

ABSTRACT
A new type of antiprion compound, Gly-9, was found to inhibit abnormal prion protein formation in prion-infected neuroblastoma cells, in a prion strain-independent manner, when the cells were treated for more than 1 day. It reduced the intracellular prion protein level and significantly modified mRNA expression levels of genes of two types: interferon-stimulated genes were downregulated after more than 2 days of treatment, and the phosphodiesterase 4D-interacting protein gene, a gene involved in microtubule growth, was upregulated after more than 1 day of treatment. A supplement of interferon given to the cells partly restored the abnormal prion protein level but did not alter the normal prion protein level. This interferon action was independent of the Janus activated kinase-signal transducer and activator of transcription signaling pathway. Therefore, the changes in interferon-stimulated genes might be a secondary effect of Gly-9 treatment. However, gene knockdown of phosphodiesterase 4D-interacting protein restored or increased both the abnormal prion protein level and the normal prion protein level, without transcriptional alteration of the prion protein gene. It also altered the localization of abnormal prion protein accumulation in the cells, indicating that phosphodiesterase 4D-interacting protein might affect prion protein levels by altering the trafficking of prion protein-containing structures. Interferon and phosphodiesterase 4D-interacting protein had no direct mutual link, demonstrating that they regulate abnormal prion protein levels independently. Although the in vivo efficacy of Gly-9 was limited, the findings for Gly-9 provide insights into the regulation of abnormal prion protein in cells and suggest new targets for antiprion compounds.
IMPORTANCE This report describes our study of the efficacy and potential mechanism underlying the antiprion action of a new antiprion compound with a glycoside structure in prion-infected cells, as well as the efficacy of the compound in prion-infected animals. The study revealed involvements of two factors in the compound's mechanism of action: interferon and a microtubule nucleation activator, phosphodiesterase 4D-interacting protein. In particular, phosphodiesterase 4D-interacting protein was suggested to be important in regulating the trafficking or fusion of prion protein-containing vesicles or structures in cells. The findings of the study are expected to be useful not only for the elucidation of cellular regulatory mechanisms of prion protein but also for the implication of new targets for therapeutic development.
INTRODUCTION
Prion diseases, synonymously referred to as transmissible spongiform encephalopathies, are fatal neurodegenerative disorders that include Creutzfeldt-Jakob disease, fatal familial insomnia, and Gerstmann-Sträussler-Scheinker syndrome in humans, as well as scrapie, bovine spongiform encephalopathy, and chronic wasting disease in animals. All of these diseases are characterized by the deposition of an abnormal isoform of prion protein (PrPsc), which is a main component of the “prion” pathogen and is converted from the protease-sensitive normal cellular isoform of prion protein (PrPc) in the central nervous system and lymphoreticular system (1). Both partial protease resistance and detergent-insoluble polymer formation are biochemical characteristics of PrPsc. A protease resistant core of PrPsc (PrPres) is detected by immunoblotting using anti-PrP antibody after treatment of PrPsc with proteinase K (1). The biosynthesis and metabolism of PrPc and PrPsc have been investigated intensively in prion-infected cells (2) but have not been elucidated fully. Particularly enigmatic are the endogenous factors regulating the formation of PrPsc or the conformational change from PrPc into PrPsc.
The increasing incidence of human prion diseases, which is attributable to increasing life expectancy, as well as outbreaks of acquired forms of prion diseases, such as variant diseases and iatrogenic diseases, have aroused great concern in many countries and have accelerated the development of antiprion remedies and prophylactics. Various antiprion compounds or biological materials reportedly inhibit PrPsc/res formation in vitro or in prion-infected cells (3–5). Some compounds and biological materials reportedly extend the incubation periods in prion-infected animals. Nevertheless, no compound or biological material has halted disease progression in prion-infected animals, except for PrPc depletion by conditional PrP gene knockout (6), which is not applicable to patients. Several compounds that have been used on patients with prion diseases on trial bases reportedly cannot produce significant clinical benefits (7–9).
In our efforts to obtain new clues to the enigma of PrPsc formation and to uncover new antiprion leads for remedies or prophylactics, we screened various compounds with chemical structures unrelated to those for previously reported compounds for antiprion activities in prion-infected cells or animals. We found glycoside compounds as a new type of antiprion compound. Glycoside compounds, which occur abundantly in plants, especially as pigments, and which are used in medicines, dyes, and cleansing agents, are any of various chemicals formed from monosaccharides by replacing the hydrogen atom of one of its hydroxyl groups with the bond to another biologically active molecule (10). This report describes our study of the efficacy and potential mechanism underlying the antiprion action of a representative glycoside compound, Gly-9 (4-methoxyphenyl 2-amino-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside), in prion-infected cells, as well as the efficacy of Gly-9 in prion-infected animals. We discuss the involvement of interferon (IFN) and phosphodiesterase 4D-interacting protein (Pde4dip) in PrPres formation, because Gly-9's effects on prion-infected cells were accompanied by gene expression alterations in IFN-stimulated genes and Pde4dip. Pde4dip is also called myomegalin and interacts with the cyclic nucleotide phosphodiesterase 4D. It is thought to function as a microtubule nucleation activator (11) and as an anchoring protein that sequesters members of the cyclic AMP (cAMP)-dependent pathway to the Golgi apparatus and to centrosomes (12).
MATERIALS AND METHODS
Compounds and reagents.
Thirty-eight glycoside compounds used for the screening for antiprion activity were purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). A list of the glycoside compounds used in the study is available upon request. As presented in Table 1, the compounds were named “Gly-” plus an arbitrary number added for our convenience. They were dissolved in 100% dimethyl sulfoxide (DMSO) and were used fresh. Recombinant murine IFN-α with a specific activity of 106 U/mg in phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (BSA), recombinant murine IFN-β with a specific activity of 23 × 106 U/mg in PBS containing 0.1% BSA, and recombinant murine IFN-γ with a specific activity of ≥10 × 106 U/mg in 10 mM sodium phosphate (pH 8.0) containing 0.1% BSA were purchased from PBL Interferon Source (Piscataway, NJ), Toray Industries Inc. (Tokyo, Japan), and PeproTech Inc. (Rocky Hill, NJ), respectively. A Janus activated kinase inhibitor, Jak inhibitor I, was purchased from Calbiochem Corp. (La Jolla, CA) and dissolved in DMSO.
TABLE 1.
Representative glycoside compounds and their inhibitory activities
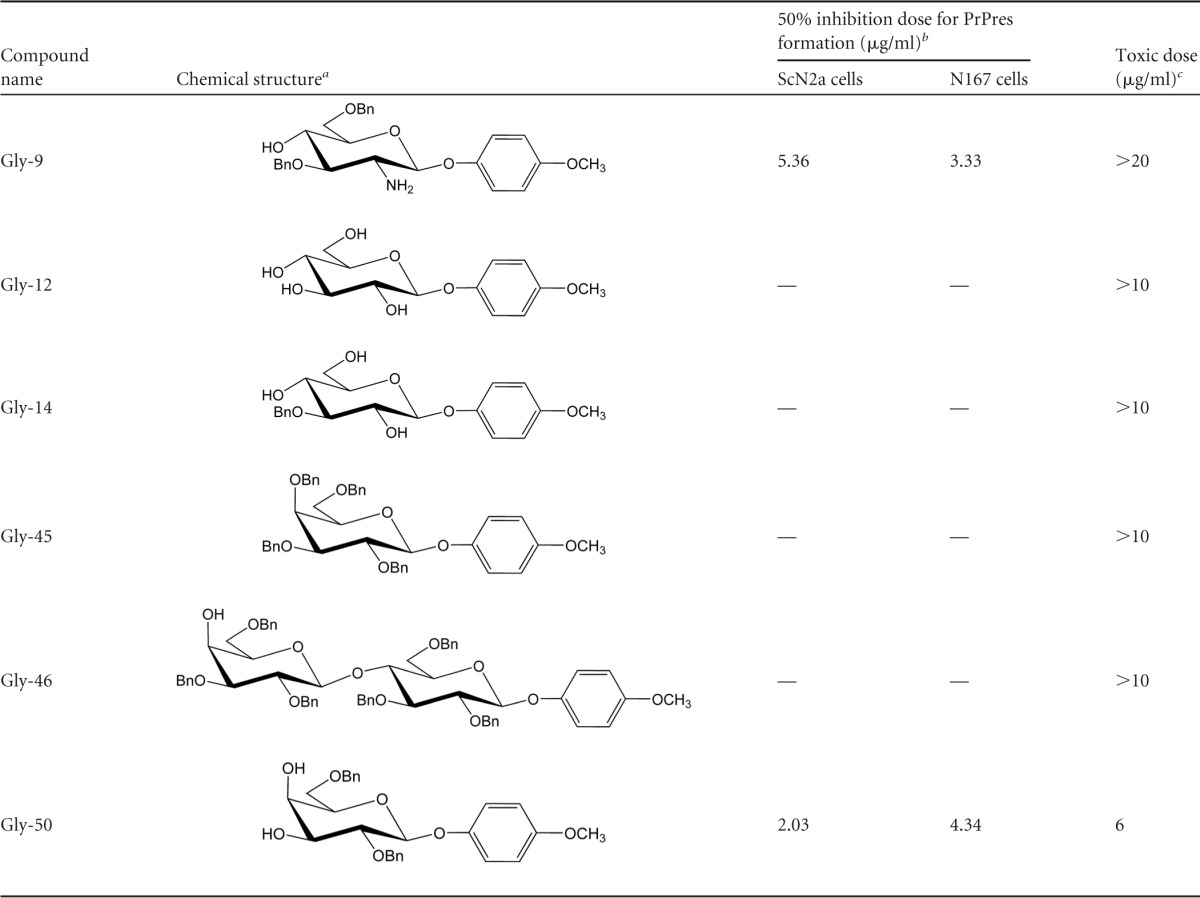
Bn, benzyl group.
—, no effectiveness at doses of <10 μg/ml.
Toxic dose was assayed in both ScN2a cells and N167 cells.
Cells and immunoblotting.
We used mouse neuroblastoma cells either left uninfected (N2a cells) or persistently infected with RML prion (ScN2a cells), 22L prion (N167 cells), or Fukuoka-1 prion (F3 cells), as previously described (13–15). Briefly, cells were cultured in the presence of test materials or their vehicles at 37°C for 3 days or a designated time. The amount of DMSO, protein, or chemical components other than test materials was equilibrated in every cell culture well by adding vehicle or vehicle components. The concentration of DMSO in the culture medium was always less than 0.2%. Test materials at the doses indicated in the study did not precipitate in the culture medium. Cells grown to confluence were washed with PBS and lysed with lysis buffer (0.5% sodium deoxycholate, 0.5% Nonidet P-40, PBS, pH 7.4). Debris was eliminated by centrifugation at 3,000 × g. The protein content of each sample was measured using the Dc protein assay reagent (Bio-Rad Laboratories Inc., Hercules, CA), with BSA as a standard. For the detection of PrPres, a cell lysate containing the same amount of protein was treated with 10 μg/ml proteinase K at 37°C for 30 min and subsequently treated with 1 mM phenylmethylsulfonyl fluoride. PrPres was precipitated by centrifugation at 10,000 × g and then suspended in a sample loading buffer. For the detection of other proteins, a cell lysate containing the same amount of protein was used without further treatments and was mixed with a concentrated loading buffer. For immunoblotting, electrophoresis on 15% SDS-PAGE gels and subsequent electrotransfer to polyvinylidene difluoride membranes were performed. After blocking with 5% nonfat dry milk in Tris-buffered saline, immunoreaction was done with anti-PrP monoclonal antibody SAF83 (1:5,000; SPI-Bio, Massy, France), anti-β-actin monoclonal antibody (1:10,000; Sigma-Aldrich, St. Louis, MO), anti-LC3 monoclonal antibody (1:5,000; NanoTools, Teningen, Germany), anti-Stat2 antibody EP1814Y (1:2,000; Epitomics, Burlingame, CA), anti-phosphorylated Stat2 antibody (1:300; Upstate Biotechnology, Lake Placid, NY), anti-Stat1 antibody EPYR2154 (1:1,000; Epitomics), or anti-phosphorylated Stat1 antibody 58D6 (1:500; Cell Signaling Technology, Danvers, MA). The secondary antibody used was from Promega Corp. (Madison, WI), Sigma-Aldrich, or Amersham (Piscataway, NJ). Immunoreactivity was visualized using CDP-Star or ECL Plus detection reagent (Amersham) and was analyzed densitometrically using the ImageJ program (National Institutes of Health, Bethesda, MD).
Flow cytometry analysis.
To analyze cell surface PrP, flow cytometry was done as described previously (14, 15). Briefly, N2a cells or N167 cells incubated in the presence of Gly-9 or vehicle (DMSO) for 3 days were dispersed using 0.1% collagenase and then washed with ice-cold 0.5% fetal calf serum in PBS. Immunoreaction was done with SAF83 (1:500) or an isotype control IgG1 (1:250) for 30 min on ice and, subsequently, with goat F(ab′)2 fragment anti-mouse IgG(H+L)–fluorescein isothiocyanate (FITC) (1:100; Beckman Coulter Inc., Brea, CA) for 30 min on ice. The cells were then analyzed using an Epics XL-ADC flow cytometer (Beckman Coulter Inc.). To analyze intracellular PrP by flow cytometry, cells were treated with phosphatidylinositol-specific phospholipase C (PIPLC) (1 U/ml; Sigma-Aldrich) for 4 h after cell dispersion treatment. The cells were then permeated with 0.1% Triton X-100 in PBS for 2 min. Subsequently, immunoreaction and analysis were performed as described earlier. To analyze the lipid raft microdomain by flow cytometry, dispersed cells were incubated with Alexa Fluor 488-conjugated cholera toxin B (1 μg/ml; Molecular Probes Inc., Grand Island, NY) for 30 min on ice, and flow cytometry analysis was then conducted.
Flotation assay.
A flotation assay of detergent-insoluble membrane complexes was completed as described in a previous report (16). Briefly, cells incubated in the presence of Gly-9 or DMSO for 3 days were washed with ice-cold PBS and then lysed on ice with 550 μl of TNET solution (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100). The lysate was added to an equal volume of ice-cold 70% Nycodenz solution (Cosmo Bio USA Inc.). Onto 800 μl of this mixed solution, 200 μl each of 25, 22.5, 20, 18, 15, 12, and 8% Nycodenz solutions in TNET was overlaid sequentially in TLS-55 ultracentrifuge tubes (Hitachi Ltd., Japan). Ultracentrifugation was then performed at 200,000 × g at 4°C for 4 h. Fractions containing 200 μl were collected from the top to the bottom and analyzed for PrPc and lipid rafts by immunoblotting with SAF83 antibody and fluorescence detection assay with Alexa Fluor 488-conjugated cholera toxin B, respectively. Fluorescence detection was performed using a Spectra Max Gemini XPS fluorometer (Molecular Devices Corp., Sunnyvale, CA). For lipid raft-associated PrPc, cholera toxin B-positive fractions were collected and analyzed by immunoblotting with SAF83 antibody.
Immunofluorescence analysis.
Immunofluorescence for detecting PrPc was performed as described below. Cells grown on glass-bottom culture dishes were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and then washed twice with PBS. The cells were then permeated with 0.4% Triton X-100 in PBS for 3 min. To block nonspecific antibody binding, cells were incubated with PBS containing 1% BSA for 20 min. Immunoreaction was done with antibody 6H4 (1:1,000; Prionics AG, Schlieren-Zurich, Switzerland) for 20 min at room temperature and, subsequently, with goat anti-mouse IgG(H+L)–Alexa Fluor 488 (1:500; Molecular Probes Inc.) for 20 min at room temperature. Nuclei were counterstained with Hoechst 33258 (Dojindo Laboratories, Tokyo, Japan). The cells were then observed by use of a confocal fluorescence microscope (magnification, ×60) (Fluoview FV300; Olympus Corp., Tokyo, Japan). To immunostain abnormal accumulations of PrPc and PrPsc in the cells, the same procedure was performed, except that the cells were incubated with 6 M guanidinium hydrochloride for 10 min between the permeation and blocking steps. Immunofluorescence for detection of Stat2 was performed similarly, using the anti-Stat2 antibody EP1814Y (1:300) and donkey anti-rabbit IgG(H+L)–Alexa Fluor 488 (1:200; Molecular Probes Inc.).
DNA microarray analysis.
N167 cells were treated with either Gly-9 or Gly-14 at a dose of 5 μg/ml for 3 days. The cells were then lysed with RNAiso-plus reagent (TaKaRa Bio Inc., Shiga, Japan). Total RNA was extracted using FastPure RNA (TaKaRa Bio Inc.). It was amplified and labeled using an Agilent Low RNA Input Fluorescent linear amplification kit (Agilent Technologies Inc., Santa Clara, CA). cRNA samples were hybridized onto whole-mouse-genome 4 × 44K 60-mer oligonucleotide arrays (G4846A; Agilent Technologies Inc.) for 17 h at 65°C in a rotator oven, with subsequent washing with wash buffers (Agilent Technologies Inc.). After the washing step, the slides were scanned using an Agilent microarray scanner (Agilent Technologies Inc.). The hybridization signals were extracted using software (Agilent Feature Extraction software; Agilent Technologies Inc.). The microarray numerical values were analyzed using GeneSpring GX software (Agilent Technologies Inc.). Data from quadruplicate samples of either Gly-9-treated cells or Gly-14-treated cells were filtered based on the flag values and, subsequently, the t test values (P < 0.05). Genes with >1.25-fold signal differences between the two treatments were then selected. The differential gene expression was confirmed by quantifying mRNA levels as described below.
Quantification of mRNA levels.
Cells were lysed with RNAiso-plus reagent. Total RNA was extracted using FastPure RNA. cDNA was then synthesized by use of a first-strand cDNA synthesis kit (TaKaRa Bio Inc.). The mRNA level was measured by real-time PCR using SYBR Premix Ex Taq II (TaKaRa Bio Inc.). The nucleotide sequences of the primers used in the study are available upon request. The fold change of gene expression was calculated using the 2−ΔΔCT method, with β-actin as an internal control.
Gene knockdown study.
Double-stranded small interfering RNAs (siRNAs) against Pde4dip and other genes were purchased from Invitrogen Corp. (Carlsbad, CA) or Thermo Scientific Dharmacon (Lafayette, CO). The sequence of the most frequently used siRNA against Pde4dip (si-Pde4dip; named si-5 in Fig. 6C) was UCGCCAAAGGGUGUCUGAAUGAGAA. The sequences of all other siRNAs used in the study are available upon request. Gene knockdown experiments were performed as described previously (17). Briefly, cells were diluted to 10% or 15% confluence with Opti MEM I (Invitrogen Corp.) including 10% fetal bovine serum. A total of 2.4 ml of each was then seeded into six-well dishes. Transfection was performed on the day after seeding. siLentFect (3.0 μl/well; Bio-Rad Laboratories, Inc.) was used for transfection of siRNA as well as for a negative control in the study. The concentrations of siRNAs used in transfections were 5 to 20 nM. The medium was changed on the day after transfection. Cells were harvested after washing with PBS 3 days after transfection.
FIG 6.

Gene knockdown effects of upregulated genes on PrPres formation. (A) Immunoblotting of PrPres in N167 cells treated with siRNAs against four upregulated genes: Tmem179, Lrrc17, Mc1r, and Srd5a1. Cells in the presence of 5 μg/ml Gly-9 (Gly-9 +) or DMSO (Gly-9 −) were treated with a mixture of four siRNAs against the above-mentioned genes (si-RNA pool) at the indicated doses for 3 days after transfection. The amount of transfection reagent was equilibrated in all cell culture wells. (B) mRNA levels of Tmem179, Lrrc17, Mc1r, and Srd5a1 in N167 cells treated with specific siRNAs. Cells in the presence of 5 μg/ml Gly-9 were treated as described for panel A and were analyzed 24 h after transfection. Data are averages and standard deviations for triplicate experimental results (*, P < 0.01 versus control [transfection reagent alone]). (C) Pde4dip coding sequences and sites of siRNAs used in the study. (D) Immunoblotting of PrPres in N167 cells treated with siRNAs against Pde4dip. Cells in the presence of 5 μg/ml Gly-9 were treated with each siRNA (si-2, si-3, si-4, or si-5) at a dose of 10 nM for 3 days after transfection. The amount of transfection reagent was equilibrated in all cell culture wells. Graphic data are averages and standard deviations for triplicate immunoblot signals (*, P < 0.01 versus control [transfection reagent alone]). (E) Pde4dip mRNA levels in N167 cells treated with siRNAs against Pde4dip. Cells in the presence of vehicle (DMSO) or 5 μg/ml Gly-9 were treated as described for panel D and were analyzed at the indicated time points after transfection. Data are averages and standard deviations for triplicate experimental results (*, P < 0.01; **, P < 0.05 versus control [transfection reagent alone] at each time point). (F) PrP mRNA levels in N167 cells treated with siRNAs against Pde4dip. Cells were treated and analyzed as described for panel E. Data are averages and standard deviations for triplicate experimental results (*, P < 0.01; **, P < 0.05 versus control [transfection reagent alone] at each time point).
Animal study.
Eight- to 10-week-old Tg7 mice overexpressing hamster PrPc (18) were used for analysis of the effectiveness of Gly-9. Using an Alzet osmotic pump (model 1004) and brain infusion kit (Durect Corp., Cupertino, CA), vehicle alone (50% DMSO, 50% polyethylene glycol 400 [PEG 400]) or test compounds at a dose of 300 μg/day were injected continually into the cerebral third ventricle for 4 weeks from 6 weeks post-intracerebral inoculation. Inoculation was done with 20 μl of 1% (wt/vol) brain homogenate from a 263K prion-infected terminally ill hamster. The animals were monitored daily until terminal disease. The survival time, which was defined in this study as the length of time from the start of injection of glycoside compounds to the onset of terminal disease, was assayed. The disease was confirmed by neuropathology and the immunohistochemistry of abnormal PrP deposition in the brain, as described previously (13, 14, 19). Briefly, for detection of abnormal PrP deposition, tissue sections were treated with a hydrolytic autoclave and incubated with anti-PrP antibody [anti-PrP(C); 1:150] (Immuno-Biological Laboratories Co. Ltd., Gunma, Japan), with subsequent incubation with EnVision/HRP labeling polymer (Dako, Glostrup, Denmark). For the detection of glial reactions, sections were incubated with anti-glial fibrillary acidic protein (anti-GFAP) antibody (1:5,500; Dako) and, subsequently, with EnVision/HRP labeling polymer. The immunoreactive product was developed with 3,3′-diaminobenzidine, and the sections were then counterstained with hematoxylin. The animal experiments described herein were performed with the approval of the Animal Experiment Ethical Committee of Tohoku University.
Statistical analysis.
Data were evaluated based on results of triplicate experiments, using one-way analysis of variance followed by the Dunnett test or Tukey-Kramer test for multiple-sample comparisons and using the t test for two-sample comparisons. Differences were considered significant if the P value was <0.05. The survival rate was calculated using the Kaplan-Meier method; its significance was evaluated using the log rank method.
Microarray data accession number.
The microarray data obtained in this study are available at the NCBI GEO repository under accession number GSE55241.
RESULTS
Inhibition of PrPres formation by glycoside compounds.
Through screening of various commercially available glycoside compounds, 4-methoxyphenyl 2-amino-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside, designated Gly-9 in this study, was found to inhibit PrPres formation in both RML prion-infected N2a cells (ScN2a cells) and 22L prion-infected N2a cells (N167 cells). Thereafter, other glycoside compounds having chemical structures similar to those of Gly-9 were examined using these prion-infected cells. Another compound, designated Gly-50, also showed antiprion activity at doses of <10 μg/ml (Table 1). These data showed that antiprion activity was related to the methoxyphenyl glycoside having two benzyl groups through ether linkage. In fact, Gly-50 had a narrower effective range than that of Gly-9. Therefore, Gly-9 was used for further study. Gly-9 was effective at inhibiting PrPres formation in all three distinct prion-infected cell models (Fig. 1A). The 50% inhibition dose of Gly-9 against PrPres formation was 3 to 8 μg/ml in each cell model. Gly-9 effects were also observed in the immunofluorescence analysis of abnormal PrP accumulation in Gly-9-treated N167 cells (Fig. 1B). Cap-like perinuclear punctate signals were diminished in Gly-9-treated N167 cells. The temporal profile of PrPres signals showed that more than 1 day of Gly-9 treatment was necessary for PrPres signal reduction in N167 cells (Fig. 1C).
FIG 1.

Gly-9 effects on PrPres formation. (A) Immunoblotting of PrPres in three distinct prion strain-infected cell lines (ScN2a, N167, and F3) treated with Gly-9. Cells were treated for 3 days with the indicated dose of Gly-9. β-Actin signals are shown as controls for the integrity of samples used for PrPres detection. Graphic data are averages and standard deviations for triplicate immunoblot signals. (B) Immunofluorescence of abnormal PrP accumulation in Gly-9-treated N167 cells. Cells were treated with 5 μg/ml Gly-9 or its vehicle (DMSO) for 3 days. Nuclei were stained with Hoechst 33258 (H33258). (C) Temporal profile of PrPres levels in Gly-9-treated N167 cells. Cells were treated with 10 μg/ml Gly-9 or its vehicle for the indicated periods. Graphic data are averages and standard deviations for triplicate immunoblot signals (n.s., not significant; *, P < 0.01).
The cell surface PrP level was reduced slightly by the treatment with Gly-9 in N167 cells but was not altered in noninfected N2a cells (Fig. 2A). In either cell, the lipid raft level detected by cholera toxin B was reduced slightly by treatment with Gly-9 (Fig. 2A). This lipid raft alteration by Gly-9 was also demonstrated by flotation assay (Fig. 2B). Both PrPc and cholera toxin B-binding lipid raft constituents were shifted to the next fractions toward the top by the treatment with Gly-9. The PrPc level of the lipid raft fraction was not altered significantly by the treatment with Gly-9 (Fig. 2C), whereas the total PrPc level and, particularly, the intracellular PrPc level were reduced remarkably by the treatment with Gly-9 (Fig. 2D and E). The PrP mRNA level, however, was not altered significantly (Fig. 2F). Immunofluorescence results demonstrated that intracellular PrPc localization was altered slightly by the treatment with Gly-9 (Fig. 2G). The PrPc signals around the cell membrane appeared to be unchanged, but intracellular scattered PrPc signals appeared to be much less prominent and tended to be assembled in the perinuclear region in Gly-9-treated cells. Therefore, alterations of PrPc levels and PrPc localization are apparently related to the reduction of PrPres signals.
FIG 2.

Gly-9 effects on PrPc, lipid rafts, and autophagy. (A) Flow cytometry of cell surface PrP and lipid rafts in either Gly-9-treated N167 cells or Gly-9-treated uninfected N2a cells. Cells were treated with 5 μg/ml Gly-9 or its vehicle (DMSO) for 3 days. Cell surface PrP and lipid rafts were labeled with anti-PrP antibody and cholera toxin B, respectively. (B) Flotation assay of PrPc and lipid rafts in Gly-9-treated N2a cells. Cells were treated and analyzed as described for panel A. S, starting material. (C) Immunoblotting of lipid raft-associated PrPc in Gly-9-treated N2a cells. Cholera toxin B-positive lipid raft fractions from panel B were collected and analyzed for their PrPc levels. Molecular size markers on the right indicate sizes in kDa. Graphic data are averages and standard deviations for triplicate immunoblot signals (n.s., not significant). (D) Immunoblotting of total PrPc in Gly-9-treated N2a cells. A vehicle (DMSO)-treated cell sample and a Gly-14-treated cell sample are shown as controls. Cells were treated as described for panel A. Molecular size markers on the right indicate sizes in kDa. Graphic data are averages and standard deviations for triplicate immunoblot signals (*, P < 0.01). (E) Flow cytometry of intracellular PrPc in Gly-9-treated N2a cells. Cells were treated as described for panel A and were then digested with PIPLC. Cells were permeated with 0.1% Triton X-100 in PBS, and intracellular PrPc was labeled with anti-PrP antibody. (F) PrP mRNA level in Gly-9-treated N167 cells. Cells were treated as described for panel A. Data are averages and standard deviations for triplicate experiments (n.s., not significant). (G) Immunofluorescence of PrPc in Gly-9-treated N2a cells. Cells were treated as described for panel A for PIPLC “−” images and as described for panel E for PIPLC “+” images. Nuclei were stained with Hoechst 33258 (H33258). (H) Immunoblotting of autophagosome-related LC3-II in Gly-9-treated N167 cells. Cells were treated for 3 days with the indicated doses of Gly-9 or trehalose. Trehalose-treated cell samples are shown as positive controls.
Recently, autophagy was reported to regulate PrPsc clearance (20). We examined whether autophagosome formation is enhanced in N167 cells treated with Gly-9. Treatment of N167 cells with Gly-9 moderately increased the signals of the autophagosomal membrane-specific phosphatidylethanolamine-conjugated form of microtubule-associated protein 1A/1B light chain 3 (LC3-II) (Fig. 2H), suggesting either enhanced autophagosome synthesis or reduced autophagosome turnover by Gly-9 treatment (21).
Modification of gene expression profiles.
The molecular mechanism of Gly-9-induced inhibition of PrPres formation was studied using DNA microarray analysis. In this analysis, Gly-14 was used as a control for Gly-9 because Gly-14 has a chemical structure similar to that of Gly-9 but has no antiprion activity. Comparison of gene expression profiles between Gly-9-treated N167 cells and Gly-14-treated N167 cells revealed two dozen differentially regulated genes with >1.25-fold signal differences (Table 2). Excluding hypothetical genes and poorly annotated genes, 10 genes, including the top 5 upregulated and top 5 downregulated genes in Gly-9-treated cells, were analyzed further. Among them, all the top 5 downregulated genes in Gly-9-treated cells were IFN-stimulated genes (Ifit1, Ccl5, Cxcl10, Oasl2, and Isg15). It was confirmed that these genes were downregulated specifically in Gly-9-treated N167 cells compared to vehicle-treated control N167 cells (Fig. 3A). We inferred that the IFN signaling pathway was downregulated in cells treated with Gly-9.
TABLE 2.
Differentially regulated genes in N167 cells treated with Gly-9
| DNA microarray probea | Gene nameb | GenBank accession no. | Fold change in microarray signals for Gly-9 relative to Gly-14 | Fold change in quantified mRNA levels |
|
|---|---|---|---|---|---|
| Gly-9 relative to Gly-14 | Gly-9 relative to DMSO | ||||
| A_55_P1984178 | Pde4dip | NM_001039376 | 1.46 | 1.47 | 1.29 |
| A_55_P2162988 | Tmem179 | NM_178915 | 1.41 | 1.46 | 1.49 |
| A_55_P2172096 | Mc1r | NM_008559 | 1.41 | 1.26 | 1.21 |
| A_51_P301930 | Lrrc17 | NM_028977 | 1.34 | 1.37 | 1.20 |
| A_55_P1999012 | LOC100046875 | XM_001476965 | 1.33 | Not examined | Not examined |
| A_55_P2347529 | 3010003L21Rik | BC106181 | 1.31 | Not examined | Not examined |
| A_55_P2132358 | None | None | 1.30 | Not examined | Not examined |
| A_55_P2072115 | AW011738 | NR_030671 | 1.30 | Not examined | Not examined |
| A_51_P420415 | Srd5a1 | NM_175283 | 1.27 | 1.22 | 0.97 |
| A_55_P1981366 | Lamc2 | NM_008485 | 1.27 | Not examined | Not examined |
| A_55_P2142072 | Synj2 | NM_011523 | 0.75 | Not examined | Not examined |
| A_52_P420504 | Acta2 | NM_007392 | 0.75 | Not examined | Not examined |
| A_55_P2065991 | S100a11 | NM_016740 | 0.75 | Not examined | Not examined |
| A_51_P367866 | Egr1 | NM_007913 | 0.72 | Not examined | Not examined |
| A_51_P454873 | Npy | NM_023456 | 0.71 | Not examined | Not examined |
| A_52_P371135 | C130050O18Rik | NM_177000 | 0.67 | Not examined | Not examined |
| A_52_P68028 | Ace | NM_009598 | 0.62 | Not examined | Not examined |
| A_55_P2103698 | Isg15 | NM_015783 | 0.62 | 0.63 | 0.64 |
| A_66_P128537 | Isg15 | NM_015783 | 0.61 | 0.63 | 0.64 |
| A_51_P387123 | Oasl2 | NM_011854 | 0.61 | 0.64 | 0.60 |
| A_66_P101942 | Gm9706 | AK019325 | 0.57 | Not examined | Not examined |
| A_55_P2016462 | Cxcl10 | NM_021274 | 0.57 | 0.52 | 0.49 |
| A_52_P638459 | Ccl5 | NM_013653 | 0.51 | 0.33 | 0.33 |
| A_51_P327751 | Ifit1 | NM_008331 | 0.32 | 0.64 | 0.57 |
The listed probes detected >1.25-fold signal differences between Gly-9-treated cells and Gly-14-treated cells.
Gene names in boldface denote the genes analyzed in this study.
FIG 3.

Downregulation of IFN-stimulated genes and effects of IFN supplements. (A) mRNA levels of IFN-stimulated genes in Gly-9-treated N167 cells. Cells were treated with 5 μg/ml Gly-9 or its vehicle (DMSO) for 3 days. Data are averages and standard deviations for triplicate experimental results (*, P < 0.01). (B) Immunoblotting of PrPres in N167 cells treated with IFN-α. Cells in the presence of vehicle (DMSO), 5 μg/ml Gly-9, or 5 μg/ml Gly-14 were treated with the indicated doses of IFN-α for 3 days. The buffer volume and protein content in dosed IFN-α solutions were equilibrated using PBS and BSA. β-Actin signals are shown as controls for the integrity of samples used for PrPres detection. Asterisks denote cell toxicity observed at the designated doses. (C) Profiles of mRNA levels of representative IFN-stimulated genes in N167 cells treated with IFN-α in the presence or absence of Gly-9. The mRNA levels of Cxcl10 and Ifit1 were analyzed in cells that were treated with 5 μg/ml Gly-9 (Gly-9 +) or DMSO (Gly-9 −) for 3 days and, simultaneously, with 5 U/ml IFN-α for the indicated times before the harvest. For “IFN-α −” samples, cells were treated with BSA-containing PBS having amounts of buffer and protein equivalent to those in a 5-U/ml IFN-α solution for 6 h before harvesting. Data are averages and standard deviations for triplicate experimental results (*, P < 0.01; n.s., not significant). (D) Immunoblotting of PrPres in N167 cells treated with IFN-β or IFN-γ. Cells were treated with IFN-β or IFN-γ as described for panel B. As a buffer of the IFN-γ solution, 10 mM sodium phosphate (pH 8.0) was used instead of PBS. (E) Immunoblotting of PrPres in N167 cells and PrPc in N2a cells treated with Gly-9 and IFN-α. Cells were treated with combinations of 5 μg/ml Gly-9 and 5 U/ml IFN-α for 3 days. Each vehicle was used as a negative control. Graphic data are averages and standard deviations for triplicate immunoblot signals (n.s., not significant; *, P < 0.01). (F) Flow cytometry of cell surface PrP and lipid rafts in N167 cells and N2a cells treated with IFN-α. Cells in the presence of 5 μg/ml Gly-9 were treated with 5 U/ml IFN-α or its vehicle (cont) for 3 days. Cell surface PrP (anti-PrP) and lipid rafts (cholera-toxin B) were labeled as described already. (G) PrP mRNA levels in N167 cells treated with IFN-α. Cells were treated with 5 μg/ml Gly-9 for 3 days and, simultaneously, with 5 U/ml IFN-α for the indicated periods before the harvest. Data are averages and standard deviations for triplicate experimental results (*, P < 0.01 versus vehicle control). (H) Immunoblotting of autophagosome-related LC3-II in N167 cells treated with Gly-9 and IFN-α. Cells were treated with combinations of 5 μg/ml Gly-9 and 5 U/ml IFN-α for 3 days. Treatment with 100 mM trehalose is shown as a positive control. Each vehicle was used as a negative control. Graphic data represent averages and standard deviations for triplicate immunoblot signals (n.s., not significant).
We investigated whether IFN supplementation of the cell medium rescues Gly-9's effects in N167 cells. Supplementation with recombinant IFN-α, which elevated gene expression levels of IFN-stimulated genes even in the presence of Gly-9, partly restored the PrPres level in Gly-9-treated N167 cells (Fig. 3B and C). Other types of recombinant IFNs, i.e., IFN-β and IFN-γ, produced similar effects on PrPres formation in Gly-9-treated N167 cells (Fig. 3D). However, supplementation with recombinant IFN-α did not recover the PrPc level (Fig. 3E). Neither the cell surface PrP level nor the cholera toxin B-labeled lipid raft level was altered by IFN-α supplementation in either Gly-9-treated N167 cells or Gly-9-treated N2a cells (Fig. 3F). The PrP mRNA expression level was reduced by the IFN-α supplement at all time points except for 1 h postsupplementation (Fig. 3G). In addition, the autophagosomal marker level was unaffected by IFN-α supplementation (Fig. 3H). Therefore, restoration of the PrPres level by IFN-α was not concordant with factors such as the PrPc level, lipid raft level, or autophagosome level.
As a representative IFN signaling pathway, the Janus activated kinase (Jak)-signal transducer and activator of transcription (Stat) signaling pathway, through which IFN-stimulated genes are regulated, was investigated in N167 cells. This signaling pathway was clearly activated by IFN-α (Fig. 4A to C) and was clearly blocked by Jak inhibitor I in the cells used in the study (Fig. 4D and E), but alteration of the PrPres level was not observed in cells treated with an inhibitor or with siRNAs against either of the key molecules of the Jak-Stat signaling pathway (Fig. 5A to D) or against the downstream IFN-induced molecules (Fig. 5E and F). Furthermore, Jak inhibitor I did not impair the IFN-α-induced restoration of the PrPres level in Gly-9-treated cells (Fig. 5G). Temporal profiles of mRNA expression levels of IFN-stimulated genes were analyzed afresh, which revealed that the alteration of mRNA expression levels of IFN-stimulated genes apparently occurred in a late phase of cell culture in the presence of Gly-9 (Fig. 5H). Collectively, these findings suggest that IFN-induced restoration of the PrPres level is not mediated by the Jak-Stat pathway and that mRNA expression changes of IFN-stimulated genes are not primarily related to the PrPres formation in Gly-9-treated cells.
FIG 4.

Jak-Stat pathway in N167 cells. (A) Immunoblotting of total Stat2 and phosphorylated Stat2 (p-Stat2) in N167 cells treated with IFN-α. Cells were treated for the indicated times with 50 U/ml IFN-α. (B) Immunoblotting of Stat proteins and their activated phosphorylated proteins (p-Stat1 and p-Stat2) in N167 cells treated with IFN-α. Cells in the presence of vehicle (DMSO), 5 μg/ml Gly-9, or 5 μg/ml Gly-14 were treated with IFN-α as described for panel A, for the indicated times. It is noteworthy that Stat proteins in N167 cells were activated by IFN-α irrespective of the presence of glycoside compounds. (C) Immunofluorescence of Stat2 in N167 cells treated with IFN-α. Cells were treated with IFN-α at the designated doses for 20 min before analysis. Nuclei were stained with Hoechst 33258 (H33258). (D) Immunoblotting of phosphorylated Stat proteins (p-Stat1 and p-Stat2) in N167 cells treated with IFN-α and a Jak inhibitor. Cells were treated with 10 U/ml IFN-α for the indicated times in the presence or absence of 5 μg/ml Gly-9 or 50 nM Jak inhibitor I (Jak inh). It is noteworthy that Jak inhibitor I blocked IFN-α-induced activation of Stat proteins irrespective of the presence of Gly-9. (E) Immunofluorescence of Stat2 in N167 cells treated with IFN-α and Jak inhibitor. Cells were treated with 5 μg/ml Gly-9 and 50 U/ml IFN-α for 20 min before analysis, in the presence or absence of 50 nM Jak inhibitor I (Jak inh). Nuclei were stained with Hoechst 33258 (H33258). It is noteworthy that Jak inhibitor I blocked IFN-α-induced translocation of Stat2 into the nucleus.
FIG 5.

Effects of alterations in Jak-Stat pathway on PrPres formation. (A) Immunoblotting of PrPres in N167 cells treated with Jak inhibitor. Cells were treated with Jak inhibitor I at the indicated doses for 3 days. The amount of DMSO was equilibrated in all cell culture wells and was less than 0.2%. (B) Immunoblotting of PrPres and Stat proteins in N167 cells treated with siRNAs against Stat genes. Cells were treated for the last 3 days before analysis with the indicated doses of either a mixture of four siRNAs against Stat1 (si-Stat1 pool) or Stat2 (si-Stat2 pool) or each of three siRNAs against Stat2 (si-9, si-10, and si-11). The amount of transfection reagent was equilibrated in all cell culture wells. (C) mRNA levels of Stat genes in N167 cells treated with siRNAs against Stat genes. Cells were treated as described for panel B and were analyzed 24 or 48 h after transfection. Data are averages and standard deviations based on results of triplicate experiments (*, P < 0.01 versus control [transfection reagent alone] at each time point). (D) mRNA levels of the PrP gene in N167 cells treated with siRNAs against Stat genes. Cells were treated and analyzed as described for panel C. Data are averages and standard deviations based on results of triplicate experiments (**, P < 0.05 versus the control [transfection reagent alone] at each time point). (E) Immunoblotting of PrPres in N167 cells treated with siRNAs against IFN-stimulated genes. Cells were treated for the last 3 days before analysis with the indicated doses of either a mixture of four siRNAs (si-RNA pool), against Cxcl10, Ifit1, Ccl5, and Isg15, or each of three siRNAs against Oasl2 (si-1, si-2, and si-3). The amount of transfection reagent was equilibrated in all cell culture wells. (F) mRNA levels of IFN-stimulated genes in N167 cells treated with siRNAs against IFN-stimulated genes. Cells were treated as described for panel E and were analyzed 48 h after transfection. Data are averages and standard deviations based on results of triplicate experiments (*, P < 0.01 versus control [transfection reagent alone]). (G) Effect of Jak inhibitor on IFN-α-induced PrPres restoration in N167 cells. Cells were treated with 5 μg/ml Gly-9, 5 U/ml IFN-α, and 50 nM Jak inhibitor I (Jak inh) for 3 days. Each vehicle was used as a negative control. Graphic data are averages and standard deviations for triplicate immunoblot signals (**, P < 0.05; n.s., not significant). (H) Temporal profiles of mRNA levels of representative IFN-stimulated genes in Gly-9-treated N167 cells. The mRNA levels of Cxcl10 and Ifit1 were analyzed in N167 cells treated with 5 μg/ml Gly-9 or its vehicle for the indicated periods. Data are averages and standard deviations for triplicate experimental results (n.s., not significant; **, P < 0.05; *, P < 0.01).
Modification of Pde4dip gene expression.
The remaining five genes selected from DNA microarray analysis were the top five upregulated genes in Gly-9-treated cells compared to Gly-14-treated cells. However, all these genes, except for the Pde4dip gene, showed a lack of restoration of the PrPres level by gene knockdown screening of Gly-9-treated N167 cells (Fig. 6A and B). Conversely, Pde4dip gene knockdown using siRNAs that never showed cytotoxicity produced restoration of PrPres levels in Gly-9-treated N167 cells (Fig. 6C to F). The Pde4dip expression alteration was not significant 24 h from the beginning of Gly-9 treatment; thereafter, it was significant (Fig. 7A). Representative gene knockdown data obtained using the si-Pde4dip siRNA (called si-5 in Fig. 6C) are depicted in Fig. 7B. The PrPres levels tended to increase in vehicle-treated N167 cells and were restored in Gly-9-treated N167 cells (Fig. 7B, top panels), whereas PrPc levels were increased in either vehicle-treated or Gly-9-treated N2a cells (Fig. 7B, bottom panels). The increases in PrPc levels were more remarkable in vehicle-treated N2a cells. The Pde4dip mRNA expression levels were reduced by si-Pde4dip, irrespective of Gly-9 treatment, whereas PrP mRNA expression levels were not modified (Fig. 7C). Consequently, these findings suggest that the restoration or increase in PrPres level by Pde4dip gene knockdown is attributable to the increase in PrPc level, which is caused by a nontranscriptional mechanism.
FIG 7.

Pde4dip involvement in PrPc and PrPres. (A) Temporal profile of Pde4dip mRNA levels in Gly-9-treated N167 cells. Cells were treated with 5 μg/ml Gly-9 or its vehicle for the indicated periods. Data are averages and standard deviations for triplicate experimental results (n.s., not significant; **, P < 0.05). (B) Immunoblotting of PrPres in N167 cells and PrPc in N2a cells treated with Gly-9 and Pde4dip gene knockdown. Cells were treated with 5 μg/ml Gly-9 or its vehicle (DMSO) for three and a half days and, simultaneously, with the indicated doses of siRNA against Pde4dip (si-Pde4dip; called si-5 in Fig. 6C) for the last 3 days before harvest. Graphic data are averages and standard deviations for triplicate immunoblot signals (n.s., not significant; *, P < 0.01; **, P < 0.05). (C) mRNA levels of Pde4dip and PrP in N167 cells treated with Gly-9 and si-Pde4dip. Cells were treated as described for panel B, and si-Pde4dip was used at a dose of 10 nM. The vehicle for Gly-9 and the transfection reagent for si-Pde4dip were used as negative controls. Data are averages and standard deviations for triplicate experimental results (*, P < 0.01; n.s., not significant). (D) Immunofluorescence of abnormal PrP accumulation in N167 cells treated with Gly-9 and si-Pde4dip. Cells were treated as described for panel C. Nuclei were stained with Hoechst 33258 (H33258). (E) Flow cytometry of cell surface PrP and lipid rafts in N2a cells treated with Gly-9 and si-Pde4dip. Cells in the presence of 5 μg/ml Gly-9 or vehicle (DMSO) were treated with transfection reagent alone (cont) or 5 nM si-Pde4dip (siRNA) for 3 days as described for panel B. Cell surface PrP (anti-PrP) and lipid rafts (cholera-toxin B) were labeled as described earlier in this report. (F) Immunoblotting of autophagosome-related LC3-II in N167 cells treated with Gly-9 and si-Pde4dip. Cells were treated as described for panel C. Treatment with 100 mM trehalose is shown as a positive control. Graphic data are averages and standard deviations for triplicate immunoblot signals (n.s., not significant).
Immunofluorescence analysis also revealed that the signals of abnormal PrP accumulation in Gly-9-treated N167 cells were increased by Pde4dip gene knockdown (Fig. 7D). In addition, localization of abnormal PrP signals was modified by Pde4dip gene knockdown in N167 cells, irrespective of Gly-9 treatment. Abnormal PrP signals, which localized in a cap-like pattern in the perinuclear area of the control cells, tended to localize in the whole cytosol of the si-Pde4dip-treated cells, in a coarse granular pattern. The cell surface PrPc level was almost unchanged by Pde4dip gene knockdown in Gly-9-treated cells but was slightly reduced in vehicle-treated cells (Fig. 7E, top panels). The lipid raft level in the cells was almost unchanged by Pde4dip gene knockdown (Fig. 7E, bottom panels). Pde4dip gene knockdown tended to reduce the LC3-II autophagosome marker level, but the change was not significant (Fig. 7F). All these findings suggest that the antiprion effect of Gly-9 is caused partly by gene expression modification of Pde4dip.
Relationship between IFN and Pde4dip.
We examined whether the downregulation of Pde4dip gene expression occurs during the IFN-α-induced restoration of the PrPres level in N167 cells. IFN-α supplementation of the medium of N167 cells did not alter Pde4dip mRNA levels in either vehicle-treated or Gly-9-treated cells (Fig. 8A, left panel), even during a long period after supplementation, except for 1 h postsupplementation, when the mRNA level increased (Fig. 8A, right panel). Next, we examined whether Pde4dip gene expression is coordinated reversely with that of IFN-stimulated genes. Pde4dip gene knockdown caused reductions of the mRNA levels of IFN-stimulated genes either at a late phase of the cell culture (Fig. 8B, Cxcl10 panel) or from an early phase of the cell culture (Fig. 8B, Ifit1 panel). These results again suggest that no apparent link exists between the IFN-α supplement effects and the Pde4dip gene knockdown effects. We then examined whether IFN-α supplementation and Pde4dip gene knockdown work synergistically or additively in N167 cells. The PrPres level of the cells treated with both was not different from that of the cells treated with Pde4dip gene knockdown alone (Fig. 8C), suggesting that IFN-α's action might be absorbed in the Pde4dip gene knockdown action or might not work under the conditions of Pde4dip gene knockdown. These findings also imply that additional factors might be involved in the antiprion action of Gly-9, because the PrPres level was not completely restored by either intervention.
FIG 8.

Relationship between IFN effects and Pde4dip effects. (A) Pde4dip mRNA levels in N167 cells treated with IFN-α. Cells were treated with 5 μg/ml Gly-9 (Gly-9 +) or vehicle (Gly-9 −) for 3 days and, simultaneously, with 5 U/ml IFN-α for the indicated periods before the harvest. Data are averages and standard deviations for triplicate experiment results (n.s., not significant; **, P < 0.05 versus vehicle control of IFN-α). (B) Temporal profiles of mRNA levels of representative IFN-stimulated genes in N167 cells treated with si-Pde4dip. The mRNA levels of Cxcl10 and Ifit1 were analyzed in cells treated with combinations of 5 μg/ml Gly-9 and 10 nM si-Pde4dip. Gly-9 treatment started half a day before transfection of si-Pde4dip. Cells were harvested at the indicated time points after transfection. The vehicle for Gly-9 and the transfection reagent for si-Pde4dip were used as negative controls. Data are averages and standard deviations for triplicate experimental results (n.s., not significant; *, P < 0.01; **, P < 0.05). (C) Immunoblotting of PrPres in N167 cells treated with combinations of si-Pde4dip and IFN-α. Cells in the presence or absence of 5 μg/ml Gly-9 were treated with 5 U/ml IFN-α for three and a half days and, simultaneously, with 10 nM si-Pde4dip for the last 3 days before harvesting. The vehicles for Gly-9 and IFN-α and the transfection reagent for si-Pde4dip were used as negative controls. Graphic data are averages and standard deviations for triplicate immunoblot signals (n.s., not significant; *, P < 0.01; **, P < 0.05).
In vivo efficacy.
To investigate the efficacy of Gly-9 in vivo, Gly-9 was administered continuously into the cerebroventricle of intracerebrally prion-infected animals as described in a previous report (19), because the blood-brain barrier permeability and pharmacokinetic parameters of Gly-9 were unknown. Injection of Gly-9 at a dose of 300 μg/day was started 6 weeks after intracerebral infection, when early features of pathology and abnormal PrP deposition in the brain are detected, as described in a previous report (19). That continued for 4 weeks. The results showed that the therapeutic efficacy of Gly-9 was marginal: the survival times from the beginning of treatment were 27.4 ± 8.9 days for Gly-9, 21.4 ± 2.5 days for Gly-14, and 22.4 ± 2.4 days for vehicle (P < 0.05 for Gly-9 versus Gly-14 or vehicle; n = 5) (Fig. 9A). Neuropathological features of the animals at the terminal disease stage were not apparently different among the three groups (Fig. 9B).
FIG 9.

Gly-9 effects on prion-infected animals. (A) Kaplan-Meier graph for animals treated with Gly-9. Tg7 mice infected intracerebrally with 263K prion were given intracerebroventricular injections of Gly-9 or Gly-14 at a dose of 300 μg/day or of vehicle continually from 6 weeks postinfection. The survival time in this study indicates the length of time from the start of injections to the onset of terminal disease. (B) Neuropathology of Gly-9-treated, terminally ill animals. Representative images stained with hematoxylin and eosin (HE) or immunolabeled for abnormal PrP deposition (PrP) or glial fibrillary acidic protein (GFAP) are shown for the brain sections of animals treated as described for panel A. The images include the deep cerebral cortex, white matter, and hippocampus pyramidal cell layer (cerebrum) or the pontine laterodorsal tegmental area (Pons). Bars, 50 μm. It is noteworthy that the cerebral sections show coarse plaque-like PrP deposition and mild glial reactions in the white matter, whereas the pontine sections show moderate spongiosis, remarkable fine granular PrP deposition, and marked glial reactions. It is also noteworthy that no apparent difference in these neuropathological features exists among the three groups.
DISCUSSION
The glycoside compounds described herein represent a new type of antiprion compound in terms of their mechanism of action and chemical structure. The representative compound Gly-9 influenced the membrane lipid raft microdomain, suggesting its similarity in action with antiprion compounds such as cholesterol modulators. These compounds are reported to disturb the membrane lipid raft microdomain, a possible site of PrP conversion or interaction between PrPc and PrPsc (22–32). However, in contrast to these compounds, Gly-9 slightly or never influenced the cell surface PrPc level and the lipid raft-associated PrPc level, but it much more prominently affected the intracellular PrPc level. In addition, the cholera toxin B-labeled lipid raft level was not concordant with the PrPres level restored in the rescue experiment by IFN-α supplementation or Pde4dip gene knockdown. Recently, Karapetyan and colleagues reported two PrPc-reducing compounds: astemizole, which stimulates autophagy but does not modify PrPc levels at a dose preventing prion replication; and tacrolimus, which reduces both membrane and intracellular PrPc levels by a nontranscriptional mechanism (33). Tacrolimus has also been reported by other researchers to suppress the neurodegenerative process of prion-infected animals as a calcineurin inhibitor (34) and to activate autophagy in both prion-infected neuroblastoma cells and animals (35). These compounds might share certain mechanisms with Gly-9 in terms of either nontranscriptional modification of the PrPc level or autophagy involvement, but the mechanism of Gly-9 action is likely to differ greatly from those of these compounds, as discussed below.
Actually, Gly-9-induced alterations in both the PrPc level and PrPc localization in cells are most likely to be linked with reduction of the PrPres level, and it is unlikely that glycoside compounds interact directly with PrPc or PrPsc molecules and consequently inhibit PrP conversion or destabilize PrPsc molecules, considering the fact that more than 1 day of treatment with Gly-9 was necessary to obtain the antiprion effect. It can thus be inferred that certain complicated mechanisms might be involved in the action of Gly-9. Analyzing gene expression profiles of Gly-9-treated cells is a reasonable course to take to search for clues to the mechanism of Gly-9 antiprion action. Genes with differentially modified expression, however, might include those which are unrelated or secondary to the antiprion action.
The DNA microarray analysis used in this study revealed some genes that were specifically modified in their mRNA expression levels in Gly-9-treated cells, but the mechanism of Gly-9 action is not necessarily explained by modification of all these gene expression levels. Actually, mRNA expression levels of IFN-stimulated genes were reduced by treatment with Gly-9, but PrPres formation was not modified by any treatment for either inhibiting the Jak-Stat signaling pathway or knocking down expression of IFN-stimulated genes. Gly-9-induced mRNA expression reductions of IFN-stimulated genes occurred at a later phase of the cell culture. Therefore, these changes might be secondary to preceding upstream events that presumably cause alteration in the metabolism of PrPc/sc molecules.
In fact, supplementation of prion-infected cells with type I IFN (IFN-α or IFN-β) or type II IFN (IFN-γ) increased the PrPres level. IFN-α supplementation lowered the PrP mRNA expression level and did not significantly modify the total or cell surface PrPc level, nor was the Gly-9-induced autophagosomal marker level modified. Therefore, increases in PrPres levels by IFNs do not seem to result from modification of either the PrPc level or the level of autophagy. It is noteworthy that the IFN-α-induced PrPres increase in prion-infected cells was mediated by a Jak-independent pathway. IFNs can activate several alternative signaling pathways, including the mitogen-activated protein kinase p38 pathway and the phosphatidylinositol 3-kinase pathway, which are, however, still dependent on the activation of Jak molecules (36). Recently, a Jak- and Stat-independent mechanism was reported for IFN signaling (37). It is hypothesized that IFN facilitates PrPres formation or slows PrPres clearance through a similar Jak- and Stat-independent mechanism. However, this hypothesis must be assessed in future studies.
It might be suspected that IFNs influence the PrPres level in a nonspecific, protein amount-dependent fashion or that IFNs block Gly-9 in a directly interacting manner. However, these are deniable based on two findings: first, the BSA used in IFN-negative controls showed no restoration of the PrPres level, even though it was used in an amount that was hundreds of times larger than the amount of IFN; and second, IFN supplementation did not reduce any Gly-9-induced effects, except for the effect on the PrPres level. Ishibashi and colleagues reported that interferon regulatory factor 3 (IRF3), a key transcription factor of the type I IFN production pathway in innate immunity, negatively regulates PrPsc formation in prion-infected cells (38). They reported that the overexpression of IRF3 decreases PrPres levels but that IRF3 gene knockdown increases PrPres levels in cells. Our observation of IFN effects in prion-infected cells is apparently inconsistent with their findings. However, given the very complicated, elaborate regulatory mechanisms of IFN production and signaling, a rational link might exist between our observations and their findings. That putative link remains to be explored.
The results of the present study also indicate that Pde4dip, whose mRNA expression was altered from the middle of the cell culture stage by the treatment with Gly-9, is likely to contribute to the Gly-9-induced inhibition of PrPres formation. The timing of the alteration of Gly-9-induced Pde4dip gene expression in the cells was parallel to that of the appearance of Gly-9's antiprion effect in the cells, which took more than 1 day of treatment. Pde4dip has been reported as a protein of the Golgi apparatus/centrosome that interacts with the cyclic nucleotide phosphodiesterase 4D (12). Apparently, Pde4dip functions as an anchor to localize components of the cAMP-dependent pathway to the Golgi apparatus/centrosomal region of the cell. Pde4dip has also been described as a microtubule nucleation activator and is related to regulating microtubule growth (11). Depletion of Pde4dip delays microtubule growth from the centrosome and Golgi apparatus. Recently, Uchiyama and colleagues reported that PrPsc is detected throughout endosomal compartments and is particularly abundant in recycling endosomes in prion-infected N2a cells (39). Therefore, Pde4dip might influence PrPres formation by altering trafficking and fusion of endosomes through the modification of microtubules. Although the Pde4dip protein expression level was not demonstrated successfully by immunoblotting or immuno-fluorescence, despite our efforts, Pde4dip gene knockdown effects were observed in the results where the localization of abnormal PrP accumulation was altered from a perinuclear cap-like pattern to a cytosolic coarse granular pattern, implying modification in the trafficking and fusion of abnormal PrP accumulation-containing structures or vesicles.
Regarding the relationship between IFN effects and Pde4dip gene knockdown effects, these two interventions produced similar outputs in this study: partial restorations of the PrPres level in Gly-9-treated prion-infected cells. Thus, there might be parts of certain pathways or factors shared by these interventions. However, no link between IFN and Pde4dip was observed. Additionally, it is noteworthy that the effects of the two interventions on the PrPc level differ completely. Consequently, it can be inferred that IFN and Pde4dip are independently involved in PrPres formation in prion-infected cells, although neither synergetic nor additive effects were demonstrated for a combination of the two interventions.
Autophagy reportedly facilitates PrPsc clearance (20) and is microtubule dependent (40). Therefore, we hypothesized that Gly-9 influences both the intracellular PrPc level and PrPres level by altering the formation and transport of autophagosomes or autolysosomes through modulating Pde4dip and subsequently altering microtubule growth. In fact, Pde4dip gene knockdown demonstrated that the autophagosome-specific LC3-II level tended to decrease, but the change was not significant. Consequently, the autophagosome formation level was not concordant with either the PrPc level or the PrPres level, suggesting that mechanisms other than autophagy regulate both PrP levels presented in this study. Similarly, for IFN, the restoration of the PrPres level was not parallel to the LC3-II level. Marzo and colleagues recently reported that 4-hydroxytamoxifen conveys PrPc molecules and PrPres molecules to lysosomes independently of autophagy (41). Consequently, Gly-9 might decrease the PrPc level and PrPres level in a similar lysosome-dependent manner. This possibility awaits further exploration.
The in vivo efficacy of Gly-9 was marginal in the animal disease model tested in this study. This result might not be surprising since Gly-9 had a complicated mechanism of action in prion-infected cells. Moreover, the pharmacokinetic parameters of Gly-9 were unknown for the animals used in the study. The pharmacophore of Gly-9 needs to be determined. Its chemical structure must be optimized to obtain much better properties of antiprion efficacy and safety. Nevertheless, as a new class of antiprion lead compounds, Gly-9 might be interesting for the therapeutic development of countermeasures against prion diseases.
Prion diseases have long incubation periods from infection to the onset of symptoms. It remains unclear whether illness invariably occurs in those who have already been infected with prions. The illness apparently occurs only in some people who are predisposed to prions in cases of acquired types of prion diseases. Therefore, it can be inferred that the replication and accumulation of prion in the body are regulated by a certain host mechanism, although this presumption remains unproven. The findings of this study on the potential molecular mechanism underlying Gly-9's action are expected to be useful not only for the implication of new targets for therapeutic development but also for the elucidation of cellular regulatory mechanisms for prion protein. The involvement of either IFN or Pde4dip in PrPres formation revealed in this study might present new clues to support the exploration of the host regulatory mechanism against prions.
ACKNOWLEDGMENTS
We thank Yuka Fujiwara, Taichi Hamanaka, and Eiji Sakai for technical assistance and Hiroshi Kurahashi for helpful discussion.
This research was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan, a grant from the Ministry of Health, Labor, and Welfare, and a grant from the National Institute of Biomedical Innovation.
Footnotes
Published ahead of print 22 January 2014
REFERENCES
- 1.Prusiner SB. 1998. Prions. Proc. Natl. Acad. Sci. U. S. A. 95:13363–13383. 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caughey B. 1991. In vitro expression and biosynthesis of prion protein. Curr. Top. Microbiol. Immunol. 172:93–107 [DOI] [PubMed] [Google Scholar]
- 3.Sim VL, Caughey B. 2009. Recent advances in prion chemotherapeutics. Infect. Disord. Drug Targets 9:81–91. 10.2174/1871526510909010081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teruya K, Kawagoe K, Kimura T, Chen CJ, Sakasegawa Y, Doh-ura K. 2009. Amyloidophilic compounds for prion diseases. Infect. Disord. Drug Targets 9:15–22. 10.2174/1871526510909010015 [DOI] [PubMed] [Google Scholar]
- 5.Trevitt CR, Collinge J. 2006. A systematic review of prion therapeutics in experimental models. Brain 129:2241–2265. 10.1093/brain/awl150 [DOI] [PubMed] [Google Scholar]
- 6.Verity NC, Mallucci GR. 2011. Rescuing neurons in prion disease. Biochem. J. 433:19–29. 10.1042/BJ20101323 [DOI] [PubMed] [Google Scholar]
- 7.Tsuboi Y, Doh-ura K, Yamada T. 2009. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology 29:632–636. 10.1111/j.1440-1789.2009.01058.x [DOI] [PubMed] [Google Scholar]
- 8.Otto M, Cepek L, Ratzka P, Doehlinger S, Boekhoff I, Wiltfang J, Irle E, Pergande G, Ellers-Lenz B, Windl O, Kretzschmar HA, Poser S, Prange H. 2004. Efficacy of flupirtine on cognitive function in patients with CJD: a double-blind study. Neurology 62:714–718. 10.1212/01.WNL.0000113764.35026.EF [DOI] [PubMed] [Google Scholar]
- 9.Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S, Rossor M, Rudge P, Siddique D, Spyer M, Thomas D, Walker S, Webb T, Wroe S, Darbyshire J. 2009. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 8:334–344. 10.1016/S1474-4422(09)70049-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kren V, Martinkova L. 2001. Glycosides in medicine: “the role of glycosidic residue in biological activity.” Curr. Med. Chem. 8:1303–1328. 10.2174/0929867013372193 [DOI] [PubMed] [Google Scholar]
- 11.Roubin R, Acquaviva C, Chevrier V, Sedjai F, Zyss D, Birnbaum D, Rosnet O. 2013. Myomegalin is necessary for the formation of centrosomal and Golgi-derived microtubules. Biol. Open 2:238–250. 10.1242/bio.20123392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verde I, Pahlke G, Salanova M, Zhang G, Wang S, Coletti D, Onuffer J, Jin SL, Conti M. 2001. Myomegalin is a novel protein of the Golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J. Biol. Chem. 276:11189–11198. 10.1074/jbc.M006546200 [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa K, Kudo Y, Nishida N, Suemoto T, Sawada T, Iwaki T, Doh-ura K. 2006. Styrylbenzoazole derivatives for imaging of prion plaques and treatment of transmissible spongiform encephalopathies. J. Neurochem. 99:198–205. 10.1111/j.1471-4159.2006.04035.x [DOI] [PubMed] [Google Scholar]
- 14.Kawasaki Y, Kawagoe K, Chen CJ, Teruya K, Sakasegawa Y, Doh-ura K. 2007. Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J. Virol. 81:12889–12898. 10.1128/JVI.01563-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamanaka T, Sakasegawa Y, Ohmoto A, Kimura T, Ando T, Doh-ura K. 2011. Anti-prion activity of protein-bound polysaccharide K in prion-infected cells and animals. Biochem. Biophys. Res. Commun. 405:285–290. 10.1016/j.bbrc.2011.01.030 [DOI] [PubMed] [Google Scholar]
- 16.Teruya K, Nishizawa K, Doh-ura K. 2010. Semisynthesis of a protein with cholesterol at the C-terminal, targeted to the cell membrane of live cells. Protein J. 29:493–500. 10.1007/s10930-010-9278-9 [DOI] [PubMed] [Google Scholar]
- 17.Kimura T, Ishikawa K, Sakasegawa Y, Teruya K, Sata T, Schatzl H, Doh-ura K. 2010. GABAA receptor subunit beta1 is involved in the formation of protease-resistant prion protein in prion-infected neuroblastoma cells. FEBS Lett. 584:1193–1198. 10.1016/j.febslet.2010.02.029 [DOI] [PubMed] [Google Scholar]
- 18.Race RE, Priola SA, Bessen RA, Ernst D, Dockter J, Rall GF, Mucke L, Chesebro B, Oldstone MB. 1995. Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron 15:1183–1191. 10.1016/0896-6273(95)90105-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doh-ura K, Ishikawa K, Murakami-Kubo I, Sasaki K, Mohri S, Race R, Iwaki T. 2004. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J. Virol. 78:4999–5006. 10.1128/JVI.78.10.4999-5006.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heiseke A, Aguib Y, Schatzl HM. 2010. Autophagy, prion infection and their mutual interactions. Curr. Issues Mol. Biol. 12:87–97 [PubMed] [Google Scholar]
- 21.Barth S, Glick D, Macleod KF. 2010. Autophagy: assays and artifacts. J. Pathol. 221:117–124. 10.1002/path.2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bate C, Salmona M, Diomede L, Williams A. 2004. Squalestatin cures prion-infected neurons and protects against prion neurotoxicity. J. Biol. Chem. 279:14983–14990. 10.1074/jbc.M313061200 [DOI] [PubMed] [Google Scholar]
- 23.Bate C, Tayebi M, Williams A. 2010. Glycosylphosphatidylinositol anchor analogues sequester cholesterol and reduce prion formation. J. Biol. Chem. 285:22017–22026. 10.1074/jbc.M110.108548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilch S, Bach C, Lutzny G, Vorberg I, Schatzl HM. 2009. Inhibition of cholesterol recycling impairs cellular PrP(Sc) propagation. Cell. Mol. Life Sci. 66:3979–3991. 10.1007/s00018-009-0158-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilch S, Nunziante M, Ertmer A, Schatzl HM. 2007. Strategies for eliminating PrP(c) as substrate for prion conversion and for enhancing PrP(Sc) degradation. Vet. Microbiol. 123:377–386. 10.1016/j.vetmic.2007.04.006 [DOI] [PubMed] [Google Scholar]
- 26.Klingenstein R, Lober S, Kujala P, Godsave S, Leliveld SR, Gmeiner P, Peters PJ, Korth C. 2006. Tricyclic antidepressants, quinacrine and a novel, synthetic chimera thereof clear prions by destabilizing detergent-resistant membrane compartments. J. Neurochem. 98:748–759. 10.1111/j.1471-4159.2006.03889.x [DOI] [PubMed] [Google Scholar]
- 27.Kumar R, McClain D, Young R, Carlson GA. 2008. Cholesterol transporter ATP-binding cassette A1 (ABCA1) is elevated in prion disease and affects PrPC and PrPSc concentrations in cultured cells. J. Gen. Virol. 89:1525–1532. 10.1099/vir.0.83358-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis V, Hooper NM. 2011. The role of lipid rafts in prion protein biology. Front. Biosci. (Landmark Ed.) 16:151–168. 10.2741/3681 [DOI] [PubMed] [Google Scholar]
- 29.Marella M, Lehmann S, Grassi J, Chabry J. 2002. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J. Biol. Chem. 277:25457–25464. 10.1074/jbc.M203248200 [DOI] [PubMed] [Google Scholar]
- 30.Pani A, Norfo C, Abete C, Mulas C, Putzolu M, Laconi S, Orru CD, Cannas MD, Vascellari S, La Colla P, Dessi S. 2007. Antiprion activity of cholesterol esterification modulators: a comparative study using ex vivo sheep fibroblasts and lymphocytes and mouse neuroblastoma cell lines. Antimicrob. Agents Chemother. 51:4141–4147. 10.1128/AAC.00524-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prior M, Lehmann S, Sy MS, Molloy B, McMahon HE. 2007. Cyclodextrins inhibit replication of scrapie prion protein in cell culture. J. Virol. 81:11195–11207. 10.1128/JVI.02559-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. 1995. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 129:121–132. 10.1083/jcb.129.1.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karapetyan YE, Sferrazza GF, Zhou M, Ottenberg G, Spicer T, Chase P, Fallahi M, Hodder P, Weissmann C, Lasmezas CI. 2013. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc. Natl. Acad. Sci. U. S. A. 110:7044–7049. 10.1073/pnas.1303510110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukherjee A, Morales-Scheihing D, Gonzalez-Romero D, Green K, Taglialatela G, Soto C. 2010. Calcineurin inhibition at the clinical phase of prion disease reduces neurodegeneration, improves behavioral alterations and increases animal survival. PLoS Pathog. 6:e1001138. 10.1371/journal.ppat.1001138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakagaki T, Satoh K, Ishibashi D, Fuse T, Sano K, Kamatari YO, Kuwata K, Shigematsu K, Iwamaru Y, Takenouchi T, Kitani H, Nishida N, Atarashi R. 2013. FK506 reduces abnormal prion protein through the activation of autolysosomal degradation and prolongs survival in prion-infected mice. Autophagy 9:1386–1394. 10.4161/auto.25381 [DOI] [PubMed] [Google Scholar]
- 36.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5:375–386. 10.1038/nri1604 [DOI] [PubMed] [Google Scholar]
- 37.Hill T, Xu C, III, Harper RW. 2010. IFNgamma mediates DUOX2 expression via a STAT-independent signaling pathway. Biochem. Biophys. Res. Commun. 395:270–274. 10.1016/j.bbrc.2010.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishibashi D, Atarashi R, Fuse T, Nakagaki T, Yamaguchi N, Satoh K, Honda K, Nishida N. 2012. Protective role of interferon regulatory factor 3-mediated signaling against prion infection. J. Virol. 86:4947–4955. 10.1128/JVI.06326-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uchiyama K, Muramatsu N, Yano M, Usui T, Miyata H, Sakaguchi S. 2013. Prions disturb post-Golgi trafficking of membrane proteins. Nat. Commun. 4:1846. 10.1038/ncomms2873 [DOI] [PubMed] [Google Scholar]
- 40.Mackeh R, Perdiz D, Lorin S, Codogno P, Pous C. 2013. Autophagy and microtubules—new story, old players. J. Cell Sci. 126:1071–1080. 10.1242/jcs.115626 [DOI] [PubMed] [Google Scholar]
- 41.Marzo L, Marijanovic Z, Browman D, Chamoun Z, Caputo A, Zurzolo C. 2013. 4-Hydroxytamoxifen engenders PrPSc clearance by conveying both PrPC and PrPSc to lysosomes independently of autophagy. J. Cell Sci. 126:1345–1354. 10.1242/jcs.114801 [DOI] [PubMed] [Google Scholar]