

ABSTRACT
Syncytin-1, a fusogenic protein encoded by a human endogenous retrovirus of the W family (HERV-W) element (ERVWE1), is expressed in the syncytiotrophoblast layer of the placenta. This locus is transcriptionally repressed in adult tissues through promoter CpG methylation and suppressive histone modifications. Whereas syncytin-1 appears to be crucial for the development and functioning of the human placenta, its ectopic expression has been associated with pathological conditions, such as multiple sclerosis and schizophrenia. We previously reported on the transactivation of HERV-W elements, including ERVWE1, during influenza A/WSN/33 virus infection in a range of human cell lines. Here we report the results of quantitative PCR analyses of transcripts encoding syncytin-1 in both cell lines and primary fibroblast cells. We observed that spliced ERVWE1 transcripts and those encoding the transcription factor glial cells missing 1 (GCM1), acting as an enhancer element upstream of ERVWE1, are prominently upregulated in response to influenza A/WSN/33 virus infection in nonplacental cells. Knockdown of GCM1 by small interfering RNA followed by infection suppressed the transactivation of ERVWE1. While the infection had no influence on CpG methylation in the ERVWE1 promoter, chromatin immunoprecipitation assays detected decreased H3K9 trimethylation (H3K9me3) and histone methyltransferase SETDB1 levels along with influenza virus proteins associated with ERVWE1 and other HERV-W loci in infected CCF-STTG1 cells. The present findings suggest that an exogenous influenza virus infection can transactivate ERVWE1 by increasing transcription of GCM1 and reducing H3K9me3 in this region and in other regions harboring HERV-W elements.
IMPORTANCE Syncytin-1, a protein encoded by the env gene in the HERV-W locus ERVWE1, appears to be crucial for the development and functioning of the human placenta and is transcriptionally repressed in nonplacental tissues. Nevertheless, its ectopic expression has been associated with pathological conditions, such as multiple sclerosis and schizophrenia. In the present paper, we report findings suggesting that an exogenous influenza A virus infection can transactivate ERVWE1 by increasing the transcription of GCM1 and reducing the repressive histone mark H3K9me3 in this region and in other regions harboring HERV-W elements. These observations have implications of potential relevance for viral pathogenesis and for conditions associated with the aberrant transcription of HERV-W loci.
INTRODUCTION
While the vast majority of members of the W family of human endogenous retroviruses (HERV-Ws) in the human genome appear to be incapable of producing coding transcripts, the ERVWE1 locus on chromosome 7q21-q22 (chr7q21-q22) is a notable exception. The env gene in this locus encodes syncytin-1 (1–3), which appears to have been functionally adopted during human evolution (4, 5). Syncytin-1 is highly expressed in the placenta and mediates the fusion and, possibly, proliferation of trophoblast cells (1, 2, 6, 7). During pregnancy, syncytin-1 may also serve other purposes, such as modulating the maternal immune system (8, 9). Ectopic expression of syncytin-1 has been associated with a range of pathological conditions, such as cancer (10, 11), multiple sclerosis (MS) (12), amyotrophic lateral sclerosis (ALS) (13), schizophrenia (14), and bipolar disorder (15). Although the involvement of syncytin-1 in the pathogenesis of any of these conditions remains to be established, detection of HERV transcripts in adult tissues is intriguing, as repetitive elements are generally considered to be transcriptionally silent. Contrary to this view, transcripts containing the HERV-W gag gene display low but diversified and tissue-specific expression (16). Moreover, experimental data from our laboratory suggest that a number of HERV-W loci, including the locus encoding syncytin-1, can be transactivated in a range of human cell lines of nonplacental origin in response to influenza A/WSN/33 virus infection (17). The mechanism underlying transactivation of ERVWE1 and other HERV-W loci in response to viral infection is unknown. Promoter studies of ERVWE1 indicate that basal promoter activity is located within the U3 region of the retroviral 5′ long terminal repeat (LTR) (18, 19). In addition, a 33-bp trophoblast-specific enhancer (TSE) region directly upstream of the 5′ LTR has been identified (19, 20). The TSE contains a binding site for the placenta-specific transcription factor glial cells missing 1 (GCM1) (19, 21, 22) and potential binding sites for additional transcription factors (19) (see Fig. 4). We previously reported upregulation of mouse Gcm1 in primary cultures of neurons or glia as well as in brain parenchyma following influenza A virus infection (23), indicating that this transcription factor can potentially be involved in the transactivation of human ERVWE1 also in nonplacental cells.
FIG 4.

CpG methylation status of the ERVWE1 enhancer (TSE) and promoter (U3) regions determined by pyrosequencing. Percent methylation of 7 CpG sites was measured in uninfected and influenza A/WSN/33 virus-infected SK-N-MC cells (MOI = 0.1 or 0.5) (A), A549 cells (MOI = 0.1, 0.5, or 1) (B), 293A cells (MOI = 0.1, 0.5, or 1) (C), and human fibroblast cultures (MOI = 0.5) (D) at 48 h after infection. Shown at the top is a schematic representation of the MaLR (LTR)-ERVWE1 (5′ LTR) region analyzed with the relative positions of the CpG sites analyzed (circles on vertical bars). Transcription starts at the U3/R (5′ LTR) boundary (arrow). CAAT and TATA boxes as well as putative and effective transcription factor binding sites (Sp-1, GATA, estrogen receptor [ER], and Oct-1) proximal to or containing CpG sites are indicated. The GCM1 binding site is underlined.
In addition to tissue-restricted expression of transcription factors, CpG methylation (24, 25) and histone modifications (26) contribute to repression of ERVWE1 and other HERV-W loci. An increasing body of literature suggests that viral infections can modify the methylation status of host DNA in cells of rat and human origin (27–30). Recent publications suggest that influenza A virus can cause CpG demethylation by interfering with DNA methyltransferase (Dnmt) activity (31, 32). In addition, a DNA methylation-independent pathway has been reported in the proviral silencing of endogenous retroviruses (ERVs) in mouse embryonic stem cells (mESCs) through trimethylation of H3K9 (33). The H3K9-specific lysine methyltransferase (KMTase) SET domain, bifurcated 1 (SETDB1), which is required for enrichment of H3K9 trimethylation (H3K9me3), appears to play a crucial role in the silencing of both endogenous and exogenous retroviruses (33). Recent studies report a role for H3K9me3 in regulating expression of repetitive regions in the mouse genome in response to exogenous stressors (34, 35). It is not known if the transactivation of ERVWE1 or other HERV-W loci by influenza A virus involves modification of CpG methylation or H3K9me3 in these regions.
The aim of the present study was therefore to investigate how influenza A virus transactivates ERVWE1 and other HERV-W loci in extraplacental cells. To this end, we examined HERV-W transcription in response to influenza A virus and the roles of GCM1, CpG methylation, and H3K9me3 in the transactivation of ERVWE1 in a range of human cell types.
MATERIALS AND METHODS
Cell culture and influenza A/WSN/33 virus infections of cultured cells.
Human neuroepithelioma (SK-N-MC, HTB-10) cells, human lung epithelial (A549) cells, human embryonic kidney (293A) cells, and human placental choriocarcinoma (JEG-3) cells were cultured in Dulbecco's modified Eagle medium. Human astrocytoma (CCF-STTG1) cells were grown in RPMI 1640 adjusted to contain 1.5 g/liter NaHCO3, 4.5 g/liter glucose, 10 mM HEPES, and 1.0 mM Na pyruvate. All cell lines were obtained from the American Type Culture Collection, Manassas, VA. All cell culture media were supplemented with 10% fetal bovine serum and penicillin-streptomycin. Primary human fibroblast cultures were established as previously described (36). Ten volunteers were recruited in this study, which was approved by the regional ethics committee at Karolinska Institutet (approval numbers 04-273/1, 2006/637-32, and 2009-06-12). Serum deprivation, poly(cytidylic-inosinic) acid [poly(I·C); Sigma-Aldrich] treatment, and influenza A/WSN/33 virus infections were performed as previously described (17).
Quantitative RT-PCR and data analysis.
RNA extraction and reverse transcription (RT) were carried out as previously described (17). Analyses of the melting temperatures (Tms) of HERV-W gag amplicons were performed as previously described (16, 37). Data on frequency distributions into Tm categories (mixing proportions) in different cultures were compared by chi-square tests (GraphPad Prism, version 3.02, software). Products from Tm analysis quantitative PCRs (qPCRs) were ligated into the pCR4-TOPO vector (Life Technologies) and sequenced at KIseq (Karolinska Institutet, Stockholm, Sweden) as previously described (17). Sequences were mapped to the human genome to identify the HERV-W loci using the February 2009 assembly of the human genome. Real-time PCRs were performed as previously described (17). Specific quantitative assays for each of the targets were designed using Primer Express (version 2.0) software. The assay for the ERVWE1 locus previously described (17) was used to determine the levels of ERVWE1 exon transcripts. Assays described previously (26) were used to determine the levels of nonspliced and spliced ERVWE1 transcripts. Threshold cycle (CT) values from the exponential phase of the qPCR amplification plot for each target transcript were normalized to those for the gene encoding β-actin (see reference 38). From these values, the fold differences in the levels of transcripts between the two groups were calculated according to the formula 2−ΔΔCT (39). Primer sequences can be obtained from the authors on request.
Plasmid cloning, siRNA, and transfection in cells.
Human GCM1 plasmid construction and transfection were carried out as previously described (23). Small interfering RNA (siRNA; Qiagen) targeted toward human GCM1 (sequence, 5′-CCGATCCAGCTATATCAAGAA-3′) was transfected into CCF-STTG1 or JEG-3 cells using the HiPerFect transfection reagent (Qiagen) according to the manufacturer's instructions.
Bisulfite conversion and pyrosequencing analysis.
Genomic DNA was extracted from cells using a DNeasy blood and tissue kit (Qiagen). Three hundred nanograms of genomic DNA was bisulfite treated using an EZ DNA Methylation-Gold kit (Zymo Research) according to the instructions from the manufacturer. PCR and sequencing primers were designed using PyroMark assay design software (version 2.0; Qiagen). One microliter of modified DNA was PCR amplified with biotin-conjugated oligonucleotides using a PyroMark PCR kit (Qiagen). The resulting PCR products were analyzed on a PyroMark Q96 MD system (Qiagen).
ChIP assays.
Chromatin immunoprecipitation (ChIP) experiments were performed using a Magna ChIP A ChIP assay kit (Millipore) according to the instructions in the kit manual. Sonicated chromatin was prepared in a Bioruptor device (UCD-200; Diagenode) using 20 cycles (high power, 30 s on and 30 s off) to obtain DNA fragments of 150 to 400 bp from CCF-STTG1 cells. Sonicated chromatin (7.5 μg) from uninfected or influenza A virus-infected cells was used in each immunoprecipitation. H3K9me3 polyclonal antibody (pAb-056-050; Diagenode), anti-histone H3 antibody (ChIP grade; catalog no. ab1791; Abcam), anti-ESET/SETDB1 antibody (catalog no. 07-1568; Millipore), rabbit polyclonal anti-WSN/33 antiserum (previously described [23]), and negative-control normal rabbit IgG (Millipore) were used. Immunoprecipitated chromatin was subjected to qPCR. The assays used for ChIP quantification of the ERVWE1 locus and HERV-W elements were as previously reported (see references 26 and 38). The GAPDH (glyceraldehyde-3-phosphate dehydrogenase) assay previously described (26) was used as a reference for normalization. Primers designed to detect LTR sequences within solo LTRs integrated in intronic regions of the ZNF277 and C9orf85 genes and the gag sequence within the proviral element of the THEM2 intron sequence were described previously (38). The amount of immunoprecipitated material was normalized to the amount of input DNA.
Statistical analyses.
The nonparametric Mann-Whitney test was used to compare groups using GraphPad Prism (version 3.02) software. A P value of <0.05 was considered significant.
RESULTS
Regulation of HERV-W transcription by influenza A/WSN/33 virus infection.
Our previous studies revealed significant quantitative and qualitative variations in transcribed HERV-W elements in primary cells from one individual (38) following influenza A/WSN/33 virus infection by using a high-resolution melting temperature (Tm) analysis method (37). To extend these previous observations, we initially investigated the influence of the viral infection on HERV-W transcripts in fibroblast cultures established from 10 volunteers. Normalized to the levels of transcripts encoding β-actin, levels of transcripts containing HERV-W gag sequences in uninfected cultures exhibited a considerable variation across individuals. Elevated levels (median, 4.6-fold; range, 1.9- to 9.4-fold) were observed in infected individual cultures (Fig. 1A). Frequency distributions and heat maps of P values obtained from pairwise chi-square comparisons of expression patterns in the different cultures are illustrated in Fig. 1B and C. Tm analyses indicated a varied pattern of the distribution of HERV-W loci across uninfected cultures that differed significantly from that observed in infected cell cultures. The patterns were very similar across individuals following the virus infection (Fig. 1C), in line with our previous observation of HERV-W gag expression changes in response to serum deprivation (16). This change was particularly evident for a Tm of 79.44°C (the orange Tm category) in Fig. 1B. The proportion of Tms in this category increased from an average of 5% to one of 37%. To identify specific HERV-W loci in this category, we cloned and sequenced representative PCR amplicons. Several of the sequenced clones could be mapped to a proviral element with a full-length 5′ LTR located in ACOT13, which corresponds to the intronic locus in THEM2 (chr6p22.2) investigated previously (38). To verify the results of the Tm analysis, we next measured transcription from the element in ACOT13 in uninfected and infected cells from 10 individuals by using a specific qPCR assay. Normalized to the level of the transcripts encoding β-actin, significantly higher levels of transcripts from this proviral intronic element were observed in infected cells (median, 12.4-fold; range, 4.6- to 27.0-fold) than uninfected cells (Fig. 1D).
FIG 1.
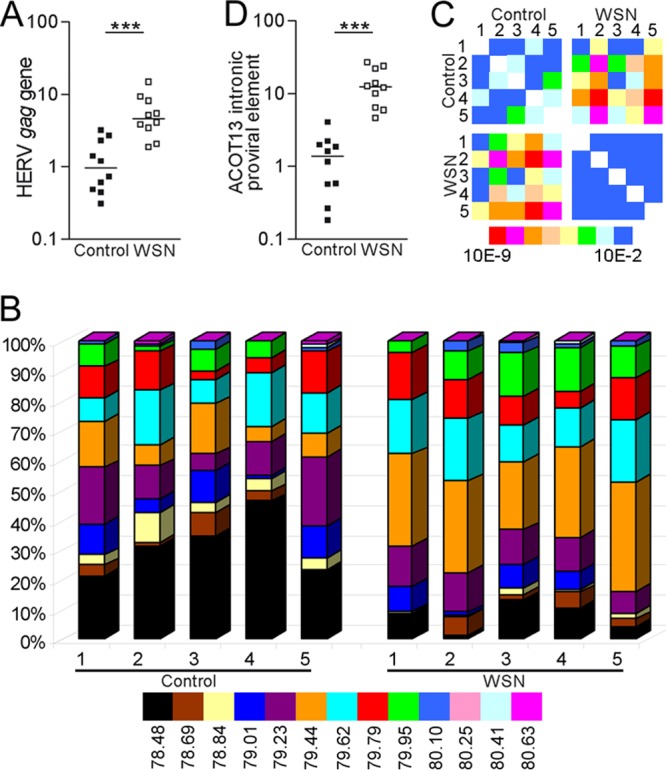
Transcription of HERV-W elements in response to virus infection. (A and B) Relative levels (A) and frequency distributions of Tm categories (B) of transcripts containing the HERV-W gag sequence in uninfected human fibroblasts (control, n = 10) and in such cells 48 h after infection with influenza A/WSN/33 virus (WSN; n = 10) at an MOI of 0.5 (data for five pairs are shown in panel B). (C) Heat maps of P values obtained from chi-square analyses of the frequency distribution diagrams. Blue is considered not significant, with P being >0.01, and P values decrease as the color moves toward red (P < 10−9). (D) Relative levels of transcripts from the proviral HERV-W element integrated in an intron of ACOT13.
ERVWE1 and GCM1 expression in vitro.
While previous observations support a role for the HERV-W LTR in directing transcription of the proviral genome, HERV-W transcripts also appear to be generated through leaky or passive transcription of surrounding genic regions. Therefore, individual HERV-W loci are potentially subject to transcriptional control by a diverse range of uncharacterized promoters. In contrast, the promoter region in the ERVWE1 locus has been characterized in great detail.
The 5′ LTR and TSE regions harbor a range of transcription factor binding sites, one of which binds GCM1, responsible for the high level of ERVWE1 transcription in the placenta (19). We therefore examined the relative levels of ERVWE1 and GCM1 transcripts in an astrocytoma cell line (CCF-STTG1) and the fibroblast cell cultures. As a reference, we also measured these levels in a choriocarcinoma cell line of placental origin (JEG-3) known to express high levels of transcripts encoding syncytin-1 (40). Normalized to β-actin gene transcript levels, both ERVWE1 and GCM1 transcripts were in excess of 100 and 1,000 times more abundant in JEG-3 cells than in CCF-STTG1 cells or primary fibroblasts, respectively (Fig. 2A). ERVWE1 and GCM1 transcripts were significantly more abundant in infected CCF-STTG1 cells than in uninfected cells (4.5- to 16.7-fold and 4.8 to 14.2-fold, respectively). Significantly more ERVWE1 (2.7- to 20.2-fold) and GCM1 (2.7- to 5.5-fold) transcripts were also observed in infected fibroblasts. In contrast, JEG-3 cells, which constitutively express high levels of ERVWE1 and GCM1 transcripts, did not harbor altered levels of either of these transcripts following viral infection (Fig. 2A). It should be noted that JEG-3 cells permitted viral replication to the same extent as nonplacental cells (i.e., all the cells harbored similar levels of transcripts encoding segment 8 of the influenza A/WSN/33 virus relative to the level of transcripts encoding β-actin; data not shown). These observations indicate the highly variable expression of HERV-W loci, including the ERVWE1 locus, across cell types and individuals. Influenza A virus infection can induce transcription of GCM1 and ERVWE1 not only in transformed cell lines but also in primary nonplacental cells.
FIG 2.
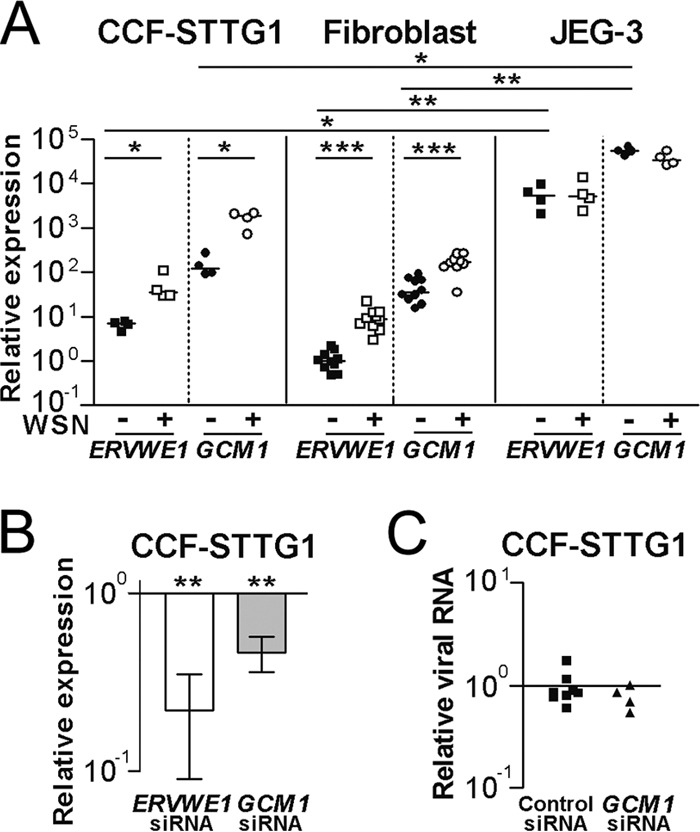
Transcription of ERVWE1 and GCM1 in response to virus infection. (A) Relative levels of ERVWE1 transcripts (env exon transcripts) and transcripts encoding GCM1 were measured in uninfected and influenza A/WSN/33 virus-infected (MOI = 0.5) CCF-STTG1 cells at 24 h postinfection, primary human fibroblasts at 48 h postinfection, and JEG-3 cells at 24 h postinfection. (B) Levels of ERVWE1 and GCM1 transcripts in CCF-STTG1 cells treated with siRNA targeted against transcripts encoding GCM1 and subsequently infected with influenza A/WSN/33 virus for 24 h relative to those in cells treated with control siRNA and infected with influenza A/WSN/33 virus. (C) Relative levels of transcripts encoding segment 8 of the influenza A/WSN/33 virus in CCF-STTG1 cells treated with scrambled control siRNA or no siRNA reagents and in cells treated with siRNA targeted against transcripts encoding GCM1. siRNA analysis data are presented as means ± SEMs from four independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Overexpression and knockdown of GCM1 in vitro.
Next, we set out to determine if ERVWE1 transcription is dependent on GCM1 in response to influenza A/WSN/33 virus infection. We initially examined the effect of overexpression of GCM1 in JEG-3 and CCF-STTG1 cells. Elevated levels of ERVWE1 transcripts following overexpression of GCM1 were observed in JEG-3 cells but not in CCF-STTG1 cells (data not shown). This observation confirms previous reports (22) that GCM1 is insufficient to increase the level of syncytin-1 in cell lines of nonplacental origin. To examine the degree to which GCM1 regulates ERVWE1 transcription, we developed siRNA against GCM1 transcripts. In JEG-3 cells, knockdown of GCM1 resulted in proportional reductions in the levels of GCM1 and ERVWE1 transcripts (data not shown). CCF-STTG1 cells transfected with siRNA targeted against GCM1 and subsequently infected with influenza A/WSN/33 virus exhibited significantly reduced levels of ERVWE1 and GCM1 transcripts compared to the levels in infected cells treated with scrambled oligonucleotides (Fig. 2B). Importantly, the siRNA targeted toward GCM1 had no effect on the levels of influenza A/WSN/33 virus mRNA detected (Fig. 2C), suggesting that the siRNA did not interfere with viral replication (by induction of a type 1 interferon response) and thereby reduce ERVWE1 transactivation. These findings suggest that GCM1 is involved in the physiological transcription of ERVWE1 in placenta-derived cells, as well as in its ectopic expression in other cell types.
Splicing of ERVWE1 transcripts.
The ERVWE1 locus can give rise to nonspliced 9.6-kb transcripts (corresponding to retroviral genomes) as well as spliced 2.8-kb env transcripts capable of encoding syncytin-1 (26). The ratio of spliced to nonspliced transcripts in choriocarcinoma cells of placental origin (BeWo cells) is reported to be 6:1 but very low in other cell types (26). We next investigated which of these transcripts is primarily generated in the uninfected and infected cells described above. As can be seen from Fig. 3A, nonspliced transcripts dominated in uninfected CCF-STTG1 and fibroblast cells, with spliced transcripts being practically undetectable. Whereas the levels of nonspliced transcripts did not change significantly, pronounced increases in the levels of spliced transcripts were detected in response to virus infection. In JEG-3 cells, neither nonspliced nor spliced transcripts differed between uninfected and infected cells. Thus, the observed upregulation of ERVWE1 transcription following influenza A/WSN/33 virus infection in nonplacental cells appears to result from the increased generation of spliced transcripts.
FIG 3.
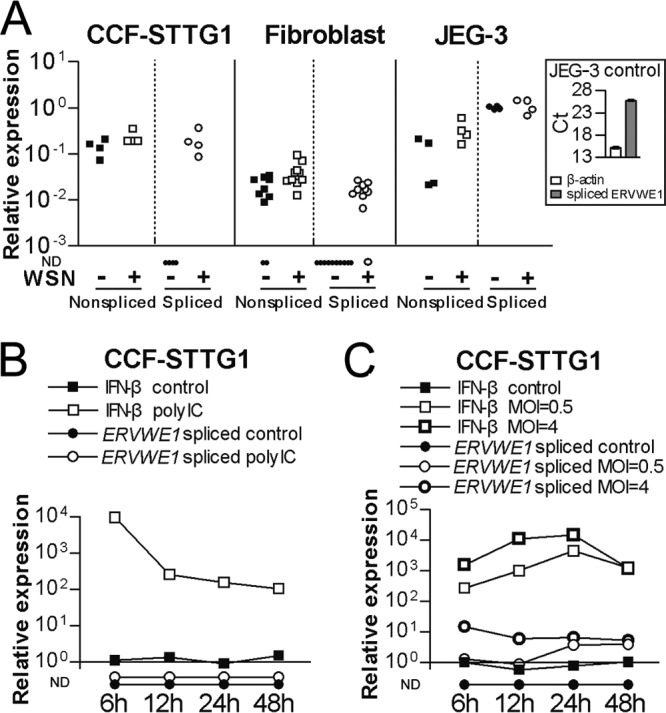
Splicing of ERVWE1 transcripts in response to stimuli. (A) Relative levels of nonspliced and spliced ERVWE1 transcripts were measured in uninfected and influenza A/WSN/33 virus-infected (MOI = 0.5) CCF-STTG1 cells at 24 h postinfection, primary human fibroblasts at 48 h postinfection, and JEG-3 cells at 24 h postinfection. (Inset) CT values of assays for transcripts encoding β-actin and spliced ERVWE1 transcripts in uninfected JEG-3 cells shown for reference. (B and C) Relative levels of spliced ERVWE1 transcripts and transcripts encoding IFN-β measured in CCF-STTG1 cells treated with 10 μg/ml poly(I·C) (B) or CCF-STTG1 cells infected with influenza A/WSN/33 virus (MOI = 0.5 or 4) (C). Samples were collected after 6 h, 12 h, 24 h, and 48 h. ND, not detectable.
To investigate if the changes in ERVWE1 transcription observed 24 to 48 h after infection were due to a cellular antiviral response or stress associated with this lytic infection (17), we next investigated the levels of spliced ERVWE1 transcripts in response to cellular starvation, double-stranded RNA, or virus infection in CCF-STTG1 cell cultures. No spliced ERVWE1 transcripts were detected in control cells or in cells deprived of serum for 24 h (data not shown). Poly(I·C)-treated cells elicited a relative 104-fold increase in the levels of transcript encoding beta interferon (IFN-β) at 6 h after stimulation, with 102-fold upregulation detected after 48 h of poly(I·C) treatment. However, no spliced ERVWE1 transcripts were observed in untreated control or poly(I·C)-treated cell cultures at any of the time points studied (Fig. 3B). We next investigated the kinetics of the generation of spliced ERVWE1 transcripts from 6 to 48 h following infection of CCF-STTG1 cells with a low titer (multiplicity of infection [MOI] = 0.5) and a higher titer (MOI = 4) of the virus. As illustrated in Fig. 3C, the levels of IFN-β gene transcripts peaked at 24 h postinfection, with the higher dose eliciting the stronger response (104-fold increase). After 48 h of infection, similar levels of IFN-β gene transcripts (103-fold) were observed regardless of the initial number of virus particles added. While spliced ERVWE1 transcripts remained undetectable in uninfected cells, such transcripts were detected at a low level at the early stages after infection at an MOI of 0.5. During these stages, approximately 16-fold higher levels of spliced ERVWE1 transcripts were detected in cells infected with influenza A virus at an MOI of 4. Similar levels of spliced transcripts were observed 48 h after infection with virus at the two different MOIs. These observations suggest that enhanced splicing of ERVWE1 transcripts is associated with influenza A virus replication rather than with the cellular antiviral response or cellular stress associated with this eventually lytic infection.
DNA methylation analysis.
Previous studies of CpGs in the promoter regions of the interleukin-6 gene (IL-6) and IL-32 suggest that these can be demethylated due to inhibition of Dnmts during influenza A virus infection (31, 32). We therefore investigated if the relative levels of transcripts encoding three different enzymes (Dnmt1, Dnmt3a, or Dnmt3b) and transcripts encoding ERVWE1, IL-6, or IL-32 were altered in response to influenza A/WSN/33 virus infection in a neuronal epithelioma cell line (SK-N-MC), a lung carcinoma cell line (A549), a kidney epithelioma cell line (293A), or primary fibroblasts. Decreases (60 to 90%) in the levels of transcripts encoding the three Dnmts were observed in the cell lines at 24 h following virus infection, while no variation in the levels of transcripts encoding Dnmt3a and Dnmt3b was detected in infected fibroblast cells. With the exception of the levels of IL-32 transcripts in SK-N-MC cells following virus infection, the levels of ERVWE1, IL-6, and IL-32 transcripts were increased in all the infected cell types investigated compared to the levels in uninfected control cells (data not shown).
Next, we examined if the observed upregulation of transcripts encoding ERVWE1, IL-6, or IL-32 was accompanied by changes in the methylation levels of CpGs in the corresponding promoters in SK-N-MC, A549, 293A, and fibroblast cells. CpG methylations in the promoter and enhancer region from positions −67 to +285 of the ERVWE1 locus were determined. Seven investigated CpG sites in this region are schematically illustrated in Fig. 4. The majority of investigated ERVWE1 CpGs exhibited high methylation levels in SK-N-MC cells (median, 89.2%; range, 30.8 to 97.0%; Fig. 4A) and 293A cells (median, 90.2%; range, 52.9 to 98.6%; Fig. 4C). Overall, a moderate methylation level was observed in A549 cells (Fig. 4B). High methylation levels were detected across the 10 fibroblast cultures, albeit with large variability among individuals (Fig. 4D). For comparison, the level of methylation of this promoter in placenta tissue has been reported to average 40% (24). No change in CpG methylation was observed in any of the infected cells compared to that in the corresponding uninfected cells (Fig. 4A to D).
We examined the methylation status of CpG sites in the promoters of IL-6 and IL-32. The methylation pattern through four CpG sites of the IL-6 promoter (range of average methylation, 29.6% to 1.9%) in A549 cells was in agreement with that obtained in previous studies using the same cell line, while these four positions exhibited extremely low methylation levels (below 5%) in SK-N-MC, 293A, and primary cell cultures (data not shown). Methylation levels were also analyzed at eight CpG sites in the IL-32 promoter in these cell types. A similar methylation pattern (varying along the promoter with an average of 91.1% methylation in the distal region that progressively declined to a 23.8% methylation level in the proximal region) was observed in A549, 293A, and fibroblast cells. However, no change in IL-6 or IL-32 promoter CpG methylation was detected in any of the cells infected with influenza A/WSN/33 virus (data not shown). Taken together, these observations indicate increased transcription of ERVWE1, IL-6, and IL-32 following virus infection, despite no change in CpG methylation in the promoter regions of these loci.
Attenuation of histone H3K9me3 associated with influenza A virus-induced transactivation of HERV-W elements.
In a previous study, differences in H3K9me3 enrichment in the 5′ LTR region of ERVWE1 between HeLa and BeWo cells were correlated with differences in the transcriptional activity of this locus (26). To investigate a potential influence of the virus infection on this repressive histone mark, we investigated enrichment of H3K9me3 in ERVWE1 in uninfected and virus-infected CCF-STTG1 cells using ChIP followed by qPCR. In uninfected CCF-STTG1 cells, H3K9me3 was highly enriched in the 5′ LTR region of ERVWE1, comparable to observations in HeLa cells (data not shown). Virus infection caused a significant decrease in the H3K9me3 level compared to the level detected in uninfected cells (Fig. 5A). High levels of H3K9me3 were also detected in the intronic and exonic regions of ERVWE1. While a significant decrease in the levels of H3K9me3 was detected in the intronic region of ERVWE1 in response to the virus infection, this change was not significantly different in the exonic region of ERVWE1. Similar levels of histone H3 were observed in infected and uninfected cells along the 5′ LTR, intronic, and exonic regions, indicating a stable occupancy of total H3 during virus infection. In light of our previous observations of transactivation of several different HERV-W loci by the virus infection, we further examined H3K9me3 in additional HERV-W loci following virus infection. We included two solo LTRs and two proviruses previously investigated (38). Compared to the levels in uninfected cells, all of the four elements exhibited a significant reduction in H3K9me3 levels but not total histone H3 levels following infection (Fig. 5B). Taken together, these results suggest a role for reduced levels of H3K9me3 in the transactivation of HERV-W elements by the virus infection.
FIG 5.

Histone H3K9me3 associated with HERV-W elements during influenza A virus infection. At 24 h postinfection, ChIP experiments were performed with rabbit IgG, antitrimethylated histone H3K9, H3, and SETDB1 antibodies on chromatin from uninfected and influenza A/WSN/33 virus-infected (MOI = 0.5) CCF-STTG1 cells. Precipitated chromatins were determined by qPCR with assays specific for ERVWE1 (A) and solo LTRs integrated in intronic regions of the ZNF277 and C9orf85 genes and proviruses integrated in intronic regions of the KYNU and ACOT13 genes (B). (C) Relative levels of transcripts encoding SETDB1 were measured in uninfected and virus-infected (MOI = 0.5) CCF-STTG1 cells at 24 h postinfection. (D) Levels of five HERV-W elements relative to the levels of GAPDH in input chromatin and in chromatin immunoprecipitated with anti-influenza A/WSN/33 virus antiserum from virus-infected (MOI = 0.5) CCF-STTG1 cells. ChIP data are presented as means ± SEMs from five independent experiments. *, P < 0.05; **, P < 0.01.
H3K9me3 is regulated by the methyltransferase SETDB1. We therefore used ChIP to analyze the occupancy of SETDB1 in the HERV-W regions described above in control and virus-infected cells. Interestingly, the level of binding of SETDB1 to the investigated regions of five HERV-W loci was high in uninfected cells but significantly reduced by 90% in infected cells (Fig. 5A and B). In addition, the levels of the transcript encoding SETDB1 were reduced in the infected CCF-STTG1 cells (Fig. 5C). Hence, the downregulation of transcripts encoding SETDB1 accompanied by its decreased occupancy in the promoter region of ERVWE1 and of other intronic HERV-W elements appears to be associated with attenuation of H3K9me3 in influenza A virus-infected CCF-STTG1 cells.
A recent study reported that the virus-encoded nucleoprotein (NP) was colocalized with H3K9me3 in transcriptionally repressed regions (41). To investigate if viral proteins were enriched in regions containing HERV-W elements compared to nonsuppressed regions such as the GAPDH region, we conducted ChIP on infected CCF-STTG1 cells using an antiserum against influenza A virus. Relative to the amount of DNA from the promoter region of GAPDH, significantly more DNA from four HERV-W loci was observed in chromatin precipitated from infected cells than in the input chromatin. Although no significant change was detected in the intronic region of chr2q22.2 provirus, the trend matched the relative enrichment of the other investigated regions (Fig. 5D).
DISCUSSION
The retroviral LTR contains the elements necessary for transcription of genomic RNA as well as spliced transcripts encoding the different viral proteins needed to generate infectious particles (42). Previous studies have suggested that the HERV-W LTR has retained at least some of its promoter activity since its integration into the germ line of human progenitors approximately 40 million years ago (43). Many of the current HERV-W loci appear to be pseudogene expansions of proviral transcripts mediated by reverse transcriptase encoded by long interspersed nuclear elements (LINEs) and lack the regulatory U3 region of the LTR (44, 45). Insofar as these are detected in the RNA population, they appear to represent leakage during transcription of genic regions (38). In addition, the human genome harbors hundreds of proviral integrations and solo LTRs that can potentially provide functional promoters. As observed here and in other studies, the activity of these LTRs appears to be low in healthy tissues (38, 46), and with the exception of the role of ERVWE1, any functional roles elude identification. The transcriptional repression appears to be a consequence of the high degree of methylation of CpGs in the promoter region of these and many other repetitive elements in most tissues (25, 47). In contrast, a low level of methylation and a high level of transcription of ERVWE1 upon binding of GCM1 to an upstream enhancer element in the human placenta are well documented (24). Our current observations expand on our previous observations (17, 38) and indicate that not only noncoding HERV-W loci but also spliced ERVWE1 transcripts capable of encoding syncytin-1 can be induced in extraplacental cells following influenza virus infection. Our current observations also suggest that GCM1 contributes to such ERVWE1 transcription. Overexpression of GCM1 has previously been reported to induce transcripts encoding syncytin-1 in placental cell lines (22). Here we used the opposite approach to show that knockdown of GCM1 during an infection resulted in a corresponding reduction in ERVWE1 transcripts. Thus, virus infection appears to induce expression from this locus through a mechanism similar to the mechanisms thought to govern the high-level, tissue-specific expression of syncytin-1 in human placenta (19, 48). Reductions of GCM1 and ERVWE1 transcripts did not affect the levels of influenza virus RNA, suggesting that these two genes are not critically involved in viral replication or in the host cell defense against influenza A virus.
The finding of spliced ERVWE1 transcripts in extraplacental cell types lends in vitro support to clinical observations of the syncytin-1 protein in the brain and blood of individuals with MS (12) and schizophrenia (14), respectively. A more recent report also indicates the presence of spliced transcripts and syncytin-1 protein in B cells and monocytes in individuals with MS as well as in some healthy blood donors (49). Our findings that enhanced splicing of ERVWE1 transcripts upon virus infection was not associated with the cellular stress response to serum deprivation or with the antiviral response to double-stranded RNA suggest a functional involvement of virus proteins mediating transactivation of ERVWE1. In addition, the detection of transactivation of ERVWE1 by influenza A virus complements earlier reports of such transactivation by the herpesviruses Epstein-Barr virus (49) and herpes simplex virus 1 (50) and potentially also by Toxoplasma gondii (51). While these persistent infections have been associated with MS (52) and schizophrenia (53), it remains to be established if they are causally involved in the etiology and/or pathogenesis of these disorders.
An increasing number of studies report changes in host genome CpG methylation following virus infection (27–30). Interestingly, a recent report also suggests that mammalian GCM proteins are directly involved in active CpG demethylation (54). The increase in ERVWE1 transcription following influenza A virus infection detected in the present study occurred despite no change in the high level of promoter methylation. This finding is in contrast to previous reports that host gene promoters (IL-6 and IL-32) can be demethylated by different influenza A virus strains (31, 32). Therefore, we also measured the levels of CpG methylation in IL-6 and IL-32 in A549 cells along with SK-N-MC, 293A, and fibroblast cells but were unable to replicate changes in methylation in these promoters in any of the investigated cells (data not shown). This apparent discrepancy could be explained by the more sensitive and accurate method of bisulfite pyrosequencing (55), which is able to detect DNA methylation differences as small as 5%, used in our study or by differences in the properties of the H1N1 WSN/33 strain used here versus the H1N1, H3N2, and H5N1 strains used in the previous studies. Thus, it remains to be established if influenza A virus can alter CpG methylation of the host cell genome.
Interactions between virus and host cells have also been reported to alter cellular chromatin (56, 57). For example, histone acetylation (58, 59) or methylation (60) has been found to play a role in regulating host gene expression following infection with various viruses in both rats and humans. By comparing HeLa and BeWo cells, Trejbalova and coworkers previously reported an inverse correlation between the levels of H3K9me3 in the 5′ LTR of ERVWE1 and transcript levels (26). We observed a similar correlation in the astrocytoma cell line CCF-STTG1 following virus infection, where the infection resulted in a decrease in H3K9me3 associated with the 5′ LTR in ERVWE1 accompanied by increased transcription. We observed similar effects on two investigated solo LTRs and two LTRs in proviral elements. The latter included an intronic element in ACOT13 for which we had evidence of increased transcription following infection. The latter observation suggests that a decrease in H3K9me3 is not restricted to a single genomic region but may occur on a more global scale. Previous studies reported that many ERV families are repressed in mESCs via KMTase-mediated H3K9 methylation (33, 61). In fact, SETDB1-mediated repression appears to be more important for silencing of murine ERVs than CpG methylation, according to data obtained in mESCs lacking KMTase or Dnmt activity (62). Our analyses demonstrate that attenuation of H3K9me3 is accompanied by a reduced occupancy of SETDB1 and transactivation of HERV-W elements in virus-infected CCF-STTG1 cells independently of CpG demethylation at ERVWE1. While reduced transcription of SETDB1 was detected in infected CCF-STTG1 cells, it remains to be established whether additional mechanisms are also involved in the reduced SETDB1 binding to chromatin at the investigated loci. Proviral silencing also involves dimethylation of H3K9 through KMTase G9a (61). In addition, du Chene et al. reported that enrichment of H3K9me3 and transcriptional repression of integrated HIV-1 LTRs involve recruitment of the KMTase SUV39H1 (63). It remains to be established whether these factors are also involved in the expression of HERV-W during virus infection.
Several influenza A virus-encoded proteins exhibit nuclear localization during different stages of the infection to provide transcription and replication of viral genes and genomes (64). The matrix protein 1 (M1), the viral ribonucleoprotein (vRNP) complex consisting of the three polymerase subunits PA, PB1, and PB2, and the nucleoprotein (NP) bind to different histone regions (41, 65). Another potential viral candidate is the multifaceted nonstructural 1 (NS1) protein, which is known to interfere with host gene transcription at many different levels (66). Our ChIP analyses suggest enrichment of viral proteins at the later stage of infection in the investigated regions containing four HERV-W elements. This observation indicates a preferential binding of viral protein to regions with HERV-W elements relative to the GAPDH promoter during virus infection. This observation supports the finding that influenza virus-encoded proteins colocalize with H3K9me3 tails during later stages of infection (41). The contributions of specific cellular and viral factors to the observed changes in the repressive H3K9me3 mark in infected cells need to be investigated in order to further our understanding of how influenza A virus interferes with the chromatin structure to alter host cell transcription.
In conclusion, we report that influenza A virus promotes transcription of members of the HERV-W family, including an intronic proviral element in ACOT13 and ERVWE1, in nonplacental cell lines and primary fibroblast cultures. Through increased levels of GCM1 transcripts, influenza A virus enhanced the transcription of spliced ERVWE1 transcripts encoding syncytin-1 in these cells. The infection had no influence on CpG methylation in the promoter region of ERVWE1, IL-6, or IL-32 but resulted in an attenuation of H3K9me3 associated with HERV-W LTRs in multiple genomic positions, including an element in ACOT13 and ERVWE1. Thus, while the present study reports observations solely made in vitro, the findings have implications of potential relevance for viral pathogenesis and for conditions associated with aberrant transcription of HERV-W loci, such as MS and schizophrenia.
ACKNOWLEDGMENTS
This study was supported by the Stanley Medical Research Institute, Hjärnfonden, and the Swedish Research Council.
We are grateful to Ou Chen for technical assistance.
Footnotes
Published ahead of print 29 January 2014
REFERENCES
- 1.Blond JL, Lavillette D, Cheynet V, Bouton O, Oriol G, Chapel-Fernandes S, Mandrand B, Mallet F, Cosset FL. 2000. An envelope glycoprotein of the human endogenous retrovirus HERV-W is expressed in the human placenta and fuses cells expressing the type D mammalian retrovirus receptor. J. Virol. 74:3321–3329. 10.1128/JVI.74.7.3321-3329.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang XY, Edouard P, Howes S, Keith JC, Jr, McCoy JM. 2000. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403:785–789. 10.1038/35001608 [DOI] [PubMed] [Google Scholar]
- 3.Voisset C, Bouton O, Bedin F, Duret L, Mandrand B, Mallet F, Paranhos-Baccala G. 2000. Chromosomal distribution and coding capacity of the human endogenous retrovirus HERV-W family. AIDS Res. Hum. Retroviruses 16:731–740. 10.1089/088922200308738 [DOI] [PubMed] [Google Scholar]
- 4.Kim HS, Takenaka O, Crow TJ. 1999. Isolation and phylogeny of endogenous retrovirus sequences belonging to the HERV-W family in primates. J. Gen. Virol. 80(Pt 10):2613–2619 [DOI] [PubMed] [Google Scholar]
- 5.Voisset C, Blancher A, Perron H, Mandrand B, Mallet F, Paranhos-Baccala G. 1999. Phylogeny of a novel family of human endogenous retrovirus sequences, HERV-W, in humans and other primates. AIDS Res. Hum. Retroviruses 15:1529–1533. 10.1089/088922299309810 [DOI] [PubMed] [Google Scholar]
- 6.Huang Q, Li J, Wang F, Oliver MT, Tipton T, Gao Y, Jiang SW. 2013. Syncytin-1 modulates placental trophoblast cell proliferation by promoting G1/S transition. Cell Signal. 25:1027–1035. 10.1016/j.cellsig.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavillette D, Marin M, Ruggieri A, Mallet F, Cosset FL, Kabat D. 2002. The envelope glycoprotein of human endogenous retrovirus type W uses a divergent family of amino acid transporters/cell surface receptors. J. Virol. 76:6442–6452. 10.1128/JVI.76.13.6442-6452.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holder BS, Tower CL, Forbes K, Mulla MJ, Aplin JD, Abrahams VM. 2012. Immune cell activation by trophoblast-derived microvesicles is mediated by syncytin 1. Immunology 136:184–191. 10.1111/j.1365-2567.2012.03568.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tolosa JM, Schjenken JE, Clifton VL, Vargas A, Barbeau B, Lowry P, Maiti K, Smith R. 2012. The endogenous retroviral envelope protein syncytin-1 inhibits LPS/PHA-stimulated cytokine responses in human blood and is sorted into placental exosomes. Placenta 33:933–941. 10.1016/j.placenta.2012.08.004 [DOI] [PubMed] [Google Scholar]
- 10.Bjerregaard B, Holck S, Christensen IJ, Larsson LI. 2006. Syncytin is involved in breast cancer-endothelial cell fusions. Cell. Mol. Life Sci. 63:1906–1911. 10.1007/s00018-006-6201-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strick R, Ackermann S, Langbein M, Swiatek J, Schubert SW, Hashemolhosseini S, Koscheck T, Fasching PA, Schild RL, Beckmann MW, Strissel PL. 2007. Proliferation and cell-cell fusion of endometrial carcinoma are induced by the human endogenous retroviral syncytin-1 and regulated by TGF-beta. J. Mol. Med. 85:23–38. 10.1007/s00109-006-0104-y [DOI] [PubMed] [Google Scholar]
- 12.Antony JM, Ellestad KK, Hammond R, Imaizumi K, Mallet F, Warren KG, Power C. 2007. The human endogenous retrovirus envelope glycoprotein, syncytin-1, regulates neuroinflammation and its receptor expression in multiple sclerosis: a role for endoplasmic reticulum chaperones in astrocytes. J. Immunol. 179:1210–1224 [DOI] [PubMed] [Google Scholar]
- 13.Oluwole SO, Yao Y, Conradi S, Kristensson K, Karlsson H. 2007. Elevated levels of transcripts encoding a human retroviral envelope protein (syncytin) in muscles from patients with motor neuron disease. Amyotroph. Lateral Scler. 8:67–72. 10.1080/17482960600864207 [DOI] [PubMed] [Google Scholar]
- 14.Perron H, Mekaoui L, Bernard C, Veas F, Stefas I, Leboyer M. 2008. Endogenous retrovirus type W GAG and envelope protein antigenemia in serum of schizophrenic patients. Biol. Psychiatry 64:1019–1023. 10.1016/j.biopsych.2008.06.028 [DOI] [PubMed] [Google Scholar]
- 15.Perron H, Hamdani N, Faucard R, Lajnef M, Jamain S, Daban-Huard C, Sarrazin S, LeGuen E, Houenou J, Delavest M, Moins-Teisserenc H, Bengoufa D, Yolken R, Madeira A, Garcia-Montojo M, Gehin N, Burgelin I, Ollagnier G, Bernard C, Dumaine A, Henrion A, Gombert A, Le Dudal K, Charron D, Krishnamoorthy R, Tamouza R, Leboyer M. 2012. Molecular characteristics of human endogenous retrovirus type-W in schizophrenia and bipolar disorder. Transl. Psychiatry 2:e201. 10.1038/tp.2012.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nellaker C, Li F, Uhrzander F, Tyrcha J, Karlsson H. 2009. Expression profiling of repetitive elements by melting temperature analysis: variation in HERV-W gag expression across human individuals and tissues. BMC Genomics 10:532. 10.1186/1471-2164-10-532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nellaker C, Yao Y, Jones-Brando L, Mallet F, Yolken RH, Karlsson H. 2006. Transactivation of elements in the human endogenous retrovirus W family by viral infection. Retrovirology 3:44. 10.1186/1742-4690-3-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng YH, Richardson BD, Hubert MA, Handwerger S. 2004. Isolation and characterization of the human syncytin gene promoter. Biol. Reprod. 70:694–701. 10.1095/biolreprod.103.023473 [DOI] [PubMed] [Google Scholar]
- 19.Prudhomme S, Oriol G, Mallet F. 2004. A retroviral promoter and a cellular enhancer define a bipartite element which controls env ERVWE1 placental expression. J. Virol. 78:12157–12168. 10.1128/JVI.78.22.12157-12168.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonnaud B, Beliaeff J, Bouton O, Oriol G, Duret L, Mallet F. 2005. Natural history of the ERVWE1 endogenous retroviral locus. Retrovirology 2:57. 10.1186/1742-4690-2-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nait-Oumesmar B, Copperman AB, Lazzarini RA. 2000. Placental expression and chromosomal localization of the human Gcm 1 gene. J. Histochem. Cytochem. 48:915–922. 10.1177/002215540004800704 [DOI] [PubMed] [Google Scholar]
- 22.Yu C, Shen K, Lin M, Chen P, Lin C, Chang GD, Chen H. 2002. GCMa regulates the syncytin-mediated trophoblastic fusion. J. Biol. Chem. 277:50062–50068. 10.1074/jbc.M209316200 [DOI] [PubMed] [Google Scholar]
- 23.Asp L, Nellaker C, Karlsson H. 2007. Influenza A virus transactivates the mouse envelope gene encoding syncytin B and its regulator, glial cells missing 1. J. Neurovirol. 13:29–37. 10.1080/13550280601103125 [DOI] [PubMed] [Google Scholar]
- 24.Gimenez J, Montgiraud C, Oriol G, Pichon JP, Ruel K, Tsatsaris V, Gerbaud P, Frendo JL, Evain-Brion D, Mallet F. 2009. Comparative methylation of ERVWE1/syncytin-1 and other human endogenous retrovirus LTRs in placenta tissues. DNA Res. 16:195–211. 10.1093/dnares/dsp011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matouskova M, Blazkova J, Pajer P, Pavlicek A, Hejnar J. 2006. CpG methylation suppresses transcriptional activity of human syncytin-1 in non-placental tissues. Exp. Cell Res. 312:1011–1020. 10.1016/j.yexcr.2005.12.010 [DOI] [PubMed] [Google Scholar]
- 26.Trejbalova K, Blazkova J, Matouskova M, Kucerova D, Pecnova L, Vernerova Z, Heracek J, Hirsch I, Hejnar J. 2011. Epigenetic regulation of transcription and splicing of syncytins, fusogenic glycoproteins of retroviral origin. Nucleic Acids Res. 39:8728–8739. 10.1093/nar/gkr562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esteki-Zadeh A, Karimi M, Straat K, Ammerpohl O, Zeitelhofer M, Jagodic M, Mehrab-Mohseni M, Sjoholm L, Rahbar A, Soderberg-Naucler C, Ekstrom TJ. 2012. Human cytomegalovirus infection is sensitive to the host cell DNA methylation state and alters global DNA methylation capacity. Epigenetics 7:585–593. 10.4161/epi.20075 [DOI] [PubMed] [Google Scholar]
- 28.Macnab JC, Adams RL, Rinaldi A, Orr A, Clark L. 1988. Hypomethylation of host cell DNA synthesized after infection or transformation of cells by herpes simplex virus. Mol. Cell. Biol. 8:1443–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan JL, Jones RJ, Kenney SC, Rivenbark AG, Tang W, Knight ER, Coleman WB, Gulley ML. 2010. Epstein-Barr virus-specific methylation of human genes in gastric cancer cells. Infect. Agents Cancer 5:27. 10.1186/1750-9378-5-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vivekanandan P, Daniel HDJ, Kannangai R, Martinez-Murillo F, Torbenson M. 2010. Hepatitis B virus replication induces methylation of both host and viral DNA. J. Virol. 84:4321–4329. 10.1128/JVI.02280-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li W, Sun W, Liu L, Yang F, Li YK, Chen YN, Fang JL, Zhang WJ, Wu JG, Zhu Y. 2010. IL-32: a host proinflammatory factor against influenza viral replication is upregulated by aberrant epigenetic modifications during influenza A virus infection. J. Immunol. 185:5056–5065. 10.4049/jimmunol.0902667 [DOI] [PubMed] [Google Scholar]
- 32.Tang BK, Zhao RH, Sun Y, Zhu Y, Zhong JA, Zhao GP, Zhu NS. 2011. Interleukin-6 expression was regulated by epigenetic mechanisms in response to influenza virus infection or dsRNA treatment. Mol. Immunol. 48:1001–1008. 10.1016/j.molimm.2011.01.003 [DOI] [PubMed] [Google Scholar]
- 33.Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y. 2010. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464:927–931. 10.1038/nature08858 [DOI] [PubMed] [Google Scholar]
- 34.Hunter RG, Murakami G, Dewell S, Seligsohn M, Baker ME, Datson NA, McEwen BS, Pfaff DW. 2012. Acute stress and hippocampal histone H3 lysine 9 trimethylation, a retrotransposon silencing response. Proc. Natl. Acad. Sci. U. S. A. 109:17657–17662. 10.1073/pnas.1215810109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maze I, Feng J, Wilkinson MB, Sun H, Shen L, Nestler EJ. 2011. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc. Natl. Acad. Sci. U. S. A. 108:3035–3040. 10.1073/pnas.1015483108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asp L, Johansson AS, Mann A, Owe-Larsson B, Urbanska EM, Kocki T, Kegel M, Engberg G, Lundkvist GB, Karlsson H. 2011. Effects of pro-inflammatory cytokines on expression of kynurenine pathway enzymes in human dermal fibroblasts. J. Inflamm. (Lond.) 8:25. 10.1186/1476-9255-8-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nellaker C, Wallgren U, Karlsson H. 2007. Molecular beacon-based temperature control and automated analyses for improved resolution of melting temperature analysis using SYBR I green chemistry. Clin. Chem. 53:98–103. 10.1373/clinchem.2006.075184 [DOI] [PubMed] [Google Scholar]
- 38.Li F, Nellaker C, Yolken RH, Karlsson H. 2011. A systematic evaluation of expression of HERV-W elements; influence of genomic context, viral structure and orientation. BMC Genomics 12:22. 10.1186/1471-2164-12-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 40.Muir A, Lever AML, Moffett A. 2006. Human endogenous retrovirus-W envelope (syncytin) is expressed in both villous and extravillous trophoblast populations. J. Gen. Virol. 87:2067–2071. 10.1099/vir.0.81412-0 [DOI] [PubMed] [Google Scholar]
- 41.Alfonso R, Lutz T, Rodriguez A, Chavez JP, Rodriguez P, Gutierrez S, Nieto A. 2011. CHD6 chromatin remodeler is a negative modulator of influenza virus replication that relocates to inactive chromatin upon infection. Cell. Microbiol. 13:1894–1906. 10.1111/j.1462-5822.2011.01679.x [DOI] [PubMed] [Google Scholar]
- 42.Rabson A, Graves B. 1997. Synthesis and processing of viral RNA. In Coffin JM, Hughes SH, Varmus HE. (ed), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 43.Schon U, Seifarth W, Baust C, Hohenadl C, Erfle V, Leib-Mosch C. 2001. Cell type-specific expression and promoter activity of human endogenous retroviral long terminal repeats. Virology 279:280–291. 10.1006/viro.2000.0712 [DOI] [PubMed] [Google Scholar]
- 44.Costas J. 2002. Characterization of the intragenomic spread of the human endogenous retrovirus family HERV-W. Mol. Biol. Evol. 19:526–533. 10.1093/oxfordjournals.molbev.a004108 [DOI] [PubMed] [Google Scholar]
- 45.Pavlicek A, Paces J, Elleder D, Hejnar J. 2002. Processed pseudogenes of human endogenous retroviruses generated by LINEs: their integration, stability, and distribution. Genome Res. 12:391–399. 10.1101/gr.216902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forsman A, Yun Z, Hu L, Uzhameckis D, Jern P, Blomberg J. 2005. Development of broadly targeted human endogenous gammaretroviral pol-based real time PCRs: quantitation of RNA expression in human tissues. J. Virol. Methods 129:16–30. 10.1016/j.jviromet.2005.04.016 [DOI] [PubMed] [Google Scholar]
- 47.Reiss D, Zhang Y, Mager DL. 2007. Widely variable endogenous retroviral methylation levels in human placenta. Nucleic Acids Res. 35:4743–4754. 10.1093/nar/gkm455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng YH, Handwerger S. 2005. A placenta-specific enhancer of the human syncytin gene. Biol. Reprod. 73:500–509. 10.1095/biolreprod.105.039941 [DOI] [PubMed] [Google Scholar]
- 49.Mameli G, Poddighe L, Mei A, Uleri E, Sotgiu S, Serra C, Manetti R, Dolei A. 2012. Expression and activation by Epstein Barr virus of human endogenous retroviruses-W in blood cells and astrocytes: inference for multiple sclerosis. PLoS One 7:e44991. 10.1371/journal.pone.0044991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruprecht K, Obojes K, Wengel V, Gronen F, Kim KS, Perron H, Schneider-Schaulies J, Rieckmann P. 2006. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: implications for multiple sclerosis. J. Neurovirol. 12:65–71. 10.1080/13550280600614973 [DOI] [PubMed] [Google Scholar]
- 51.Frank O, Jones-Brando L, Leib-Mosch C, Yolken R, Seifarth W. 2006. Altered transcriptional activity of human endogenous retroviruses in neuroepithelial cells after infection with Toxoplasma gondii. J. Infect. Dis. 194:1447–1449. 10.1086/508496 [DOI] [PubMed] [Google Scholar]
- 52.Giovannoni G, Cutter GR, Lunemann J, Martin R, Munz C, Sriram S, Steiner I, Hammerschlag MR, Gaydos CA. 2006. Infectious causes of multiple sclerosis. Lancet Neurol. 5:887–894. 10.1016/S1474-4422(06)70577-4 [DOI] [PubMed] [Google Scholar]
- 53.Torrey EF, Yolken RH. 2003. Toxoplasma gondii and schizophrenia. Emerg. Infect. Dis. 9:1375–1380. 10.3201/eid0911.030143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hitoshi S, Ishino Y, Kumar A, Jasmine S, Tanaka KF, Kondo T, Kato S, Hosoya T, Hotta Y, Ikenaka K. 2011. Mammalian Gcm genes induce Hes5 expression by active DNA demethylation and induce neural stem cells. Nat. Neurosci. 14:957–964. 10.1038/nn.2875 [DOI] [PubMed] [Google Scholar]
- 55.Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. 2003. Sensitive and quantitative universal pyrosequencing methylation analysis of CpG sites. Biotechniques 35:146–150 http://www.biotechniques.com/multimedia/archive/00011/03351md01_11443a.pdf [DOI] [PubMed] [Google Scholar]
- 56.Adhya D, Basu A. 2010. Epigenetic modulation of host: new insights into immune evasion by viruses. J. Biosci. 35:647–663. 10.1007/s12038-010-0072-9 [DOI] [PubMed] [Google Scholar]
- 57.Lieberman PM. 2006. Chromatin regulation of virus infection. Trends Microbiol. 14:132–140. 10.1016/j.tim.2006.01.001 [DOI] [PubMed] [Google Scholar]
- 58.Alkhalil A, Hammamieh R, Hardick J, Ichou MA, Jett M, Ibrahim S. 2010. Gene expression profiling of monkeypox virus-infected cells reveals novel interfaces for host-virus interactions. Virol. J. 7:173. 10.1186/1743-422X-7-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iseki H, Shimizukawa R, Sugiyama F, Kunita S, Iwama A, Onodera M, Nakauchi H, Yagami K. 2005. Parvovirus nonstructural proteins induce an epigenetic modification through histone acetylation in host genes and revert tumor malignancy to benignancy. J. Virol. 79:8886–8893. 10.1128/JVI.79.14.8886-8893.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tian Y, Ni D, Yang W, Zhang Y, Zhao K, Song J, Mao Q, Tian Z, van Velkinburgh JC, Yang D, Wu Y, Ni B. 2014. Telbivudine treatment corrects HBV-induced epigenetic alterations in liver cells of patients with chronic hepatitis B. Carcinogenesis 35:53–61. 10.1093/carcin/bgt317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leung DC, Dong KB, Maksakova IA, Goyal P, Appanah R, Lee S, Tachibana M, Shinkai Y, Lehnertz B, Mager DL, Rossi F, Lorincz MC. 2011. Lysine methyltransferase G9a is required for de novo DNA methylation and the establishment, but not the maintenance, of proviral silencing. Proc. Natl. Acad. Sci. U. S. A. 108:5718–5723. 10.1073/pnas.1014660108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, Lorincz MC. 2011. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8:676–687. 10.1016/j.stem.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M. 2007. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 26:424–435. 10.1038/sj.emboj.7601517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Engelhardt OG, Fodor E. 2006. Functional association between viral and cellular transcription during influenza virus infection. Rev. Med. Virol. 16:329–345. 10.1002/rmv.512 [DOI] [PubMed] [Google Scholar]
- 65.Zhirnov OP, Klenk HD. 1997. Histones as a target for influenza virus matrix protein M1. Virology 235:302–310. 10.1006/viro.1997.8700 [DOI] [PubMed] [Google Scholar]
- 66.Hale BG, Randall RE, Ortin J, Jackson D. 2008. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89:2359–2376. 10.1099/vir.0.2008/004606-0 [DOI] [PubMed] [Google Scholar]