

ABSTRACT
We recently demonstrated that a soluble protein, Gas6, can facilitate viral entry by bridging viral envelope phosphatidylserine to Axl, a receptor tyrosine kinase expressed on target cells. The interaction between phosphatidylserine, Gas6, and Axl was originally shown to be a molecular mechanism through which phagocytes recognize phosphatidylserine exposed on dead cells. Since our initial report, several groups have confirmed that Axl/Gas6, as well as other phosphatidylserine receptors, facilitate entry of dengue, West Nile, and Ebola viruses. Virus binding by viral envelope phosphatidylserine is now a viral entry mechanism generalized to many families of viruses. In addition to Axl/Gas6, various molecules are known to recognize phosphatidylserine; however, the effects of these molecules on virus binding and entry have not been comprehensively evaluated and compared. In this study, we examined most of the known human phosphatidylserine-recognizing molecules, including MFG-E8, TIM-1, -3, and -4, CD300a, BAI1, and stabilin-1 and -2, for their abilities to facilitate virus binding and infection. Using pseudotyped lentiviral vectors, we found that a soluble phosphatidylserine-binding protein, MFG-E8, enhances transduction. Cell surface receptors TIM-1 and -4 also enhance virus binding/transduction. The extent of enhancement by these molecules varies, depending on the type of pseudotyping envelope proteins. Mutated MFG-E8, which binds viral envelope phosphatidylserine without bridging virus to cells, but, surprisingly, not annexin V, which has been used to block phagocytosis of dead cells by concealing phosphatidylserine, efficiently blocks these phosphatidylserine-dependent viral entry mechanisms. These results provide insight into understanding the role of viral envelope phosphatidylserine in viral infection.
IMPORTANCE Envelope phosphatidylserine has previously been shown to be important for replication of various envelope viruses, but details of this mechanism(s) were unclear. We were the first to report that a bifunctional serum protein, Gas6, bridges envelope phosphatidylserine to a cell surface receptor, Axl. Recent studies demonstrated that many envelope viruses, including vaccinia, dengue, West Nile, and Ebola viruses, utilize Axl/Gas6 to facilitate their entry, suggesting that the phosphatidylserine-mediated viral entry mechanism can be shared by various enveloped viruses. In addition to Axl/Gas6, various molecules are known to recognize phosphatidylserine; however, the effects of these molecules on virus binding and entry have not been comprehensively evaluated and compared. In this study, we examined most human phosphatidylserine-recognizing molecules for their abilities to facilitate viral infection. The results provide insights into the role(s) of envelope phosphatidylserine in viral infection, which can be applicable to the development of novel antiviral reagents that block phosphatidylserine-mediated viral entry.
INTRODUCTION
Viral envelope phosphatidylserine (PtdSer) has previously been shown to be important for enveloped virus replication, but details of this mechanism(s) were unclear. Several studies demonstrated that vaccinia virus envelope PtdSer mediates binding to target cells (1–4) and that this binding elicits signaling that facilitates postbinding steps of viral entry. HIV-1 can also use viral envelope PtdSer for its replication (5). In addition, anti-PtdSer antibodies were shown to inhibit Pichinde virus replication (6). Although these studies demonstrated that viral envelope PtdSer plays an important role in enveloped virus replication, most likely during virus attachment and entry, the cellular molecules involved were not known. Because of the presence of envelope protein (Env)-mediated virus binding in virus infection, it was difficult to independently study PtdSer-mediated virus binding and identify its molecular mechanisms.
We reported the first identification of a molecular mechanism of PtdSer-dependent virus binding by using targeting lentiviral vectors that specifically transduce the desired cell types (7). Targeting lentiviral vectors eliminate the original receptor-binding activity of the pseudotyping Envs (8, 9). With the original receptor-binding activity of Envs lacking, we found that lentiviral vectors can bind certain cell types by mechanisms that are independent of the interactions between Envs and their receptors. Instead, this binding is mediated by the soluble protein Gas6, which bridges target cells to vectors. The N-terminal domain of Gas6 binds to PtdSer, a lipid exposed on the viral envelope, and the C-terminal domain binds to Axl, a receptor tyrosine kinase expressed on phagocytic cells. This divalent binding activity enables Gas6 to bridge virus to cells, thereby increasing viral transduction to a level comparable to that for virions bearing wild-type Envs.
By investigating vectors pseudotyped with various Envs, we found that Gas6 increased the infectious titers of lentiviral vectors pseudotyped with Envs from Sindbis virus (Sindbis), Ross River virus (RRV), and baculovirus (gp64). We also found that Gas6 and Axl mediate PtdSer-dependent entry of vaccinia virus. Subsequently, other research groups reported that dengue, West Nile, and Ebola viruses can use the same viral entry pathway (10–13). These results demonstrate that the PtdSer-mediated viral entry mechanism is a general mechanism shared by various enveloped viruses to facilitate viral entry and infection.
Gas6 is known to mediate phagocytosis of dead cells by bridging the PtdSer exposed on dead cells to the Axl expressed on phagocytes (Fig. 1A) (14, 15). PtdSer is normally present in the inner cell membrane layer of living cells (16). When cells undergo apoptosis or necrosis, PtdSer moves to the outer layer of the membrane and is thereby exposed on the cell surface. Phagocytes recognize these dead cells by binding to the exposed PtdSer and then remove the dead cells by phagocytosis (17, 18). Therefore, virus entry using viral envelope PtdSer has been termed “apoptotic mimicry” (1, 2, 4).
FIG 1.
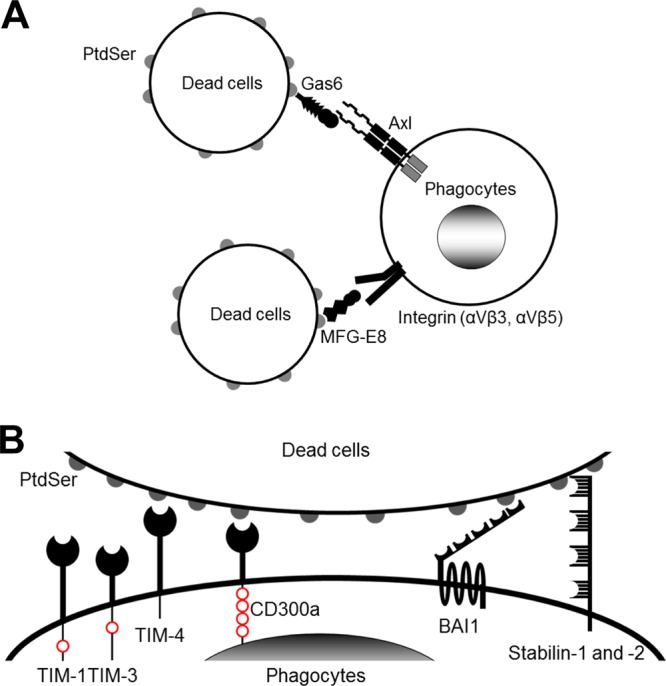
Molecular mechanisms of phagocytosis of dead cells. (A) Molecular mechanisms of phagocytosis of dead cells mediated by soluble molecules that bridge the PtdSer exposed on dead cells to receptors on phagocytes. (B) Molecular mechanisms of phagocytosis of dead cells mediated by receptors that directly recognize PtdSer. TIM-1, -3, and -4 and CD300a bind PtdSer via their N-terminal IgV domains. TIM-1 and -4 each have one cytoplasmic tyrosine residue and CD300a has four cytoplasmic tyrosine residues; these residues are phosphorylated and mediate signaling after binding to PtdSer (red circles). BAI1 is a seven-transmembrane receptor. Its extracellular N terminus has five thrombospondin type 1 repeat domains that bind PtdSer. The N termini of stabilin-1 and -2 each have four clusters of epidermal growth factor-like domain repeats that bind PtdSer. The cytoplasmic domains of BAI1 and stabilin-1 and -2 can activate Rac1 GTPase. In addition to these receptors, RAGE has recently been identified to bind PtdSer; however, its PtdSer-binding domain(s) has not yet been identified.
Phagocytes recognize PtdSer on dead cells by two broadly categorized molecular mechanisms. One is mediated by soluble molecules that bridge phagocytes to dead cells (Fig. 1A). In addition to Gas6 and Axl, MFG-E8 can also act as a bridge by binding to PtdSer on dead cells and to integrin αVβ3 and/or integrin αVβ5 on phagocytes (19). The other mechanism mediates binding through receptors expressed on phagocytes that directly bind PtdSer (Fig. 1B). In humans, TIM-1, -3, and -4, CD300a, BAI1, and stabilin-1 and -2 have been known to bind PtdSer directly (Table 1) (20–27). In addition, RAGE (receptor for advanced glycation end products) was recently shown to bind PtdSer (28, 29).
TABLE 1.
Human PtdSer-binding molecules investigated in this study
| Moleculea | Molecular mass (kDa) | Expression |
|---|---|---|
| MFG-E8 | 46 | Mammary epithelial cells, macrophages |
| TIM-1 | 100 | Th2 cells, mast cells, regulatory B cells, kidney epithelial cells |
| TIM-3 | 60 | Subset of activated CD4 and CD8 cells, Th1 cells, monocytes, microglia |
| TIM-4 | 60 | Tingible-body macrophages |
| CD300a | 60 | NK cells, T cells, B cells, neutrophils, plasmacytoid dendritic cells, mast cells, eosinophils |
| BAI1 | 200 | Neuron, glia, macrophages |
| Stabilin-1 | 280 | Noncontinuous endothelial cells, macrophages |
| Stabilin-2 | 280 | Sinusoidal endothelial cells, macrophages |
MFG-E8 and CD300a can bind PtdEtr, in addition to PtdSer.
Among these PtdSer-recognizing molecules, recent studies showed that TIM-1 can mediate infection/transduction of murine leukemia virus (MLV) and feline immunodeficiency virus vectors pseudotyped with various types of Env, as well as replication-competent viruses, including West Nile, yellow fever, dengue, Tacaribe, Ross River, and Sindbis viruses, baculovirus, and replication-competent vesicular stomatitis virus (VSV) harboring the Ebola virus Env (10, 30, 31). TIM-4 also mediates infection/transduction, albeit less effectively. Although these results indicated that various PtdSer-binding molecules can support infection by enveloped viruses, these studies also showed that BAI1 and RAGE cannot support viral infection (10, 30). These results indicate that not all PtdSer-binding molecules can mediate virus binding and entry. Knowing which PtdSer-recognizing molecular mechanisms can support virus binding/entry is essential to further elucidation of the role of viral envelope PtdSer in viral replication in vitro and in vivo.
PtdSer-mediated binding occurs independently of viral Envs, but Envs are required for the subsequent fusion step (7). Therefore, our lentiviral vector pseudotyped with a mutant Env that possesses no or minimal receptor-binding activity while still maintaining fusion activity is an ideal means to dissect the mechanisms of PtdSer-mediated viral infection/transduction. In the studies described herein, we examined the abilities of most of the known PtdSer-binding molecules to mediate virus binding and viral transduction using lentiviral vectors pseudotyped with our targeting Env, followed by analysis with the vectors pseudotyped with various other Envs.
MATERIALS AND METHODS
Plasmids.
The cDNAs of human Gas6 (hGas6), Axl, TIM-1, -3, and -4, CD300a, and BAI1 were purchased from DNASU (Tempe, AZ), the cDNA of human stabilin-1 was purchased from Promega (Madison, WI), and the cDNA of human stabilin-2 was provided by Edward Harris (University of Nebraska, Lincoln, NE) (32). The CD8 leader and Flag tag sequences were inserted into the cDNAs of TIM-1, -3, and -4, CD300a, BAI1, and stabilin-1 and -2 at the 5′ end by replacing each leader sequence as described previously (21, 33–36). The cDNA of human Axl and cDNAs of human TIM-1, -3, and -4, CD300a, or BAI1 with or without the Flag tag sequence were inserted into the lentiviral vector plasmid pLVU/myc (Addgene, Cambridge, MA) (37). The cDNA of stabilin-1 or -2 with or without the Flag tag sequence was inserted into pcDNA6.2 DEST (Invitrogen, Carlsbad, CA). The 3′ end of the cDNA of hGas6 was conjugated with the Flag tag sequence and cloned into a mammalian expression vector, pCI (Promega). The expression vectors of mouse MFG-E8 (mMFG-E8) and a mutant mouse MFG-E8 (D89E) were provided by Shigekazu Nagata (Kyoto University, Kyoto, Japan) (19).
Lentiviral vectors.
All lentiviral vectors were produced in 293T cells by a previously described calcium phosphate transfection method (38). Enhanced green fluorescent protein (EGFP)-expressing lentiviral vectors pseudotyped with various Envs were produced by transfecting 293T cells with an Env expression vector (Sindbis, 2.2, 2.2 1L1L, VSV envelope G protein [VSV-G], gp64, or RRV), the packaging plasmid PAX2 (Addgene), and a lentiviral vector (cppt2e) containing EGFP as its transgene (39). The 2.2 1L1L, VSV-G, and gp64 pseudotypes labeled with EGFP were generated by transfecting 293T cells with PAX2 and p-gagEGFP (NIH AIDS Research and Reference Reagent Program, Germantown, MD), in addition to expression vectors for each Env (40). The lentiviral vectors expressing hGas6, Axl, TIM-1, -3, and -4, CD300a, and BAI1 with or without the Flag tag sequence were generated by transfecting 293T cells with the VSV-G, PAX2, and pLVU vectors containing cDNA of the corresponding receptors. After transfection, the cells were cultured in AIM-V medium (Invitrogen). The supernatant was harvested and filtered at 48 h posttransfection. The lentiviral vectors were purified by ultracentrifugation, using a sucrose cushion (phosphate-buffered saline [PBS] containing 25% sucrose and 1 mM EGTA), and the pellets were resuspended in PBS. The concentrations of lentiviral vectors were quantitated by enzyme-linked immunosorbent assay (ELISA) by measuring the amounts of viral capsid protein p24. The concentrated virus was frozen at −70°C.
Proteins.
Human MFG-E8 (hMFG-E8) was purchased from R&D Biosystems (Minneapolis, MN). Annexin V (ANX V) was purchased from three different companies (eBioscience, San Diego, CA; ARCOsystems, Beijing, China; and Adipogen, Liestal, Switzerland) to confirm its effect on lentiviral transduction. The results presented in Fig. 4 and 8 were obtained using ANX V purchased from eBioscience.
FIG 4.
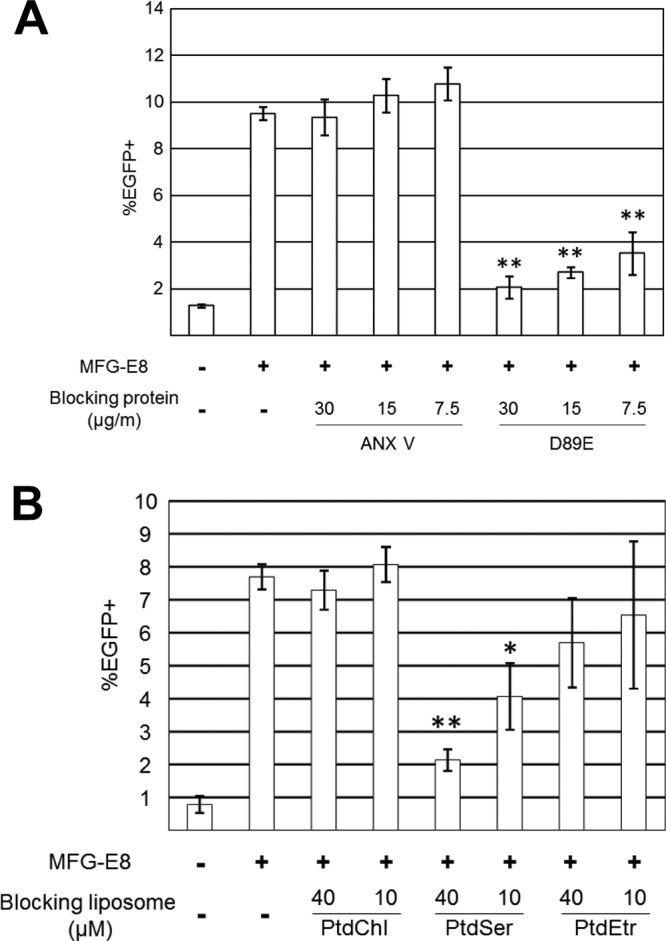
Role of viral envelope PtdSer in MFG-E8-mediated lentiviral transduction. (A) Blocking of MFG-E8-mediated transduction of HMVECs with the 2.2 1L1L pseudotype by ANX V or D89E. HMVECs were incubated with hMFG-E8 (3 μg/ml) for 4 h prior to transduction. The 2.2 1L1L pseudotype (500 ng p24/ml) was preincubated with ANX V or D89E for 1 h at the concentrations shown in the figure prior to transduction. HMVECs were incubated for 2 h with the 2.2 1L1L pseudotype in the presence of ANX V or D89E. This experiment was repeated twice in singlicate and twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. (B) Blocking of MFG-E8-mediated transduction of HMVECs with the 2.2 1L1L pseudotype (100 ng p24/ml) by liposomes. HMVECs were incubated with hMFG-E8 (3 μg/ml) for 4 h, followed by incubation for 1 h with liposomes consisting of either PtdChl only (PtdChl), 75% PtdChl and 25% PtdSer (PtdSer), or 75% PtdChl and 25% PtdEtr (PtdEtr). The cells were then transduced with the 2.2 1L1L pseudotype for 2 h in the presence of the same liposomes. This experiment was repeated twice in singlicate and twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. For panels A and B, significance was calculated using a two-sample Student t test by comparing the MFG-E8-mediated transduction without blocking reagents to that with the blocking reagents indicated in the figure (*, P < 0.05; **, P < 0.01).
FIG 8.

Role of viral envelope PtdSer in TIM-1- and TIM-4-mediated lentiviral transduction. (A) Blocking of TIM-1-mediated 2.2 1L1L pseudotype transduction by ANX V or D89E. The 2.2 1L1L pseudotype (50 ng p24/ml) was incubated with ANX V or D89E for 1 h prior to transduction at the concentrations indicated in the figure. The vectors were then used for transduction of 293T and TIM-1 293T cells. (B) Blocking of TIM-4-mediated gp64 pseudotype transduction by ANX V or D89E. The gp64 pseudotype (25 ng p24/ml) was incubated with ANX V or D89E for 1 h prior to transduction at the concentrations indicated in the figure. The vectors were then used for the transduction of 293T and TIM-4 293T cells. (C) Blocking of TIM-1-mediated 2.2 1L1L pseudotype transduction by liposomes. TIM-1 293T cells were incubated for 1 h with the liposomes indicated in the figure. The cells were then transduced with the 2.2 1L1L pseudotype (50 ng p24/ml) for 2 h in the presence of the same liposomes. (D) Blocking of TIM-4-mediated gp64 pseudotype transduction by liposomes. TIM-4 293T cells were incubated for 1 h with the liposomes indicated in the figure. The cells were then transduced with the gp64 pseudotype (25 ng p24/ml) for 2 h in the presence of the same liposomes. (E) Blocking by D89E of HLA class I-mediated transduction of 293T cells with the 2.2 pseudotype. The 2.2 pseudotype (100 ng p24/ml) was conjugated with an anti-HLA class I antibody (Ab), followed by 1 h of incubation with D89E at the concentrations indicated in the figure. The vectors were then used for transduction of 293T cells. The experiments whose results are presented in panels A, B, and E were repeated twice in singlicate and twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. The experiments whose results are presented in panels C and D were repeated twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. For panels A to D, significance was calculated by comparing the results of TIM-1- or TIM-4-mediated transduction without blocking reagents to those with blocking reagents using a two-sample Student t test (*, P < 0.05; **, P < 0.01).
Recombinant hGas6, mMFG-E8, and D89E were produced by transfecting 293T cells with plasmids expressing each protein. To facilitate γ-carboxylation of hGas6 during production, the cells were also transfected with a plasmid expressing human γ-glutamylcarboxylase (Origene, Rockville, MD), and cells were cultured in medium containing 10 μg/ml vitamin K1 (Sigma-Aldrich, St. Louis, MO) before, during, and after transfection. Following transfection, the cells were washed twice with serum-free medium and cultured in Opti-SFM medium (Invitrogen). At 4 days posttransfection, the supernatants of the transfected cells were harvested and filtered. Supernatants were subjected to ammonium sulfate precipitation at 20% saturation to remove contaminating molecules and 50% saturation to precipitate the target proteins. The ammonium sulfate precipitates were subjected to gel filtration in order to exchange the buffer to Hanks' balanced salt solution (HBSS), followed by purification using a FLAG M purification kit (Sigma-Aldrich) according to the manufacturer's protocol. The fractions containing target proteins were consolidated and dialyzed into HBSS (Invitrogen). The purity and amounts of the proteins were confirmed by SDS-PAGE, SYPRO Ruby staining, and imaging and analysis by an FX imager (Bio-Rad, Hercules, CA). The purity of all purified proteins was >95%.
Cells.
Human microvascular endothelial cells (HMVECs) were cultured in an EGM-2 bullet kit (Lonza, Walkersville, MD). 293T cells stably expressing Flag-tagged Axl, TIM-1, -3, or -4, CD300a, BAI1, or stabilin-1 or -2 were designated Axl 293T, TIM-1 293T, TIM-3 293T, TIM-4 293T, CD300a 293T, BAI1 293T, stabilin-1 293T, and stabilin-2 293T, respectively. 293T cells stably expressing TIM-1, -3, or -4, CD300a, BAI1, or stabilin-1 or -2 without the Flag tag were designated Flag tag-negative (Flag−) TIM-1 293T, Flag− TIM-3 293T, Flag− TIM-4 293T, Flag− CD300a 293T, Flag− BAI1 293T, Flag− stabilin-1 293T, and Flag− stabilin-2 293T, respectively. Axl, TIM-1, -3, and -4, CD300a, and BAI1 were expressed by transducing 293T cells with lentiviral vectors expressing the corresponding cDNAs at a multiplicity of infection (MOI) of 0.1. Stabilin-1 and -2 were expressed by transfecting 293T cells with linearized plasmids expressing stabilin-1 or -2. The transduced/transfected cells were cultured for 1 to 2 weeks in Iscove's modified Dulbecco's medium (IMDM; Sigma-Aldrich) containing 10% fetal calf serum (FCS; Sigma-Aldrich) and blasticidin (Invitrogen). When blasticidin could not completely eliminate nontransduced cells, the cells expressing transduced receptors were purified using a FACSAria cell sorter (BD Bioscience, San Jose, CA). Three clones of stabilin-2 293T cells, STAB 2-15, 2-17, and 2-20 293T cells, were generated by limiting dilutions of stabilin-2 293T cells. STAB 2-5 and 2-8 293T cells were generated by limiting dilutions of Flag− stabilin-2 293T cells. 293T cells were cultured in IMDM containing 10% FCS and antibiotics; the cells expressing transduced cDNA were cultured in the same medium supplemented with blasticidin.
Flow cytometry analysis of expression of transduced cDNA.
Expression of Axl and integrins αVβ3 and αVβ5 was analyzed by staining with anti-Axl (R&D Biosystems) and with anti-integrin αVβ3 (clone 27.1) and αVβ5 (clone P1F6) antibodies (Chemicon, Temucula, CA), respectively. The primary staining was followed by secondary staining with Alexa 488-conjugated goat anti-mouse IgG antibody (Life Technologies, Carlsbad, CA). Murine IgG1 isotype control antibody (eBioscience, San Diego, CA) was used as a control. Expression of Axl, TIM-1, and CD300a was analyzed by phycoerythrin (PE)-conjugated anti-Axl (R&D Biosystems), anti-TIM-1 (Biolegend, San Diego, CA), and anti-CD300a (Sigma-Aldrich) antibodies, respectively. Appropriate isotype control antibodies conjugated with PE were purchased from eBioscience. Expression of TIM-3 was analyzed by staining the cells with anti-TIM-3 (R&D Biosystems) or rat IgG2a isotype control (eBioscience) antibodies, followed by staining with Alexa 488-conjugated goat anti-rat antibody (Invitrogen). Expression of TIM-4 was analyzed by staining 293T and TIM-4 293T cells with goat anti-TIM-4 antibody (R&D Biosystems) or control goat IgG (Sigma-Aldrich), followed by staining with PE-conjugated donkey anti-goat IgG antibody (Abcam, Cambridge, England). Expression of BAI1 on 293T and BAI1 293T cells was analyzed by staining the cells with anti-BAI1 (R&D Biosystems) or mouse IgG1 isotype control (eBioscience) antibodies, followed by Alexa 647-conjugated goat anti-mouse IgG antibody (Invitrogen). Expression of stabilin-2 on 293T and atabilin-2, STAB 2-15, 2-17, and 2-20 293T cells was analyzed by staining the cells with anti-stabilin-2 antibodies (provided by Edward Harris) (41) or mouse IgG2b isotype control antibodies (eBioscience), followed by staining with Alexa 488-conjugated goat anti-mouse IgG antibody (Invitrogen). To analyze the expression of stabilin-1, 293T cells were first fixed and permeabilized using a FIX & PERM kit (Invitrogen) according to the manufacturer's protocol. The cells were then stained with sheep anti-stabilin-1 antibody (R&D Biosystems) or control sheep antibody (R&D Biosystems), followed by Alexa 488-conjugated goat antisheep antibody (Jackson ImmunoResearch, Inc., West Grove, PA).
Expression of Flag-tagged receptors was analyzed by staining cells with allophycocyanin (APC)-conjugated anti-Flag tag antibody (Columbia Bioscience, Frederick, MD) and APC-conjugated mouse IgG1 isotype control antibody (eBioscience).
All flow cytometry analyses in this study were performed using an LSRFortessa flow cytometer (BD Bioscience).
Analysis of binding of hGas6, mMFG-E8, and D89E to cells.
HMVECs (5 × 105) were incubated with hGas6, mMFG-E8, or D89E at 3 μg/ml for 1 h at 37°C in the presence of 0.1% sodium azide. 293T and Axl 293T cells (2 × 105) were incubated with hGas6 at 3 μg/ml for 1 h at 37°C in the presence of 0.1% sodium azide. The cells were then fixed using the FIX & PERM kit, followed by staining with APC-conjugated anti-Flag tag antibody.
Transduction of cells.
HMVECs (1 × 104 cells/well) were seeded into 48-well plates 2 days before transduction for all transduction experiments. The volumes of reagents and viruses used for all transduction experiments of this study were 150 μl unless otherwise indicated. The cells were incubated for 4 h with PBS+ (PBS with 1 mM CaCl2, 0.5 mM MgCl2, and 0.3% bovine serum albumin [Sigma-Aldrich]) or PBS+ containing various concentrations of hGas6, hMFG-E8, mMFG-E8, or D89E. These reagents were replaced with lentiviral vectors pseudotyped with various types of Envs resuspended in PBS+. The cells and virus were then incubated for 2 h, followed by replacement of virus with EGM bullet kit medium.
293T cells and 293T cells ectopically expressing one of the PtdSer receptors were seeded into 48-well plates (1.25 × 104 cells/well) in IMDM containing 2% FCS 2 days prior to transduction. Axl 293T cells were incubated for 4 h with 1 μg/ml of hGas6 before transduction. The cells were incubated with lentiviral vectors pseudotyped with various Envs for 2 h, followed by replacement of virus with medium. EGFP expression was analyzed by flow cytometry at 3 days postinfection.
Virus binding assay.
HMVECs (1 × 105) were incubated for 1 h with hGas6, hMFG-E8, or D89E at 1 μg/ml in 200 μl of PBS+. The cells were then incubated for 2 h with 200 μl of the 2.2 1L1L pseudotype labeled with EGFP (1,000 ng p24/ml), followed by fixation using the FIX & PERM kit. Cells (2 × 105), including 293T cells and Axl 293T cells incubated with 1 μg/ml hGas6 in 200 μl of PBS+ for 1 h and TIM-1, -3, and -4 293T, CD300a, BAI1, stabilin-1 and -2, and STAB 2-15, 2-17, and 2-20 293T cells, were incubated for 2 h with 200 μl of the 2.2 1L1L pseudotype labeled with EGFP (500 ng p24/ml). Cells were then fixed with the FIX & PERM kit. Virus binding was analyzed by flow cytometry.
Blocking of lentiviral transduction using ANX V and D89E.
The 2.2 1L1L pseudotype (500 ng p24/ml) incubated with various concentrations of ANX V or D89E in PBS+ for 1 h at 37°C was used to transduce HMVECs that had been preincubated with hMFG-E8 (3 μg/ml) in PBS+ for 4 h at 37°C. HMVECs were then incubated with the virus for 2 h at 37°C, and EGFP expression was analyzed at 3 days postransduction.
The 2.2 1L1L pseudotype (50 ng p24/ml) or gp64 pseudotype (25 ng p24/ml) that had been incubated with various concentrations of ANX V or D89E in PBS+ for 1 h at 37°C was used to transduce 293T and TIM-1 and -4 293T cells. Cells were then incubated with the virus for 2 h at 37°C, and EGFP expression was analyzed at 3 days postransduction.
Blocking of lentiviral transduction using liposomes.
Phosphatidylcholine (PtdChl), PtdSer, and phosphatidylethanolamine (PtdEtr) in chloroform were purchased from Avanti Polar (Alabaster, AL). PtdChl liposomes consisted only of PtdChl, whereas PtdSer and PtdEtr liposomes consisted of 75% PtdChl and 25% PtdSer or PtdEtr, respectively. The lipids in chloroform were mixed, dried, resuspended in PBS, sonicated, and filtered. Liposomes were aliquoted and stored at −80°C until use.
HMVECs were preincubated with hMFG-E8 (3 μg/ml) in PBS+ for 4 h at 37°C, followed by incubation with various concentrations of liposomes for 1 h at 37°C. The cells were then transduced with the 2.2 1L1L pseudotype (500 ng p24/ml), and transduction efficiencies were analyzed as previously described.
TIM-1 and -4 293T cells preincubated with various concentrations of liposomes for 1 h at 37°C were transduced with the 2.2 1L1L pseudotype (50 ng p24/ml) and gp64 pseudotype (25 ng p24/ml), respectively. Transduction and analyses of transduction efficiencies were performed as previously described.
RESULTS
Targeting lentiviral vector system used for initial investigation of viral envelope PtdSer-mediated viral transduction.
We used lentiviral vectors pseudotyped with modified Sindbis virus Envs to initially investigate viral envelope PtdSer-mediated virus binding and transduction (Fig. 2A). Sindbis virus has two types of Envs, E1 and E2. E2 mediates binding to its receptors, including heparan sulfate, the laminin receptor, and other unknown receptors (42–45). E1 mediates fusion between the target cell membrane and the viral envelope (46).
FIG 2.

Binding of lentiviral vectors mediated by soluble PtdSer-recognizing molecules. (A) Schematic representation of chimeric Sindbis virus Envs. The Sindbis virus Env is first synthesized as a polypeptide and subsequently cleaved by cellular proteases to generate E3, E2, 6K, and E1 proteins. E1 and E2 are incorporated into the viral envelope. Both the 2.2 and 2.2 1L1L pseudotypes are derived from the wild-type Sindbis virus Env with four mutations (red lines) to reduce their binding to original receptors while maintaining fusion activity. In addition, 2.2 1L1L has two flexible linkers (Gly-Gly-Gly-Gly-Ser) at amino acid (a.a.) 70 of the E2 protein, and 2.2 contains the IgG-binding domain of protein A (ZZ), which was inserted into the E2 region at amino acid 70. (B) Expression levels of Axl (red line), integrin αVβ3 (blue line), and integrin αVβ5 (green line) were analyzed by flow cytometry after staining with antibodies specific to each antigen. The isotype of all antibodies was IgG1; thus, an IgG1 isotype control antibody was used for negative-control staining (black line). (C) Schematic representation of recombinant hGas6, hMFG-E8, mMFG-E8, and D89E, a mutant of MFG-E8 that cannot bind integrins. Gla, γ-carboxyglutamic acid domain; EGF, E1, and E2, epidermal growth factor-like domains; SHBG, sex hormone-binding globulin domain; C1 and C2, discoid domains; P/T, proline/threonine-enriched motif. The γ-carboxyglutamic acid domain of Gas6 and the C2 domain of MFG-E8 bind PtdSer. (D) Staining of HMVECs with an APC-conjugated anti-Flag tag antibody after incubation with hGas6 (red line), mMFG-E8 (blue line), or D89E (green line). The black line represents staining without prior incubation with any ligand. (E) Binding of the EGFP-labeled 2.2 1L1L pseudotype to HMVECs after incubation with hGas6, hMFG-E8, or mMFG-E8. Black line, HMVECs without virus; red line, HMVECs with virus with or without the bridging molecules indicated in the figure. The MFI values correspond to those for EGFP signals, which increase upon binding of EGFP-labeled virus. This experiment was repeated twice in singlicate and once in triplicate, and the MFI values shown are the averages and standard deviations of the triplicate experiment; representative flow cytometry histograms are shown. Significance was calculated by comparing the value of virus binding without a bridging molecule to the value of virus binding in the presence of the bridging molecules indicated in the figure using a two-sample Student t test (**, P < 0.01).
The 2.2 1L1L pseudotype is a mutated Env derived from the wild-type Sindbis virus Env containing mutations at its receptor-binding regions (38, 46–50). 2.2 1L1L cannot mediate efficient binding to target cells due to the mutations; however, it still maintains the intact fusion activity of E1. While the 2.2 1L1L pseudotype cannot bind and transduce target cells, it can transduce target cells efficiently when the virus binding is mediated by viral envelope PtdSer. The 2.2 1L1L pseudotype was previously used to investigate Axl/Gas6-mediated infection through binding to PtdSer. Therefore, the 2.2 1L1L pseudotype can be used to study host PtdSer-recognizing molecules for their ability to mediate virus binding and transduction with no/minimum interference from Env-mediated binding.
MFG-E8 enhances transduction by lentiviral vectors.
We first determined whether MFG-E8 can increase lentivirus binding and transduction by bridging virus and cells. Using HMVECs that express Axl (Fig. 2B), we identified Gas6 as a protein that bridges virus to cells. HMVECs also express integrins αVβ3 and αVβ5, which bind MFG-E8 (Fig. 2B); thus, this cell type was used to investigate whether MFG-E8 can mediate lentiviral transduction.
We used recombinant human and mouse MFG-E8 (hMFG-E8 and mMFG-E8, respectively) and a mutant mouse MFG-E8 (D89E) that contained a mutation within its integrin-binding RGD motif (Fig. 2C) (19). hGas6, mMFG-E8, and D89E were fused with a C-terminal Flag tag. We analyzed the binding of these Flag-tagged molecules to HMVECs by staining cells with an APC-conjugated anti-Flag tag antibody after incubation with each molecule (Fig. 2D). Flow cytometry analysis revealed that mMFG-E8 binds to HMVECs more efficiently than hGas6. The mutation at the integrin-binding region decreased the binding of mMFG-E8, confirming that binding of mMFG-E8 to HMVECs is primarily mediated by the interaction between the integrins and the RGD motif.
We used the 2.2 1L1L pseudotype labeled with EGFP to measure virus binding by flow cytometry. The binding of virus was quantified as the mean fluorescence intensity (MFI) of the FL1 (EGFP) signal. In the absence of any bridging molecule, the 2.2 1L1L pseudotype minimally bound to HMVECs (Fig. 2E). As we previously reported, preincubation of HMVECs with hGas6 drastically increased binding of 2.2 1L1L (7). However, we could not detect a significant enhancement of virus binding by MFG-E8 with this assay. These results demonstrated that the enhancement of virus binding by MFG-E8 is not as strong as that by hGas6.
We next investigated whether MFG-E8 can increase the transduction of the 2.2 1L1L pseudotype. We incubated HMVECs with hGas6, human or mouse MFG-E8, or D89E prior to transduction with the 2.2 1L1L pseudotype carrying EGFP as its transgene. Because enhancement of viral transduction by hGas6 is very strong, we initially used low MOIs (100 ng p24/ml) to avoid saturation of viral transduction enhancement. Both human and murine MFG-E8 enhanced viral transduction only at high concentrations (1 to10 μg/ml), while hGas6 increased transduction even at the lowest concentrations used (10 ng/ml) (Fig. 3A). MFG-E8-mediated enhancement of transduction was observed at multiple MOIs (Fig. 3B). Consistent with the results of virus binding assays (Fig. 2E), the enhancement by MFG-E8 was lower (5- to 10-fold) than that by hGas6 (200-fold).
FIG 3.

Lentiviral transduction mediated by MFG-E8. (A) Enhancement of HMVEC transduction with the 2.2 1L1L pseudotype (100 ng p24/ml) after incubation with hGas6, hMFG-E8, mMFG-E8, or D89E at the concentrations shown on the x axis. The fold enhancement was calculated by the following formula: percentage of EGFP-expressing cells preincubated with each ligand divided by percentage of EGFP-expressing cells preincubated with buffer only. This experiment was repeated twice in singlicate and twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. The proportion of EGFP-expressing cells preincubated with buffer only was 0.12% ± 0.05% (average ± standard deviation). Significance was calculated by comparing the value for the no-protein samples to the value for the samples with 10 μg/ml proteins, using a two-sample Student t test (*, P < 0.05; **, P < 0.01). (B) Transduction of HMVECs with the 2.2 1L1L pseudotype with or without preincubation with hMFG-E8 at high (2,500 ng p24/ml), medium (500 ng p24/ml), and low (100 ng p24/ml) MOIs. This experiment was repeated twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. (C) Transduction of HMVECs with lentiviral vectors pseudotyped with Sindbis (100 ng p24/ml), RRV (400 ng p24/ml), gp64 (400 ng p24/ml), or VSV-G (8 ng p24/ml) after preincubation with hMFG-E8, mMFG-E8, or D89E at 3 μg/ml. This experiment was repeated once in singlicate and twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. Significance was calculated by comparing the value of transduction obtained with the no-protein samples to the value of transduction obtained with the proteins indicated in the figure using a two-sample Student t test (*, P < 0.05; **, P < 0.01).
We next investigated whether MFG-E8 can enhance the transduction of other pseudotypes. We found that it enhanced the transduction of lentiviral vectors pseudotyped with Sindbis, RRV, and gp64 (Fig. 3C). D89E does not enhance viral transduction (Fig. 3A and C), which demonstrated that the interaction between the integrins and the RGD motif is important for MFG-E8-mediated viral transduction. Our previous study showed that transduction by the VSV-G pseudotype was not as strongly enhanced by hGas6 as that by other pseudotypes (7). MFG-D8 also did not enhance transduction by the VSV-G pseudotype (Fig. 3C).
Role of viral envelope PtdSer in MFG-E8-mediated lentiviral transduction.
We investigated whether transduction by MFG-E8 is mediated by its interaction with viral envelope PtdSer. Previous studies have shown that binding of MFG-E8 to apoptotic cells is inhibited by incubating the apoptotic cells with ANX V, which specifically binds and conceals exposed PtdSer (19). We preincubated the 2.2 1L1L pseudotype with ANX V to block MFG-E8-mediated viral transduction. Our previous biochemical studies demonstrated that ANX V binds to the 2.2 1L1L pseudotype in a typical calcium-dependent manner (7). Surprisingly, ANX V could not block MFG-E8-mediated viral transduction (Fig. 4A). We confirmed this result, using recombinant ANX V from three companies and a very high concentration of ANX V (100 μg/ml) (data not shown). The recombinant ANX V used in this study specifically blocked binding of PE-conjugated ANX V to apoptotic HMVECs at the same concentrations used to block transduction, confirming that the recombinant ANX V used in this study can bind to PtdSer (data not shown). Since the affinity between ANX V and PtdSer is strong (Kd [dissociation constant], 5 nM) (51) and MFG-E8 does not bind virus strongly (Fig. 2E), it is difficult to explain the inability of ANX V to block MFG-E8-mediated transduction by the weak affinity of ANX V for viral envelope PtdSer.
There are two possible explanations for this phenomenon: (i) MFG-E8 binds to molecules other than PtdSer present on the viral envelope, or (ii) ANX V cannot completely occlude all PtdSer on the viral envelope.
In addition to binding to PtdSer, MFG-E8 is known to bind PtdEtr, although its affinity for PtdEtr is not as strong as that for PtdSer (19). Similar to PtdSer, PtdEtr is specifically present in the inner layer of the cell membrane and becomes exposed on the cell surface upon cell death (52). Although exposure of PtdEtr on the viral envelope has not been reported, we investigated the possibility that PtdEtr plays a role in MFG-E8-mediated transduction.
Currently, there is no reagent that specifically conceals PtdEtr without toxicity (53); thus, we used liposomes consisting of PtdEtr to block MFG-E8-mediated transduction (Fig. 4B). The liposome consisting of PtdChl did not block MFG-E8-mediated lentiviral transduction, but the liposome consisting of PtdSer did so efficiently. The liposome consisting of PtdEtr cannot significantly inhibit MFG-E8-mediated transduction. These results are consistent with the known affinities of MFG-E8 for PtdChl, PtdSer, and PtdEtr (19). Although the very weak inhibition mediated by PtdEtr indicated a role for viral envelope PtdEtr in MFG-E8-mediated transduction, the strong inhibition by the PtdSer liposome suggests that binding of virus to MFG-E8 is mainly mediated by viral envelope PtdSer. The results of this experiment did not eliminate the possibility that ANX V cannot conceal all of the viral envelope PtdSer.
Ectopic expression of PtdSer receptors on 293T cells.
We examined seven types of human receptors that can directly bind PtdSer, including TIM-1, -3, and -4, CD300a, BAI1, and stabilin-1 and -2 (Fig. 1B) (20–27). For the TIM receptor family, it is known that the affinity of TIM-3 for PtdSer is very weak (54). Although these PtdSer receptors are known to mediate binding of phagocytes and dead cells, CD300a-mediated binding is known to inhibit, rather than facilitate, phagocytosis (23, 24, 55, 56).
Because 293T cells do not express any of these PtdSer receptors, we analyzed the functions of these PtdSer receptors by stably expressing them on 293T cells, using lentiviral or plasmid vectors. We also expressed Axl on 293T cells and compared the degree of enhancement of transduction mediated by these PtdSer receptors to that mediated by Axl and hGas6.
Axl, TIM-1,-3, and -4, CD300a, and BAI1 were expressed on 293T cells using lentiviral vectors (Fig. 5A). The lentiviral vector contained the ubiquitin C promoter to express inserted cDNAs and the simian virus 40 (SV40) promoter to express the blasticidin resistance gene. Because the cDNAs of stabilin-1 and -2 are very large (8 kb) and exceed the capacity of lentiviral vectors (57), we employed plasmid vectors for their expression (Fig. 5A). The plasmid vector contains the cytomegalovirus (CMV) promoter to express the inserted cDNA and the SV40 promoter to express the blasticidin-resistant gene. Previous studies (21, 33–36) from other research groups used Flag tag-conjugated TIM-1, -3, and -4, CD300a, and BAI1 to examine their functions. We also conjugated the Flag tag with all of the PtdSer receptors except Axl in the same manner as the previous studies.
FIG 5.

Ectopic expression of PtdSer receptors on 293T cells. (A) Schematic representation of lentiviral vectors expressing TIM-1, -3, and -4, CD300a, or BAI1 and plasmid vectors expressing stabilin-1 and -2. SIN LTR, self-inactivating long terminal repeat; Ubc pro, ubiquitin C promoter; SV40 pro, SV40 promoter; TK, thymidine kinase. (B) Expression of Axl, TIM-1, -3, and -4, CD300a, BAI1, and stabilin-1 and -2 on 293T cells ectopically expressing each of these receptors analyzed by the use of antibodies specific to each receptor. Black line, staining by control antibodies; red line, staining by antibodies specific to each PtdSer receptors. (C) (Left column and top four panels of right column) Expression of TIM-1, -3, and -4, CD300a, BAI1, and stabilin-1 and -2 on 293T cells ectopically expressing each of these receptors and parental 293T cells analyzed by the antibody specific to the Flag tag. Black line, staining by APC-conjugated isotype control antibody; red line, staining by APC-conjugated anti-Flag tag antibody. (Bottom two panels of right column) Expression of Axl was analyzed by incubating 293T and Axl 293T cells with hGas6, followed by staining with the anti-Flag tag antibody. Black line, staining by APC-conjugated anti-Flag tag antibody without preincubation with hGas6; red line, staining by APC-conjugated anti-Flag tag antibody after incubation with hGas6.
The cells expressing these receptors were selected by culturing in medium containing blasticidin. When necessary, the cells expressing the transgenes were sorted by flow cytometry.
Staining of stably transduced or transfected 293T cells with antibodies specific to each PtdSer receptor confirmed that 293T cells do not naturally express these PtdSer receptors but do so after stable transduction of the cDNA of each PtdSer receptor (Fig. 5B). The expression of stabilin-2 decreased over passages of cells, even after sorting for the stabilin-2-positive population and culturing of the cells in the presence of blasticidin. This decrease in expression seemed to be caused by the slower growth of cells that express stabilin-2 at high levels. Therefore, we generated three clones of stabilin-2 293T cells, designated STAB 2-15, 2-17, and 2-20 293T cells (Fig. 5B).
Staining of these cells with antibodies specific to each PtdSer receptor confirmed expression of each PtdSer receptor on transduced or transfected 293T cells. However, antibodies specific to each PtdSer receptor cannot indicate the relative expression levels of different PtdSer receptors because of the different affinity of each antibody to each PtdSer. To compare these, we stained the cells with an anti-Flag tag antibody (Fig. 5C). Staining of the Flag tag demonstrated that all PtdSer receptors are expressed abundantly, except in stabilin-2 293T cells. In the case of Axl 293T cells, hGas6 bound to Axl serves as a PtdSer-binding molecule; therefore, Axl 293T cells preincubated with hGas6, which contains the Flag tag in its C terminus, were stained by the anti-Flag tag antibody. The results showed that hGas6 specifically binds to Axl 293T cells in an Axl-dependent manner, but the signal of Flag tag staining was not as strong as that of staining of other PtdSer receptors. This indicated that Axl 293T cells preincubated with hGas6 might not have as many PtdSer-binding sites as cells expressing PtdSer receptors.
TIM-1 and -4 and CD300a increase binding to lentiviral vectors.
We determined whether expression of each PtdSer receptor can increase the binding of the EGFP-labeled 2.2 1L1L pseudotype. The virus binds to 293T cells at a low level when PtdSer is not ectopically expressed. Expression of Axl, along with incubation of hGas6, increased the signal caused by virus binding (Fig. 6A), demonstrating that the increased FL1 (EGFP) signals can be used to determine the ability of each PtdSer receptor to mediate virus binding. Using this assay, we examined virus binding to all types of cells expressing various PtdSer receptors. The binding of virus was quantified as the MFI of the FL1 (EGFP) signal (Fig. 6B). Among all the types of PtdSer molecules tested, Axl/hGas6 increased virus binding the most strongly, followed by TIM-1. TIM-4 and CD300a also increased virus binding, but to an extent less than that achieved with TIM-1.
FIG 6.

Binding of the 2.2 1L1L pseudotype mediated by PtdSer receptors. (A) Representative flow cytometry analysis data for the binding of the EGFP-labeled 2.2 1L1L pseudotype (100 ng p24) to 293T cells and to Axl 293T cells preincubated with hGas6. Black line, 293T cells without virus; red line, 293T cells with virus; blue line, Axl 293T cells preincubated with hGas6 without virus; green line, Axl 293T cells preincubated with hGas6 with virus. (B) Quantitation of EGFP-labeled 2.2 1L1L pseudotype binding to 293T cells, Axl 293T cells preincubated with hGas6, and TIM-1, -3, or -4, CD300a, BAI1, stabilin-1 or -2, STAB 2-15, STAB 2-17, and STAB 2-20 293T cells. The values on the y axis correspond to the MFIs of the EGFP signals, which increase by binding of EGFP-labeled virus. This experiment was repeated once in singlicate and twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. Significance was calculated by comparing the value for 293T cells to the values for the cells expressing each type of PtdSer receptor using a two-sample Student t test (**, P < 0.01).
TIM-1 and -4 enhance transduction of lentiviral vectors.
We next investigated whether transduction by various pseudotypes is enhanced by the expression of PtdSer receptors. Axl/Gas6 and TIM-1 increased the transduction of lentiviral vectors pseudotyped with all Envs tested, including 2.2 1L1L (Fig. 7A), Sindbis (Fig. 7B), RRV (Fig. 7C), gp64 (Fig. 7D), and VSV-G (Fig. 7E). TIM-4 enhanced transduction of the RRV, gp64, and VSV-G pseudotypes but not that of the 2.2 1L1L and Sindbis pseudotypes. Although VSV-G pseudotype transduction was enhanced by Axl/Gas6 and TIM-1 and -4, the extent of enhancement was much less than for the other pseudotypes.
FIG 7.

Lentiviral transduction mediated by PtdSer receptors. Percentage of EGFP-expressing 293T cells; Axl 293T cells preincubated with hGas6, and TIM-1, -3, or -4, CD300a, BAI1, stabilin-1 or -2, STAB 2-15, STAB 2-17, or STAB 2-20 293T cells after transduction with lentiviral vectors pseudotyped with 2.2 1L1L (50 ng p24/ml) (A), Sindbis (20 ng p24/ml) (B), RRV (40 ng p24/ml) (C), gp64 (25 ng p24/ml) (D), or VSV-G (10 ng p24/ml) (E). EGFP expression was analyzed at 3 days postransduction. The results are the averages of triplicate experiments and are shown with standard deviations. (F) Percentage of EGFP-expressing 293T cells and Flag− TIM-1, -3, or -4, Flag− CD300a, Flag− BAI1, Flag− stabilin-1 or -2, STAB 2-5, and STAB 2-8 293T cells after transduction with the 2.2 1L1L pseudotype (50 ng p24/ml). (G) Titers (EGFP transduction units [TU]/ml) of various pseudotypes with 293T cells, Axl 293T cells preincubated with hGas6, and TIM-1 293T cells as target cells. The titers came from concentrated virus stocks that contained 60 μg p24/ml. The experiments whose results are presented in panels A and F were repeated three times in singlicate and twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. The experiments whose results are presented in panels B to E were repeated twice in triplicate, and the results shown are the averages and standard deviations of one representative triplicate experiment. For panels A to F, significance was calculated by comparing the transduction efficiencies of 293T cells to those of PtdSer receptor-expressing cells using a two-sample Student t test (*, P < 0.05; **, P < 0.01).
Of note, we observed that the efficiencies of infection of stabilin-1 and STAB 2-17 293T cells with the VSV-G pseudotype and the efficiencies of infection of STAB 2-15 and 2-17 293T cells with the Sindbis pseudotype were decreased compared to the efficiencies of infection of 293T cells with the same pseudotypes. We are uncertain whether these decreases were caused by inhibition at the viral entry step due to interaction of viral envelope PtdSer and stabilin-1 or -2 because (i) the cells which highly express stabilin-1 or -2 grow slower than conventional 293T cells and (ii) we could not observe virus binding mediated by stabilin-1 or -2. Therefore, we did not further pursue the mechanisms of this inhibition of viral infection.
The data presented in Fig. 7A to E were generated using the cells expressing the Flag-tagged PtdSer receptors (21, 33–36); however, we also generated 293T cells expressing untagged PtdSer receptors and cell clones expressing untagged stabilin-2 (STAB 2-5 and STAB 2-8 293T cells) and confirmed that the data for 2.2 1L1L pseudotype transduction obtained using Flag-tagged PtdSer receptors were the same as those obtained using untagged receptors (Fig. 7F).
The extent of enhancement by Axl/Gas6 and TIM-1 varies depending on the type of pseudotyping Envs.
The titers of lentiviral vectors are usually determined using 293T cells as target cells (58, 59). With this titration method, the VSV-G pseudotype usually has the highest titers among various pseudotypes. Since the effects of Axl/Gas6 and TIM-1 on lentiviral titers differ among various pseudotypes, the pseudotype with the highest titer changes when transduction is supported by Axl/Gas6 and/or TIM-1. We calculated the titers of each pseudotype when 293T cells, Axl 293T cells preincubated with hGas6, and TIM-1 293T cells were used for titration (Fig. 7G). When 293T cells were used as target cells, the VSV-G pseudotype showed the highest titers. However, since transduction of the gp64 pseudotype was drastically enhanced by Axl/Gas6 and TIM-1, the titers of the gp64 pseudotype were the highest among those of all pseudotypes tested in Axl 293T cells preincubated with hGas6 and TIM-1 293T cells.
Blocking of TIM-1-, TIM-4-, and MFG-E8-mediated lentiviral transduction by D89E.
We next investigated whether TIM-1 and -4 mediate lentiviral transduction by binding to viral envelope PtdSer. We attempted to block TIM-1- and TIM-4-mediated transduction by concealing viral envelope PtdSer, using ANX V (Fig. 8A and B). ANX V could not inhibit transduction mediated by TIM-1 or -4. Unlike MFG-E8, TIM-1 and -4 are known to specifically bind PtdSer (20, 21, 54). Consistent with the known specificities of TIM-1 and -4, transduction mediated by TIM-1 and -4 was inhibited only by the liposomes consisting of PtdSer (Fig. 8C and D). These results demonstrate that ANX V cannot prevent the binding of TIM-1 or -4 to viral envelope PtdSer.
We attempted to use a different protein that conceals viral envelope PtdSer and potentially inhibits binding to TIM-1 and -4. D89E has been used as a dominant negative mutant to block PtdSer-dependent phagocytosis of dead cells (21, 60–62) because it can bind PtdSer, whereas its binding to integrins is impaired. We attempted to block TIM-1- and TIM-4-mediated lentiviral transduction by incubating the virus with D89E prior to transduction.
As a control viral vector that uses a PtdSer-independent entry pathway, we used a lentiviral vector pseudotyped with the modified Sindbis virus Env, 2.2, which contains the Fc-binding region of protein A (Fig. 2A) (7). The 2.2 pseudotype can be conjugated with monoclonal antibodies that mediate binding of the virus to target cells. 293T cells were minimally transduced when the 2.2 pseudotype was not conjugated with antibodies (Fig. 8E). When the virus was conjugated with the antibody that recognizes 293T cells (anti-HLA class I antibody), the virus efficiently transduced the cells using HLA class I as a viral receptor. Transduction by the 2.2 pseudotype conjugated with anti-HLA class I antibody was not inhibited by preincubation of the virus with D89E (Fig. 8E), confirming that D89E does not inhibit PtdSer-independent viral entry.
In contrast, D89E completely blocked TIM-1- and TIM-4-mediated lentiviral transduction (Fig. 8A and B). These results suggest that TIM-1 and -4 mediate lentiviral transduction by binding to PtdSer. We also found that transduction of HMVEC with the 2.2 1L1L pseudotype mediated by MFG-E8 can also be inhibited by D89E (Fig. 4A). These results demonstrate that D89E can block multiple PtdSer-dependent viral entry mechanisms.
DISCUSSION
The studies described herein examined most of the currently known human PtdSer-recognizing molecules for their abilities to support virus binding and lentiviral transduction. We found that a soluble protein, MFG-E8, that bridges integrins to PtdSer can enhance transduction by pseudotyped virus. We also found that three types of cell surface receptors, TIM-1 and -4 and CD300a, enhance virus binding. TIM-1 increased the transduction of all pseudotypes tested. TIM-4 increased transduction only with the RRV and gp64 pseudotypes. CD300a could not enhance transduction with any pseudotype. ANX V, which has been used to conceal PtdSer and block phagocytosis of dead cells, could not block viral transduction mediated by MFG-E8 and TIM-1 and -4. A mutant of MFG-E8 which can conceal PtdSer on virus blocked the viral transduction mediated by MFG-E8, as well as TIM-1 and -4.
MFG-E8 could enhance transduction with vectors pseudotyped with various Envs, including 2.2 1L1L, Sindbis, RRV, and gp64, by 5- to 10-fold (Fig. 3C). As shown in Fig. 2E and 3A, the 2.2 1L1L pseudotype did not bind or transduce HMVECs without support from soluble PtdSer-binding molecules. This low level of Env-dependent binding, as well as the abundant expression of Axl and integrins αVβ3 and αVβ5, enabled us to identify Gas6 and MFG-E8 as virus-cell bridging molecules and to investigate the details of viral entry mediated solely by viral envelope PtdSer.
The results of 2.2 1L1L transduction demonstrated that enhancement of transduction by MFG-E8 (5- to 10-fold) is not as efficient as that by Gas6 (>200-fold) (Fig. 3A). This could be explained by the results which demonstrated that virus binding mediated by MFG-E8 is much less efficient than that mediated by Gas6 (Fig. 2E). Unlike Axl and TIM-1 and -4, integrin αVβ3 and/or αVβ5 is expressed on a wide variety of phagocytic and nonphagocytic cell types (47, 63–68). Integrins/MFG-E8 play a role in viral infection/transduction of wider varieties of cell types than other PtdSer-recognizing molecules do. Since MFG-E8 is a component of milk (69, 70), investigation of the role of MFG-E8 in mother-to-child transmission of enveloped viruses may provide a new target for prevention of mother-to-child virus spread.
Viral envelope PtdSer-mediated binding helps to elucidate the molecular mechanisms of TIM-1- and Axl-mediated viral infection, which were previously unclear (71, 72). Integrins are also known to support infection of various enveloped viruses, including human cytomegalovirus (HCMV) and hantavirus (66, 68, 73). For entry of HCMV, Env of HCMV (gB) binds to integrin αVβ3 (66). This interaction mediates the postentry steps of these viruses by inducing the signals required for postbinding steps of viral replication rather than mediating initial binding of the virus. Binding of MFG-E8 to integrin αVβ3 and/or αVβ5 has been shown to induce signaling (74). Therefore, the interaction between viral envelope PtdSer, MFG-E8, and integrin αVβ3 and/or αVβ5 may also support viral infection by facilitating postbinding steps by inducing signals.
TIM-4 enhances transduction by the RRV, gp64, and VSV-G pseudotypes but does not enhance transduction by the 2.2 1L1L and Sindbis pseudotypes (Fig. 7A to D). Analysis of virus binding showed that TIM-4 can enhance the binding of the 2.2 1L1L pseudotype (Fig. 6B). These results indicate that binding of the 2.2 1L1L pseudotype via TIM-4 does not result in transduction. A similar phenomenon was observed in a study by another research group which investigated the ability of TIM-4 to mediate phagocytosis of apoptotic cells (62). Both TIM-1 and TIM-4 have been shown to mediate binding of phagocytes to apoptotic cells, but only TIM-1 and not TIM-4 can mediate phagocytosis (20, 62). TIM-1 has a signaling tyrosine motif, but TIM-4 does not (Fig. 1B) (54); thus, the investigators proposed that TIM-4 cannot mediate phagocytosis of dead cells due to the lack of signals. The 2.2 1L1L and Sindbis pseudotypes might also require signals from TIM receptors for efficient endocytosis of virus. The RRV, gp64, and VSV-G pseudotypes may not require such signals because they can induce endocytosis by binding of the Envs to their receptors.
CD300a increased virus binding but did not enhance the transduction of any pseudotype (Fig. 6B and 7A to F). CD300a is known to bind to apoptotic cells; however, this binding does not induce phagocytosis of dead cells (23, 24, 56). Rather, the signals elicited by CD300a suppress phagocytosis. Therefore, the resulting signals from the binding of CD300a to virus might also suppress viral endocytosis, which would cause abortive virus infection.
The results obtained with TIM-4 and CD300a suggest that binding of viral envelope PtdSer to its receptors elicits signals that play a role in postbinding steps of virus infection. The role of such signaling was intensively studied for Axl/Gas6-mediated virus infection. One study demonstrated that the signals elicited by the interactions between PtdSer, Gas6, and Axl dampen type I interferon signaling of target cells to promote infection of West Nile virus and lentiviral vectors (11). Another study demonstrated that Axl signaling induces macropinocytosis to facilitate endocytosis of feline immunodeficiency virus and VSV pseudotyped with the Ebola virus Env (13). These results suggest that viral envelope PtdSer can facilitate multiple steps of viral replication by interaction with PtdSer-recognizing molecules. Since the signaling pathways of each PtdSer-recognizing molecule are different, the effects of the signaling on postbinding steps of viral infection will differ for each PtdSer-recognizing molecule.
Analyzing the infectivity of the 2.2 1L1L and Sindbis pseudotypes enabled us to compare the efficiency of Env-mediated infection and PtdSer-mediated infection (Fig. 7A and B). One nanogram of p24 of the Sindbis pseudotype could transduce 1.4 × 103 293T cells. The same amount of the 2.2 1L1L pseudotype transduced the same number of Axl 293T cells (1.5 × 103 cells) when hGas6 was present. These results demonstrate that PtdSer can mediate lentiviral transduction as efficiently as Envs. Another group demonstrated that the interaction between PtdSer and TIM-1 can mediate dengue virus infection as efficiently as the interaction between Env and DC-SIGN (10). These results demonstrate that viral envelope PtdSer can mediate virus binding/entry as efficiently as Envs, and this is likely dependent on the host cell type. One study demonstrated that dengue virus infection of cells which endogenously express TIM-1 and Axl was almost completely blocked by using both anti-TIM-1 and anti-Axl antibodies (10). This result indicated that viral envelope PtdSer-mediated virus binding is indispensable for infection of certain combinations of virus and cells, at least in cell culture.
A previous study using MLV vectors pseudotyped with Ebola or Marburg virus Envs demonstrated that TIM-1 cannot mediate infection by these viruses when Niemann-Pick type C1 (NPC1) (75, 76), a specific receptor of Ebola and Marburg virus Envs, is absent on target cells (31). These results suggested that the binding and endocytosis mediated by PtdSer-recognizing molecules would not be sufficient to trigger fusion if the Envs need to bind particular molecules to induce their conformational changes. However, PtdSer-binding molecules can efficiently support entry of the virus containing Sindbis virus Envs that have mutations in their receptor-binding regions. Therefore, the binding and endocytosis mediated by PtdSer-binding molecules would be sufficient for entry of viruses containing Envs that only require exposure to low pH to trigger their fusion activity (i.e., Sindbis and Semliki forest virus Envs) (77–80).
Our results, as well those from other studies, showed that the extent of enhancement varies according to the type of pseudotyping Envs (30, 31). Although the reasons for these differences are not clear, it is possible that the levels of exposed PtdSer on the viral envelope vary according to the type of pseudotyping Envs, which changes the avidity of the virions for PtdSer-binding molecules. Envs that effectively induce the death of 293T cells during viral production are likely to expose more PtdSer on the viral envelope. The levels of PtdSer exposed on the viral envelope will also affect the replication efficiency of replication-competent viruses that use PtdSer-binding molecules for their replication. Importantly, replication-competent viruses harvested from animals (VSV harboring the Ebola virus Env and Ross River, Sindbis, and Tacaribe viruses) were shown to use TIM-1 for cellular entry in in vitro settings (30, 31), suggesting that exposure of viral envelope PtdSer also occurs in vivo.
The significance of the roles of viral envelope PtdSer in replication-competent viral infection and lentiviral transduction has not been established in in vivo settings because it was not clear which PtdSer-recognizing molecules could mediate viral entry. Our results will allow us to focus on viral entry routes via Axl/Gas6, MFG-E8/integrins, and TIM-1 and -4 when studying the role of viral envelope PtdSer in virus infection and transduction in vivo.
Mutation analysis of TIM-1 in previous studies suggested that viral envelope PtdSer is the ligand for TIM-1 during infection by enveloped viruses (30). ANX V has been used to occlude PtdSer on dead cells and block binding to MFG-E8 (19) or phagocytosis of apoptotic cells (81); however, ANX V cannot block viral entry via MFG-E8 and TIM-1 and -4. On the other hand, we found that a mutant of MFG-E8, D89E, can block MFG-E8-, TIM-1-, and TIM-4-mediated lentiviral transduction. The reason why D89E, but not ANX V, can efficiently block PtdSer-dependent viral entry is still not clear. Since the ligands of MFG-E8 and TIM receptors are well studied and the common ligand of TIM receptors and MFG-E8 is only PtdSer (54, 82, 83), it is unlikely that D89E conceals an unidentified molecule(s) that also plays a role in virus binding via MFG-E8 and TIM-1 and -4. One possible explanation is that D89E can completely conceal exposed PtdSer on the viral envelope, while ANX V cannot due to its different access to PtdSer upon binding. Another possible explanation is that ANX V binds to PtdSer at a site different from where MFG-E8 and TIM-1 and -4 bind. Elucidating the mechanisms of D89E-mediated inhibition of viral entry via MFG-E8 and TIM-1 and -4 may lead to novel antiviral reagents that block the entry of all of the various families of enveloped viruses that use the PtdSer-mediated route of infection.
ACKNOWLEDGMENTS
We thank Shigekazu Nagata and Edward Harris for providing reagents, Juanjuan Du and Jing Wei for advice on liposome preparation, Jerome A. Zack, Benhur Lee, TingTing Wu, Rikinari Hanayama, and Gene-Errol Ringpis for discussion, and Wendy Aft and Jamie Ngo for proofreading of the manuscript. We thank the UCLA CFAR Virology Core Laboratory for the p24 ELISA and UCLA JCCC flow cytometry core for cell sorting.
This work was supported by UCLA CTSI grant UL1TR000124, the UCLA AIDS Institute and the UCLA Center for AIDS Research (AI28697), and U.S. National Institutes of Health grants R21AI095004 and R01AI108400.
Footnotes
Published ahead of print 29 January 2014
REFERENCES
- 1.Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535. 10.1126/science.1155164 [DOI] [PubMed] [Google Scholar]
- 2.Mercer J, Knebel S, Schmidt FI, Crouse J, Burkard C, Helenius A. 2010. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc. Natl. Acad. Sci. U. S. A. 107:9346–9351. 10.1073/pnas.1004618107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt FI, Bleck CK, Helenius A, Mercer J. 2011. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J. 30:3647–3661. 10.1038/emboj.2011.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laliberte JP, Moss B. 2009. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc. Natl. Acad. Sci. U. S. A. 106:17517–17521. 10.1073/pnas.0909376106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Callahan MK, Popernack PM, Tsutsui S, Truong L, Schlegel RA, Henderson AJ. 2003. Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J. Immunol. 170:4840–4845 [DOI] [PubMed] [Google Scholar]
- 6.Soares MM, King SW, Thorpe PE. 2008. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat. Med. 14:1357–1362. 10.1038/nm.1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morizono K, Xie Y, Olafsen T, Lee B, Dasgupta A, Wu AM, Chen IS. 2011. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 9:286–298. 10.1016/j.chom.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morizono K, Chen IS. 2005. Targeted gene delivery by intravenous injection of retroviral vectors. Cell Cycle 4:854–856. 10.4161/cc.4.7.1789 [DOI] [PubMed] [Google Scholar]
- 9.Morizono K, Chen IS. 2011. Receptors and tropisms of envelope viruses. Curr. Opin. Virol. 1:13–18. 10.1016/j.coviro.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E, Lemke G, Schwartz O, Amara A. 2012. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 12:544–557. 10.1016/j.chom.2012.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharyya S, Zagorska A, Lew ED, Shrestha B, Rothlin CV, Naughton J, Diamond MS, Lemke G, Young JA. 2013. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 14:136–147. 10.1016/j.chom.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brindley MA, Hunt CL, Kondratowicz AS, Bowman J, Sinn PL, McCray PB, Jr, Quinn K, Weller ML, Chiorini JA, Maury W. 2011. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 415:83–94. 10.1016/j.virol.2011.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunt CL, Kolokoltsov AA, Davey RA, Maury W. 2011. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 85:334–347. 10.1128/JVI.01278-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, Mattsson K, Fisher J, Gies DR, Jones PF, Masiakowski P, Ryan TE, Tobkes NJ, Chen DH, DiStefano PS, Long GL, Basilico C, Goldfarb MP, Lemke G, Glass DJ, Yancopoulos GD. 1995. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 80:661–670. 10.1016/0092-8674(95)90520-0 [DOI] [PubMed] [Google Scholar]
- 15.Ishimoto Y, Ohashi K, Mizuno K, Nakano T. 2000. Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J. Biochem. 127:411–417. 10.1093/oxfordjournals.jbchem.a022622 [DOI] [PubMed] [Google Scholar]
- 16.Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. 2013. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341:403–406. 10.1126/science.1236758 [DOI] [PubMed] [Google Scholar]
- 17.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. 1992. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148:2207–2216 [PubMed] [Google Scholar]
- 18.Nagata S, Hanayama R, Kawane K. 2010. Autoimmunity and the clearance of dead cells. Cell 140:619–630. 10.1016/j.cell.2010.02.014 [DOI] [PubMed] [Google Scholar]
- 19.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. 2002. Identification of a factor that links apoptotic cells to phagocytes. Nature 417:182–187. 10.1038/417182a [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. 2007. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 27:927–940. 10.1016/j.immuni.2007.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. 2007. Identification of Tim4 as a phosphatidylserine receptor. Nature 450:435–439. 10.1038/nature06307 [DOI] [PubMed] [Google Scholar]
- 22.DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, Karisola P, Pichavant M, Kaplan GG, Umetsu DT, Freeman GJ, Casasnovas JM. 2010. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol. 184:1918–1930. 10.4049/jimmunol.0903059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakahashi-Oda C, Tahara-Hanaoka S, Honda S, Shibuya K, Shibuya A. 2012. Identification of phosphatidylserine as a ligand for the CD300a immunoreceptor. Biochem. Biophys. Res. Commun. 417:646–650. 10.1016/j.bbrc.2011.12.025 [DOI] [PubMed] [Google Scholar]
- 24.Simhadri VR, Andersen JF, Calvo E, Choi SC, Coligan JE, Borrego F. 2012. Human CD300a binds to phosphatidylethanolamine and phosphatidylserine, and modulates the phagocytosis of dead cells. Blood 119:2799–2809. 10.1182/blood-2011-08-372425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. 2007. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450:430–434. 10.1038/nature06329 [DOI] [PubMed] [Google Scholar]
- 26.Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, Kwon TH, Park RW, Kim IS. 2008. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 15:192–201. 10.1038/sj.cdd.4402242 [DOI] [PubMed] [Google Scholar]
- 27.Park SY, Jung MY, Lee SJ, Kang KB, Gratchev A, Riabov V, Kzhyshkowska J, Kim IS. 2009. Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J. Cell Sci. 122:3365–3373. 10.1242/jcs.049569 [DOI] [PubMed] [Google Scholar]
- 28.He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, Yamada M, Yamaya M, Maekawa T, Yamamoto Y, Yamamoto H. 2011. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 12:358–364. 10.1038/embor.2011.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friggeri A, Banerjee S, Biswas S, de Freitas A, Liu G, Bierhaus A, Abraham E. 2011. Participation of the receptor for advanced glycation end products in efferocytosis. J. Immunol. 186:6191–6198. 10.4049/jimmunol.1004134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moller-Tank S, Kondratowicz AS, Davey RA, Rennert PD, Maury W. 2013. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J. Virol. 87:8327–8341. 10.1128/JVI.01025-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jemielity S, Wang JJ, Chan YK, Ahmed AA, Li W, Monahan S, Bu X, Farzan M, Freeman GJ, Umetsu DT, Dekruyff RH, Choe H. 2013. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 9:e1003232. 10.1371/journal.ppat.1003232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris EN, Kyosseva SV, Weigel JA, Weigel PH. 2007. Expression, processing, and glycosaminoglycan binding activity of the recombinant human 315-kDa hyaluronic acid receptor for endocytosis (HARE). J. Biol. Chem. 282:2785–2797. 10.1074/jbc.M607787200 [DOI] [PubMed] [Google Scholar]
- 33.de Souza AJ, Oriss TB, O'Malley KJ, Ray A, Kane LP. 2005. T cell Ig and mucin 1 (TIM-1) is expressed on in vivo-activated T cells and provides a costimulatory signal for T cell activation. Proc. Natl. Acad. Sci. U. S. A. 102:17113–17118. 10.1073/pnas.0508643102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. 2006. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem. Biophys. Res. Commun. 351:571–576. 10.1016/j.bbrc.2006.10.079 [DOI] [PubMed] [Google Scholar]
- 35.Martinez-Barriocanal A, Comas-Casellas E, Schwartz S, Jr, Martin M, Sayos J. 2010. CD300 heterocomplexes, a new and family-restricted mechanism for myeloid cell signaling regulation. J. Biol. Chem. 285:41781–41794. 10.1074/jbc.M110.140889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koh JT, Lee ZH, Ahn KY, Kim JK, Bae CS, Kim HH, Kee HJ, Kim KK. 2001. Characterization of mouse brain-specific angiogenesis inhibitor 1 (BAI1) and phytanoyl-CoA alpha-hydroxylase-associated protein 1, a novel BAI1-binding protein. Brain Res. Mol. Brain Res. 87:223–237. 10.1016/S0169-328X(01)00004-3 [DOI] [PubMed] [Google Scholar]
- 37.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J. 2010. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142:387–397. 10.1016/j.cell.2010.06.036 [DOI] [PubMed] [Google Scholar]
- 38.Morizono K, Bristol G, Xie YM, Kung SK, Chen IS. 2001. Antibody-directed targeting of retroviral vectors via cell surface antigens. J. Virol. 75:8016–8020. 10.1128/JVI.75.17.8016-8020.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morizono K, Ku A, Xie Y, Harui A, Kung SK, Roth MD, Lee B, Chen IS. 2010. Redirecting lentiviral vectors pseudotyped with Sindbis virus-derived envelope proteins to DC-SIGN by modification of N-linked glycans of envelope proteins. J. Virol. 84:6923–6934. 10.1128/JVI.00435-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hermida-Matsumoto L, Resh MD. 2000. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J. Virol. 74:8670–8679. 10.1128/JVI.74.18.8670-8679.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou B, Weigel JA, Fauss L, Weigel PH. 2000. Identification of the hyaluronan receptor for endocytosis (HARE). J. Biol. Chem. 275:37733–37741. 10.1074/jbc.M003030200 [DOI] [PubMed] [Google Scholar]
- 42.Wang KS, Kuhn RJ, Strauss EG, Ou S, Strauss JH. 1992. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J. Virol. 66:4992–5001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Byrnes AP, Griffin DE. 1998. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 72:7349–7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klimstra WB, Heidner HW, Johnston RE. 1999. The furin protease cleavage recognition sequence of Sindbis virus PE2 can mediate virion attachment to cell surface heparan sulfate. J. Virol. 73:6299–6306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myles KM, Pierro DJ, Olson KE. 2003. Deletions in the putative cell receptor-binding domain of Sindbis virus strain MRE16 E2 glycoprotein reduce midgut infectivity in Aedes aegypti. J. Virol. 77:8872–8881. 10.1128/JVI.77.16.8872-8881.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanz MA, Rejas MT, Carrasco L. 2003. Individual expression of Sindbis virus glycoproteins. E1 alone promotes cell fusion. Virology 305:463–472. 10.1006/viro.2002.1771 [DOI] [PubMed] [Google Scholar]
- 47.Morizono K, Pariente N, Xie Y, Chen IS. 2009. Redirecting lentiviral vectors by insertion of integrin-targeting peptides into envelope proteins. J. Gene Med. 11:549–558. 10.1002/jgm.1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morizono K, Ringpis GE, Pariente N, Xie Y, Chen IS. 2006. Transient low pH treatment enhances infection of lentiviral vector pseudotypes with a targeting Sindbis envelope. Virology 355:71–81. 10.1016/j.virol.2006.07.015 [DOI] [PubMed] [Google Scholar]
- 49.Morizono K, Xie Y, Helguera G, Daniels TR, Lane TF, Penichet ML, Chen IS. 2009. A versatile targeting system with lentiviral vectors bearing the biotin-adaptor peptide. J. Gene Med. 11:655–663. 10.1002/jgm.1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morizono K, Xie Y, Ringpis GE, Johnson M, Nassanian H, Lee B, Wu L, Chen IS. 2005. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat. Med. 11:346–352. 10.1038/nm1192 [DOI] [PubMed] [Google Scholar]
- 51.Rand ML, Wang H, Pluthero FG, Stafford AR, Ni R, Vaezzadeh N, Allison AC, Kahr WH, Weitz JI, Gross PL. 2012. Diannexin, an annexin A5 homodimer, binds phosphatidylserine with high affinity and is a potent inhibitor of platelet-mediated events during thrombus formation. J. Thromb. Haemost. 10:1109–1119. 10.1111/j.1538-7836.2012.04716.x [DOI] [PubMed] [Google Scholar]
- 52.Emoto K, Toyama-Sorimachi N, Karasuyama H, Inoue K, Umeda M. 1997. Exposure of phosphatidylethanolamine on the surface of apoptotic cells. Exp. Cell Res. 232:430–434. 10.1006/excr.1997.3521 [DOI] [PubMed] [Google Scholar]
- 53.Choung SY, Kobayashi T, Inoue J, Takemoto K, Ishitsuka H, Inoue K. 1988. Hemolytic activity of a cyclic peptide Ro09-0198 isolated from Streptoverticillium. Biochim. Biophys. Acta 940:171–179 [DOI] [PubMed] [Google Scholar]
- 54.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. 2010. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 235:172–189. 10.1111/j.0105-2896.2010.00903.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeBell KE, Simhadri VR, Mariano JL, Borrego F. 2012. Functional requirements for inhibitory signal transmission by the immunomodulatory receptor CD300a. BMC Immunol. 13:23. 10.1186/1471-2172-13-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakahashi-Oda C, Tahara-Hanaoka S, Shoji M, Okoshi Y, Nakano-Yokomizo T, Ohkohchi N, Yasui T, Kikutani H, Honda S, Shibuya K, Nagata S, Shibuya A. 2012. Apoptotic cells suppress mast cell inflammatory responses via the CD300a immunoreceptor. J. Exp. Med. 209:1493–1503. 10.1084/jem.20120096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar M, Keller B, Makalou N, Sutton RE. 2001. Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene Ther. 12:1893–1905. 10.1089/104303401753153947 [DOI] [PubMed] [Google Scholar]
- 58.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15:871–875. 10.1038/nbt0997-871 [DOI] [PubMed] [Google Scholar]
- 59.Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, Trono D. 1998. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 72:9873–9880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S. 2005. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 437:754–758. 10.1038/nature03964 [DOI] [PubMed] [Google Scholar]
- 61.Asano K, Miwa M, Miwa K, Hanayama R, Nagase H, Nagata S, Tanaka M. 2004. Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J. Exp. Med. 200:459–467. 10.1084/jem.20040342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Toda S, Hanayama R, Nagata S. 2012. Two-step engulfment of apoptotic cells. Mol. Cell. Biol. 32:118–125. 10.1128/MCB.05993-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thumshirn G, Hersel U, Goodman SL, Kessler H. 2003. Multimeric cyclic RGD peptides as potential tools for tumor targeting: solid-phase peptide synthesis and chemoselective oxime ligation. Chemistry 9:2717–2725. 10.1002/chem.200204304 [DOI] [PubMed] [Google Scholar]
- 64.Cao Q, Li ZB, Chen K, Wu Z, He L, Neamati N, Chen X. 2008. Evaluation of biodistribution and anti-tumor effect of a dimeric RGD peptide-paclitaxel conjugate in mice with breast cancer. Eur. J. Nucl. Med. Mol. Imaging 35:1489–1498. 10.1007/s00259-008-0744-y [DOI] [PubMed] [Google Scholar]
- 65.Mousa SA. 2005. Alpha v integrin affinity/specificity and antiangiogenesis effect of a novel tetraaza cyclic peptide derivative, SU015, in various species. J. Cardiovasc. Pharmacol. 45:462–467. 10.1097/01.fjc.0000159044.27618.be [DOI] [PubMed] [Google Scholar]
- 66.Wang X, Huang DY, Huong SM, Huang ES. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat. Med. 11:515–521. 10.1038/nm1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruoslahti E, Pierschbacher MD. 1987. New perspectives in cell adhesion: RGD and integrins. Science 238:491–497. 10.1126/science.2821619 [DOI] [PubMed] [Google Scholar]
- 68.Stewart PL, Nemerow GR. 2007. Cell integrins: commonly used receptors for diverse viral pathogens. Trends Microbiol. 15:500–507. 10.1016/j.tim.2007.10.001 [DOI] [PubMed] [Google Scholar]
- 69.Larocca D, Peterson JA, Urrea R, Kuniyoshi J, Bistrain AM, Ceriani RL. 1991. A Mr 46,000 human milk fat globule protein that is highly expressed in human breast tumors contains factor VIII-like domains. Cancer Res. 51:4994–4998 [PubMed] [Google Scholar]
- 70.Hanayama R, Nagata S. 2005. Impaired involution of mammary glands in the absence of milk fat globule EGF factor 8. Proc. Natl. Acad. Sci. U. S. A. 102:16886–16891. 10.1073/pnas.0508599102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shimojima M, Takada A, Ebihara H, Neumann G, Fujioka K, Irimura T, Jones S, Feldmann H, Kawaoka Y. 2006. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 80:10109–10116. 10.1128/JVI.01157-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kondratowicz AS, Lennemann NJ, Sinn PL, Davey RA, Hunt CL, Moller-Tank S, Meyerholz DK, Rennert P, Mullins RF, Brindley M, Sandersfeld LM, Quinn K, Weller M, McCray PB, Jr, Chiorini J, Maury W. 2011. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. U. S. A. 108:8426–8431. 10.1073/pnas.1019030108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER. 1998. Beta3 integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. U. S. A. 95:7074–7079. 10.1073/pnas.95.12.7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Akakura S, Singh S, Spataro M, Akakura R, Kim JI, Albert ML, Birge RB. 2004. The opsonin MFG-E8 is a ligand for the alphavbeta5 integrin and triggers DOCK180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Exp. Cell Res. 292:403–416. 10.1016/j.yexcr.2003.09.011 [DOI] [PubMed] [Google Scholar]
- 75.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343. 10.1038/nature10348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cote M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. 2011. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477:344–348. 10.1038/nature10380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bron R, Wahlberg JM, Garoff H, Wilschut J. 1993. Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J. 12:693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gibbons DL, Ahn A, Chatterjee PK, Kielian M. 2000. Formation and characterization of the trimeric form of the fusion protein of Semliki Forest virus. J. Virol. 74:7772–7780. 10.1128/JVI.74.17.7772-7780.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thongthai W, Weninger K. 2009. Photoinactivation of Sindbis virus infectivity without inhibition of membrane fusion. Photochem. Photobiol. 85:801–806. 10.1111/j.1751-1097.2008.00475.x [DOI] [PubMed] [Google Scholar]
- 80.Corver J, Bron R, Snippe H, Kraaijeveld C, Wilschut J. 1997. Membrane fusion activity of Semliki Forest virus in a liposomal model system: specific inhibition by Zn2+ ions. Virology 238:14–21. 10.1006/viro.1997.8799 [DOI] [PubMed] [Google Scholar]
- 81.Kenis H, van Genderen H, Deckers NM, Lux PA, Hofstra L, Narula J, Reutelingsperger CP. 2006. Annexin A5 inhibits engulfment through internalization of PS-expressing cell membrane patches. Exp. Cell Res. 312:719–726. 10.1016/j.yexcr.2005.11.023 [DOI] [PubMed] [Google Scholar]
- 82.Aziz M, Jacob A, Matsuda A, Wang P. 2011. Review: milk fat globule-EGF factor 8 expression, function and plausible signal transduction in resolving inflammation. Apoptosis 16:1077–1086. 10.1007/s10495-011-0630-0 [DOI] [PubMed] [Google Scholar]
- 83.Rodriguez-Manzanet R, DeKruyff R, Kuchroo VK, Umetsu DT. 2009. The costimulatory role of TIM molecules. Immunol. Rev. 229:259–270. 10.1111/j.1600-065X.2009.00772.x [DOI] [PMC free article] [PubMed] [Google Scholar]