAb A6.2 Fabs with ∼10-fold-weaker affinities and were slightly less sensitive to MAb A6.2 in in vitro neutralization assays. Interestingly, the Q298E, G300F, G300R, A381F, and A382P mutants bound antibody in ELISAs and/or Western blots, but binding was not observed by plasmon resonance. This may be due to variations in the sensitivities of the assays. For example, the intact IgG avidity to this mutant may be strong enough to react in Western blots, ELISAs, and neutralization assays, but the intrinsic affinity of the Fab alone as measured by plasmon resonance may be too weak to detect binding. Interestingly, Q298S virus did not react in an ELISA and reacted weakly in the Western blot but bound well in the plasmon resonance experiment. This might be due to protein instability when blotted to nitrocellulose or the plastic ELISA plates, while stability is maintained in solution, supporting binding in the context of the nickel affinity chips. Nevertheless, these three very different assays are highly complementary in that they cover a broad range of affinities and look at different types of antigen presentation. While results from binding assays with recombinant protein were not in all cases a faithful predictor of neutralization activity in vitro, all of the viruses with mutations that mediated escape from neutralization were unable to bind to MAb A6.2 by plasmon resonance. One possible explanation is that subtle changes in folding occur between the P domains of native virions versus bacterially expressed P domains. Thus, while the bacterially expressed WT P domain folds into a biologically relevant conformation and can block binding of MNV-1 to macrophages (27), this may not be true for all mutant P domains. Finally, the specific conformation of a tightly bound epitope-Ab complex in a biologically native environment could potentially adversely impact the successful engagement of the virus with the cell receptor responsible for initiating the infection, despite high-affinity binding.
The atomic structure of the MAb A6.2 Fab shows a very hydrophobic CDR3 loop.
To better understand the interaction of MAb A6.2 with its antigen, the structure of the A6.2 Fab was resolved to ∼2.5 Å (Fig. 7A and Table 2). Typically, the heavy chain, in particular the third hypervariable loop CDR3, makes the majority of the contact with the epitope of the antigen (42). However, when the molecular surface of the Fab A6.2 paratope was assigned a hydrophobic index range from nonpolar (blue) to charged (red), a striking hydrophobicity was observed for the CDR3 loop (Fig. 7B). The strong hydrophobic (blue) patch is mainly comprised of the CDR3 loop, which has the sequence YFYALDYW.
FIG 7.

Crystal structure of anti-MNV-1 Fab A6.2. (A) Shown here is a stereo diagram of a typical example of the final electron density of the refined structure of Fab A6.2 to an ∼2.5-Å resolution. The carbon, nitrogen, oxygen, and sulfur atoms are colored white, blue, red, and yellow, respectively. (B) In this stereo figure, the molecular surface of the Fab MAb A6.2 paratope is colored according to the hydrophobic index, ranging from blue for nonpolar to red for charged. The main chain atoms for the whole Fab were assigned a relatively neutral hydrophobicity index corresponding to the color green. The white line denotes the border between the heavy and light chains. The approximate location of the very hydrophobic CDR3 loop is also indicated.
Improved fitting of atomic resolution structures into the cryo-TEM electron density.
Previously, we determined the cryo-TEM structure of MNV-1 bound to the A6.2 Fab fragment (22) and subsequently fit the atomic structure of the P domain into the density map (27) to approximately determine the paratope-epitope contact interface. This modeling was performed with a randomly selected antibody structure since the sequence and structure of A6.2 were unknown. The atomic structure of A6.2 not only shows the chemical nature of the paratope but also shows the true elbow angle between variable and constant domains. With the atomic structures of both the antigen and the antibody determined, the fitting process was revisited. First, the inner core of the MNV/Fab electron density, representing the shell domain of the capsid and the inner RNA interior, was removed. Two copies of the Fab A6.2 structure and the P domain were then roughly fitted into the electron density. This time, the elbow angle was recognized and used during the fitting process. Once placed into approximate positions, the program COLLAGE in the SITUS package (43) was used to refine the orientations and positions of the three components in an unbiased manner (Fig. 8). Surprisingly, the paratope-epitope interface had far fewer molecular clashes in the A subunit than the B subunit. In the B subunit, the E′F′ and A′B′ loops are parallel to each other and stick straight up from the top of the P domain, causing the E′F′ loop to clash with the CDR3 loop of the heavy chain. In the A subunit, the E′F′ and A′B′ loops are splayed apart and the CDR3 loop inserts between these two loops. This places the hydrophobic CDR3 loop of MAb A6.2 directly into a hydrophobic cleft between the A′B′ and E′F′ loops in the A conformation. In contrast, the same residues are deeply buried under the tips of the A′B′ and E′F′ loops in the B conformation and are not accessible to MAb A6.2. Taken together, the modeling data strongly suggest that MAb A6.2 prefers the A conformation over the B conformation both in terms of structural complementarity and with regard to hydrophobicity.
FIG 8.

Refitting MAb A6.2 Fab structure into the cryo-TEM for Fab-virus interaction. With the crystal structures of both the Fab and the P domain determined, the fitting of the structures into the molecular envelopes from the cryo-TEM image reconstructions was revisited. (Top) As detailed in Materials and Methods, the program package SITUS was used to refit the structures in an unbiased manner. In the previous crystal structure (27), there are two conformations for the P domain that were designated subunits A (green) and B (brown). The heavy and light chains for the Fab are shown in red and blue, respectively. (Bottom) Details of the fitted ensemble. The two loops that comprise the epitope are splayed apart in the A conformation compared to the B conformation. This causes severe clashes in the case of the B conformation (left side) compared to the A conformation (right side). Furthermore, the splayed conformation of A exposes a hydrophobic patch that complements the similarly nonpolar CDR3 heavy-chain loop.
Mutant viruses escape MAb A6.2 neutralization by stabilization of the B conformation.
Using the new model of the paratope-epitope interface, we analyzed the location of the amino acids shown to mediate escape. At least three of the escape mutations, L386F, A382K, and A382R, may block antibody binding by stabilizing the B conformation, while the escape mutations A382P and V378F likely affected the overall structure of the loops. L386 lies between the A′B′ and E′F′ loops (Fig. 9A and B). In the B conformation, the L386F escape mutant remained buried and did not contact the bound antibody. In the A conformation, the L386F mutation would place the larger Phe side chain directly in contact with the bound antibody. Exposure of so much of this hydrophobic residue is energetically unfavorable. This implied that the L386F mutation favors the B conformation, which was not bound by MAb A6.2. Therefore, L386F may be a conformational escape mutation in that it stabilizes the conformation that is not recognized by the antibody (i.e., B conformation). A similar observation was made for A382R, because the A382R mutation in the B conformation likely places the Arg into the solvent and thus should be the favorable structure (Fig. 9C). In contrast, in the A conformation, the A382R mutation would place the Arg side chain into a cluster of basic residues. This would destabilize the A conformation and favor the B conformation to which the antibody cannot bind. A similar mechanism of stabilizing the nonbinding conformation is likely for A382K, which also mediated escape from neutralization. In case of A382P and V378F, the mutation likely disrupted the loop structure to prevent MAb A6.2 binding. On the other hand, other amino acid mutations at positions 378 and 382 (i.e., V378A, V378L, and A382G) are still neutralized. The mutations do not affect antibody binding because they are smaller in size and have the same charge to maintain flexibility of the E′F′ loop. Therefore, the antigen remains in the A conformation to which the antibody can bind.
FIG 9.

MNV-1 P domain mutant viruses may escape MAb A6.2 neutralization by stabilization of the B conformation. (A) The location of the L386F mutation in the MNV-1 P domain. In the B conformation (blue), L386 lies between the two antigenic loops and does not contact the antibody. (B) As shown in this surface rendering of the top of the P domain, L386 (mauve) is fully exposed in the A conformation (tan). This suggests that L366F mutation will stabilize B over A and block antibody binding since the A conformation appears to be preferred for antibody binding. (C) Similarly, the A382R mutation may destabilize the A conformation since it would place an Arg residue in the middle of a highly basic patch of the P domain.
The reason for a loss in MAb neutralization in case of the escape mutation D385G was less apparent, but the overall conformation of the epitope might also be affected. The glycine mutation may give the loop more flexibility or favor a different overall conformation to which MAb A6.2 does not as effectively bind. Alternatively, escape could be mediated by a loss in charge at this position. The fact that the D385G mutation appeared so quickly during bulk passaging (Fig. 1B) may be due to the fact that having a Gly on the tip of this loop has the lowest impact on protein folding compared to its ability to abrogate antibody binding. Furthermore, the mutation was already present at the start of the experiment and was detectable in our viral stock at a level of ∼1% based on the deep sequencing analysis.
In support of the proposed stabilization model described above, the A381F mutation did not mediate escape from neutralization, despite the large, hydrophobic side chain, while V378F and L386F escaped neutralization. With both the V378F and the L386F mutations, the hydrophobicity of a residue between the two antigenic loops is enhanced such that the B conformation may be stabilized over the A conformation. The A381F mutation affects antibody binding (Table 5) but not to the extent needed to result in escape. This intermediary effect could be due to competing effects on antibody binding. The side chain for the A381F mutation is likely pointing into the solvent in the unbound structure. Unlike the L386F mutation, it does not appear to stabilize either the A or B conformation but likely distorts the E′F′ loop. In the A conformation, the side chain in the A381F mutation could interact with the hydrophobic patch on the heavy-chain CDR3 loop when the antibody binds. Therefore, while the deformation caused by the A381F mutation likely affects antibody binding (Table 5), the enhanced hydrophobic interactions with the antibody may partially compensate for the E′F′ loop distortion.
Taken together, these results support a model whereby MAb A6.2 binds to the A conformation and mutations that push the structural equilibrium of the P domain toward the B conformation mediate escape by indirectly affecting antibody binding.
Conclusions.
In the past few years, progress has been made in norovirus vaccine development (44). However, one of the problems facing development is the high adaptability of the virus. Understanding the mechanism of neutralization escape provides insight into development of effective neutralizing antibody vaccines. The E′F′ loop at the tip of the P domain is a region of high mobility (27) that forms at least two conformations, A and B. Using the atomic structure of the MAb A6.2 Fab fragment determined here and revisiting the previous cryoelectron microscopy image reconstruction, structural modeling overwhelmingly supports the notion that MAb A6.2 prefers binding to the A conformation. In this conformation, a large hydrophobic patch is exposed when the A′B′ and E′F′ loops are splayed apart, complementing the markedly hydrophobic CDR3 loop in the heavy chain.
Analysis of the escape mutants demonstrates that MAb A6.2 drives evolution of MNV-1 and that residues in the E′F′ loop of the MNV-1 P domain are necessary for MAb A6.2 neutralization. A number of the amino acid changes that mediate escape from antibody neutralization are the kinds of mutations that are typically observed with escapes and represent steric or charge interference with antibody binding (e.g., A382K, A382R, D385G, and D385E). However, the other mutations that are distal to the antibody contact region can only be explained by effects on the structural equilibrium between the A and B conformations of the P domain (e.g., L386F). This is quite different from, as an example, human rhinovirus (HRV), where the epitope is structurally rigid and held closely to the surface of the virion (45). In the case of HRV, the rigidity of the area around the epitope greatly limits the repertoire of escape mutants in that many of the possible mutations greatly affect viability. In case of MNV, by being able to change the structural equilibrium at residues outside the antibody contact region, the virus potentially has a larger palette of mutations to work with. Recent studies investigating antibody responses toward the human norovirus Norwalk virus in the chimpanzee infection model have demonstrated that the E′F′ loop is involved in the binding of at least some of the neutralizing antibodies (46). This commonality of antibody epitopes in human and murine noroviruses raises the possibility that the principles identified in the present study using MNV are applicable to human norovirus antibody recognition and evolution. By understanding the potential diversity of escape mutants, it thus may be possible to better predict the human norovirus strain that will cause the next pandemic.
ACKNOWLEDGMENTS
This work was supported by Defense Advanced Research Projects Agency (DARPA) contract HR0011-11-C-0093. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank Elizabeth Smith at the University of Michigan Hybridoma Core for growing hybridoma cells and Ian Goodfellow (Cambridge University, United Kingdom) for help with the RNA-based reverse-genetics system. Finally, we would like to acknowledge the use of the National Resource for Automated Molecular Microscopy (NRAMM) for cryo-TEM structure determination of the MNV/Fab complex.
Footnotes
Published ahead of print 5 February 2014
REFERENCES
- 1.Holland J, Spindler K, Horodyski F, Grabau E, Nichol S, VandePol S. 1982. Rapid evolution of RNA genomes. Science 215:1577–1585. 10.1126/science.7041255 [DOI] [PubMed] [Google Scholar]
- 2.Schneider WL, Roossinck MJ. 2001. Genetic diversity in RNA virus quasispecies is controlled by host-virus interactions. J. Virol. 75:6566–6571. 10.1128/JVI.75.14.6566-6571.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lauring AS, Frydman J, Andino R. 2013. The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 11:327–336. 10.1038/nrmicro3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall AJ, Lopman BA, Payne DC, Patel MM, Gastanaduy PA, Vinje J, Parashar UD. 2013. Norovirus disease in the United States. Emerg. Infect. Dis. 19:1198–1205. 10.3201/eid1908.130465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debbink K, Lindesmith LC, Donaldson EF, Costantini V, Beltramello M, Corti D, Swanstrom J, Lanzavecchia A, Vinje J, Baric RS. 2013. Emergence of new pandemic GII.4 Sydney norovirus strain correlates with escape from herd immunity. J. Infect. Dis. 208:1877–1887. 10.1093/infdis/jit370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matthews JE, Dickey BW, Miller RD, Felzer JR, Dawson BP, Lee AS, Rocks JJ, Kiel J, Montes JS, Moe CL, Eisenberg JN, Leon JS. 2012. The epidemiology of published norovirus outbreaks: a review of risk factors associated with attack rate and genogroup. Epidemiol. Infect. 140:1161–1172. 10.1017/S0950268812000234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanton LH, Adams SM, Beard RS, Wei G, Bulens SN, Widdowson MA, Glass RI, Monroe SS. 2006. Molecular and epidemiologic trends of caliciviruses associated with outbreaks of acute gastroenteritis in the United States, 2000–2004. J. Infect. Dis. 193:413–421. 10.1086/499315 [DOI] [PubMed] [Google Scholar]
- 8.Siebenga JJ, Vennema H, Duizer E, Koopmans MP. 2007. Gastroenteritis caused by norovirus GGII.4, The Netherlands, 1994–2005. Emerg. Infect. Dis. 13:144–146. 10.3201/eid1301.060800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siebenga JJ, Vennema H, Renckens B, de Bruin E, van der Veer B, Siezen RJ, Koopmans M. 2007. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 81:9932–9941. 10.1128/JVI.00674-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Payne DC, Vinje J, Szilagyi PG, Edwards KM, Staat MA, Weinberg GA, Hall CB, Chappell J, Bernstein DI, Curns AT, Wikswo M, Shirley SH, Hall AJ, Lopman B, Parashar UD. 2013. Norovirus and medically attended gastroenteritis in U.S. children. N. Engl. J. Med. 368:1121–1130. 10.1056/NEJMsa1206589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindesmith LC, Beltramello M, Donaldson EF, Corti D, Swanstrom J, Debbink K, Lanzavecchia A, Baric RS. 2012. Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLoS Pathog. 8:e1002705. 10.1371/journal.ppat.1002705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindesmith LC, Costantini V, Swanstrom J, Debbink K, Donaldson EF, Vinje J, Baric RS. 2013. Emergence of a norovirus GII.4 strain correlates with changes in evolving blockade epitopes. J. Virol. 87:2803–2813. 10.1128/JVI.03106-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taube S, Kolawole AO, Hohne M, Wilkinson JE, Handley SA, Perry JW, Thackray LB, Akkina R, Wobus CE. 2013. A mouse model for human norovirus. mBio 4(4):e00450–13. 10.1128/mBio.00450-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaudhry Y, Skinner MA, Goodfellow IG. 2007. Recovery of genetically defined murine norovirus in tissue culture by using a fowlpox virus expressing T7 RNA polymerase. J. Gen. Virol. 88:2091–2100. 10.1099/vir.0.82940-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW., IV 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578. 10.1126/science.1077905 [DOI] [PubMed] [Google Scholar]
- 16.Ward VK, McCormick CJ, Clarke IN, Salim O, Wobus CE, Thackray LB, Virgin HW, IV, Lambden PR. 2007. Recovery of infectious murine norovirus using pol II-driven expression of full-length cDNA. Proc. Natl. Acad. Sci. U. S. A. 104:11050–11055. 10.1073/pnas.0700336104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW., IV 2004. Replication of norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2:e432. 10.1371/journal.pbio.0020432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wobus CE, Thackray LB, Virgin HW., IV 2006. Murine norovirus: a model system to study norovirus biology and pathogenesis. J. Virol. 80:5104–5112. 10.1128/JVI.02346-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290. 10.1126/science.286.5438.287 [DOI] [PubMed] [Google Scholar]
- 20.Prasad BV, Hardy ME, Jiang X, Estes MK. 1996. Structure of Norwalk virus. Arch. Virol. Suppl. 12:237–242 [DOI] [PubMed] [Google Scholar]
- 21.Prasad BV, Rothnagel R, Jiang X, Estes MK. 1994. Three-dimensional structure of baculovirus-expressed Norwalk virus capsids. J. Virol. 68:5117–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katpally U, Wobus CE, Dryden K, Virgin HW, IV, Smith TJ. 2008. Structure of antibody-neutralized murine norovirus and unexpected differences from viruslike particles. J. Virol. 82:2079–2088. 10.1128/JVI.02200-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi JM, Hutson AM, Estes MK, Prasad BV. 2008. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc. Natl. Acad. Sci. U. S. A. 105:9175–9180. 10.1073/pnas.0803275105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tan M, Hegde RS, Jiang X. 2004. The P domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J. Virol. 78:6233–6242. 10.1128/JVI.78.12.6233-6242.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donaldson EF, Lindesmith LC, Lobue AD, Baric RS. 2010. Viral shape-shifting: norovirus evasion of the human immune system. Nat. Rev. Microbiol. 8:231–241. 10.1038/nrmicro2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nilsson M, Hedlund KO, Thorhagen M, Larson G, Johansen K, Ekspong A, Svensson L. 2003. Evolution of human calicivirus RNA in vivo: accumulation of mutations in the protruding P2 domain of the capsid leads to structural changes and possibly a new phenotype. J. Virol. 77:13117–13124. 10.1128/JVI.77.24.13117-13124.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taube S, Rubin JR, Katpally U, Smith TJ, Kendall A, Stuckey JA, Wobus CE. 2010. High-resolution X-ray structure and functional analysis of the murine norovirus 1 capsid protein protruding domain. J. Virol. 84:5695–5705. 10.1128/JVI.00316-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thackray LB, Wobus CE, Chachu KA, Liu B, Alegre ER, Henderson KS, Kelley ST, Virgin HW., IV 2007. Murine noroviruses comprising a single genogroup exhibit biological diversity despite limited sequence divergence. J. Virol. 81:10460–10473. 10.1128/JVI.00783-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Hernandez MB, Bragazzi Cunha J, Wobus CE. 2012. Plaque assay for murine norovirus. J. Vis. Exp. 66:e4297. 10.3791/4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lochridge VP, Hardy ME. 2007. A single-amino-acid substitution in the P2 domain of VP1 of murine norovirus is sufficient for escape from antibody neutralization. J. Virol. 81:12316–12322. 10.1128/JVI.01254-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yunus MA, Chung LM, Chaudhry Y, Bailey D, Goodfellow I. 2010. Development of an optimized RNA-based murine norovirus reverse genetics system. J. Virol. Methods 169:112–118. 10.1016/j.jviromet.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 34.Fox DA, Smith E. 2013. Quantitative production of monoclonal antibodies. In Howard GC, Kaser MR. (ed), Making and using antibodies: a practical handbook, 2nd ed. CRC Press, Boca Raton, FL [Google Scholar]
- 35.Bailey S. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystllogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 36.Pelkmans L, Kartenbeck J, Helenius A. 2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 3:473–483. 10.1038/35074539 [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 38.Heymann JB. 2001. Bsoft: image and molecular processing in electron microscopy. J. Struct. Biol. 133:156–169. 10.1006/jsbi.2001.4339 [DOI] [PubMed] [Google Scholar]
- 39.Sanjuan R, Nebot MR, Chirico N, Mansky LM, Belshaw R. 2010. Viral mutation rates. J. Virol. 84:9733–9748. 10.1128/JVI.00694-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailey D, Thackray LB, Goodfellow IG. 2008. A single amino acid substitution in the murine norovirus capsid protein is sufficient for attenuation in vivo. J. Virol. 82:7725–7728. 10.1128/JVI.00237-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith TJ. 2011. Structural studies on antibody recognition and neutralization of viruses. Curr. Opin. Virol. 1:150–156. 10.1016/j.coviro.2011.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis MM. 2004. The evolutionary and structural ‘logic’ of antigen receptor diversity. Semin. Immunol. 16:239–243. 10.1016/j.smim.2004.08.003 [DOI] [PubMed] [Google Scholar]
- 43.Wriggers W. 2010. Using Situs for the integration of multi-resolution structures. Biophys. Rev. 2:21–27. 10.1007/s12551-009-0026-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richardson C, Bargatze RF, Goodwin R, Mendelman PM. 2013. Norovirus virus-like particle vaccines for the prevention of acute gastroenteritis. Expert Rev. Vaccines 12:155–167. 10.1586/erv.12.145 [DOI] [PubMed] [Google Scholar]
- 45.Smith TJ, Chase ES, Schmidt TJ, Olson NH, Baker TS. 1996. Neutralizing antibody to human rhinovirus 14 penetrates the receptor-binding canyon. Nature 383:350–354. 10.1038/383350a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Z, Sosnovtsev SV, Bok K, Parra GI, Makiya M, Agulto L, Green KY, Purcell RH. 2013. Development of Norwalk virus-specific monoclonal antibodies with therapeutic potential for the treatment of Norwalk virus gastroenteritis. J. Virol. 87:9547–9557. 10.1128/JVI.01376-13 [DOI] [PMC free article] [PubMed] [Google Scholar]