

ABSTRACT
Lymphocyte colonization by gammaherpesviruses (γHVs) is an important target for cancer prevention. However, how it works is not clear. Epstein-Barr virus drives autonomous B cell proliferation in vitro but in vivo may more subtly exploit the proliferative pathways provided by lymphoid germinal centers (GCs). Murid herpesvirus 4 (MuHV-4), which realistically infects inbred mice, provides a useful tool with which to understand further how a γHV colonizes B cells in vivo. Not all γHVs necessarily behave the same, but common events can with MuHV-4 be assigned an importance for host colonization and so a potential as therapeutic targets. MuHV-4-driven B cell proliferation depends quantitatively on CD4+ T cell help. Here we show that it also depends on T cell-independent survival signals provided by the B cell-activating factor (BAFF) receptor (BAFF-R). B cells could be infected in BAFF-R−/− mice, but virus loads remained low. This corresponded to a BAFF-R-dependent defect in GC colonization. The close parallels between normal, antigen-driven B cell responses and virus-infected B cell proliferation argue that in vivo, γHVs mostly induce infected B cells into normal GC reactions rather than generating large numbers of autonomously proliferating blasts.
IMPORTANCE γHVs cause cancers by driving the proliferation of infected cells. B cells are a particular target. Thus, we need to know how virus-driven B cell proliferation works. Controversy exists as to whether viral genes drive it directly or less directly orchestrate the engagement of normal, host-driven pathways. Here we show that the B cell proliferation driven by a murid γHV requires BAFF-R. This supports the idea that γHVs exploit host proliferation pathways and suggests that interfering with BAFF-R could more generally reduce γHV-associated B cell proliferation.
INTRODUCTION
Gammaherpesviruses (γHVs) persist in lymphocytes and cause lymphocytic cancers. Therefore, it is important to understand how their lymphocyte infections work. The standard model for many years has been in vitro B cell transformation by Epstein-Barr virus (EBV) (1), which is independent of normal lymphoid tissue organization. However, in vivo EBV persists not in proliferating blasts but in resting memory B cells (2, 3) that show evidence of passage through germinal centers (GCs) (4). Thus, there is discrepancy between in vitro and in vivo virus behaviors. In vitro infections place viral genes outside the context in which they have evolved to function, but at the same time, human analyses are limited in their sampling and capacity to establish cause and effect. Therefore, resolving the discrepancy is not straightforward.
Related γHVs provide another source of information. Those that infect experimentally tractable mammals are particularly useful for establishing cause and effect in a realistic context. Murid herpesvirus 4 (MuHV-4) is a well-characterized example. Despite immortalizing only fetal B cells in vitro (5), it colonizes adult lymphoid GCs in vivo (6) to establish a persistent infection of memory B cells (7–9). The Kaposi's sarcoma-associated herpesvirus (KSHV) also colonizes B cells in vivo (10) and fails to transform them in vitro. Thus, EBV, KSHV, and MuHV-4 differ in vitro but remain strikingly similar in host colonization. MuHV-4 therefore provides an opportunity to understand functionally in inbred laboratory mice how many γHVs may interact with B cells in vivo (11–13). There is no guarantee that every γHV acts in the same way, but with MuHV-4 we can establish a relatively complete functional framework onto which the more fragmented information about human infections can be mapped.
MuHV-4 drives B cell activation and proliferation greatly in excess of antigen-specific responses (14, 15). However, both depend on CD4+ T cells (16), CD40 ligand (17), and CD40 (18), implying a similar need for T cell-derived survival signals. Antigen-specific responses also require T cell-independent survival signals, of which those delivered by B cell-activating factor (BAFF) through its main receptor (BAFF-R) have central importance (19, 20). The BAFF-R-deficient phenotype was defined first in AsWyn/J mice (21), in which C-terminal receptor disruption creates a dominant negative mutant (22): transitional B cells developing in the bone marrow fail to survive or undergo T1 to T2 maturation. BAFF-R is also required for follicular B cell survival. Thus, competition for limiting amounts of BAFF regulates circulating B cell numbers. B1 B cells are preserved without BAFF-R, but B2 numbers are severely reduced and marginal-zone B cells are essentially absent (23). IgM responses are still made, but GCs form only transiently and IgG responses are weak (24, 25). Targeted BAFF-R (26) and BAFF knockouts show similar phenotypes (20).
BAFF-R signaling works in part through the induction of antiapoptotic bcl-2 family members (27). γHVs encode bcl-2 homologs and inhibit mitochondrial apoptosis pathways (28), so infected B cells might be expected to show independence of BAFF-R-mediated homeostatic control; conversely, extensive reliance on normal B cell physiology (29) would keep virus-driven lymphoproliferation BAFF-R dependent. Therefore, to understand better how γHV host colonization works, we determined the extent to which it depends on BAFF-R.
MATERIALS AND METHODS
Mice.
C57BL/6J (Harlan U.K.) and BAFF-R−/− mice (26) (kindly provided by Andrew Sage and Lauren Baker, Division of Cardiovascular Medicine, Cambridge University Medical School) were maintained at the Cambridge University Department of Pathology animal unit and infected with MuHV-4 when 6 to 12 weeks old, either intranasally (i.n.) in 30 μl of Dulbecco's modified Eagle's medium (DMEM) under isoflurane anesthesia (104 PFU) or intraperitoneally (i.p.) in 100 μ1 of DMEM (105 PFU). All animal experiments were approved by the Cambridge University Ethical Review Board and by the 1986 Animal Scientific Procedures Act (project license 80/2538).
Cells and viruses.
BHK-21 cells (American Type Culture Collection CCL-10) and 3T3-ORF50 cells (30) were grown in Dulbecco's modified Eagle's medium, 2 mM glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% fetal calf serum (PAA Laboratories). Wild-type (WT) and EF1α-eGFP MuHV-4 (31) were grown on BHK-21 cells, and their titers were determined. ORF50-deficient MuHV-4 was grown on and its titer determined on 3T3-ORF50 cells (30). Virions were harvested from infected cell supernatants by ultracentrifugation (35,000 × g; 90 min), and cell debris was removed by low-speed centrifugation (500 × g; 10 min).
Infectivity assays.
Infectious virus was measured by plaque assay. Virus stocks or organ homogenates were incubated with BHK-21 cells (2 h; 37°C) and then overlaid with 0.3% carboxymethylcellulose. Four days later, the cells were fixed (4% formaldehyde) and stained (0.1% toluidine blue) for plaque counting. Latent plus infectious virus was measured by infectious center assay: single-cell suspensions of explanted spleens, lymph nodes (LNs), and peritoneal washes were obtained postmortem and cocultured with BHK-21 cell monolayers that were then fixed and stained after 4 days. Typically, <5% of the total virus recoverable from lymphoid tissue or peritoneal washes is detected by plaque assay, so the infectious center assay measures mainly latency. Statistical comparisons were by Student's 2-tailed unpaired t test unless stated otherwise.
Viral genome quantitation.
MuHV-4 genomic coordinates 4166 to 4252 were amplified by PCR from 50 to 80 ng DNA of organ homogenates (Rotor-Gene 3000; Corbett Research). PCR products were quantitated by hybridization with a TaqMan probe (genomic coordinates 4218 to 4189) and converted to genome copies by comparison with a standard curve of cloned plasmid template amplified in parallel. Cellular DNA was quantitated in the same reaction by amplifying part of the adenosine phosphoribosyl transferase (APRT) gene, again with TaqMan probe hybridization and template dilutions amplified in parallel. Viral DNA loads were then normalized by the cellular genome copy number of each sample (32).
Immunohistochemistry and in situ hybridization.
Spleens were fixed in phosphate-buffered saline (PBS)–4% formaldehyde (24 h; 4°C), dehydrated in 70% ethanol, and embedded in paraffin. Seven-micrometer sections were dewaxed in xylene and hydrated in graded ethanol solutions. Endogenous peroxidase activity was quenched in PBS–3% H2O2 (10 min; 23°C). Sections were then blocked with an avidin/biotin blocking kit (Vector Laboratories) and PBS–2% bovine serum albumin (BSA)–2% rabbit serum (1 h; 23°C). B cells were detected with anti-B220 (RA3-6B2; Abcam), biotinylated goat anti-rabbit IgG polyclonal antibody (pAb) (Vector Laboratories), and the Vectastain Elite ABC peroxidase system. All antibody incubations were for 1 h at room temperature, and the sections were washed 3 times in PBS after each incubation. Detection was with ImmPACT DAB substrate (5 min; 23°C; Vector). Viral tRNA/microRNAs (miRNAs) 1 to 4 were detected by in situ hybridization (6). After dewaxing and rehydration, fixed sections were treated with proteinase K (100 μg/ml; 10 min; 37°C), acetylated with 25% acetic anhydride in 0.1 M triethanolamine, and then hybridized in 50% formamide–10 mM Tris, pH 7.5 (58°C; 18 h), with a digoxigenin-labeled riboprobe complementary to the tRNA/miRNA transcripts generated by T7 transcription of pEH1.4. The hybridized probe was detected with alkaline phosphatase-conjugated antidigoxigenin Fab fragments (Boehringer Ingelheim) and BCIP–NBT (5-bromo-4-chloro-3-indolylphosphate–Nitro Blue Tetrazolium) substrate. Sections were then counterstained with Mayer's hemalum, dehydrated in ethanol, and mounted in DPX (BDH).
Immunofluorescence.
Organs were fixed in 1% formaldehyde–10 mM sodium periodate–75 mM l-lysine (24 h; 4°C), equilibrated in 30% sucrose (18 h; 4°C), and then frozen in optimal cutting temperature (OCT) matrix compound. Nine-micrometer sections were cut and then air dried (1 h; 23°C), blocked with 0.3% Triton X-100–5% normal goat serum (1 h; 23°C), and incubated (18 h; 4°C) with antibodies to enhanced green fluorescent protein (eGFP) (rabbit pAb; Abcam), macrophage receptor with collagenous structure (MARCO) (ED31; Serotec), IgM (biotin-conjugated goat pAb; Southern Biotech), IgD (11-26c; Southern Biotech), B220 (RA3-6B2; Abcam), and CD169 (3D6.112; Serotec).
GC B cell staining was with fluorescein-conjugated peanut agglutinin (PNA) (BD Biosciences). Sections were washed 3 times in PBS, incubated (1 h; 23°C) with Alexa 633-conjugated goat anti-rat IgG pAb, streptavidin-conjugated Alexa 568, and Alexa 488- or 568-conjugated goat anti-rabbit IgG pAb (Invitrogen), washed 3 times in PBS, and mounted in Prolong Gold plus DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen). Fluorescence was visualized with a Leica TCS SP5 confocal microscope and analyzed with ImageJ.
RESULTS
Intranasal infection.
We infected BAFF-R−/− or wild-type (WT) mice intranasally (i.n.) with MuHV-4 and determined virus titers at day 7, the peak of lytic infection, and at day 13, the peak of latent infection (Fig. 1a). The day 7 lytic virus titers in BAFF-R−/− mouse lungs were significantly higher than those in lungs of WT mice, possibly due to the immunodeficiency of BAFF-R−/− mice. By contrast, latent viral titers in BAFF-R−/− lymph nodes (LNs) and spleens were reduced 10- to 100-fold. BAFF-R−/− lymphoid infections were not completely ablated, and spleen titers increased from day 7 to day 13, but the absolute amounts of infection remained low. Viral DNA loads were also significantly lower in BAFF-R−/− lymphoid tissue than in that of WT mice (Fig. 1b), implying that the lower infectious center assay titers reflected fewer infected cells rather than impaired virus reactivation. After 30 days (Fig. 1c), BAFF-R−/− spleen colonization remained significantly below that of WT mice. Thus, there was a long-term defect in lymphoid infection after i.n. infection rather than just a delay.
FIG 1.

Impaired colonization of BAFF-R−/− mice by i.n. MuHV-4. (a) C57BL/6J (WT) or BAFF-R−/− mice were infected i.n. with MuHV-4 (104 PFU). Seven days later, titers of infectious virus in lungs were detemined by plaque assay. Circles show individual mice; crosses show means. BAFF-R−/− lung titers were significantly higher than those of WT mice (P < 0.01). (b) Mice were infected as for panel a, and virus loads in mediastinal lymph nodes and spleens were determined 7 and 13 days later by infectious center assay. Circles show individual mice; crosses show means. The dashed lines indicate the lower limit of assay sensitivity. At day 13, WT titers were significantly higher than those of BAFF-R−/− mice in both lymph nodes (P < 0.001) and spleens (P < 0.0001). (c) Mice were infected as for panel a, and virus loads in spleens were determined 30 days later by infectious center assay. Circles show individual mice; crosses show means. The dashed line indicates the lower limit of assay sensitivity. WT titers remained significantly higher than those of BAFF-R−/− mice (P < 0.05). (d) The organs of mice for panel b were assayed for viral DNA by Q-PCR. Viral genome loads (M2) are normalized by the cellular genome load of each sample (APRT). At day 13, viral DNA loads were significantly higher in WT spleens and lymph nodes than in those of BAFF-R−/− mice (P < 0.003).
Intraperitoneal infection.
MuHV-4 infection causes marked splenomegaly, and this is where lymphoid infection has been studied in most detail. However, splenic infection by i.n. virus depends on it first reaching LNs (31). Therefore, to determine how much of the BAFF-R−/− spleen infection defect shown in Fig. 1 was specific to this site rather than a consequence of impaired LN infection, we tested splenic colonization more directly by intraperitoneal (i.p.) virus inoculation (Fig. 2). Early on (day 3), BAFF-R−/− mice showed higher virus titers than WT, again possibly due to their immunodeficiency. However BAFF-R−/− titers declined with time, whereas WT titers increased, and by day 8 WT titers were significantly higher. Therefore, despite substantial early virus seeding, spleen infection was poorly amplified in the absence of BAFF-R.
FIG 2.
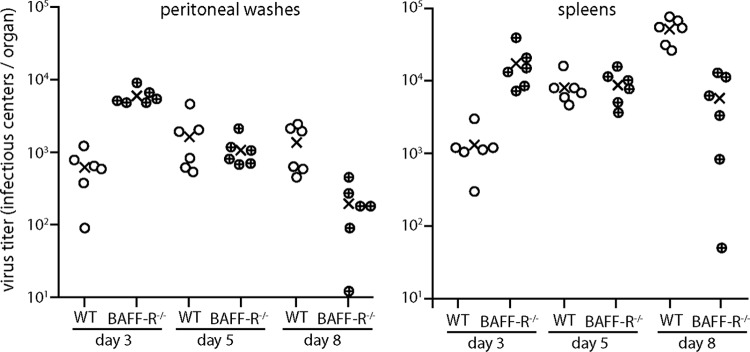
Impaired colonization of BAFF-R−/− mice by i.p. MuHV-4. C57BL/6J (WT) or BAFF-R−/− mice were infected i.p. with MuHV-4 (105 PFU). Virus in peritoneal washes and spleens was then titered by infectious center assay. Circles show individual mice; crosses show means. At day 3, BAFF-R−/− titers were significantly higher in both peritoneal washes (P < 0.0001) and spleens (P < 0.01). At day 8, WT titers were signifiantly higher in both spleens (P < 0.001) and peritoneal washes (P < 0.02).
Infectious center assays over the next 3 weeks (Fig. 3a) showed that BAFF-R−/− spleen colonization remained significantly below that of WT mice. Thus, again there was a fundamental, BAFF-R-dependent infection defect that did not correct with time. The same applied to splenic virus genome loads, measured by quantitative PCR (Q-PCR) (Fig. 3b). BAFF-R−/− mice showed a greater reduction in infectious centers than in viral genome loads. No difference in plaque size was observed, as might be expected for a reactivation defect. Rather, the antibody deficiency of BAFF-R−/− mice may have slowed their clearance of infected cell debris more than that of infectivity, which is achieved mainly by T cells.
FIG 3.
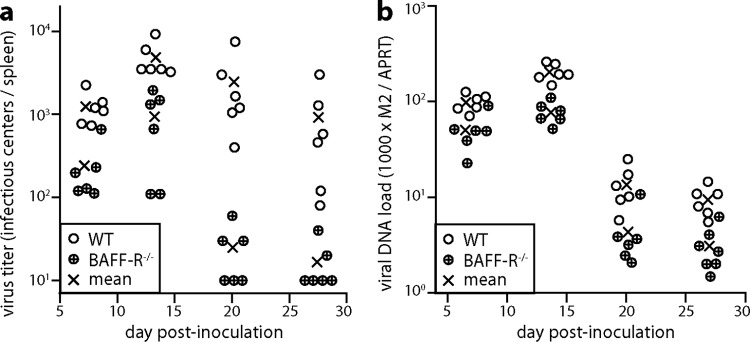
MuHV-4 colonization of i.p.-inoculated BAFF-R−/− mice does not recover with time. (a) We infected C57BL/6J (WT) or BAFF-R−/− mice i.p. with MuHV-4 (105 PFU) and determined the titers of recoverable virus in peritoneal washes and spleens by infectious center assay 7, 13, 20, and 27 days later. Circles show individual mice; crosses show means. BAFF-R−/− titers were significantly lower at all time points (P < 0.05). (b) The same samples as for panel a were assayed for viral genome load (M2) relative to cellular genome load (APRT) by Q-PCR. Normalized viral genome loads were significantly lower in BAFF-R−/− mice at all time points (P < 0.03).
Defective formation of infected B cell follicles in BAFF-R−/− mice.
Staining BAFF-R−/− spleens for the B cell marker B220 revealed reduced numbers and sizes of B cell follicles compared to those in WT spleens (Fig. 4a), consistent with BAFF-R being required for GC maintenance as well as mature B cell survival (20). This phenotype was maintained after MuHV-4 infection. Therefore, neither viral gene expression nor the immune stimulation associated with infection compensated for the BAFF-R-dependent GC defect.
FIG 4.

Defective GC formation in MuHV-4-infected BAFF-R−/− mice. (a) Spleen sections from C57BL/6J (WT) and BAFF-R−/− mice were stained for B220 (brown) either before (naive) or 20 days after i.p. MuHV-4 (105 PFU) and counterstained with Mayer's hemalum. The scale bar shows 500 μm. The number of B220 follicles per section was significantly lower in BAFF-R−/− (mean ± standard deviation [SD], 4.7 ± 1.4; n = 6) than in WT (14.2 ± 1.5) spleens, both before and after infection (P < 10−6). (b) We infected C57BL/6J (WT) and BAFF-R−/− mice i.p. with MuHV-4 (105 PFU) and then detected viral tRNA/miRNA transcripts by in situ hybridization (dark staining). Examples of positive follicles are shown. B220 staining of the same spleens (brown) relates the extent of tRNA/miRNA staining to the extent of the white pulp occupied by B cells. Scale bars show 100 μm. (c) tRNA/miRNA+ cells were counted for spleen sections from 3 mice per group, and follicles were scored as uninfected or infected at a low (1 to 5 cells per section), medium (5 to 10 cells per section), or high (>10 cells per section) level. Bars show means ± SD, counting 20 to 30 follicles per mouse. At both day 6 and day 20, WT spleens contained significantly fewer tRNA/miRNA+ follicles than BAFF-R−/− spleens (P < 0.0001 by 2-tailed Fisher's exact test), and among the positive follicles, the number of tRNA/miRNA+ cells was significantly higher in WT spleens (P < 0.01 by Student's unpaired 2-tailed t test).
In situ hybridization for viral tRNA expression showed perifollicular colonization of WT spleens at day 6 and more extensive follicular colonization at day 20 (Fig. 4b). BAFF-R−/− spleens also showed perifollicular viral tRNA expression at day 6. However, there was much less subsequent colonization of follicles, commensurate with the reduced B220 staining. Quantitation across multiple sections (Fig. 4c) established that BAFF-R−/− spleens contained both a significantly higher number of negative follicles and, among the positive follicles, a significantly lower mean number of tRNA+ cells. This result confirmed that MuHV-4 could not effectively colonize or amplify the transient GCs of BAFF-R−/− mice.
Infection tracked by viral eGFP expression.
We then tracked spleen infection by lytic cycle-independent eGFP expression from a viral EF1α promoter. Four days after i.p. infection of WT mice (Fig. 5), eGFP was seen mainly in CD169+ and MARCO+ marginal-zone macrophages; very little was in IgM+ or IgD+ B cells. After 8 days, relatively few macrophages were eGFP+; infection localized instead to B220+ B cell follicles, with many eGFP+ IgM+ and eGFP+ IgD+ cells. Thus, WT infection started in splenic macrophages and then spread to B cells in GCs. BAFF-R−/− spleens (Fig. 6) also showed marginal-zone macrophage infection after 4 days, and eGFP+ Ig+ cells were associated with B220+ follicles at day 8. However, the follicles were smaller and less numerous than in WT spleens, consistent with the in situ hybridization data (Fig. 4b), and the eGFP+Ig+ cells were more commonly IgM+ than IgD+ (Fig. 7). Impaired B cell maturation from IgMhi IgDlo (hi indicates high; lo indicates low) to IgMlo IgDhi is well described for BAFF and BAFF-R deficient mice (21, 24). Therefore, infected BAFF-R−/− B cells remained bound by their genetic maturation defect.
FIG 5.

Colonization of C57BL/6J splenic follicles by eGFP+ MuHV-4. We infected C57BL/6J mice i.p. with MuHV-4 expressing eGFP from an intergenic EF1α promoter (105 PFU) and 4 or 8 days later identified infection by staining splenic sections for eGFP (green) plus IgM (red; mainly marginal-zone B cells) and, as a further cell type marker, either B220 (mainly follicular B cells), MARCO (marginal-zone macrophages), IgD (mainly follicular B cells), or CD169 (marginal-zone metallophilic macrophages) (white). Nuclei were counter-stained with DAPI (blue). The boxed regions of the left-hand overviews (scale bars show 100 μm) are shown at higher power in the right-hand zoomed images, with either merged or individual channels. Arrows show examples of eGFP+ cells.
FIG 6.

Colonization of BAFF-R−/− splenic follicles by eGFP+ MuHV-4. We infected BAFF-R−/− mice i.p. with eGFP+ MuHV-4 as for Fig. 5 and 4 or 8 days later stained splenic sections for eGFP (green) plus IgM (red) and, as a further cell type marker, either B220, MARCO, IgD, or CD169 (white). Nuclei were counter-stained with DAPI (blue). The boxed regions of the left-hand overviews (scale bars shows 100 μm) are shown at higher power in the right-hand zoomed images, with either merged or individual channels. Arrows show examples of eGFP+ cells.
FIG 7.
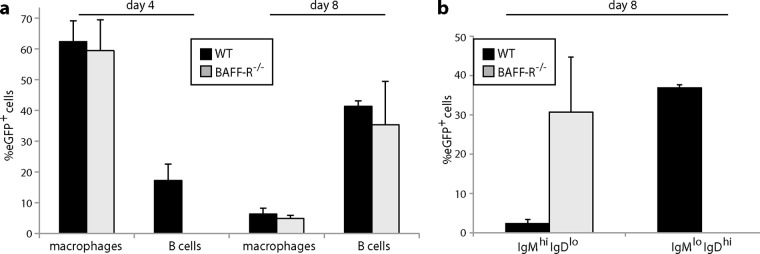
Quantitation of splenic colonization by eGFP+ MuHV-4. (a) We infected C57BL/6J (WT) or BAFF-R−/− mice i.p. with eGFP+ MuHV-4 as for Fig. 5 and 6 and after 4 or 8 days typed eGFP+ cells as macrophages (CD169+ or MARCO+) or B cells (IgM+ or IgD+). Each bar shows the mean ± SD for 3 sections from each of 3 mice. Macrophage infection was not significantly different between the mouse groups, but WT mice showed significantly more B cell infection at day 4 (P < 0.001). (b) EGFP+ B cells at day 8 were further subdivided into IgMhi IgDlo (predominantly marginal-zone) and IgMlo IgDhi (predominantly follicular) populations. The eGFP+ B cells of WT mice were significantly higher in IgMlo IgDhi and significantly lower in IgMhi IgDlo than those of BAFF-R−/− mice (P < 0.001).
Peanut agglutinin (PNA) staining at 8 days postinfection (Fig. 8) showed PNA+ eGFP+ cells in WT but not BAFF-R−/− spleens. The eGFP+ WT cells tended to be PNAint (int indicates intermediate) rather than PNAhi, and far from all were PNA+, but there was a clear difference from the eGFP+ BAFF-R−/− cells, which also did not localize to PNA+ areas of the spleen (Fig. 8a and b). These data were consistent with MuHV-4 infecting BAFF-R−/− B cells but then failing to overcome their defect in GC formation.
FIG 8.

Colonization of GC B cells by eGFP+ MuHV-4. (a) We infected C57BL/6J (WT) or BAFF-R−/− mice i.p. with eGFP+ MuHV-4 (105 PFU) and 8 days later stained splenic sections for eGFP (green) and PNA (red). Nuclei were stained with DAPI (blue). Arrows show eGFP+ PNA+ cells. The boxed areas in the left-hand overview images (scale bar = 100 μm) are shown at higher magnification, with separate and merged channels in the right-hand images. No eGFP+ PNA+ cells were seen in BAFF-R−/− spleens. (b) Mice were infected as for panel a, and eGFP+ cells were scored as inside or outside PNA+ follicles. Each bar shows the mean ± SD for 3 sections from each of 3 mice. In WT mice, a significantly higher proportion of the eGFP+ cells were inside follicles than in BAFF-R−/− mice (P < 0.0001).
IgG response to MuHV-4 infection.
We measured antigen-specific B cell responses in infected WT and BAFF-R−/− mice by enzyme-linked immunosorbent assay (ELISA) for MuHV-4-specific serum IgG (Fig. 9). BAFF-R−/− responses were 10-fold lower than those of WT controls. Thus, BAFF-R−/− mice showed quantitatively similar impairments of antigen-specific B cell responses and virus-driven lymphoproliferation, consistent with these processes having a similar requirement for BAFF-R signaling. To allow for differences in virus load possibly affecting antibody responses, we also tested the response of WT and BAFF-R−/− mice to ORF50-deficient MuHV-4 (Fig. 9c), which fails to replicate without complementation and so elicits virion antigen-specific antibodies only as input virus. Responses were lower than with WT MuHV-4, but again WT mice had a substantially greater response than BAFF-R−/− mice.
FIG 9.
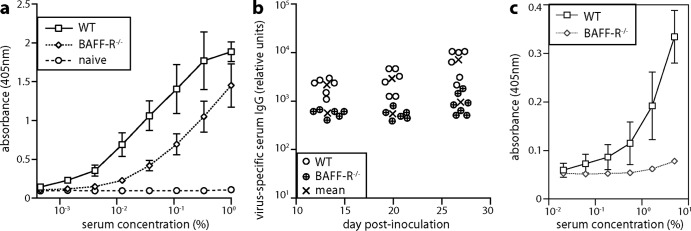
MuHV-4-specific antibody response. (a) We infected C57BL/6J (WT) or BAFF-R−/− mice i.p. with MuHV-4 (105 PFU). Sera taken 27 days later were assayed for MuHV-4-specific IgG by ELISA. Each line shows mean ± SD absorbance readings of sera from 6 mice. naive, uninfected controls (2 mice). (b) Mice were infected as for panel a and bled 13, 20, and 27 days later, and MuHV-4-specific serum IgG was measured by ELISA. Titers were normalized by comparison with a pooled standard immune serum. Circles show individual titers; crosses show means. Although IgG responses were readily detectable in BAFF-R−/− mice, WT titers were significantly higher at each time point (P < 0.01). (c) We infected C57BL/6J (WT) or BAFF-R−/− mice i.p. with ORF50-deficient MuHV-4 (106 PFU), which cannot replicate without complementation. Sera taken 14 days later were assayed for MuHV-4-specific IgG by ELISA. Each line shows mean ± SD absorbance readings of sera from 4 mice.
DISCUSSION
EBV analyses have produced two contrasting models of how γHVs behave in B cells: viral genes driving autonomous B cell proliferation (33) and infected B cells hitchhiking along host pathways of proliferation and differentiation (29). EBV certainly can transform B cells, but the in vitro cultures in which this is the predominant infection outcome may be unrealistic. MuHV-4 provides functional in vivo data to supplement in vitro and descriptive in vivo analyses of EBV. It drives B cell proliferation poorly in CD4+ T cell-deficient (16) and B cell receptor transgenic (34) mice. We showed here that BAFF-R is also important. Thus, host colonization required both T cell and non-T cell host survival signals, consistent with the hitchhiking model. In vitro and in vivo B cell infections differ in the latter being more exposed to immune attack. Therefore, host-dependent B cell proliferation may predominate in vivo because reducing viral gene expression makes lymphoproliferation immunologically less conspicuous.
Infected B cells must still be driven into GCs. They lack obvious antigen specificity (14, 15), so the signal presumably comes from viral genes. The only MuHV-4 genes with clear latency expression profiles are ORF73 and M2 (35). ORF73 encodes the viral episome maintenance protein (EBNA-1 in EBV, LANA in KSHV) (36), while M2 regulates B cell receptor signaling (like LMP-2A in EBV and K1 in KSHV) (37). The MuHV-4 latency program of ± ORF73 ± M2 is therefore analogous to the EBV latency maintenance program of ± EBNA-1 ± LMP-2A (29). This would suggest that as with antigen-driven responses, the stimulus for MuHV-4-infected B cells to enter GCs comes from without.
MuHV-4-driven B cell activation exceeding both antigen-specific responses and B cell infection rates (14, 15) suggests the possibility of superantigen-like B cell activation. This has precedents in murine retroviruses using superantigens to colonize B cells (38), in herpesvirus saimiri producing a T cell superantigen (39), and in EBV inducing a retroviral superantigen (40) and promoting VH4-34+ B cell expansion (41). Splenic myeloid infection preceding B cell infection implied abundant opportunity for myeloid-derived proteins to promote B cell colonization (42). Infected myeloid cells transcribe M1, M3, and M4, a set of homologous genes for secreted viral glycoproteins (8). M1 drives a superantigen-like Vβ4+ CD8+ T cell stimulation (43, 44), and both the M3 chemokine binding protein (45) and M4 promote B cell infection (46–48). Thus, lytically infected myeloid cells could drive latently infected B cells into GCs.
Transgenic bcl-2 expression rescues B cell numbers in BAFF-R-deficient mice (27). MuHV-4—like EBV and KSHV—expresses an antiapoptotic bcl-2 homolog (M11) (49) but failed to overcome the limits on B cell proliferation set by BAFF-R deficiency. MuHV-4 lacking M11 (M11− MuHV-4) has a latency establishment defect (50), but M11 may not be expressed at a suitable stage of the viral life cycle to substitute for BAFF-R signaling. The myeloid/B cell infection model again provides a possible explanation: M11 is transcribed in splenic macrophages and dendritic cells rather than follicular or GC B cells (8) and i.p.-inoculated, M11-deficient MuHV-4 shows impaired macrophage infection (51). Thus, the latency defect of M11− MuHV-4 could be an indirect consequence of impaired myeloid infection rather than a direct consequence of B cell apoptosis. A similar knock-on effect would explain why disrupting the major histocompatibility complex (MHC) class I evasion protein K3—another MuHV-4 lytic gene product (52) that functions in myeloid cells (53)—impairs B cell infection (54). Thus, we hypothesize that myeloid-expressed viral genes foster a suitable microenvironment for host-driven—and therefore BAFF-R-dependent—infected B cell proliferation.
The B1 B cells present in BAFF-R−/− mice could not substitute for B2 B cells in providing a normal reservoir of long-term infection. Nor could the remaining B2 B cells of BAFF-R−/− mice (approximately 10% of WT numbers) proliferate to form a normal reservoir, even though MuHV-4 normally infects only 1 to 2% of mature B cells and any infected B cells managing to traverse transient BAFF-R−/− GCs should then become BAFF-R independent (20). This was consistent with a strong viral reliance on host-driven lymphoproliferation. The implication is that BAFF-R might be a viable target for therapeutic intervention when infection-associated lymphoproliferation causes disease, aiming to reduce lymphoproliferation and the chance of oncogenic host mutations in GCs without the profound immunosuppression of memory B cell depletion.
ACKNOWLEDGMENTS
B.F. was supported by the Portuguese Foundation for Science and Technology. This work was also supported by grants from the BBSRC (BB/J014419/1), ARC (FT130100138), NHMRC (1064015 and 1060138), and Belspo (BelVir).
Footnotes
Published ahead of print 5 February 2014
REFERENCES
- 1.Brinkmann MM, Schulz TF. 2006. Regulation of intracellular signalling by the terminal membrane proteins of members of the Gammaherpesvirinae. J. Gen. Virol. 87:1047–1074. 10.1099/vir.0.81598-0 [DOI] [PubMed] [Google Scholar]
- 2.Babcock GJ, Decker LL, Freeman RB, Thorley-Lawson DA. 1999. Epstein-Barr virus-infected resting memory B cells, not proliferating lymphoblasts, accumulate in the peripheral blood of immunosuppressed patients. J. Exp. Med. 190:567–576. 10.1084/jem.190.4.567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hochberg D, Souza T, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. 2004. Acute infection with Epstein-Barr virus targets and overwhelms the peripheral memory B-cell compartment with resting, latently infected cells. J. Virol. 78:5194–5204. 10.1128/JVI.78.10.5194-5204.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Souza TA, Stollar BD, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. 2005. Peripheral B cells latently infected with Epstein-Barr virus display molecular hallmarks of classical antigen-selected memory B cells. Proc. Natl. Acad. Sci. U. S. A. 102:18093–18098. 10.1073/pnas.0509311102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang X, Paden CR, Morales FM, Powers RP, Jacob J, Speck SH. 2011. Murine gamma-herpesvirus immortalization of fetal liver-derived B cells requires both the viral cyclin D homolog and latency-associated nuclear antigen. PLoS Pathog. 7:e1002220. 10.1371/journal.ppat.1002220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowden RJ, Simas JP, Davis AJ, Efstathiou S. 1997. Murine gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during latency. J. Gen. Virol. 78:1675–1687 [DOI] [PubMed] [Google Scholar]
- 7.Flaño E, Kim IJ, Woodland DL, Blackman MA. 2002. Gamma-herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J. Exp. Med. 196:1363–1372. 10.1084/jem.20020890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marques S, Efstathiou S, Smith KG, Haury M, Simas JP. 2003. Selective gene expression of latent murine gammaherpesvirus 68 in B lymphocytes. J. Virol. 77:7308–7318. 10.1128/JVI.77.13.7308-7318.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willer DO, Speck SH. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 77:8310–8321. 10.1128/JVI.77.15.8310-8321.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambroziak JA, Blackbourn DJ, Herndier BG, Glogau RG, Gullett JH, McDonald AR, Lennette ET, Levy JA. 1995. Herpes-like sequences in HIV-infected and uninfected Kaposi's sarcoma patients. Science 268:582–583. 10.1126/science.7725108 [DOI] [PubMed] [Google Scholar]
- 11.Nash AA, Sunil-Chandra NP. 1994. Interactions of the murine gammaherpesvirus with the immune system. Curr. Opin. Immunol. 6:560–563. 10.1016/0952-7915(94)90141-4 [DOI] [PubMed] [Google Scholar]
- 12.Stevenson PG, Simas JP, Efstathiou S. 2009. Immune control of mammalian gamma-herpesviruses: lessons from murid herpesvirus-4. J. Gen. Virol. 90:2317–2330. 10.1099/vir.0.013300-0 [DOI] [PubMed] [Google Scholar]
- 13.Barton E, Mandal P, Speck SH. 2011. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu. Rev. Immunol. 29:351–397. 10.1146/annurev-immunol-072710-081639 [DOI] [PubMed] [Google Scholar]
- 14.Stevenson PG, Doherty PC. 1999. Non-antigen-specific B-cell activation following murine gammaherpesvirus infection is CD4 independent in vitro but CD4 dependent in vivo. J. Virol. 73:1075–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sangster MY, Topham DJ, D'Costa S, Cardin RD, Marion TN, Myers LK, Doherty PC. 2000. Analysis of the virus-specific and nonspecific B cell response to a persistent B-lymphotropic gammaherpesvirus. J. Immunol. 164:1820–1828 [DOI] [PubMed] [Google Scholar]
- 16.Usherwood EJ, Ross AJ, Allen DJ, Nash AA. 1996. Murine gammaherpesvirus-induced splenomegaly: a critical role for CD4 T cells. J. Gen. Virol. 77:627–630. 10.1099/0022-1317-77-4-627 [DOI] [PubMed] [Google Scholar]
- 17.Brooks JW, Hamilton-Easton AM, Christensen JP, Cardin RD, Hardy CL, Doherty PC. 1999. Requirement for CD40 ligand, CD4+ T cells, and B cells in an infectious mononucleosis-like syndrome. J. Virol. 73:9650–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim IJ, Flaño E, Woodland DL, Lund FE, Randall TD, Blackman MA. 2003. Maintenance of long term gamma-herpesvirus B cell latency is dependent on CD40-mediated development of memory B cells. J. Immunol. 171:886–892 [DOI] [PubMed] [Google Scholar]
- 19.Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML. 2001. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science 293:2111–2114. 10.1126/science.1061964 [DOI] [PubMed] [Google Scholar]
- 20.Rickert RC, Jellusova J, Miletic AV. 2011. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol. Rev. 244:115–133. 10.1111/j.1600-065X.2011.01067.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller DJ, Hayes CE. 1991. Phenotypic and genetic characterization of a unique B lymphocyte deficiency in strain A/WySnJ mice. Eur. J. Immunol. 21:1123–1130. 10.1002/eji.1830210506 [DOI] [PubMed] [Google Scholar]
- 22.Lentz VM, Cancro MP, Nashold FE, Hayes CE. 1996. Bcmd governs recruitment of new B cells into the stable peripheral B cell pool in the A/WySnJ mouse. J. Immunol. 157:598–606 [PubMed] [Google Scholar]
- 23.Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, Schneider P, Sizing ID, Mullen C, Strauch K, Zafari M, Benjamin CD, Tschopp J, Browning JL, Ambrose C. 2001. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science 293:2108–2111. 10.1126/science.1061965 [DOI] [PubMed] [Google Scholar]
- 24.Rahman ZS, Rao SP, Kalled SL, Manser T. 2003. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J. Exp. Med. 198:1157–1169. 10.1084/jem.20030495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vora KA, Wang LC, Rao SP, Liu ZY, Majeau GR, Cutler AH, Hochman PS, Scott ML, Kalled SL. 2003. Cutting edge: germinal centers formed in the absence of B cell-activating factor belonging to the TNF family exhibit impaired maturation and function. J. Immunol. 171:547–551 [DOI] [PubMed] [Google Scholar]
- 26.Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. 2004. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J. Immunol. 173:2245–2252 [DOI] [PubMed] [Google Scholar]
- 27.Rahman ZS, Manser T. 2004. B cells expressing Bcl-2 and a signaling-impaired BAFF-specific receptor fail to mature and are deficient in the formation of lymphoid follicles and germinal centers. J. Immunol. 173:6179–6188 [DOI] [PubMed] [Google Scholar]
- 28.Lagunoff M, Carroll PA. 2003. Inhibition of apoptosis by the gamma-herpesviruses. Int. Rev. Immunol. 22:373–399. 10.1080/08830180305218 [DOI] [PubMed] [Google Scholar]
- 29.Thorley-Lawson DA. 2001. Epstein-Barr virus: exploiting the immune system. Nat. Rev. Immunol. 1:75–82. 10.1038/35095584 [DOI] [PubMed] [Google Scholar]
- 30.Milho R, Frederico B, Efstathiou S, Stevenson PG. 2012. A heparan-dependent herpesvirus targets the olfactory neuroepithelium for host entry. PLoS Pathog. 8:e1002986. 10.1371/journal.ppat.1002986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frederico B, Milho R, May JS, Gillet L, Stevenson PG. 2012. Myeloid infection links epithelial and B cell tropisms of murid herpesvirus-4. PLoS Pathog. 8:e1002935. 10.1371/journal.ppat.1002935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaspar M, May JS, Sukla S, Frederico B, Gill MB, Smith CM, Belz GT, Stevenson PG. 2011. Murid herpesvirus-4 exploits dendritic cells to infect B cells. PLoS Pathog. 7:e1002346. 10.1371/journal.ppat.1002346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rickinson AB, Yao QY, Wallace LE. 1985. The Epstein-Barr virus as a model of virus-host interactions. Br. Med. Bull. 41:75–79 [DOI] [PubMed] [Google Scholar]
- 34.McClellan KB, Gangappa S, Speck SH, Virgin HW. 2006. Antibody-independent control of gamma-herpesvirus latency via B cell induction of anti-viral T cell responses. PLoS Pathog. 2:e58. 10.1371/journal.ppat.0020058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Husain SM, Usherwood EJ, Dyson H, Coleclough C, Coppola MA, Woodland DL, Blackman MA, Stewart JP, Sample JT. 1999. Murine gammaherpesvirus M2 gene is latency-associated and its protein a target for CD8(+) T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 96:7508–7513. 10.1073/pnas.96.13.7508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madureira PA, Matos P, Soeiro I, Dixon LK, Simas JP, Lam EW. 2005. Murine gamma-herpesvirus 68 latency protein M2 binds to Vav signaling proteins and inhibits B-cell receptor-induced cell cycle arrest and apoptosis in WEHI-231 B cells. J. Biol. Chem. 280:37310–37318. 10.1074/jbc.M507478200 [DOI] [PubMed] [Google Scholar]
- 37.Habison AC, Beauchemin C, Simas JP, Usherwood EJ, Kaye KM. 2012. Murine gammaherpesvirus 68 LANA acts on terminal repeat DNA to mediate episome persistence. J. Virol. 86:11863–11876. 10.1128/JVI.01656-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acha-Orbea H, Finke D, Attinger A, Schmid S, Wehrli N, Vacheron S, Xenarios I, Scarpellino L, Toellner KM, MacLennan IC, Luther SA. 1999. Interplays between mouse mammary tumor virus and the cellular and humoral immune response. Immunol. Rev. 168:287–303. 10.1111/j.1600-065X.1999.tb01299.x [DOI] [PubMed] [Google Scholar]
- 39.Yao Z, Maraskovsky E, Spriggs MK, Cohen JI, Armitage RJ, Alderson MR. 1996. Herpesvirus saimiri open reading frame 14, a protein encoded by T lymphotropic herpesvirus, binds to MHC class II molecules and stimulates T cell proliferation. J. Immunol. 156:3260–3266 [PubMed] [Google Scholar]
- 40.Sutkowski N, Conrad B, Thorley-Lawson DA, Huber BT. 2001. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity 15:579–589. 10.1016/S1074-7613(01)00210-2 [DOI] [PubMed] [Google Scholar]
- 41.Chapman C, Spellerberg M, Hamblin T, Stevenson F. 1995. Pattern of usage of the VH4-21 gene by B lymphocytes in a patient with EBV infection indicates ongoing mutation and class switching. Mol. Immunol. 32:347–353. 10.1016/0161-5890(94)00162-T [DOI] [PubMed] [Google Scholar]
- 42.Stevenson PG. 2004. Immune evasion by gamma-herpesviruses. Curr. Opin. Immunol. 16:456–462. 10.1016/j.coi.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 43.Tripp RA, Hamilton-Easton AM, Cardin RD, Nguyen P, Behm FG, Woodland DL, Doherty PC, Blackman MA. 1997. Pathogenesis of an infectious mononucleosis-like disease induced by a murine gamma-herpesvirus: role for a viral superantigen? J. Exp. Med. 185:1641–1650. 10.1084/jem.185.9.1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evans AG, Moser JM, Krug LT, Pozharskaya V, Mora AL, Speck SH. 2008. A gammaherpesvirus-secreted activator of Vbeta4+ CD8+ T cells regulates chronic infection and immunopathology. J. Exp. Med. 205:669–684. 10.1084/jem.20071135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parry CM, Simas JP, Smith VP, Stewart CA, Minson AC, Efstathiou S, Alcami A. 2000. A broad spectrum secreted chemokine binding protein encoded by a herpesvirus. J. Exp. Med. 191:573–578. 10.1084/jem.191.3.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bridgeman A, Stevenson PG, Simas JP, Efstathiou S. 2001. A secreted chemokine binding protein encoded by murine gammaherpesvirus-68 is necessary for the establishment of a normal latent load. J. Exp. Med. 194:301–312. 10.1084/jem.194.3.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans AG, Moorman NJ, Willer DO, Speck SH. 2006. The M4 gene of gammaHV68 encodes a secreted glycoprotein and is required for the efficient establishment of splenic latency. Virology 344:520–531. 10.1016/j.virol.2005.08.020 [DOI] [PubMed] [Google Scholar]
- 48.Geere HM, Ligertwood Y, Templeton KM, Bennet I, Gangadharan B, Rhind SM, Nash AA, Dutia BM. 2006. The M4 gene of murine gammaherpesvirus 68 modulates latent infection. J. Gen. Virol. 87:803–807. 10.1099/vir.0.81577-0 [DOI] [PubMed] [Google Scholar]
- 49.Wang GH, Garvey TL, Cohen JI. 1999. The murine gammaherpesvirus-68 M11 protein inhibits Fas- and TNF-induced apoptosis. J. Gen. Virol. 80:2737–2740 [DOI] [PubMed] [Google Scholar]
- 50.de Lima BD, May JS, Marques S, Simas JP, Stevenson PG. 2005. Murine gammaherpesvirus 68 bcl-2 homologue contributes to latency establishment in vivo. J. Gen. Virol. 86:31–40. 10.1099/vir.0.80480-0 [DOI] [PubMed] [Google Scholar]
- 51.Gangappa S, van Dyk LF, Jewett TJ, Speck SH, Virgin HW. 2002. Identification of the in vivo role of a viral bcl-2. J. Exp. Med. 195:931–940. 10.1084/jem.20011825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coleman HM, Brierley I, Stevenson PG. 2003. An internal ribosome entry site directs translation of the murine gammaherpesvirus 68 MK3 open reading frame. J. Virol. 77:13093–13105. 10.1128/JVI.77.24.13093-13105.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith CM, Gill MB, May JS, Stevenson PG. 2007. Murine gammaherpesvirus-68 inhibits antigen presentation by dendritic cells. PLoS One 2:e1048. 10.1371/journal.pone.0001048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stevenson PG, May JS, Smith XG, Marques S, Adler H, Koszinowski UH, Simas JP, Efstathiou S. 2002. K3-mediated evasion of CD8(+) T cells aids amplification of a latent gamma-herpesvirus. Nat. Immunol. 3:733–740. 10.1038/ni818 [DOI] [PubMed] [Google Scholar]