

ABSTRACT
Severe acute respiratory syndrome coronavirus (SARS-CoV) and Ebola, Hendra, and Nipah viruses are members of different viral families and are known causative agents of fatal viral diseases. These viruses depend on cathepsin L for entry into their target cells. The viral glycoproteins need to be primed by protease cleavage, rendering them active for fusion with the host cell membrane. In this study, we developed a novel high-throughput screening assay based on peptides, derived from the glycoproteins of the aforementioned viruses, which contain the cathepsin L cleavage site. We screened a library of 5,000 small molecules and discovered a small molecule that can inhibit the cathepsin L cleavage of all viral peptides with minimal inhibition of cleavage of a host protein-derived peptide (pro-neuropeptide Y). The small molecule inhibited the entry of all pseudotyped viruses in vitro and the cleavage of SARS-CoV spike glycoprotein in an in vitro cleavage assay. In addition, the Hendra and Nipah virus fusion glycoproteins were not cleaved in the presence of the small molecule in a cell-based cleavage assay. Furthermore, we demonstrate that the small molecule is a mixed inhibitor of cathepsin L. Our broad-spectrum antiviral small molecule appears to be an ideal candidate for future optimization and development into a potent antiviral against SARS-CoV and Ebola, Hendra, and Nipah viruses.
IMPORTANCE We developed a novel high-throughput screening assay to identify small molecules that can prevent cathepsin L cleavage of viral glycoproteins derived from SARS-CoV and Ebola, Hendra, and Nipah viruses that are required for their entry into the host cell. We identified a novel broad-spectrum small molecule that could block cathepsin L-mediated cleavage and thus inhibit the entry of pseudotypes bearing the glycoprotein derived from SARS-CoV or Ebola, Hendra, or Nipah virus. The small molecule can be further optimized and developed into a potent broad-spectrum antiviral drug.
INTRODUCTION
Severe acute respiratory syndrome coronavirus (SARS-CoV), Ebola virus (EBOV), Hendra virus (HeV), and Nipah virus (NiV) are highly infectious zoonotic viruses, with various species of bat as the identified natural reservoir (1–6), and are the causative agents of severe acute respiratory syndrome (7), severe acute hemorrhagic fever (EBOV) (8), and fatal encephalitis (HeV and NiV) (9, 10).
Enveloped viruses enter their target cells by fusion of the viral envelope with the host cell membrane, delivering the viral genome to the cytoplasm (11). SARS-CoV, the Middle East respiratory syndrome coronavirus (MERS-CoV), EBOV, HeV, and NiV are enveloped viruses that critically require cathepsin L (CatL), a host intracellular lysosomal protease, for their glycoprotein processing and cleavage, which are required for virus fusion and entry into the host cells (12–16). SARS-CoV and EBOV infect the target cells after cleavage of their fusion glycoproteins by CatL in the endocytic vesicles (14, 15). The fusion (F0) protein of HeV and NiV is translocated to the membrane during viral assembly and then internalized, allowing for CatL-mediated cleavage into the F1 and F2 subunits. The processed F protein is then incorporated into the viral particle (12, 13). The high virulence of these viruses and the absence of effective therapies or vaccines pose an ongoing threat to the public health. Developing a broad-spectrum antiviral drug that can inhibit the aforementioned viral infections would represent a first but potent step for the protection of the public.
Antiviral drugs can be broadly divided into 4 major classes. One group consists of nucleoside analogs that interfere with replication of the viral genome and includes the first successful antiviral drug, acyclovir, which is effective against herpesvirus infections and can delay HIV-1 progression (17–19). The first antiviral drug to be approved for treating HIV, zidovudine (AZT), is also a nucleoside analog that blocks reverse transcriptase (20). However, these nucleoside analogs can target only viruses that use their own polymerases for genome replication, such as HIV and herpesviruses, as they have lower affinity to cellular DNA polymerase.
The second class of antivirals includes inhibitors of viral proteases, which are involved in the processing of viral proteins required for the final viral assembly and release. Since HIV assembly requires a similar protease, considerable effort has been made to develop protease inhibitors to treat HIV. Although protease inhibitors became available in the 1990s and have proven effective, they have shown side effects (21, 22). Other limitations of protease inhibitor use include the inability to target a wide range of viruses, as they are highly specific in their action and the protease is present in only certain viruses.
A third class of antivirals includes inhibitors of virus uncoating (23). These agents act on virus penetration and uncoating and include amantadine and rimantadine, which were introduced to combat influenza (23). According to the U.S. Centers for Disease Control and Prevention (CDC), 100% of seasonal H3N2 and 2009 H1N1 samples tested have shown resistance to amantadine, which is no longer recommended for treatment of influenza (24).
The final stage in the virus life cycle is the release of matured viruses from the host cell, and a fourth class of antivirals has targeted this step. Two drugs, named zanamivir (Relenza) and oseltamivir (Tamiflu), which have been recently introduced to treat influenza, prevent the release of viral particles by blocking neuraminidase found on the surfaces of influenza viruses (25, 26). However, the use of these neuraminidase inhibitors is restricted to neuraminidase-containing viruses, and development of resistance to this class of drugs is common (27).
In this study, we made use of a high-throughput screening assay (HTSA), optimized in our laboratory, to identify and characterize lead candidates from libraries of small molecules that can block the cleavage of the viral glycoproteins without inhibiting CatL itself, thus preserving its critical host protease function. We identified a candidate molecule in the HTSA that inhibited the cleavage of peptides derived from SARS-CoV, EBOV, HeV, and NiV glycoproteins, with minimal inhibition of the cleavage of the host pro-neuropeptide Y (pro-NPY)-derived peptide. Moreover, the identified molecule inhibited the entry of all four types of pseudotyped viruses into mammalian cells. Additionally, mechanistic studies indicated that the candidate molecule is a mixed inhibitor of CatL activity. These results demonstrate the utility of our HTSA in the identification of a potential broad-spectrum antiviral lead molecule that can block SARS-CoV, EBOV, HeV, and NiV entry into cells.
MATERIALS AND METHODS
Cells.
293FT cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Cell gro) supplemented with l-glutamine (2 mM) (Invitrogen, Grand Island, NY), sodium pyruvate (1 mM) (Invitrogen), minimal essential medium (MEM) nonessential amino acids (Invitrogen), and 10% fetal bovine serum (FBS) (Invitrogen). The cells were used for the preparation and entry inhibition assays of different pseudotypes bearing SARS-CoV-S, EBOV-GP, HeV-G and -F, NiV-G and -F, and vesicular stomatitis virus G (VSV-G) glycoproteins. The 293FT cells transiently transfected with the human ACE2 expression plasmid (a gift from Michael Farzan) were used for the SARS-CoV-S pseudotype entry inhibition experiments.
Virus- and host-derived peptide synthesis.
Ten-amino-acid peptides derived from the glycoproteins of SARS-CoV (S, amino acids 674 to 683) (28), EBOV (GP, amino acids 196 to 205) (29), HeV (F0, amino acids 105 to 114) (12, 30), and NiV (F0, amino acids 105 to 114) (13, 31), the host pro-neuropeptide Y (pro-NPY) (amino acids 34 to 43) (32), and host peptide F (Pep F) (amino acids 1 to 10) (33) containing the CatL cleavage sites were synthesized and labeled on the N terminus with 5-carboxytetramethylrhodamine (TAMRA) as a light quencher and on the C terminus by 5-carboxyfluorescein (5-FAM) as a light emitter in the Protein Research Laboratory at the University of Illinois at Chicago (UIC). A similarly designed peptide derived from mouse hepatitis virus (MHV) glycoprotein (S) (amino acids 713 to 722) was used as a control (28). The labeled peptides were purified using reversed-phase high-performance liquid chromatography (HPLC).
Mass spectrometry.
The SARS-CoV-S-, EBOV-GP-, HeV-F0-, NiV-F0-, host pro-NPY-, and host Pep F-derived labeled and unlabeled peptides (1 μM) were incubated with 1 μg/ml of human CatL (Sigma-Aldrich, St. Louis, MO) for 1 h at room temperature (RT) in ammonium acetate buffer (pH 5.5) containing 4 mM EDTA and 8 mM dithiothreitol (DTT). The cleavage products were analyzed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry in the UIC Protein Research Laboratory.
Optimization of the HTSA.
The high-throughput screening assay (HTSA) used in this study is a fluorescence resonance energy transfer (FRET)-based assay. The labeled SARS-CoV-S protein-derived peptide was used as a substrate in the primary screen. The assay was optimized in black 384-well plates (Thermo Scientific, Pittsburgh, PA) by adding 1 μg/ml human CatL (Sigma-Aldrich) to 3 μM labeled SARS-CoV-S peptide and by adding 0.25, 0.5, and 1 μg/ml CatL to 1 μM SARS-CoV-S peptide in a 50-μl total volume of ammonium acetate (NH4Ac) buffer (pH 5.5) supplemented with 4 mM EDTA and 8 mM DTT, followed by incubation at room temperature. The fluorescence was measured over time, at 535 nm after excitation at 485 nm, using a fluorescence reader (Tecan Infinite F200 Pro; Tecan Group Ltd., Morrisville, NC) at the UIC HTS facility. Similarly, the EBOV-GP, HeV-F0, NiV-F0, MHV-S, host pro-NPY, and host Pep F peptides were tested by incubating 1 μM peptide with different concentrations of CatL (0.25, 0.5, and 1 μg/ml). The rate of the reaction was measured from the slope of the curve. The quality of the screening assay (Z factor) was determined as follows: Z factor = 1 − (3[σp + σn]/|μp − μn|), where σp = standard deviation of the positive signal, σn = standard deviation of the negative signal, μp = mean of the positive signal, and μn = mean of the negative signal. Z factors between 0.5 and 1 were considered excellent.
Screening for inhibitors of CatL-mediated cleavage of viral peptides.
A library of 5,000 small molecules (Chembridge Diverset Library), at a 40 μM concentration, obtained from Chembridge Corporation (San Diego, CA) was tested in duplicates for inhibiting CatL-mediated cleavage. The peptide at a 1 μM concentration mixed with the small-molecule library was incubated with 0.25 μg/ml CatL in 50 μl of reaction buffer at pH 5.5 for 45 min at room temperature. The reaction was stopped with 10 μl 0.5 M acetic acid, and the fluorescence intensity was measured using a fluorescence reader (Tecan Infinite F200 Pro; Tecan Group Ltd.). The percent inhibition was calculated using the following formula: fluorescence signal in the absence of the small molecule − fluorescence signal in the presence of the small molecule × 100/fluorescence signal in the absence of the small molecule − fluorescence signal in the absence of the enzyme. The top 50 hits that inhibited the cleavage of SARS-CoV-S-derived peptide at a cutoff of 61% inhibition were screened in duplicates for the inhibition of cleavage of EBOV-GP, HeV-F0, and NiV-F0 as well as cleavage of pro-NPY-derived labeled peptides.
Preparation of pseudotyped viruses.
Pseudotyped viruses (EBOV-GP, SARS-CoV-S, HeV, NiV, and VSV-G) were generated by cotransfecting 2 × 106 293FT cells (grown in DMEM with 10% FBS) with pHIV-GFP-luc expression vector (18 μg), pgagpol HIV vector (1.8 μg), pHIV-Rev (360 ng), and pHIV-TAT (360 ng) (34), along with the pcDNA3.1-S plasmid (10 μg) coding for the SARS-CoV-S glycoprotein, the pcDNA3.1-GP plasmid (10 μg) coding for the EBOV-GP glycoprotein, or the pcDNA3.1-VSVG plasmid (1 μg) coding for the VSV glycoprotein (G) using calcium phosphate transfection according to a previously described protocol (35). For the production of HIV/ΔE, only HIV coding vectors were used for transfection. For HeV and NiV pseudotyped viruses, pCAGG expression plasmids coding for G (15 μg) and F (5 μg) protein of HeV or NiV were transfected along with HIV vectors as described above. The culture medium was changed the following morning and the supernatant collected 24 and 48 h later and pooled. The virus stocks were frozen at −80°C until used.
Pseudovirus inhibition assay.
Different pseudotyped viruses (EBOV-GP, SARS-CoV-S, and VSV-G as a negative control), normalized for equal infectivity using a HIV-1 p24 enzyme-linked immunosorbent assay (ELISA) kit (Express Biotech International, Thurmont, MD), were added to 293FT cells in the presence of a 10 μM concentration of the candidate inhibitory small molecules identified in the HTS. For the SARS-CoV-S pseudotyped virus inhibition assays, the 293FT cells were transiently transfected with human ACE2 expression plasmid (0.4 μg/well), using Effectene transfection reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. On the following morning, the medium was changed, and 4 h later, the cells were treated. The virus or virus-molecule mixtures were added to 2 × 105 293FT cells/well seeded in 6-well plates. Seventy-two hours later, the cells were lysed and the luciferase expression was determined using a luciferase assay kit (Promega, Madison, WI) according to the manufacturer's instructions. For HeV and NiV, the small molecule 5705213 and its derivative 7402683 were used at a final concentration of 100 μM during both pseudotyped virus preparation and transduction. A commercial CatL inhibitor I, Z-Phe-Tyr-CHO (catalog no. 219421; Calbiochem, Billerica, MA) (10 μM concentration) and dimethyl sulfoxide (DMSO) were used as positive and negative controls, respectively. The percent entry inhibition of the candidate small molecule on different pseudotypes was calculated using the following formula: luciferase reading of mock-treated virus − luciferase reading of small-molecule-treated virus × 100/luciferase reading of mock treated virus − luciferase reading of HIV/ΔE.
IC50 determination.
Pseudotypes bearing Ebola virus-GP or SARS-CoV-S were added to 293FT cells seeded at a density of 2 × 105 cells/well in the presence of different concentrations of the small molecule 5705213 and its derivative 7402683 (1 to 200 μM). Seventy-two hours later, cells were lysed and the luciferase expression was determined using a luciferase assay kit (Promega). The small-molecule concentration was plotted against the percent inhibition of viral entry, and the concentration of the small molecule that inhibited the viral entry by 50% (IC50) was determined. Cathepsin L inhibitor III (catalog no. 219427; Calbiochem) was used as a control.
Cytotoxicity assay (MTT assay).
293FT cells were seeded at a density of 104 cells/well in 96-well plates. The following day, the cells were treated with different concentrations (10, 30, 50, and 100 μM) of the selected small molecule, and the cytotoxic effect of the small molecule was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT reagent) (Roche, Indianapolis, IN) according to the manufacturer's instructions. Briefly, following 24, 48, and 72 h of incubation with each small molecule, the cells were washed once with phosphate-buffered saline (PBS), and 100 μl/well of fresh DMEM, without phenol red, was added. Subsequently, 10 μl of MTT reagent was added to each well and incubated for 4 h at 37°C, and then 100 μl of 10% SDS in 0.01 M HCl was added to each well with vigorous mixing and further incubated for 4 h at 37°C. The optical density (OD) was measured at 595 nm, and the percent viability was calculated relative to the untreated control after subtracting the background. For determining the 50% cytotoxic concentration (CC50) of the small molecule 5705213 and its derivative 7402683, the 293FT cells were incubated with increasing concentrations (3.9 μM to 2 mM) of each small molecule, and the cytotoxicity was measured at 72 h postincubation as mentioned above. The CC50 (concentration of the small molecule that killed 50% of cells) was determined from the linear portion of the curve. Cathepsin L inhibitor III was used as a control, and its CC50 was determined as described above.
Enzyme kinetics.
Different concentrations of the SARS-CoV-S-, EBOV-GP-, HeV-F0-, and NiV-F0-derived labeled peptide (2 to 64 μM) were incubated with a fixed concentration of CatL (0.5 μg/ml) in the absence or presence of the small molecule 5705213 for 40 min at room temperature. The reaction was stopped with 10 μl 0.5 M acetic acid, after which the fluorescence was read, at 535 nm after excitation at 485 nm, with a fluorescence reader (Tecan Infinite F200 Pro; Tecan Group Ltd.). The velocity of the reaction at different substrate concentrations was calculated (fluorescence units/minute) and plotted versus substrate concentration. The inverse velocity was further plotted versus the inverse substrate concentration (Lineweaver-Burk plot), from which the Km and Vmax were calculated.
SARS-CoV-S recombinant protein expression and purification.
A cDNA encoding the SARS-CoV-S ectodomain (amino acids 12 to 1184) was amplified using the pcDNA3.1-S expression plasmid, expressing full-length SARS-CoV-S protein, as a template. The forward primer with a 5′ NheI 6×His tag and a reverse primer with a 5′ BamHI Flag tag were used in the PCR. The PCR product was digested with NheI and BamHI (New England BioLabs, Ipswich, MA) independently and cloned into the pET-11b bacterial expression vector (EMD4 Biosciences, Billerica, MA). The recombinant plasmid with His-S-Flag DNA was transformed into DE3 BL21 cells (Agilent Technologies, Santa Clara, CA). The transformed cells were induced, at an OD of 0.8, with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 2 h, after which the cells were lysed with lysis buffer (80 mM Tris-HCl [pH 6.8], 15% SDS, 0.006% bromophenol blue, and 15% glycerol). The protein expression was detected by Coomassie blue staining following separation on 4 to 15% SDS-polyacrylamide gels and confirmed by Western blotting using monoclonal anti-Flag mouse antibody (Sigma-Aldrich) and secondary anti-mouse horseradish peroxidase (HRP)-conjugated antibody (Promega). The His-S-Flag inclusion bodies were purified and dissolved in 100 mM Tris-HCl buffer with 8 M urea (pH 8.8), and the protein was refolded by gradual dilution in a refolding buffer (0.1 M Tris [pH 8], 0.4 M arginine, 0.1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM EDTA, 5 mM glutathione [GSH], and 0.5 mM glutathione disulfide [GSSG]).
SARS-CoV-S protein cleavage inhibition assay.
Purified recombinant SARS-CoV-S protein was incubated for 4 h with 2 μg/ml CatL in ammonium acetate buffer (pH 5.5), containing 4 mM EDTA and 8 mM DTT, in the absence or presence of increasing concentrations of the small molecule 5705213 and its derivative 7402683. The cleavage of the protein was detected by Western blotting using anti-Flag mouse monoclonal antibody (Sigma-Aldrich).
Inhibition assay of endogenous cleavage of Hendra and Nipah virus fusion protein.
293FT cells were plated at a density of 2 × 105 cells/well in 6-well plates and transfected with 2 μg of Nipah or Hendra virus F0 expression plasmid using Polyfect transfection reagent (Qiagen). Four hours later, they were treated with the small molecules (5705213 and its derivative 7402683) at a 100 μM concentration. The commercial CatL inhibitor I, Z-Phe-Tyr-CHO (Calbiochem), was used as a positive control at a 10 μM concentration. The cells were lysed 48 h later using a lysis buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40, and 0.1% SDS), and F0 processing was determined by Western blotting using cross-reactive anti-Nipah and -Hendra virus F protein monoclonal antibody.
SARS-CoV entry in the presence of TMPRSS2.
293FT cells were plated at a density of 2 × 105 cells/well in 6-well plates and transfected with the human SARS-CoV receptor ACE2 plasmid alone (1 μg) or with the receptor plus increasing amounts of transmembrane protease/serine subfamily member 2 (TMPRSS2) protease plasmid (10 to 800 ng) using the Polyfect transfection reagent (Qiagen). Twenty-four hours later, the cells were transduced with the SARS-CoV-S pseudotyped virus. HIV/ΔE, which does not express the SARS-CoV-S protein, was used as a negative control. At 72 h postransduction, the entry was quantified by measuring the luciferase expression, as relative light units (RLU), in cell lysates. The entry of SARS-CoV-S pseudotyped virus into 293FT cells, transfected with either the receptor alone (ACE2 plasmid) or the receptor plus 10 ng of TMPRSS2 plasmid, was measured in the presence or absence of different inhibitors. Camostat mesylate was used as a TMPRSS2 protease inhibitor (Tocris Bioscience, Bristol, United Kingdom) and Z-FY(t-Bu)-DMK as a CatL inhibitor (cathepsin L inhibitor III; Calbiochem catalog no. 219427). All inhibitors were used at a 10 μM concentration except the small molecule 5705213, which was used at a 50 μM final concentration.
CatB inhibition assay.
The small molecule 5705213 and its derivative 7402683 were incubated at different concentrations (50 to 200 μM) with cathepsin B (CatB) and its fluorogenic substrate (Sensolyte 520 cathepsin B assay kit; Anaspec Inc., catalog no. 72164) for 30 min at room temperature according to the manufacturer's instructions. The reaction was stopped using 10 μl 0.5 M acetic acid, and the fluorescence was read at 535 nm after excitation at 485 nm (Tecan Infinite F200 Pro; Tecan Group Ltd.). The percent inhibition was calculated as before. The CatB inhibitor supplied in the kit was used as a control at concentrations of 10 nM, 100 nM, and 1 μM.
RESULTS
Synthesis of virus- and host protein-derived peptides containing CatL cleavage sites.
Peptides derived from the amino acid sequences of glycoproteins of SARS-CoV, EBOV, HeV, and NiV, the host pro-neuropeptide Y (pro-NPY), and the host peptide F (Pep F), which contained the naturally conserved CatL cleavage sites, were synthesized in the protein research laboratory at UIC (Fig. 1A to F). The peptide derived from MHV-S glycoprotein was used as a negative control (Fig. 1G). The peptides were labeled on the N terminus with 5-carboxytetramethylrhodamine (TAMRA) as a quencher and on the C terminus with 5-carboxyfluorescein (5-FAM) as an emitter. With the intact peptide, no fluorescence emission can be detected at 535 nm on excitation of 5-FAM at 485 nm due to the quenching by TAMRA. In contrast, on cleavage of the peptide by CatL, an emission of light at 535 nm can be detected. As determined by mass spectrometry, all labeled and unlabeled peptides were cleaved at the expected sites (data not shown), confirming that the fluorophores did not affect the CatL-mediated cleavage.
FIG 1.
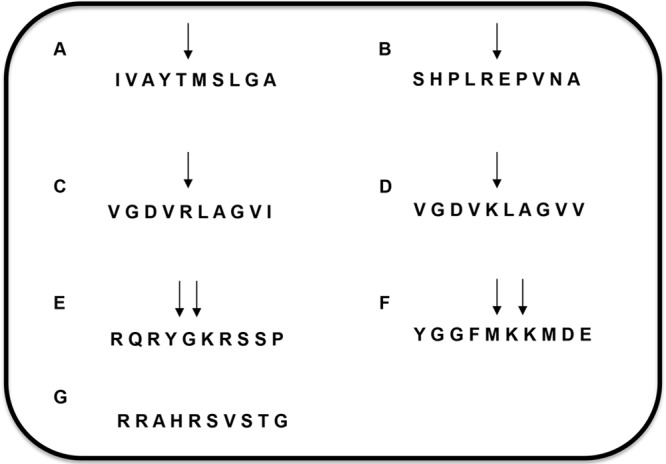
Host- and virus-derived peptides. Peptides composed of 10 amino acids derived from the glycoproteins of viruses and human pro-neuropeptide Y and peptide F were synthesized. (A) SARS-CoV spike (S) protein-derived peptide (amino acids 674 to 683); (B) EBOV-GP protein-derived peptide (amino acids 196 to 205); (C) NiV fusion protein (F0)-derived peptide (amino acids 105 to 114); (D) HeV fusion protein (F0)-derived peptide (amino acids 105 to 114); (E) human pro-neuropeptide Y (pro-NPY)-derived peptide (amino acids 34 to 43); (F) human peptide F-derived peptide (amino acids 1 to 10); (G) control MHV-S glycoprotein-derived peptide (amino acids 713 to 722). Arrows indicate the cathepsin L cleavage sites in the corresponding peptides.
Optimization of the HTSA.
The SARS-CoV-S-derived labeled peptide was used initially to optimize the high-throughput screening assay (HTSA). The labeled SARS-CoV-S-derived peptide was treated with CatL, and the cleavage was measured in the form of increased fluorescence over time, with no increase in fluorescence in CatL-untreated peptide (Fig. 2A). Although different viral peptides showed different rates of cleavage in a dose-dependent manner, the host peptides, particularly the host pro-NPY-derived peptide, was cleaved at a higher rate than the viral peptides (Fig. 2B). However, CatL failed to cleave MHV-S-derived control peptide (Fig. 2B). The validity of the assay was determined based on the Z factor, which was calculated for different peptides incubated with 0.5 μg/ml CatL after stopping the reaction with 0.5 M acetic acid. The Z factor was found to be between 0.5 and 1.0 and thus validated the assay.
FIG 2.
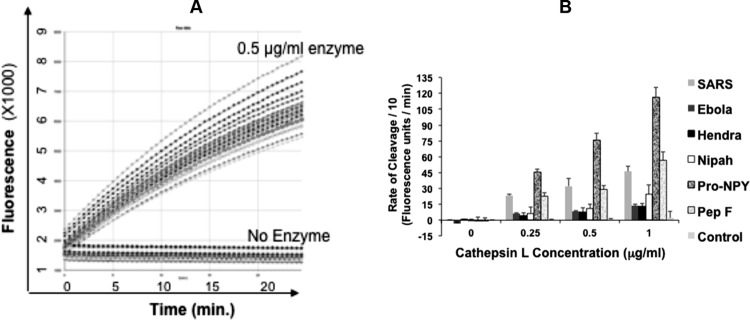
Screening assay optimization. (A) Cathepsin L (0.5 μg/ml) was incubated with 1 μM labeled SARS-CoV-derived peptide at RT, and the fluorescence emitted was measured over time compared to that for untreated peptide (16 replicates each). (B) Cleavage rates for different viral peptides, human pro-neuropeptide Y (pro-NPY), and peptide F (Pep F), shown as fluorescence units/min calculated from the slope of the curve at different enzyme concentrations. Mouse hepatitis virus (MHV) glycoprotein (S)-derived peptide was used as a control. The bars show the averages from an experiment done in triplicates and repeated twice with similar results, and error bars represent standard deviations (SD).
HTS of a small-molecule library identifies potential inhibitors of CatL-mediated cleavage.
A library of 5,000 small molecules from Chembridge Corporation (Chembridge Diverset Library) was tested against the labeled SARS-CoV-S-derived peptide. We identified 50 small molecules that inhibited the CatL cleavage of SARS peptide at a cutoff of 61% inhibition (data not shown). The validity of the HTSA for small-molecule screening was reconfirmed using the means and standard deviations of positive and negative values. This yielded a Z factor value of 0.61 and confirmed the reliability of the CatL-mediated cleavage inhibition assay.
Rescreening of the top 50 hits as potential broad-spectrum inhibitors of CatL-mediated cleavage of viral peptides.
The top 50 hits were tested for their ability to inhibit CatL-mediated cleavage of EBOV-, HeV-, and NiV-derived peptides and the host pro-NPY-derived peptide. Twelve small molecules out of 50 showed inhibition of cleavage of all viral peptides relative to a much lower level of inhibition of the cleavage of the pro-NPY-derived peptide (Table 1).
TABLE 1.
Screening of the top 50 hits against all virus- and host-derived peptides
| Compound | % Inhibition of peptide cleavagea |
||||
|---|---|---|---|---|---|
| EBOV | NiV | HeV | SARS-CoV | Pro-NPY | |
| 6874634 | 44.6 | 39.55 | 24.79 | 91.98 | 3.6 |
| 5219666 | 32.6 | 102.26 | 31.07 | 98.65 | 22.5 |
| 5172420 | 6.1 | 55.68 | 8.20 | 42.29 | 4.1 |
| 7665576 | −3.8 | 63.70 | 37.87 | 60.65 | 2.0 |
| 7928055 | − 16.0 | 18.03 | −0.67 | −0.15 | −0.8 |
| 5152606 | 31.8 | 121.65 | 108.40 | 82.15 | 70.2 |
| 7923236 | 31.8 | 94.08 | 39.17 | 88.99 | −2.7 |
| 7909513 | −1.6 | 5.15 | 5.24 | 24.06 | 12.0 |
| 5669125 | −1.8 | 23.57 | −2.70 | 57.39 | 3.3 |
| 7798500 | 66.3 | 53.73 | 40.40 | 77.83 | 21.4 |
| 7938158 | −12.6 | 15.92 | −14.40 | 1.34 | −5.3 |
| 7793889 | −3.4 | 51.07 | −7.77 | 76.53 | 6.1 |
| 7951692 | 10.2 | 16.58 | 0.34 | 14.51 | 0.7 |
| 7950181 | −8.6 | 6.45 | −9.26 | 8.55 | −7.0 |
| 7888659 | 66.9 | 80.96 | 73.49 | 79.93 | 64.8 |
| 7808526 | 9.6 | 40.80 | −4.08 | 69.24 | 1.2 |
| 6572698 | 34.0 | 31.81 | 7.92 | 65.51 | −12.1 |
| 5182554 | 11.6 | 44.08 | 42.12 | 80.76 | 20.5 |
| 7924029 | 45.7 | 55.7 | 32.88 | 65.73 | 10.1 |
| 7914021 | 10.1 | 63.91 | 24.49 | 78.34 | 4.8 |
| 7927865 | −3.4 | 27.96 | 0.86 | 52.46 | 6.4 |
| 7946410 | 5.7 | 18.33 | 6.50 | 24.34 | 8.8 |
| 7946204 | −24.6 | 12.33 | −7.50 | 39.61 | 9.6 |
| 5169083 | 13.1 | 49.78 | 35.73 | 44.31 | 29.9 |
| 5175089 | 6.4 | 2.06 | −2.85 | 38.88 | 10.0 |
| 7948109 | 6.2 | 46.11 | 37.33 | 38.54 | 14.3 |
| 7931205 | 10.7 | 81.01 | 22.44 | 50.22 | 5.3 |
| 7786009 | 2.1 | 58.07 | 37.22 | 51.28 | 10.5 |
| 7945999 | −0.6 | 55.23 | 33.41 | 39.55 | 4.9 |
| 7948190 | −2.6 | 45.06 | 7.38 | 33.84 | 11.8 |
| 7928214 | −6.1 | 12.87 | −0.01 | 23.55 | 0.9 |
| 5705213 | 82.4 | 88.36 | 90.92 | 77.79 | 18.9 |
| 7910528 | 33.4 | 57.76 | 12.10 | 56.45 | 10.7 |
| 7976237 | 25.4 | 31.98 | 21.93 | 47.55 | 15.1 |
| 7927434 | 10.5 | −8.16 | −8.72 | −3.09 | 1.3 |
| 7948346 | 25.2 | 49.25 | 17.70 | 72.37 | 10.2 |
| 7914488 | 17.4 | 30.96 | 23.22 | 47.94 | 2.2 |
| 7557708 | 26.8 | 39.35 | 23.55 | 53.23 | 2.1 |
| 7927879 | −6.6 | 30.39 | 3.55 | 28.07 | 7.9 |
| 7947992 | 20.9 | 97.85 | 32.65 | 25.60 | 3.4 |
| 7905966 | −12.1 | 9.77 | 3.23 | 37.16 | 6.4 |
| 7940158 | 12.3 | 42.76 | 25.12 | 61.47 | 3.7 |
| 7790059 | 1.8 | 41.82 | −11.51 | 66.62 | 1.1 |
| 7760561 | 1.1 | 27.20 | 5.70 | 32.44 | 15.2 |
| 7945787 | −0.3 | −11.7 | −10.47 | 30.53 | −2.5 |
| 7963087 | −1.5 | 46.45 | −2.95 | 47.39 | 4.5 |
| 5728323 | −15.9 | 13.74 | −9.09 | 49.81 | −0.7 |
| 7946700 | −11.5 | 15.09 | −0.72 | 51.17 | 11.5 |
| 7924129 | −11.7 | −0.50 | −10.37 | 41.66 | −1.2 |
| 7944514 | 8.05 | 12.40 | 3.26 | 40.85 | 8.1 |
Values are average percent inhibitions of the cleavage of viral and host peptides by different compounds (40 μM) tested in duplicates. Results for the 12 small molecules out of 50 that showed inhibition of cleavage of all viral peptides relative to a much lower level of inhibition of the cleavage of the pro-NPY-derived peptide are in bold.
HTSA-selected inhibitors differentially inhibit host cell entry of pseudotyped viruses.
Nine uncolored small molecules out of the 12 identified from the HTSA, which showed higher inhibitions of viral peptide cleavage while minimally inhibiting the cleavage of pro-NPY-derived peptide, were tested for inhibiting the entry of pseudotypes bearing the SARS-CoV-S or EBOV-GP glycoprotein. Four (i.e., 7910528, 7914021, 5705213, and 5182554) (Fig. 3) of the 9 small molecules were found to inhibit the entry of both pseudotypes bearing the EBOV-GP and SARS-CoV-S glycoproteins (Fig. 4). The small molecules 7910528, 7914021, 5705213, and 5182554 showed 23.3% and 30.3%, 51.5% and 27.1%, 39.8% and 64.7%, and 60.45% and 49.3% inhibition of EBOV-GP and SARS-CoV-S glycoprotein-bearing pseudotypes, respectively. Other tested small molecules surprisingly enhanced the entry of pseudotypes bearing the SARS-CoV-S glycoprotein. When we tested derivatives of different small molecules, only the small molecule 7402683 showed higher inhibition than its parent 5705213 (53.3% and 68.3% inhibition for EBOV-GP and SARS-CoV-S pseudotypes, respectively) (Fig. 4). As expected, DMSO did not affect the entry of different pseudotyped viruses. Similarly, the entry of VSV-G pseudotyped virus was not affected by any of the small molecules. These results showed that the identified small molecules could specifically inhibit the entry of viruses that utilize CatL for entry into the target cells.
FIG 3.
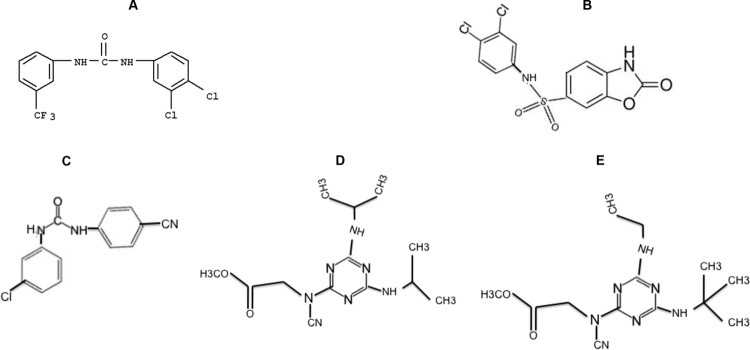
Chemical structures of the small molecules identified by pseudovirus inhibition assay. Four small molecules showed inhibition of both EBOV and SARS-CoV pseudotyped virus entry. (A) Compound 5182554 {N-(3,4-dichlorophenyl)-N′-[3-(trifluoromethyl)phenyl]urea}; (B) compound 7910528 [N-(3,4-dichlorophenyl)-2-oxo-2,3-dihydro-1,3-benzoxazole-6-sulfonamide]; (C) compound 7914021 [N-(3-chlorophenyl)-N′-(4-cyanophenyl)urea]; (D and E) Compound 5705213 {methyl-N-[4,6-bis(isopropylamino)-1,3,5-triazin-2-yl]-N-cyanoglycinate} (D) and (E) its derivative 7402683 {methyl-N-[4-(tert-butylamino)-6-(ethylamino)-1,3,5-triazin-2-yl]-N-cyanoglycinate}.
FIG 4.
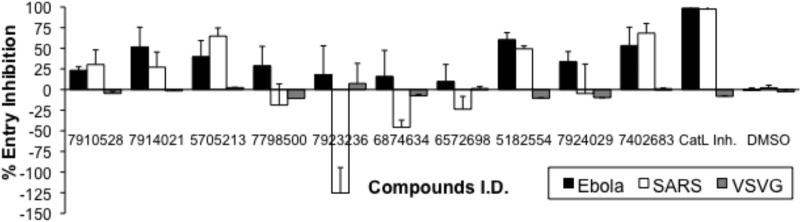
Pseudovirus entry inhibition assay. Selected compounds were tested for inhibiting the entry of pseudotyped viruses bearing SARS-CoV-S or EBOV-GP. Ten nanograms of HIV/p24-equivalent pseudoviruses were treated with a 10 μM concentration of each compound for 1 h at RT. Subsequently, 293FT cells or 293FT cells transfected with human ACE2 expression plasmid were transduced with Ebola virus-GP and SARS-CoV-S pseudoviruses, respectively. The luciferase expression was determined at 72 h postransduction, and percentages of inhibitions were calculated. VSV-G pseudotyped virus and DMSO-treated viruses were used as negative controls, and cathepsin L inhibitor I (CatL Inh.)-treated cells served as a positive control. Error bars represent SD from a representative experiment performed using triplicates.
The small molecule 5705213 and its derivative are not cytotoxic, and their actions are specific.
An MTT-based cell cytotoxicity assay was performed at different time points to test whether the EBOV and SARS-CoV pseudotyped virus entry inhibition was due to specific effects of the tested small molecules on CatL cleavage or due to toxic effects on cell viability or proliferation. The small molecules 7914021 and 5182554 were found to be toxic to the 293FT cells when treated at concentrations of 10 μM or above. The cytotoxic effect was dramatic for the two small molecules at concentrations above 10 μM after 1 day of incubation with 293FT cells (20 to 100% cytotoxicity). This cytotoxicity increased over the next 2 days even at a 10 μM concentration (Fig. 5A). In contrast, the small molecule 5705213 and its derivative 7402683 did not show significant cytotoxic effects (cell viability was over 80%) on the 293FT cells at the 10 to 100 μM concentration range over 3 days of incubation (Fig. 5A). This further confirmed the specific inhibition of CatL cleavage of EBOV-GP and SARS-CoV-S on the surface of pseudotypes by the small molecule 5705213 and its derivative. To further ensure the specific inhibitory effect of the small molecules on the viruses that utilize CatL for entry, the pseudotyped virus bearing VSV-G was treated with different concentrations of the small molecule 5705213 and its derivative, and the luciferase activity was measured at 72 h postransduction. The small molecules did not show any significant inhibitory effect on the entry of VSV-G pseudotyped virus even at concentrations up to 100 μM (Fig. 5B). While the small molecule 5705213 showed IC50s of 9 μM (Fig. 6A) and 15 μM (Fig. 6B), the derivative showed higher potency, with IC50s of 6 μM (Fig. 6C) and 10 μM (Fig. 6D) against SARS-CoV-S and EBOV-GP pseudotyped viruses, respectively. The commercial cathepsin L inhibitor III, a commonly used specific cathepsin L inhibitor (36–38), showed IC50s of 5 and 7 μM against SARS-CoV-S and EBOV-GP pseudotypes, respectively (Fig. 6E and F). The CC50s for the small molecule 5705213, its derivative, and the commercial CatL inhibitor III were found to be 400 μM, 350 μM, and 420 μM, respectively (Fig. 6G, H, and I). The small molecule 5705213 and its derivative 7402683 showed selectivity indices (SI) (CC50/IC50) of 26.7 and 35 against pseudotyped virus bearing EBOV-GP, respectively, while they showed SI of 44.4 and 58.3 against SARS-CoV-S pseudotyped virus, respectively. The calculated SI for CatL inhibitor III were 84 and 60 against SARS-CoV-S and EBOV-GP pseudotypes, respectively.
FIG 5.
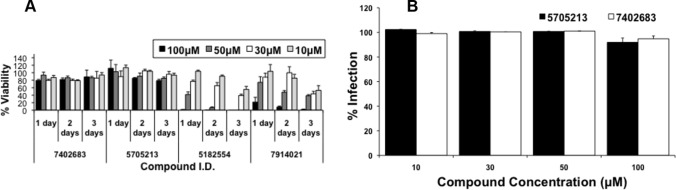
Small molecule 5705213 and its derivative are not cytotoxic, and their actions are specific. (A) Four molecules identified in the pseudovirus entry inhibition assay, with the highest inhibitory effect on both EBOV-GP and SARS-CoV-S pseudotyped viruses, were tested for their cytotoxic effect on 293FT cells. An MTT assay was performed over 3 days using different concentrations of each compound (10 to 100 μM) to assess cell viability. OD was measured at 595 nm. The cell viability calculated was normalized to the untreated control after subtracting the background. Error bars represent the SD from an experiment performed using triplicates. (B) Different concentrations of the selected noncytotoxic small molecule and its derivative were tested against the VSV-G pseudotyped virus, and the infection normalized to the untreated virus was calculated. Error bars represent SD from a representative experiment performed using triplicates.
FIG 6.

IC50s and CC50s of small molecules and commercial CatL inhibitor III. The small molecule 5705213 and its derivative 7402683 were tested for dose-dependent inhibition of the entry of pseudotypes bearing SARS-CoV-S and EBOV-GP. The concentration causing 50% entry inhibition was determined from the linear portion of the curve. The CC50 was determined by MTT assay. (A) IC50 of 5705213 against SARS-CoV-S pseudotypes; (B) IC50 of 5705213 against EBOV-GP pseudotypes; (C) IC50 of 7402683 against SARS-CoV-S pseudotypes; (D) IC50 of 7402683 against EBOV-GP pseudotypes; (E) IC50 of the commercial CatL inhibitor III against SARS-CoV-S pseudotypes; (F) IC50 of the commercial CatL inhibitor III against EBOV-GP pseudotypes; (G) CC50 of 5705213; (H) CC50 of 7402683; (I) CC50 of the commercial CatL inhibitor III.
The small molecule 5705213 and its derivative 7402683 inhibit CatL-mediated cleavage of SARS-CoV-S-Flag recombinant protein in vitro.
To further test the inhibitory effects of the small molecules 5705213 and 7402683, we expressed the SARS-CoV-S-Flag recombinant protein in Escherichia coli BL21 cells and purified the protein. We found that there was a dose-dependent cleavage of the recombinant SARS-CoV-S-Flag protein by CatL (Fig. 7A), which was inhibited by the small molecule 5705213 (Fig. 7B) and its derivative 7402683 (Fig. 7C) in a dose-dependent manner at the tested concentrations.
FIG 7.

Small molecule 5705213 and its derivative 7402683 inhibit in vitro CatL-mediated cleavage of SARS-CoV-S-Flag recombinant protein. (A) Dose-dependent cleavage of recombinant SARS-CoV-S-Flag protein by CatL. The SARS-CoV-S-Flag protein was incubated with 0.5, 1, and 2 μg/ml of CatL for 4 h at RT, and the cleavage of the protein was detected by Western blotting using anti-Flag mouse monoclonal antibody. (B and C) Inhibition of the CatL-mediated cleavage of SARS-CoV-S-Flag protein by the small molecules 5705213 and 7402683, respectively. The SARS-CoV-S-Flag protein was incubated with 2 μg/ml of CatL for 4 h at RT in absence or in the presence of increasing concentration of the small molecules (10 to 320 μM). The control (Ctrl) lane represents the S-Flag protein in the absence of the enzyme and inhibitor. CatL inhibitor I (CatL inh.) was used as a positive control of inhibition. Asterisks represent nonspecific bands.
The small molecule 5705213 and its derivative 7402683 inhibit the endogenous processing of Nipah and Hendra virus fusion glycoproteins and the entry of pseudotyped viruses.
Next, we tested the inhibitory effect of small molecules 5705213 and 7402683 on the endogenous processing of NiV-F0 and HeV-F0 proteins in 293FT cells transiently expressing the fusion proteins. We found that both molecules and the commercial CatL inhibitor I were able to efficiently inhibit the endogenous cleavage by CatL (Fig. 8A and B). The two small molecules as well as the control CatL inhibitor I inhibited the entry of HeV-F0 and NiV-F0 pseudotyped viruses into 293FT cells by 80 to 100% (Fig. 8C). As expected, the VSV-G pseudotyped virus entry was unaffected by the small molecules (Fig. 8C).
FIG 8.
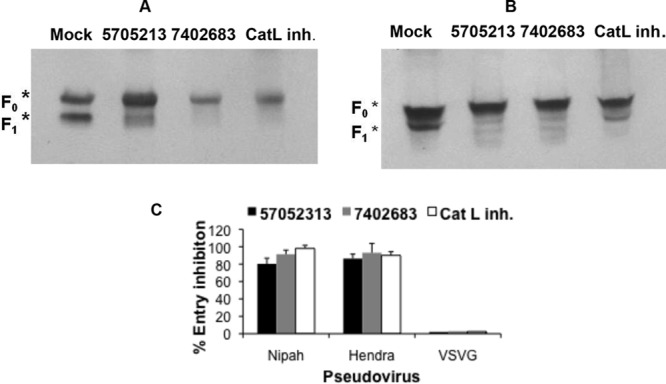
Endogenous processing of Nipah and Hendra virus fusion (F0) proteins and entry of pseudotyped viruses are inhibited by the small molecule 5705213 and its derivative 7402683. (A and B) Endogenous processing of Nipah and Hendra virus F0 proteins, respectively, in the presence or absence of the small molecules 5705213 and 7402683. 293FT cells were transfected with Nipah or Hendra virus F0 expression plasmid and then treated with the small molecules, at a concentration of 100 μM, 4 h posttransfection. The cathepsin L inhibitor I (CatL inh.) was used as a control. The cells were lysed 48 h later, and F0 processing was determined by Western blotting using cross-reactive anti-Nipah and -Hendra virus F protein monoclonal antibody. F0 represents uncleaved fusion protein, while F1 is the fusion subunit of the F0 protein. (C) Pseudovirus entry inhibition assay. Nipah and Hendra pseudotyped virus entry into 293FT cells in the presence of a 100 μM concentration of each molecule was quantified by measuring luciferase expression at 72 h postransduction. VSV-G pseudotyped virus was used as a negative control and Cat L inh. I as a positive control. Error bars are SD from a representative experiment performed using triplicates.
A combination of the small molecule 5705213 and a TMPRSS2 inhibitor can more dramatically inhibit SARS-CoV-S pseudotyped virus entry.
The transmembrane protease/serine subfamily member 2 (TMPRSS2) has been shown to activate the entry of several viruses, including influenza virus (39, 40), MERS-CoV (41), respiratory parainfluenza virus (42), and human coronavirus 229E (43) and SARS-CoV natural infection and entry into pneumocytes, which is also activated by endosomal cathepsin L (14, 44, 45). Therefore, we tested the entry of the pseudotypes bearing SARS-CoV-S glycoprotein into 293FT cells expressing ACE2 receptor with or without TMPRSS2. We found a 1.5-fold increase in pseudotyped virus entry into cells expressing the TMPRSS2 membrane protease relative to the entry in cells with no TMPRSS2 expression. However, the viral entry decreased upon increasing the amount of TMPRSS2 expression (Fig. 9A). This may be due to the nonspecific proteolytic effect of highly expressed TMPRSS2 protease.
FIG 9.

SARS-CoV pseudotyped virus entry is more inhibited by a combination of the small molecule 5705213 and protease inhibitor in cells expressing TMPRSS2 protease. (A) Entry of pseudotyped SARS-CoV into 293FT cells transfected with the ACE2 plasmid either alone or in combination with increasing amounts of TMPRSS2 protease plasmid was measured. HIV/ΔE not expressing the SARS-CoV spike (S) protein was used as a negative control. Entry was quantified by measuring the luciferase expression as relative light units (RLU) in cell lysates prepared at 72 h postransduction. Error bars are SD from a representative experiment performed using triplicates. (B) Entry of pseudotyped SARS-CoV into 293FT cells, transfected with either the ACE2 receptor plasmid or receptor plus 10 ng of TMPRSS2 plasmid, in the presence or absence (mock) of different inhibitors. Cathepsin L Inhibitor III (CatL Inh. III) and camostat mesylate as a serine protease inhibitor (Protease Inh.) were used at 10 μM, while the small molecule 5705213 was used at 50 μM. Error bars are SD from a representative experiment performed using triplicates.
Next, we tested the inhibitory effect of the small molecule 5705213, a commercial protease inhibitor (camostat) (36, 41), and a combination of both in 293FT cells expressing human ACE2, with or without the expression of TMPRSS2. As expected, the small molecule 5705213 and the commercial CatL inhibitor III, unlike the protease inhibitor, were able to inhibit the entry by 75 and 60%, respectively, in the absence of the TMPRSS2. In the presence of TMPRSS2, a rescue of inhibition by the small molecule 5705213 and the commercial CatL inhibitor III was observed (Fig. 9B). The protease inhibitor inhibited the entry by 48%, while a combination of our small molecule and the protease inhibitor completely blocked the entry of pseudotypes bearing SARS-CoV-S. Our results are consistent with previously published data that showed complete blockage of SARS-CoV entry by a combination of CatL inhibitor and TMPRSS2 inhibitor (36). This suggested that our identified small molecule 5705213 may be used in combination with a commercial protease inhibitor to completely block SARS-CoV natural lung infection.
The small molecule 5705213 is a mixed inhibitor for CatL.
We sought to determine the nature of the lead small molecule inhibitor 5705213 using an enzyme assay based on Michaelis-Menten kinetics. The Lineweaver-Burk plot provides a graphical way for analysis of the Michaelis-Menten equation, V = Vmax [S]/Km + [S]; taking the reciprocal gives 1/V = Km + [S]/Vmax[S] = Km/Vmax · 1/[S] + 1/Vmax (where V is the reaction velocity, Km is the Michaelis-Menten constant, Vmax is the maximum reaction velocity, [S] is the substrate concentration, and Km/Vmax is the slope). Accordingly, different concentrations (2 to 64 μM) of labeled peptides were incubated with 0.5 μg/ml of CatL in the presence or absence of the small molecule 5705213. From the Lineweaver-Burk plot, we found that there was a dose-dependent increase of Km and a decrease in Vmax in the presence of the small molecule (Fig. 10A to D). We additionally found that our small molecule 5705213 bound to the free enzyme with a higher affinity than to the enzyme-substrate complex (Fig. 10E). These results suggested that the identified small molecule is a mixed inhibitor based on currently established kinetics.
FIG 10.

Small molecule 5705213 is a mixed enzyme inhibitor and has more affinity to free enzyme than to enzyme-substrate complex. (A to D) Lineweaver-Burk plots. Different concentrations (2 to 64 μM) of labeled SARS-CoV-derived peptide (A), labeled Ebola virus-derived peptide (B), labeled Hendra virus-derived peptide (C), and labeled Nipah virus-derived peptide (D) were incubated with 0.5 μg/ml of CatL for 40 min in the presence or absence of the small molecule 5705213. The reaction was stopped with 0.5 M acetic acid, after which the fluorescence was measured at 535 nm after excitation at 485 nm. The velocity of the reaction was calculated as fluorescence units/min. The reciprocal of substrate concentration was plotted against the reciprocal of velocity to get the Lineweaver-Burk plot. The Km was calculated from the y intercept (Vmax) and slope (Km/Vmax) in absence and in the presence of the inhibitor. (E) Binding affinity of the small molecule 5705213. The small molecule at a 50 or 100 μM concentration was mixed with the substrate SARS-CoV-derived peptide, the enzyme was added and mixed, and then the mixture was added to the substrate, or the inhibitor was added after the enzyme was added to the substrate. The percentages of inhibition were calculated. Asterisks indicate a significant difference with a P value of 0.0002 to 0.006. ns, nonsignificant.
The small molecule 5705213 and its derivative 7402683 are more specific inhibitors of CatL than CatB.
Next we used a commercial kit from Anaspec Inc. for measuring cathepsin B (CatB) activity and to test if our identified small molecules can inhibit CatB as well. Compared to the results of a CatL inhibition assay using our SARS-CoV-S-derived labeled peptide, we found that the small molecule 5705213 and its derivative 7402683 inhibited the cathepsin B activity by 16.4% and 38%, respectively, at a 50 μM concentration. The inhibition was 43.5% and 65%, respectively, at a 200 μM concentration (Fig. 11). The inhibitions observed for CatB was lower than those for CatL at the same concentrations of the small molecules (ranging from 64 to 95%) (Fig. 11). These results suggest that our small molecules are more specific and have higher affinity to CatL than to CatB.
FIG 11.
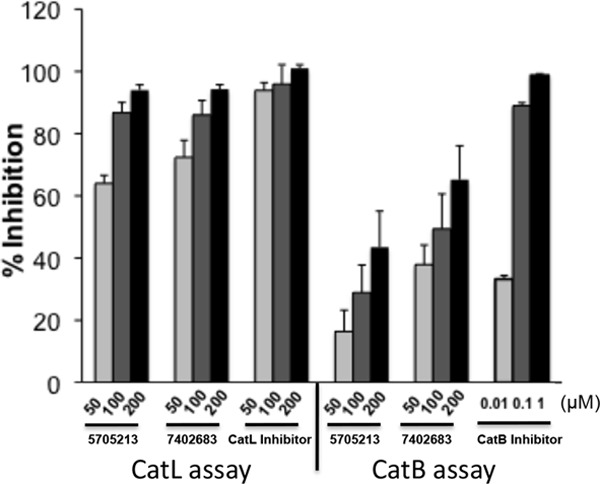
The small molecules 5705213 and 7402683 are more specific inhibitors of CatL than CatB. The small molecules were tested in different concentrations (50 to 200 μM) for inhibition of CatB activity in an in vitro activity assay (Anaspec Inc.) The percentages of inhibition were calculated for each inhibitor. The inhibition of CatB activity was compared to the inhibition of CatL activity against SARS-CoV-S-derived peptide in the presence of a 50 to 200 μM concentration of the small molecules 5705213 and 7402683. The CatL inhibitor III and CatB inhibitor were used as controls in the CatL and CatB assays, respectively. The experiment was performed in triplicates and repeated thrice with similar results. Error bars represent SD from a representative experiment.
DISCUSSION
There is no effective therapy for infections with SARS-CoV, EBOV, HeV, and NiV, which are highly infectious zoonotic viruses with high case-fatality rates. SARS-CoV belongs to family Coronaviridae (7), EBOV belongs to family Filoviridae (8), and NiV and HeV are closely related and belong to the genus Henipaviruses within the Paramyxoviridae family (9, 10, 46, 47). Several studies have described inhibitors for different viruses, including monoclonal antibodies and small molecules (48–51). In this study, we screened a library of small molecules using a novel HTSA that we developed, and we discovered a small molecule that could inhibit the entry of the above-mentioned pseudotyped viruses into mammalian cells by blocking the cleavage of the viral glycoproteins. Additionally, we show that this broad-spectrum antiviral small molecule is a mixed inhibitor of CatL and appears to be a suitable candidate for future optimization and development into a potent broad-spectrum antiviral against SARS-CoV, EBOV, HeV, and NiV.
There have been several attempts to discover potent inhibitors of SARS-CoV, EBOV, HeV, and NiV. For example, some novel inhibitors of papain-like protease which inhibited live SARS-CoV infection of Vero E6 cells were described (52). Another group has described a small molecule named oxocarbazate which blocks human CatL, thus inhibiting the entry of pseudotypes bearing SARS-CoV-S or EBOV-GP glycoproteins into human embryonic kidney 293T cells (53). In another study, highly potent inhibitors of human CatL were identified by screening combinatorial pentapeptide amide collections (54). However, whether these inhibitors affect the processing of host protein substrates of CatL has not yet been evaluated, and thus the potential undesirable side effects are unknown. CatL is an important host protease involved in processing and biosynthesis of neuropeptides such as proenkephalin, pro-neuropeptide Y (pro-NPY), prodynorphin, and proganalin (32, 33). However, this enzyme action has been hijacked by different viruses to gain entry into the target cells. Thus, directly blocking CatL enzymatic activity is likely to have undesirable side effects on human health. However, the novel approach presented in this study allowed us to identify lead small molecules that can selectively inhibit CatL-mediated cleavage of several viral glycoproteins and thus prevent viral entry into the cell without affecting its ability to process host proteins. It is unlikely that the identified inhibitors directly bound to the peptide substrates because they are short and likely lack tertiary structure. A lower binding affinity of CatL to viral peptides than to host peptides could be responsible for the selective inhibition of CatL cleavage of viral proteins and not the host proteins by the identified small molecules.
This speculation was supported by the finding that the rates of cleavage of viral peptides were substrate dependent and lower than the rate of host pro-NPY cleavage. This was further validated by testing the cleavage of viral, host peptide F, and pro-NPY peptides using different CatL concentrations (0.25, 0.5, and 1 μg/ml). Although the SARS-CoV-S-derived peptide was more rapidly cleaved than the other viral peptides, interestingly, the rate of cleavage of the pro-NPY-derived peptide was higher than the rate of cleavage of all viral peptides. This difference in the cleavage rate may be due to differences in the amino acids flanking the CatL cleavage sites in the viral peptides, which are hydrophobic, while they are hydrophilic in pro-NPY. The rapid cleavage of pro-NPY-derived peptide relative to viral peptides suggests that the viruses have evolved to exploit the host machinery to establish infection.
The results from the pseudotyped virus entry inhibition assays showed higher inhibition to pseudotyped virus bearing SARS-CoV-S glycoprotein by the small molecule 5705213 and its derivative 7402683. Other small molecules showed unexpected enhancement of SARS-CoV-S pseudotyped virus entry. The reason for such enhancement is not known. The MTT-based cell cytotoxicity assay showed that small molecule 5705213 and its derivative 7402683 (unlike small molecules 7914021 and 5182554) are not cytotoxic even at the highest concentration used, which thus indicated that their inhibitory effect was specific. Moreover, these small molecules inhibited the SARS-CoV-S recombinant protein cleavage in vitro and the endogenous processing of the HeV and NiV F0 proteins. The specificity of their action was further confirmed when these small molecules inhibited the entry of pseudotypes bearing the EBOV-GP and SARS-CoV-S but not the entry of VSV-G pseudotyped virus.
Several studies have described the role of TMPRSS2 membrane protease in SARS-CoV entry (44, 45, 55). The TMPRSS2 protease is expressed at multiple sites in the human respiratory and gastrointestinal tracts (56). Thus, the entry of SARS-CoV in a typical infection would involve either a plasma membrane or endocytic pathway. Accordingly, a complete inhibition of SARS-CoV entry should involve blockage of both pathways. Therefore, we tested our identified inhibitor individually and in combination with a commercially available protease inhibitor. It was interesting to see a complete inhibition of the entry of the pseudotyped virus bearing the SARS-CoV-S into 293FT cells expressing both ACE2 receptor and TMPRSS2 protease. Our results are consistent with a previous study which showed complete inhibition of SARS-CoV entry with a combination of CatL inhibitor and camostat (36) and confirmed the efficacy of our small molecule as an inhibitor of the CatL-mediated cleavage of viral proteins. They also showed the possibility of combining our small molecule with serine protease inhibitors to completely inhibit SARS-CoV infection.
In determining the mechanism of action of the identified inhibitor, we employed an enzyme assay based on Michaelis-Menten kinetics. Different enzyme inhibitors include (i) competitive, (ii) noncompetitive, (iii) uncompetitive, and (iv) mixed inhibitors. The type of the inhibitor is determined based on the change in enzyme reaction parameters (Vmax and Km). For competitive enzyme inhibitors, there is an increase in Km and no change in Vmax. For noncompetitive inhibitors, a decrease in Vmax and no change in Km are observed. For uncompetitive inhibitors, a decrease in both Km and Vmax are noted. A mixed enzyme inhibitor causes an increase in Km and a decrease in Vmax. The small molecule 5705213 is most likely a mixed inhibitor because it increased Km and decreased Vmax. Moreover, our small-molecule inhibitors appear to be more specific to CatL than to CatB. The future testing of these inhibitors against live viruses would be important and beneficial in developing them into potent broad-spectrum inhibitors. Taken together, the present findings appear novel in that we identified small molecules that can be potentially developed into broad-spectrum antiviral drugs against a range of highly pathogenic viruses with minimal undesirable side effects.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant 1U01AI082296.
We thank Thomas M. Gallagher at the Department of Microbiology and Immunology, Loyola University Stritch School of Medicine, for providing the TMPRSS2 expression plasmid. We thank Lijun Rong at the Department of Microbiology and Immunology, University of Illinois at Chicago, for providing the EBOV-GP expression plasmid.
Footnotes
Published ahead of print 5 February 2014
REFERENCES
- 1.Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102:14040–14045. 10.1073/pnas.0506735102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679. 10.1126/science.1118391 [DOI] [PubMed] [Google Scholar]
- 3.Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Delicat A, Paweska JT, Gonzalez JP, Swanepoel R. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438:575–576. 10.1038/438575a [DOI] [PubMed] [Google Scholar]
- 4.Field HE, Mackenzie JS, Daszak P. 2007. Henipaviruses: emerging paramyxoviruses associated with fruit bats. Curr. Top. Microbiol. Immunol. 315:133–159. 10.1007/978-3-540-70962-6_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halpin K, Hyatt AD, Fogarty R, Middleton D, Bingham J, Epstein JH, Rahman SA, Hughes T, Smith C, Field HE, Daszak P, Henipavirus Ecology Research G 2011. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: a comprehensive experimental study of virus transmission. Am. J. Trop. Med. Hyg. 85:946–951. 10.4269/ajtmh.2011.10-0567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Middleton DJ, Weingartl HM. 2012. Henipaviruses in their natural animal hosts. Curr. Top. Microbiol. Immunol. 359:105–121. 10.1007/82_2012_210 [DOI] [PubMed] [Google Scholar]
- 7.Peiris JS, Guan Y, Yuen KY. 2004. Severe acute respiratory syndrome. Nat. Med. 10:S88–S97. 10.1038/nm1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seah SK. 1978. Lassa, Marburg and Ebola: newly described African fevers. Can. Med. Assoc. J. 118:347–348 [PMC free article] [PubMed] [Google Scholar]
- 9.Chua KB, Goh KJ, Wong KT, Kamarulzaman A, Tan PS, Ksiazek TG, Zaki SR, Paul G, Lam SK, Tan CT. 1999. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 354:1257–1259. 10.1016/S0140-6736(99)04299-3 [DOI] [PubMed] [Google Scholar]
- 10.Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, Gubler DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels P, Ling AE, Peters CJ, Anderson LJ, Mahy BW. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432–1435. 10.1126/science.288.5470.1432 [DOI] [PubMed] [Google Scholar]
- 11.Kielian M, Jungerwirth S. 1990. Mechanisms of enveloped virus entry into cells. Mol. Biol. Med. 7:17–31 [PubMed] [Google Scholar]
- 12.Pager CT, Dutch RE. 2005. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 79:12714–12720. 10.1128/JVI.79.20.12714-12720.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pager CT, Craft WW, Jr, Patch J, Dutch RE. 2006. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology 346:251–257. 10.1016/j.virol.2006.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 102:11876–11881. 10.1073/pnas.0505577102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaletsky RL, Simmons G, Bates P. 2007. Proteolysis of the Ebola virus glycoproteins enhances virus binding and infectivity. J. Virol. 81:13378–13384. 10.1128/JVI.01170-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qian Z, Dominguez SR, Holmes KV. 2013. Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS-CoV) in virus entry and syncytia formation. PLoS One 8:e76469. 10.1371/journal.pone.0076469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lisco A, Vanpouille C, Tchesnokov EP, Grivel JC, Biancotto A, Brichacek B, Elliott J, Fromentin E, Shattock R, Anton P, Gorelick R, Balzarini J, McGuigan C, Derudas M, Götte M, Schinazi RF, Margolis L. 2008. Acyclovir is activated into a HIV-1 reverse transcriptase inhibitor in herpesvirus-infected human tissues. Cell Host Microbe 4:260–270. 10.1016/j.chom.2008.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMahon MA, Siliciano JD, Lai J, Liu JO, Stivers JT, Siliciano RF, Kohli RM. 2008. The antiherpetic drug acyclovir inhibits HIV replication and selects the V75I reverse transcriptase multidrug resistance mutation. J. Biol. Chem. 283:31289–31293. 10.1074/jbc.C800188200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mujugira A, Magaret AS, Celum C, Baeten JM, Lingappa JR, Morrow RA, Fife KH, Delany-Moretlwe S, de Bruyn G, Bukusi EA, Karita E, Kapiga S, Corey L, Wald A. 2013. Daily acyclovir to decrease herpes simplex virus type 2 (HSV-2) transmission from HSV-2/HIV-1 coinfected persons: a randomized controlled trial. J. Infect. Dis. 208:1366–1374. 10.1093/infdis/jit333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith RA, Gottlieb GS, Anderson DJ, Pyrak CL, Preston BD. 2008. Human immunodeficiency virus types 1 and 2 exhibit comparable sensitivities to zidovudine and other nucleoside analog inhibitors in vitro. Antimicrob. Agents Chemother. 52:329–332. 10.1128/AAC.01004-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menéndez-Arias L, Tozsér J. 2008. HIV-1 protease inhibitors: effects on HIV-2 replication and resistance. Trends Pharmacol. Sci. 29:42–49. 10.1016/j.tips.2007.10.013 [DOI] [PubMed] [Google Scholar]
- 22.Reyskens KM, Essop MF. 2014. HIV protease inhibitors and onset of cardiovascular diseases: a central role for oxidative stress and dysregulation of the ubiquitin-proteasome system. Biochim. Biophys. Acta 1842:256–268. 10.1016/j.bbadis.2013.11.019 [DOI] [PubMed] [Google Scholar]
- 23.Vanderlinden E, Naesens L. 25 June 2013. Emerging antiviral strategies to interfere with influenza virus entry. Med. Res. Rev. 10.1002/med.21289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Govorkova EA, Baranovich T, Seiler P, Armstrong J, Burnham A, Guan Y, Peiris M, Webby RJ, Webster RG. 2013. Antiviral resistance among highly pathogenic influenza A (H5N1) viruses isolated worldwide in 2002–2012 shows need for continued monitoring. Antiviral Res. 98:297–304. 10.1016/j.antiviral.2013.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, Van Phan T, Smythe ML, White HF, Oliver SW. 1993. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 363:418–423. 10.1038/363418a0 [DOI] [PubMed] [Google Scholar]
- 26.Woods JM, Bethell RC, Coates JA, Healy N, Hiscox SA, Pearson BA, Ryan DM, Ticehurst J, Tilling J, Walcott SM. 1993. 4-Guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid is a highly effective inhibitor both of the sialidase (neuraminidase) and of growth of a wide range of influenza A and B viruses in vitro. Antimicrob. Agents Chemother. 37:1473–1479. 10.1128/AAC.37.7.1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dapat C, Kondo H, Dapat IC, Baranovich T, Suzuki Y, Shobugawa Y, Saito K, Saito R, Suzuki H. 2013. Neuraminidase inhibitor susceptibility profile of pandemic and seasonal influenza viruses during the 2009-2010 and 2010-2011 influenza seasons in Japan. Antiviral Res. 99:261–269. 10.1016/j.antiviral.2013.06.003 [DOI] [PubMed] [Google Scholar]
- 28.Bosch BJ, Bartelink W, Rottier PJ. 2008. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J. Virol. 82:8887–8890. 10.1128/JVI.00415-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hood CL, Abraham J, Boyington JC, Leung K, Kwong PD, Nabel GJ. 2010. Biochemical and structural characterization of cathepsin L-processed Ebola virus glycoprotein: implications for viral entry and immunogenicity. J. Virol. 84:2972–2982. 10.1128/JVI.02151-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michalski WP, Crameri G, Wang L, Shiell BJ, Eaton B. 2000. The cleavage activation and sites of glycosylation in the fusion protein of Hendra virus. Virus Res. 69:83–93. 10.1016/S0168-1702(00)00169-6 [DOI] [PubMed] [Google Scholar]
- 31.Moll M, Diederich S, Klenk HD, Czub M, Maisner A. 2004. Ubiquitous activation of the Nipah virus fusion protein does not require a basic amino acid at the cleavage site. J. Virol. 78:9705–9712. 10.1128/JVI.78.18.9705-9712.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Funkelstein L, Toneff T, Hwang SR, Reinheckel T, Peters C, Hook V. 2008. Cathepsin L participates in the production of neuropeptide Y in secretory vesicles, demonstrated by protease gene knockout and expression. J. Neurochem. 106:384–391. 10.1111/j.1471-4159.2008.05408.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Funkelstein L, Beinfeld M, Minokadeh A, Zadina J, Hook V. 2010. Unique biological function of cathepsin L in secretory vesicles for biosynthesis of neuropeptides. Neuropeptides 44:457–466. 10.1016/j.npep.2010.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiffen JC, Bailey CG, Ng C, Rasko JE, Holst J. 2010. Luciferase expression and bioluminescence does not affect tumor cell growth in vitro or in vivo. Mol. Cancer 9:299. 10.1186/1476-4598-9-299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coughlin M, Lou G, Martinez O, Masterman SK, Olsen OA, Moksa AA, Farzan M, Babcook JS, Prabhakar BS. 2007. Generation and characterization of human monoclonal neutralizing antibodies with distinct binding and sequence features against SARS coronavirus using XenoMouse. Virology 361:93–102. 10.1016/j.virol.2006.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. 2012. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 86:6537–6545. 10.1128/JVI.00094-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gnirss K, Kühl A, Karsten C, Glowacka I, Bertram S, Kaup F, Hofmann H, Pöhlmann S. 2012. Cathepsins B and L activate Ebola but not Marburg virus glycoproteins for efficient entry into cell lines and macrophages independent of TMPRSS2 expression. Virology 424:3–10. 10.1016/j.virol.2011.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. 2006. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 80:4174–4178. 10.1128/JVI.80.8.4174-4178.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pöhlman S, Schughart K. 2013. TMPRSS2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 9:e1003774. 10.1371/journal.ppat.1003774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peitsch C, Klenk HD, Garten W, Böttcher-Friebertshäuser E. 2014. Activation of influenza A viruses by host proteases from swine airway epithelium. J. Virol. 88:282–291. 10.1128/JVI.01635-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shirato K, Kawase M, Matsuyama S. 2013. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 87:12552–12561. 10.1128/JVI.01890-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abe M, Tahara M, Sakai K, Yamaguchi H, Kanou K, Shirato K, Kawase M, Noda M, Kimura H, Matsuyama S, Fukuhara H, Mizuta K, Maenaka K, Ami Y, Esumi M, Kato A, Takeda M. 2013. TMPRSS2 is an activating protease for respiratory parainfluenza viruses. J. Virol. 87:11930–11935. 10.1128/JVI.01490-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, Welsch K, Winkler M, Schneider H, Hofmann-Winkler H, Thiel V, Pöhlmann S. 2013. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 87:6150–6160. 10.1128/JVI.03372-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. 2011. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 85:873–882. 10.1128/JVI.02062-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. 2010. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 84:12658–12664. 10.1128/JVI.01542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Selvey L, Sheridan J. 1995. Outbreak of severe respiratory disease in humans and horses due to a previously unrecognized paramyxovirus. J. Travel Med. 2:275. 10.1111/j.1708-8305.1995.tb00679.x [DOI] [PubMed] [Google Scholar]
- 47.Selvey LA, Wells RM, McCormack JG, Ansford AJ, Murray K, Rogers RJ, Lavercombe PS, Selleck P, Sheridan JW. 1995. Infection of humans and horses by a newly described morbillivirus. Med. J. Aust. 162:642–645 [DOI] [PubMed] [Google Scholar]
- 48.Elshabrawy HA, Coughlin MM, Baker SC, Prabhakar BS. 2012. Human monoclonal antibodies against highly conserved HR1 and HR2 domains of the SARS-CoV spike protein are more broadly neutralizing. PLoS One 7:e50366. 10.1371/journal.pone.0050366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adedeji AO, Severson W, Jonsson C, Singh K, Weiss SR, Sarafianos SG. 2013. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J. Virol. 87:8017–8028. 10.1128/JVI.00998-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gagnon H, Beauchemin S, Kwiatkowska A, Couture F, D'Anjou F, Levesque C, Dufour F, Desbiens AR, Vaillancourt R, Bernard S, Desjardins R, Malouin F, Dory YL, Day R. 10 December 2013. Optimization of furin inhibitors to protect against the activation of influenza hemagglutinin H5 and Shiga toxin. J. Med. Chem. 10.1021/jm400633d [DOI] [PubMed] [Google Scholar]
- 51.Yu X, Sainz B, Jr, Petukhov PA, Uprichard SL. 2012. Identification of hepatitis C virus inhibitors targeting different aspects of infection using a cell-based assay. Antimicrob. Agents Chemother. 56:6109–6120. 10.1128/AAC.01413-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghosh AK, Takayama J, Rao KV, Ratia K, Chaudhuri R, Mulhearn DC, Lee H, Nichols DB, Baliji S, Baker SC, Johnson ME, Mesecar AD. 2010. Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein-ligand X-ray structure and biological evaluation. J. Med. Chem. 53:4968–4979. 10.1021/jm1004489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shah PP, Wang T, Kaletsky RL, Myers MC, Purvis JE, Jing H, Huryn DM, Greenbaum DC, Smith AB, III, Bates P, Diamond SL. 2010. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 78:319–324. 10.1124/mol.110.064261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brinker A, Weber E, Stoll D, Voigt J, Muller A, Sewald N, Jung G, Wiesmuller KH, Bohley P. 2000. Highly potent inhibitors of human cathepsin L identified by screening combinatorial pentapeptide amide collections. Eur. J. Biochem. 267:5085–5092. 10.1046/j.1432-1327.2000.01570.x [DOI] [PubMed] [Google Scholar]
- 55.Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K, Niemeyer D, Schneider H, Drosten C, Pohlmann S. 2011. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 85:4122–4134. 10.1128/JVI.02232-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P, Lucas JM, Nelson PS, Pohlmann S, Soilleux EJ. 2012. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS One 7:e35876. 10.1371/journal.pone.0035876 [DOI] [PMC free article] [PubMed] [Google Scholar]