

ABSTRACT
The tailed double-stranded DNA (dsDNA) bacteriophage ϕ29 packages its 19.3-kbp genome into a preassembled procapsid structure by using a transiently assembled phage-encoded molecular motor. This process is remarkable considering that compaction of DNA to near-crystalline densities within the confined space of the capsid requires that the packaging motor work against significant entropic, enthalpic, and DNA-bending energies. The motor consists of three phage-encoded components: the dodecameric connector protein gp10, an oligomeric RNA molecule known as the prohead RNA (pRNA), and the homomeric ring ATPase gp16. Although atomic resolution structures of the connector and different pRNA subdomains have been determined, the mechanism of self-assembly and the resulting stoichiometry of the various motor components on the phage capsid have been the subject of considerable controversy. Here a subnanometer asymmetric cryoelectron microscopy (cryo-EM) reconstruction of a connector-pRNA complex at a unique vertex of the procapsid conclusively demonstrates the pentameric symmetry of the pRNA and illuminates the relative arrangement of the connector and the pRNA. Additionally, a combination of biochemical and cryo-EM analyses of motor assembly intermediates suggests a sequence of molecular events that constitute the pathway by which the motor assembles on the head, thereby reconciling conflicting data regarding pRNA assembly and stoichiometry. Taken together, these data provide new insight into the assembly, structure, and mechanism of a complex molecular machine.
IMPORTANCE Viruses consist of a protein shell, or capsid, that protects and surrounds their genetic material. Thus, genome encapsidation is a fundamental and essential step in the life cycle of any virus. In dsDNA viruses, powerful molecular motors essentially pump the viral DNA into a preformed protein shell. This article describes how a viral dsDNA packaging motor self-assembles on the viral capsid and provides insight into its mechanism of action.
INTRODUCTION
An essential step in the life cycle of any virus is the encapsidation of the viral chromosome within the confines of a protective protein shell (capsid). In double-stranded DNA (dsDNA) viruses, such as herpesviruses, adenoviruses, and the tailed dsDNA bacteriophages, a relatively rigid, icosahedral protein shell (the procapsid, or prohead), is first assembled, followed by insertion of a single copy of the replicated viral genome into the head (1, 2, 3). Among the challenges encountered during genome packaging are the needs to recognize the viral genome among the multitude of other nucleic acids in the infected cell and to overcome the considerable entropic, enthalpic, and DNA-bending energies that oppose the compaction of the viral genome within the capsid (4, 5).
In order to accomplish these tasks, dsDNA viruses encode complex molecular motors that self-assemble on the precursor capsid and utilize energy derived from ATP hydrolysis to package the viral genome to near-crystalline densities (2, 3). These force-generating motors are assembled at a unique vertex of the head that contains the dodecameric head-tail connector (portal) ring (6, 7, 8) and serves as the binding site for the force-generating ATPase component of the motor. To date, the ATPase component in packaging complexes has been visualized in situ as pentameric rings (8, 9, 10), although both hexameric and tetrameric ATPase rings have also been suggested (11, 12). Furthermore, little is known regarding the molecular events that guide the assembly and disassembly of these transiently assembled motor complexes on the viral capsid. Thus, visualization of these multicomponent molecular machines at various stages during their assembly is critical to understanding how they self-assemble as a functional motor complex on the virus capsid.
Bacteriophage ϕ29 is an excellent model system for exploring motor assembly and for mechanistic studies of DNA packaging (4, 5, 13). Due to the small size, relative simplicity, and highly efficient in vitro assembly of bacteriophage ϕ29, a wealth of genetic, biochemical, structural, and single-molecule data is available. Like other dsDNA phages, ϕ29 is assembled via a well-defined morphogenetic pathway that includes the formation of the prohead, the assembly of a packaging motor complex on the head, ATP-driven translocation, and motor disassembly at the completion of packaging (4, 14) (Fig. 1). Unlike the other well-studied dsDNA phages, ϕ29 has an additional essential motor component, an oligomeric ring of RNA (termed pRNA), that binds to proheads and bridges the connector and ATPase components (8, 9, 15, 16, 17) (Fig. 1 and 2). The formation of the RNA ring is accompanied by intermolecular base pairing between complementary loops of adjacent pRNAs. Understanding how the pRNA assembles on the prohead via interactions with the connector and the head shell, as well as how it provides a structural scaffold for the assembly of the functional ATPase ring, is essential to determining how the coordinated activity of these components results in the translocation of the genomic DNA. Since the pRNA ring is thought to functionally substitute for a protein domain(s) found in the other dsDNA packaging motors (3), characterization of the assembly events and complex symmetry mismatches in the ϕ29 system will likely inform us as to general principles of motor assembly and function in this broad class of molecular machines.
FIG 1.

Assembly pathway of bacteriophage ϕ29.
FIG 2.

Structures of the ϕ29 connector and pRNA. (A) Side view of the connector. Eleven of the 12 monomers are shown in green, and one monomer is colored by domain, with the wide, central, and narrow domains shown in blue, red and yellow, respectively. (B) Single monomer of the connector structure, shown in the same color scheme and orientation as in panel A, with central-domain α-helices α-1, α-3, and α-5 labeled at their N-terminal ends. (C) Secondary structural diagram of the pRNA. The A-, D-, and E-helices are labeled, along with the CE-, D-, and E-loops and the U-rich 3-way junction (3wj; bases shown in green). (D) Atomic structure of the prohead-binding domain of the pRNA (PDB 3R4F) (18), labeled and colored as in panel C. (Reprinted from the Proceedings of the National Academy of Sciences of the United States of America [18] with permission of the publisher.)
The assembly pathway of pRNA has been the subject of much controversy (8, 9, 15, 17, 18, 19, 20, 21), and there are multiple and sometimes contradictory reports regarding various aspects of motor assembly (9, 15, 17, 22, 23). For example, N-terminal residues of the 12-fold connector, including a critical RKR motif, have been shown to be essential for pRNA binding (24, 25). However, fitting atomic structures of the pRNA prohead binding domain into cryoelectron microscopy (cryo-EM) structures of proheads showed that pRNA interacts primarily with the 5-fold capsid rather than with the connector (9, 18, 19). Specifically, the E-loop of pRNA (Fig. 2C and D) appears to make contact with the head shell, and mutagenesis has confirmed that the E-loop is indeed required for prohead binding (18). Additionally, intermolecular base pairing between pRNA molecules had been identified as necessary for binding (15, 17, 22, 23). Initially, since pRNA readily dimerizes in solution and since mutation of the pseudoknot residues that prevents dimerization correlated with a loss of DNA packaging, the dimer had been proposed as the functional binding unit (15, 17, 22, 23). However, more recent studies have shown that dimerization is not essential, as evidenced by the fact that mutant pRNAs that are monomeric in solution yet retain wild-type (wt) pseudoknot sequences still bind efficiently and competitively to the prohead and are active in packaging (26). Thus, the intermolecular interaction is essential only when pRNA is bound to the prohead. Notwithstanding, the culmination of these many events is an extraordinarily stable pRNA assembly at the connector vertex of the prohead, since the bound pRNA ring does not readily exchange off the head (27).
Here we describe a series of structural and biochemical experiments designed to visualize the packaging motor vertex at high resolution, reconcile conflicting data on pRNA stoichiometry, and probe the sequence of molecular events that occur during motor assembly. A subnanometer asymmetric cryo-EM reconstruction of the connector-pRNA vertex of proheads shows pentameric symmetry for the pRNA and illuminates both pRNA-connector and pRNA-capsid interactions. Additionally, cryo-EM structures of ϕ29 proheads before and after pRNA attachment document conformational changes in the procapsid that must occur to create the primary binding site for pRNA attachment during motor assembly. We further demonstrate that intermolecular base pairing between neighboring subunits in the pRNA pentamer results in stable ring formation wherein individual subunits are effectively locked into place. Taken together, these results not only suggest a defined sequence of molecular events that constitute part of the assembly pathway of a complex molecular motor but also shed light on basic biological processes, such as molecular recognition, self-assembly, and coordinated protein-protein and protein-nucleic acid interactions.
MATERIALS AND METHODS
Production of wild-type and mutant pRNAs.
ΔCCA, F6/F7, F6, and F7 mutant pRNAs had been constructed previously (17, 28, 29). F6/F7ΔCCA, F6ΔCCA, and F7ΔCCA pRNAs were created using inverse PCR, based on the method of Wang and Wilkinson (30), in the pRT72 plasmid system that encodes pRNA (31). Plasmid DNA was sequenced to verify the presence of the desired mutations. Wild-type and mutant pRNAs were produced by in vitro transcription using T7 RNA polymerase and were purified by denaturing urea polyacrylamide gel electrophoresis as described previously (31). The purified RNAs were refolded by heating to 80°C for 3 min and then cooled to room temperature, and 1/10 volume of 10× TM (500 mM Tris [pH 7.6], 50 mM MgCl2) was added prior to use.
Production and purification of packaging components.
Fiberless proheads, DNA-gp3, and the packaging ATPase gp16 were produced as described previously (29, 32). Briefly, proheads were produced by infection of Bacillus subtilis 12A (Sup−) with the sus8.5(900)-sus16(300)-sus14(1241) ϕ29 mutant (defective in head fibers and the packaging ATPase) (33). Infected cells were harvested at 65 min and were concentrated 100-fold prior to lysis. Proheads were purified on 10-to-30% (wt/vol) sucrose gradients in 1× TMS buffer (50 mM Tris [pH 7.8], 10 mM MgCl2, 100 mM NaCl). The particle band was removed, and particles were concentrated by centrifugation. A second round of sucrose gradient purification and concentration by centrifugation was performed. The particles were resuspended in 1× TMS. The packaging ATPase was produced in Bacillus subtilis from plasmid pSACB-gp16 and was purified by chromatography, as described previously (29). DNA-gp3 was extracted from CsCl-purified phage and was isolated on a CsCl gradient as described previously (32).
Invasion of various prohead-pRNA complexes by ΔCCA pRNA.
RNA-free proheads were produced by digestion of pRNA from fiberless proheads with RNase A treatment, followed by repurification of the RNA-free particles by ultracentrifugation as described previously (29). Purified pRNA-free proheads (2.25 × 1012) were incubated with either 120-base pRNA, F6/F7 pRNA, or F6+F7 pRNA (in a molar ratio of 1:20) in 60 μl in 0.5× TMS for 20 min at room temperature. To remove unbound pRNA, the particles were layered on top of 5 ml of 5% (wt/vol) sucrose in 0.5× TMS buffer and were pelleted in a SW55 rotor at 35,000 rpm for 2.5 h at 4°C. The reconstituted proheads were resuspended in 0.5× TMS. These purified prohead-pRNA complexes (1 × 1011) were then challenged with pRNAs containing the lethal ΔCCA mutation and the desired pseudoknot sequences (wtΔCCA, F6/F7ΔCCA, F6ΔCCA, or F7ΔCCA pRNA; molar ratio, 1:10) for 20 min at room temperature. These particles were then used in the DNA packaging assay.
DNA packaging assay.
The in vitro DNA packaging assay is based on a DNase protection assay and was performed as described previously (29, 34). Briefly, reconstituted proheads (8.3 nM), DNA-gp3 molecules (4.2 nM), and gp16 molecules (166 to 208 nM) were mixed together in 0.5× TMS buffer in 20 μl and were incubated for 5 min at room temperature. ATP was then added to 0.5 mM to initiate packaging. After 15 min of incubation, the mixture was treated with DNase I (final concentration, 1 μg/ml) and was incubated for 10 min to digest the unpackaged DNA. An EDTA-proteinase K mixture was then added to the reaction mixture (final concentrations, 25 mM and 500 μg/ml, respectively), and the mixture was incubated for 30 min at 65°C to inactivate the DNase I and release the protected, packaged DNA from particles. The packaged DNA was analyzed by agarose gel electrophoresis. Packaging efficiency was calculated by densitometry using a UVP gel documentation system. Input DNA was used as the standard to determine the percentage of DNA packaging, and packaging efficiencies in the absence and presence of challenger ΔCCA pRNA were then compared.
Production of pRNA-naïve proheads from the cloned gene construct expressed in Escherichia coli.
ϕ29 prohead particles were assembled in E. coli from the pAR7-8-8.5-10 plasmid (35), which encodes the prohead genes, and were isolated and purified as described previously (36) with the following modifications. Briefly, 2 h postinduction, cells were concentrated 100× for lysis, and the prohead particles were then isolated in 10-to-30% (wt/vol) linear sucrose density gradients containing TMS buffer in the SW55 rotor at 35,000 rpm for 2 h. The prohead band was removed and particles pelleted in the SW55 rotor at 35,000 rpm for 2 h at 4°C. The pellet was resuspended in TMS buffer and was purified through a second round of sucrose gradients and concentration by ultracentrifugation. The purified particles were resuspended in TMS buffer.
Cryo-EM of E. coli proheads with pRNA.
For the sample of proheads before pRNA binding, pRNA-naïve proheads obtained from overexpression of the pAR7-8-8.5-10 plasmid in E. coli (35) were directly applied to grids prior to plunge freezing. For the sample of prohead particles after pRNA binding, pRNA-naïve particles were incubated with recombinantly expressed 120-base pRNA (molar ratio, 1:10; prepared as described above) for 10 min. In both cases, prohead particles were flash-frozen on holey carbon grids in liquid ethane using the FEI Vitrobot. Images were recorded at a magnification of ×78,000 using a 300-kV FEI TF30 transmission electron microscope equipped with a Gatan 4k by 4k charge-coupled device (CCD) camera, with electron dose levels of approximately 25 e−/Å2. Micrographs were binned to give an effective pixel size of 3.01 Å. Defocus ranges of 1.2 to 3.5 μm were used for collection of data on E. coli proheads before and after incubation with pRNA. Model-based reconstructions were performed with SPIDER (37) based on a modified ϕ29 map from the Electron Microscopy Data Bank (EMDB 1419) (19). The numbers of particles incorporated into the reconstructions of E. coli proheads before and after incubation with pRNA were 10,004, and 10,142, respectively. The resolutions of these reconstructions were determined by the Fourier shell correlation (FSC) method using a correlation coefficient of 0.5 between two independent half-data sets as the cutoff criterion (Table 1).
TABLE 1.
Final resolution and symmetry imposed for cryo-EM reconstructions
| Particle/component analyzed | Symmetry imposed | Final resolution (Å) |
|---|---|---|
| Connector-pRNA complex | C1 | 10 |
| Proheads produced in E. coli prior to addition of exogenously produced pRNA | C5 | 13 |
| Proheads produced in E. coli after addition of exogenously produced pRNA | C5 | 15 |
Asymmetric reconstruction of the connector-pRNA complex.
The asymmetric reconstruction of the connector-pRNA complex was calculated by computationally extracting the density corresponding to the connector-pRNA complex from cryo-EM images of ϕ29 proheads (20, 38) and then using these modified images for subclassification and image reconstruction. To subtract the capsid density from the cryo-EM micrographs of the proheads, images of proheads were initially used to calculate a 3-dimensional (3D) reconstruction of the entire prohead to a resolution of ∼10 Å. The data used for this reconstruction were the same data used to calculate a previously published 12.5-Å structure of the prohead (19); the improved resolution was obtained primarily by unbinning the data, resulting in a 2.12-Å pixel size, and improving the fitted parameters of the contrast transfer function. Next, the previously determined molecular envelopes of the connector and pRNA (9) were combined and were used to make a 3D mask encompassing both components. Voxel values within this mask were then replaced with zeros except at mask edges, where a smooth Gaussian falloff was imposed to minimize masking artifacts. Thus, the residual density after masking included only the density corresponding to the capsid. This capsid-only density was projected in the known orientations of each 2D experimental image and was then translated by the known translational parameters using bilinear interpolation for nonintegral shifts. After scaling for density magnitude, each capsid-only projection was subtracted from the corresponding cryo-EM image of the prohead such that the remaining density in the resulting “difference image” corresponded to only the pRNA and the connector. In order to minimize the effect of residual capsid density remaining from scaling errors after subtraction, the 3D mask encompassing the pRNA and the connector described above was inverted (i.e., voxels inside and outside the mask were set to values of one and zero, respectively) and was then projected onto the “difference images,” again using the known orientation and translation corresponding to each image. Next, all pixels outside the area circumscribed by the projected mask were set to zero except at the mask edges, where a soft Gaussian falloff was imposed to minimize masking artifacts. Note that prior to mask projection, the mask was circularly symmetrized around the long axis of the motor to remove any features that might bias the alignment of the masked images. The difference images were then recentered based on the center-of-mass of the projected mask by using bilinear interpolation for nonintegral shifts, and the box size was reduced to encompass only the residual motor density. Subsequent subclassification was thus restricted to masked areas corresponding to the motor and a small portion of the capsid at the motor vertex. In this way, the contribution of the capsid during classification was minimized and the contribution of the motor maximized, and the reduced box size greatly reduced the computational expense. For subclassification, we used our previously published approach, which is now commonly used to calculate asymmetric reconstructions of viruses (20, 38, 39). These methods take advantage of the dominant pseudosymmetry of virus particles (typically 5-fold or icosahedral) to save computational effort by significantly reducing the orientational search space and by making use of realistic restraints with regard to the relative orientation of the motor and the capsid. All image-processing steps were performed using either freely available software (37, 40, 41) or software developed in the Morais laboratory (20, 38; also unpublished data). Although the resolution of the resulting reconstruction is better than 8.5 Å by the FSC method, this number may partially reflect masking and/or subtraction artifacts. Based on the appearance of the map, and the more reliably determined FSC-based resolution of the entire prohead reconstruction prior to subtraction and masking, we have interpreted the map as if it were at a resolution of ∼10 Å (Table 1).
RESULTS AND DISCUSSION
Asymmetric reconstruction of the connector-pRNA vertex.
Although several high-resolution asymmetric reconstructions of mature, tailed phage particles have been reported (39, 42, 43, 44, 45, 46), to date it has not been possible to image motor complexes bound to proheads at a resolution sufficient to visualize the correct symmetries of all the various components. For tailed particles, the relatively large mass of the tail provides sufficient signal to fully resolve the inherent 5–6 symmetry mismatch between the capsid and the tail. In contrast, phage packaging motors are typically much smaller than phage tails, and thus, resolving any symmetry mismatches present in the motor during image processing is challenging. For example, asymmetric reconstructions of ϕ29 and T4 have shown pentamers of the pRNA (ϕ29) or packaging ATPase (T4) (10, 20). However, in neither case was the resolution of the reconstructions sufficient to resolve the 12-fold symmetry of the connector. As a result, there is some question as to whether possible symmetry mismatches in the motor were fully resolved. Hence, the ability to reconstruct the known dodecameric structure of the connector would provide a definitive internal control demonstrating that an image-processing procedure was able to faithfully reconstruct symmetry mismatches associated with dsDNA packaging motors and to determine if a preferred orientation/alignment between mismatched components existed.
To properly resolve the symmetry mismatches in dsDNA packaging motors, it is necessary to detect signals from components whose mass is less than 5% of the relatively massive 5-fold head shell (see reference 20 for a comprehensive discussion of challenges associated with reconstructing symmetry mismatches). To minimize the contribution of capsid density and simultaneously maximize the contribution of the motor, we have first subtracted the density corresponding to the capsid shell from cryo-EM images of ϕ29 proheads and then masked out any residual capsid density not accounted for by the subtraction. The resulting difference images were then analyzed and aligned by virtue of the remaining motor components, with minimum bias from the massive head, yielding a subnanometer resolution reconstruction of the connector-pRNA complex as it exists on the prohead (Fig. 3). The resulting structures of the connector and pRNA are described below.
FIG 3.

Asymmetric reconstruction of the connector-pRNA vertex. (A through C) Top (A), bottom (B), and side (C) views of the asymmetric reconstruction of connector-pRNA complex are shown. Densities corresponding to the capsid, connector, and pRNA are rendered in gray, green, and magenta, respectively. The fitted X-ray structure of the connector is shown in yellow. A model of the pRNA based on fitting the X-ray structure of the prohead-binding domain of the pRNA (18), the NMR structure of the CCA bulge (29), and an ideal RNA helix as the A-helix was also fitted; the five subunits are colored red, green, blue, yellow, and purple. (D) Reconstruction of the connector-pRNA vertex rendered at a higher contour level (∼4 standard deviations above the mean), where it is possible to recognize α-helices in the central region of the connector. Central domain α-helices α-1, α-3, and α-5 labeled in one magenta colored connector subunit. (E) A rendering of the density corresponding to the connector at a lower contour level superimposed on the rendering of the complex presented in panel D shows extra density corresponding to the crown region of the connector (blue). (F) Density corresponding to the pRNA rendered at a very high contour level (magenta), indicating that pRNA bases involved in the intermolecular Watson-Crick base pairing are highly ordered in the reconstruction. The arrow indicates an RNA superhelix that spans two pRNA subunits, and the contributing E-, D-, and A-helical elements are labeled.
Connector structure.
The dodecameric symmetry of the connector is clearly visible in the asymmetric reconstruction of the difference images (Fig. 3A and B). In addition to resolving individual subunits within the dodecamer, we can also clearly identify distinct domains within monomers, as well as some secondary structural elements, including the long α-helices that traverse the middle region of the connector (Fig. 3D). Fitting an X-ray crystal structure of the connector (8) into its corresponding EM density indicates good overall agreement between the two structures (Fig. 3). Visual inspection shows that the density corresponding to the wide end of the connector agrees particularly well with the crystal structure, whereas the central domain region exhibited less agreement. However, clearly visible α-helical density corresponding to helix α-5 (8) (residues 208 to 226) (Fig. 2B and 3) in the reconstruction indicated how the crystal structure could easily be adapted to cryo-EM density. Although the density corresponding to helix α-3 (residues 130 to 156) (Fig. 2B and 3) can be identified, it is not as clearly α-helical in character as that for helix α-5, possibly suggesting that this helix is less well-ordered. Further, the absence of any clear density for the N-terminal helix α-1 (residues 13 to 38) (Fig. 2B and 3) indicates that this helix may be mobile, disordered in situ, or differently ordered in individual connecter monomers such that its density is degraded upon interparticle averaging. The observed structural differences between the crystal structure of the connector and the EM density derived from prohead-embedded connectors suggests that interactions with the capsid and the pRNA may modulate the structure of the connector. We also observed density corresponding to the segments of the connector that were not resolved in the crystal structure: the C-terminal domain (residues 287 to 309) (Fig. 3E) that protrudes into the prohead cavity and the channel-loop structures (residues 229 to 246) that project into the lumen of the connector. Similar features were present in the crystal structures of the connectors of phages SPP1 and P22 (6, 7). Taken together, visualization of all of these known structural elements validates our method for successfully determining the structure and symmetry of this complex motor assemblage at a high resolution.
pRNA structure.
The structure of each of the individual pRNA monomers in the pRNA ring is consistent with previously obtained structural results from X-ray crystallography and nuclear magnetic resonance (NMR) (18, 47, 48, 49), and their quaternary organization agrees with previous cryo-EM results (8, 9, 19, 20). Importantly, despite the absence of any imposed symmetry, the pRNA visualized in the reconstruction is clearly pentameric (Fig. 3B), in agreement with our earlier structural results (8, 9, 19, 20). Previously, it had been argued that 5-fold symmetry could be unintentionally imposed by virtue of the influence of the massive 5-fold head shell on particle alignment (21). Here this effect is mitigated by subtraction of the head shell density from individual cryo-EM images, and the dodecameric connector provides an internal control to verify that symmetry mismatches have been resolved in cryo-EM reconstructions of ϕ29 proheads. In particular, the ability to resolve 12 connector monomers, as well as secondary structural elements within these monomers, demonstrates that even relatively small, nonpentameric components are properly aligned and averaged in the asymmetric prohead reconstruction. Therefore, arguments that 5-fold symmetry could be imposed unintentionally due to the influence of the massive 5-fold head shell on particle alignment are now rendered invalid.
In addition to conclusively demonstrating the oligomeric state of the pRNA, our improved map of the pRNA allowed us to evaluate previous models proposed for the pRNA oligomer. Several structural models of the oligomeric pRNA have been put forth. Early models were based almost entirely on computational modeling using restraints obtained from genetic, biochemical, and biophysical experiments (17, 50). Later models included additional low-resolution structural information from cryo-EM structures of ϕ29 proheads (8, 19). More recently, X-ray and NMR structures of various pRNA fragments have inspired a new generation of models (18, 47, 49). Some of these models rely on fitting known high-resolution structures of pRNA fragments into low-resolution, 5-fold averaged cryo-EM structures of proheads (18, 47), whereas others have simply replaced part of a previous computational model of the oligomer with the corresponding experimentally determined pRNA fragments (49).
Comparison of these models to our pRNA density indicated that a model proposed previously by Harjes et al. (47), incorporating X-ray, NMR, and low-resolution 5-fold averaged cryo-EM information, agreed best with the current structural results. This model was originally obtained by fitting structures of the prohead-binding/oligomerization domain (bases 25 to 95; obtained via X-ray crystallography) (18) (PDB 3R4F) (Fig. 2C and D), the CCA bulge (bases 12 to 24 and 96 to 105; obtained via NMR) (47) (PDB 2LQZ) (Fig. 2C), and the distal portion of the A-helix (bases 1 to 11 and 106 to 117; obtained via modeling of the A-helix as an ideal RNA helix) (Fig. 2C) into a 12.5-Å resolution 5-fold-symmetric cryo-EM reconstruction of the ϕ29 prohead. It should be noted that in a recent crystal structure of the pRNA three-way junction (3WJ) (49) (PDB 4KZ2), the relative orientations of the RNA helices within a monomer do not agree with our map. This may be due to the absence of the base-pairing CE- and D-loops in this construct, and the resulting lack of biologically relevant intermolecular interactions.
To optimize the fit and account for deviations from 5-fold symmetry present in the asymmetric reconstruction, the five monomers of the pRNA model were first fitted separately, followed by adjustment of the relative orientations of the three structural components described above (Fig. 3B, C, and F). The resulting model of the pRNA is consistent with several previously reported structural features, including the following: (i) bases 22 to 84 form a pentameric, ring-like structure that binds to the prohead (18); (ii) five pRNA A-helices (bases 1 to 28 and 92 to 120) extend as spokes from this central ring (9); (iii) the orientation of the A-helix is influenced by the CCA bulge (bases 18 to 20) (29, 47); (iv) intermolecular Watson-Crick base pairing between the C-E loop of one monomer (bases 45 to 48) and the D-loop of another (bases 82 to 85) mediates ring formation (15, 17, 18); (v) the E-loop of the pRNA (bases 53 to 58) makes extensive contact with the head shell (18, 47); and (vi) multiple helical elements from adjacent pRNA monomers stack to form an RNA superhelix that spans two pRNA subunits (18) (Fig. 3F).
Additional insight into the organization and relative flexibility of various structural elements of the pRNA can be obtained by analyzing the cryo-EM density magnitude at various points in the map (Fig. 3C and F). The density corresponding to the central U-rich junction (Fig. 2C and D) (bases 28 to 30, 71 to 75, 91, and 92) that joins three helical elements (3WJ) is relatively weak (density magnitude, <2 standard deviations above the mean), consistent with the high-temperature factors reported for this region in the crystal structure of the prohead-binding domain of the pRNA (18). Hence, despite its central location within a pRNA monomer, the U-rich junction is likely somewhat flexible. In contrast, some of the strongest density in the map corresponds to the RNA superhelix (Fig. 2D and 3F) that is comprised of helical elements from adjacent pRNA monomers via costacking of the A- and D-helices from one subunit (bases 1 to 28 and 92 to 100 and bases 75 to 80 and 86 to 91, respectively) with the E-helix from an adjacent subunit (bases 49 to 52 and 59 to 62) (18), indicating that although two distinct pRNA subunits contribute to this structural feature, it is highly ordered. Within the RNA superhelix, the density corresponding to the intermolecular base pairing between the CE- and D-loops is especially strong (density magnitude, >4 standard deviations above the mean) (Fig. 3F). Taken together, these observations suggest that the basic “structural” unit of the pRNA (i.e., the RNA superhelix) may be different from the topological unit (i.e., the pRNA monomer), an arrangement that could mediate coordination and communication between adjacent subunits in the motor, possibly via intermolecular base pairing. Furthermore, the density corresponding to the E-loop that binds to the capsid shell is also high, suggesting that this is a strong, well-ordered interaction that anchors the pRNA on the prohead.
Connector-pRNA interactions.
Twelve interactions between the narrow end of the connector and the inner lumen of the pentameric pRNA that can only be visualized in an asymmetric reconstruction are present in the map of the pRNA-connector complex (Fig. 3B and C). Because of the 5–12 symmetry mismatch between these two components, these interactions are necessarily nonequivalent. Hence, although the interactions are somewhat weak overall, some appear stronger than others, as indicated by their increased relative density magnitudes. Although the resolution of the present reconstruction is not sufficient to identify individual bases or amino acid side chains, the fitted structures of the connector and the pRNA model suggest which residues from the connector interact with particular bases in the pRNA (Fig. 3A). Due to the 5–12 symmetry mismatch, distances between residues in any particular connector subunit and bases from the nearest pRNA subunits will differ. Nevertheless, connector residues 167 to 179 are invariably closest in space to bases 50 through 56 (corresponding to the E-helix) from one pRNA and bases 83 and 84 from the neighboring pRNA, and density connecting the connector and pRNA at these two regions is apparent. These bases either are part of, or flank, the intermolecular pseudoknot. Additionally, the ability to simultaneously align both connectors and pRNAs when one is averaging over thousands of particles indicates that the relative orientations of these two components are fixed and that thus, the connector has a preferred orientation and is not free to rotate, in agreement with the findings of biochemical and single-particle experiments (51, 52). If this were not the case, it would be impossible to simultaneously align both components unless different angular orientations were applied to subimage areas corresponding to connectors and pRNAs during image reconstruction (see reference 20 for further discussion).
Assembly of pRNA on procapsids.
In addition to pRNA symmetry, the related question of how pRNA assembles on the head has also been controversial. On the one hand, as stated above, it has been shown that pRNA binds to the N-terminal RKR residues of the connector, suggesting that the pRNA interacts primarily with the connector. On the other hand, structural data show that the pRNA interacts extensively with the capsid, suggesting that the capsid provides the primary binding site for pRNA. Additionally, it has been shown that pRNA remains bound to particles in which connectors have been displaced, again suggesting that the pRNA interacts primarily with the capsid rather than the connector. Thus, there must be other essential pRNA-protein interactions on the head that stabilize the functional quaternary structure of the pRNA on the prohead.
An initial clue regarding a plausible pRNA assembly pathway that reconciles these conflicting data came from the pseudoatomic structure of the prohead obtained by fitting homologues of ϕ29 structural components into cryo-EM reconstructions of proheads (19). This structure clearly showed that the pRNA makes extensive contacts with the capsid that would be sterically impossible if a subdomain of the head shell protein (the C-terminal Ig-like domain) surrounding the pRNA were not rotated ∼180° from its expected position (9, 18, 19) (Fig. 4A), suggesting that this rotation would be an essential event in motor assembly. Hence, an assembly scenario compatible with all available data is one where the pRNA initially recognizes and binds to the RKR residues at the N terminus of the connector, followed by rotation of the Ig-like C-terminal domains in capsid proteins surrounding the pRNA (19), which would uncover the tight binding site for the pRNA E-loop on the capsid. Once bound to the procapsid, adjacent pRNA subunits would then be properly positioned to form intermolecular base pairs that close the pentameric ring and essentially lock the pRNA on the prohead.
FIG 4.

Cryo-EM reconstructions of proheads produced in E. coli before and after the addition of in vitro-transcribed pRNA. (A) Pseudoatomic structure of the prohead. Pentameric subunits are colored blue, and quasiequivalent alternating subunits in hexamers are shown in red and green. (Reprinted from Molecular Cell [19] with permission of the publisher.) (Inset) Zoomed-in view of the connector-pRNA vertex looking from the bottom of the head at a slightly skewed angle. The connector is shown in yellow and the pRNA in magenta. One of the five hexamers circumscribing the vertex is also shown, with HK97 domains colored green, observed positions of Ig-like domains shown in blue, and the “expected” positions of Ig-like domains shown in cyan, demonstrating that if the domain had not moved, it would have clashed with pRNA. The approximately 180° degree rotation is indicated by the arrow connecting two circled Ig-like domains. (B) (Left) Cryo-EM reconstruction of a pRNA-naïve E. coli-produced prohead, with Ig-like domains colored blue. The Ig-like domain that rotates upon the addition of pRNA is circled. (Center) Reconstruction of E. coli-produced proheads after incubation with in vitro-transcribed pRNA. Ig-like domains are colored blue. The rotated Ig-like domain is circled. (Right) Superposition of structures of proheads before (yellow) and after (green) the addition of in vitro-transcribed pRNA. The approximately 180° rotation is indicated by the arrow connecting two circled Ig-like domains. The pRNA is shown in magenta.
Cryo-EM reconstructions of proheads before and after pRNA binding.
To test whether the conformational changes in the pRNA assembly scenario proposed above occur, we first determined cryo-EM structures of proheads assembled in the absence of pRNA. These particles are produced in an E. coli overexpression system that codes only for the protein components of the prohead; such particles have been shown previously to have full biological activity when mixed with in vitro-transcribed pRNA and recombinantly expressed ATPase (35). Since these particles are pRNA naïve, it was predicted that they would not have undergone the conformational change necessary for binding pRNA, and hence the C-terminal Ig-like domain would not have rotated from the conformation observed throughout the rest of the head shell. This is indeed the case. A ∼13-Å-resolution reconstruction of these pRNA-naïve particles shows that all the C-terminal Ig-like domains in capsomers circumscribing the connector vertex do indeed sit at capsomeric vertices, like the other 230 capsid proteins in the head (Fig. 4B, left). To test the prediction that pRNA binding induces rotation of the Ig-like domain, we then calculated a ∼15-Å-resolution reconstruction of the same RNA-naïve particles after incubation with in vitro-transcribed pRNA. The resulting map showed that, as predicted, after pRNA addition, the Ig-like domain rotates ∼180° from its original position to accommodate pRNA binding and now occupies the position seen in prohead-pRNA complexes assembled in vivo (Fig. 4B, center and right). This result demonstrates how the prohead structure is modulated to facilitate pRNA binding and validates an essential contact between the E-loop of pRNA and the capsid shell. It also dictates that any model of assembly must include a role for the shell, in addition to the previously characterized connector interaction.
Genetic and biochemical analysis of pRNA assembly.
In addition to the head-shell contact, intermolecular base pairing between adjacent molecules in the pRNA ring have been shown to be essential for pRNA-prohead assembly (15, 17). Although the pRNA ring is normally composed of identical pRNA molecules paired via the intermolecular interaction, it has also been shown that functional rings can be assembled from combinations of two different mutant pRNAs engineered to have complementary pseudoknots (15, 17, 23). Specifically, the CE- and D-loops involved in the intermolecular interaction (Fig. 2C) can be mutated such that an individual RNA molecule is no longer self-complementary, thereby preventing ring formation, and hence cannot support DNA packaging. However, when this molecule is mixed with a second, complementary mutant pRNA, DNA packaging activity is restored. This experimental design allows for an ordering of pRNA on the head alternating between the two mutant pRNAs (Fig. 5A). Given the pentameric symmetry of pRNA, this would result in four base-paired interfaces and a single mismatched interface, thus providing an opportunity to further probe the role of the intermolecular interaction. It has been reported that prohead-pRNA complexes challenged with an excess of free RNA do not readily exchange RNA (31). We hypothesized that the intermolecular interaction confers this stability, preventing exchange of RNAs by creating a closed, fully base paired pRNA ring. To test this, RNA-free proheads were incubated with an excess of either (i) wild-type pRNA, (ii) the mutant F6/F7, in which both pseudoknot loops are mutated, but the mutant sequences are self-complementary, thereby permitting ring closure, or (iii) the ordered dimer pair F6+F7 pRNAs, in which each mutant RNA has one wild-type loop and one mutated loop (the mutated loops are complementary) (17) (Fig. 5A). These proheads were then challenged with pRNAs of various pseudoknot sequence that also contained an additional mutation that is lethal for DNA packaging (i.e., deletion of the essential CCA bulge [(ΔCCA] of the A-helix [Fig. 3C] [27, 29]). The various particles were then assayed for the ability to support DNA packaging in the in vitro DNA-packaging system. Previously, it has been shown that incorporation of even a single pRNA mutant impacts DNA packaging (53). Here, incorporation of a ΔCCA- pRNA into the ring would result in the loss of DNA-packaging activity, thus acting as a reporter for the ability of the ring to be invaded via an open, non-base-paired interface.
FIG 5.
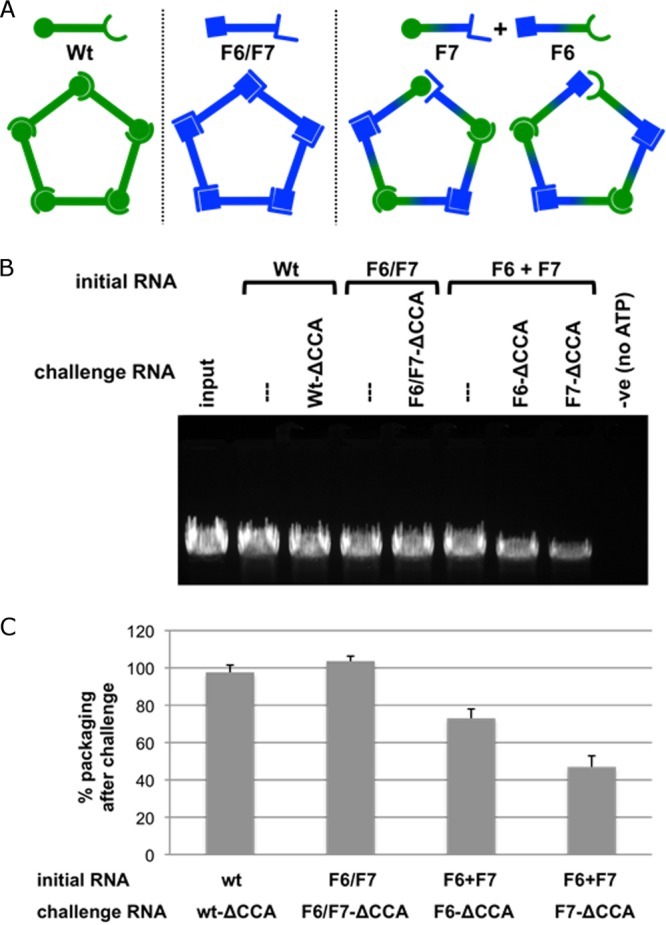
Effect of a mismatched interface in the pRNA ring on pRNA assembly and DNA packaging. (A) Schematic illustrating pentameric pRNA ring formation. The wt and the double mutant F6/F7 generate closed pRNA rings, while the dimer pair F6+F7 generates a single mismatched interface. The open symbol end represents the CE-loop of pRNA, and the closed symbol end represents the D-loop. The loop sequences (CE-45–48 and D-82–85 loops) for the pRNAs are as follows: for the wt, AACC and GGUU, respectively; for F6/F7, GCGA and UCGC, respectively; for F6, GCGA and GGUU, respectively; and for F7, AACC and UCGC, respectively) (17). (B) Effect on DNA packaging using assembled pRNA-prohead complexes challenged with pRNA containing the lethal ΔCCA mutation. The initial RNA is the pRNA form used to reconstitute RNA-free proheads. The challenge RNA (ΔCCA mutation-containing pRNA in corresponding pseudoknot sequence backgrounds) is then added to the prohead-pRNA complexes, and after incubation, the mixture is assessed in the in vitro DNA-packaging assay (see Materials and Methods). After the assay is completed, unpackaged DNA is digested by the addition of DNase I. The packaged, protected DNA is then extracted and analyzed by agarose gel electrophoresis (34) and the effect on DNA packaging assessed (see Materials and Methods). Vertical lines above lanes indicate nonchallenged samples. (C) Histogram showing packaging efficiency in the presence of the challenge pRNA containing the ΔCCA mutation compared to that for the nonchallenged sample (taken as 100%). For wt pRNA challenged with wt-ΔCCA, the packaging efficiency was 97.6% ± 3.9% (compare the second and third lanes); for the F6/F7 double mutant challenged with F6/F7-ΔCCA, it was 103.6% ± 2.7% (compare the fourth and fifth lanes); and for the dimer pair F6 +F7, it was 73.0% ± 5.0% for F6-ΔCCA challenge (compare the sixth and seventh lanes) and 47.0% ± 5.9% for F7-ΔCCA challenge (compare the sixth and eight lanes). n = 3 experiments; standard deviations are reported, and a single representative gel is shown.
Proheads with wild-type pRNA bound had similar packaging activities even when challenged with the lethal ΔCCA pRNA, demonstrating that no pRNA exchange occurs (Fig. 5B, second and third lanes). Similarly, the double mutant F6/F7, which creates a closed ring from mutant sequences, also retained high packaging activity in the presence of the lethal ΔCCA pRNA, indicating there is no pRNA exchange (Fig. 5B, fourth and fifth lanes). In contrast, with the ordered dimer pair F6+F7 pRNA, which would leave a mismatched intersubunit interface, addition of F6- or F7-ΔCCA pRNA decreased packaging by ∼25% to 50% from that for the unchallenged sample, indicating that there was exchange of bound pRNA with the free ΔCCA pRNA (Fig. 5B, seventh and eighth lanes versus sixth lane). This effect was specific to proheads with the assembled ordered dimer pair F6+F7 rings; addition of the challenger RNA F6- or F7-ΔCCA to proheads with assembled wild-type or double mutant F6/F7 rings did not impact DNA packaging efficiency (data not shown). Thus, the self-complementary wt and F6/F7 pRNAs that would yield a closed ring are unaffected by the addition of ΔCCA-pRNA, while for the dimer pair F6+F7 pRNA, the lack of ring closure due to the mismatched interface creates an opportunity for RNA exchange. Of note, if the pRNA were organized as a hexamer on the prohead (or any even-numbered oligomer), the dimer pair F6+F7 would create a closed ring and thus would be expected to display packaging behavior similar to that of the self-complementary wt and F6/F7 rings, which can create closed rings for any given symmetry. This is clearly not the case. Thus, the reduction in packaging observed with F6+F7 is consistent only with the pentameric ring containing a mismatched interface. Taken together, these results strongly support a pentameric RNA ring on the head and assign function to the intermolecular interaction as the event causing ring closure that “locks” the pRNA on the head. They also suggest that a single mismatched interface can be tolerated in DNA packaging. It has been shown recently that there is functional asymmetry in ϕ29 motor operation (54). During each motor cycle in packaging, only four of the ATPase subunits couple ATP hydrolysis to DNA translocation, while the fifth ATPase subunit is thought to function as a regulatory subunit that serves to align the DNA and motor in each cycle. This inherent asymmetry in motor operation may explain how the motor can accommodate some types of asymmetry in the other motor components, in this case, a single mismatched interface in the pentameric pRNA ring.
Conclusion.
The oligomerization state of the pRNA on the prohead has been a source of considerable controversy, and extensive cases for both hexameric and pentameric pRNAs have been made. Most recently, a model of a hexameric pRNA was built by incorporating the X-ray crystal structure of a monomeric fragment (3WJ) of the prohead-binding domain of the pRNA into a previous computational model of a hexameric pRNA that was based on constraints from genetic, biochemical, and biophysical data (49, 50). As noted above, the crystallization construct this model used did not include the CE- and D-loops, which are necessary for the essential intermolecular base pairing and ring formation. Indeed, the structure had only a monomer in the asymmetric unit, and application of crystallographic I4 space group symmetry builds only a tetramer. Hence, the X-ray data presented by Zhang et al. (49) do not provide evidence for a hexameric pRNA. Further, the model fit takes into account only interactions with an isolated connector, which we have shown to be relatively weak, whereas the clearly demonstrated extensive interactions between pRNA and the capsid are not taken into account.
The density corresponding to the pRNA in the asymmetric reconstruction of the connector-pRNA vertex presented here is clearly pentameric. Given the ability to resolve the known symmetry of the connector, this result is incompatible with a hexameric pRNA on the prohead. Furthermore, the excellent fit of the X-ray structure of the prohead-binding/oligomerization domain of the pRNA (18) and the NMR structure of the CCA bulge (47) to the cryo-EM structure demonstrates that several independent methods give rise to essentially identical structures. It is difficult to account for such a result if the structures are not truly similar, suggesting that the observed cryo-EM density corresponding to the pRNA reflects its true structure and organization on the prohead. This fitting also indicates that although the pRNA is close to the connector, it makes much more extensive contacts with the 5-fold symmetric head than with the 12-fold symmetric connector. Here we also show a domain in the capsid protein undergoing a conformational change necessary to accommodate binding of the pRNA E-loop. This result explains a previous observation that pRNA can bind to proheads even when connectors have been dislodged from the unique vertex (9), indicating that the pRNA's final binding site is primarily on the 5-fold symmetric capsid rather than on the 12/6-fold symmetric connector. Additionally, the genetic and biochemical experiments described above to probe ring closure are consistent with only an odd-numbered pRNA ring.
The availability of a well-defined packaging system that allows the generation and manipulation of various motor assembly intermediates, combined with the ability to visualize such intermediates, provides a powerful means of dissecting a complex molecular machine. The data presented here suggest a coordinated assembly pathway for the pentameric pRNA on ϕ29 procapsids wherein (i) the pRNA initially recognizes and interacts with the N terminus of the connector; (ii) rotation of the Ig-like domain in capsid proteins circumscribing the connector pRNA vertex uncovers a tight binding site for the E-loop of the pRNA to attach to the prohead; and (iii) formation of intermolecular pseudoknots between adjacent subunits closes the pRNA ring and locks the pRNA on the prohead, possibly providing a route of communication between ATPase subunits in the motor.
The ability to visualize a packaging motor at a high resolution opens a new critical avenue of investigation, applicable for any of the dsDNA phages, since it complements recent structural and single-molecule advances. Recently, several atomic structures of various motor components have been generated, and single-molecule studies probing the dynamics of packaging have shown highly coordinated motor action and a division of labor among motor subunits, as both translocating and regulatory subunits are suggested (5, 54). Visualization of the motor in various stages of assembly and, importantly, the motor in action will document the relationship between motor components and the movement of protein subdomains in DNA translocation that will provide key information for the continued development of mechanistic models of DNA packaging.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants GM095516 (to S.G. and M.C.M.) and GM059604 (to S.G.) from the National Institutes of Health.
We also acknowledge the Sealy Center for Structural and Molecular Biophysics for support of the UTMB cryo-EM core facility. We also thank the Characterization Facility, College of Science and Engineering, University of Minnesota, for use of the Tecnai T30 TEM.
Footnotes
Published ahead of print 8 January 2014
REFERENCES
- 1.Earnshaw WC, Casjens SR. 1980. DNA packaging by the double-stranded DNA bacteriophages. Cell 21:319–331. 10.1016/0092-8674(80)90468-7 [DOI] [PubMed] [Google Scholar]
- 2.Johnson JE, Chiu W. 2007. DNA packaging and delivery machines in tailed bacteriophages. Curr. Opin. Struct. Biol. 17:237–243. 10.1016/j.sbi.2007.03.011 [DOI] [PubMed] [Google Scholar]
- 3.Rao VB, Feiss M. 2008. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 42:647–681. 10.1146/annurev.genet.42.110807.091545 [DOI] [PubMed] [Google Scholar]
- 4.Grimes S, Jardine PJ, Anderson D. 2002. Bacteriophage ϕ29 DNA packaging. Adv. Virus Res. 58:255–294. 10.1016/S0065-3527(02)58007-6 [DOI] [PubMed] [Google Scholar]
- 5.Morais MC. 2012. The dsDNA packaging motor in bacteriophage ϕ29. Adv. Exp. Med. Biol. 726:511–547. 10.1007/978-1-4614-0980-9_23 [DOI] [PubMed] [Google Scholar]
- 6.Lebedev AA, Krause MH, Isidro AL, Vagin AA, Orlova EV, Turner J, Dodson EJ, Tavares P, Antson AA. 2007. Structural framework for DNA translocation via the viral portal protein. EMBO J. 26:1984–1994. 10.1038/sj.emboj.7601643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olia AS, Prevelige PE, Jr, Johnson JE, Cingolani G. 2011. Three-dimensional structure of a viral genome-delivery portal vertex. Nat. Struct. Mol. Biol. 18:597–603. 10.1038/nsmb.2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simpson AA, Tao Y, Leiman PG, Badasso MO, He Y, Jardine PJ, Olson NH, Morais MC, Grimes S, Anderson DL, Baker TS, Rossmann MG. 2000. Structure of the bacteriophage ϕ29 DNA packaging motor. Nature 408:745–750. 10.1038/35047129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morais MC, Koti JS, Bowman VD, Reyes-Aldrete E, Anderson DL, Rossmann MG. 2008. Defining molecular and domain boundaries in the bacteriophage ϕ29 DNA packaging motor. Structure 16:1267–1274. 10.1016/j.str.2008.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun S, Kondabagil K, Draper B, Alam TI, Bowman VD, Zhang Z, Hegde S, Fokine A, Rossmann MG, Rao VB. 2008. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell 135:1251–1262. 10.1016/j.cell.2008.11.015 [DOI] [PubMed] [Google Scholar]
- 11.Andrews BT, Catalano CE. 2012. The enzymology of a viral genome packaging motor is influenced by the assembly state of the motor subunits. Biochemistry 51:9342–9353. 10.1021/bi300890y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz C, De Donatis GM, Fang H, Guo P. 2013. The ATPase of the ϕ29 DNA packaging motor is a member of the hexameric AAA+ superfamily. Virology 443:20–27. 10.1016/j.virol.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo P, Grimes S, Anderson D. 1986. A defined system for in vitro packaging of DNA-gp3 of the Bacillus subtilis bacteriophage ϕ29. Proc. Natl. Acad. Sci. U. S. A. 83:3505–3509. 10.1073/pnas.83.10.3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson DL, Reilly BE. 1993. Morphogenesis of bacteriophage ϕ29, p 859–867 In Hoch JA, Losick R, Sonenshein A. (ed), Bacillus subtilis and other gram-positive bacteria: physiology, biochemistry and molecular genetics. ASM Press, Washington, DC [Google Scholar]
- 15.Guo P, Zhang C, Chen C, Garver K, Trottier M. 1998. Inter-RNA interaction of phage ϕ29 pRNA to form a hexameric complex for viral DNA transportation. Mol. Cell 2:149–155. 10.1016/S1097-2765(00)80124-0 [DOI] [PubMed] [Google Scholar]
- 16.Guo PX, Erickson S, Anderson D. 1987. A small viral RNA is required for in vitro packaging of bacteriophage ϕ29 DNA. Science 236:690–694. 10.1126/science.3107124 [DOI] [PubMed] [Google Scholar]
- 17.Zhang F, Lemieux S, Wu X, St-Arnaud D, McMurray CT, Major F, Anderson D. 1998. Function of hexameric RNA in packaging of bacteriophage ϕ29 DNA in vitro. Mol. Cell 2:141–147. 10.1016/S1097-2765(00)80123-9 [DOI] [PubMed] [Google Scholar]
- 18.Ding F, Lu C, Zhao W, Rajashankar KR, Anderson DL, Jardine PJ, Grimes S, Ke A. 2011. Structure and assembly of the essential RNA ring component of a viral DNA packaging motor. Proc. Natl. Acad. Sci. U. S. A. 108:7357–7362. 10.1073/pnas.1016690108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morais MC, Choi KH, Koti JS, Chipman PR, Anderson DL, Rossmann MG. 2005. Conservation of the capsid structure in tailed dsDNA bacteriophages: the pseudoatomic structure of ϕ29. Mol. Cell 18:149–159. 10.1016/j.molcel.2005.03.013 [DOI] [PubMed] [Google Scholar]
- 20.Morais MC, Tao Y, Olson NH, Grimes S, Jardine PJ, Anderson DL, Baker TS, Rossmann MG. 2001. Cryoelectron-microscopy image reconstruction of symmetry mismatches in bacteriophage ϕ29. J. Struct. Biol. 135:38–46. 10.1006/jsbi.2001.4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shu D, Zhang H, Jin J, Guo P. 2007. Counting of six pRNAs of ϕ29 DNA-packaging motor with customized single-molecule dual-view system. EMBO J. 26:527–537. 10.1038/sj.emboj.7601506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C, Sheng S, Shao Z, Guo P. 2000. A dimer as a building block in assembling RNA. A hexamer that gears bacterial virus ϕ29 DNA-translocating machinery. J. Biol. Chem. 275:17510–17516. 10.1074/jbc.M909662199 [DOI] [PubMed] [Google Scholar]
- 23.Chen C, Zhang C, Guo P. 1999. Sequence requirement for hand-in-hand interaction in formation of RNA dimers and hexamers to gear ϕ29 DNA translocation motor. RNA 5:805–818. 10.1017/S1355838299990350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atz R, Ma S, Gao J, Anderson DL, Grimes S. 2007. Alanine scanning and Fe-BABE probing of the bacteriophage ϕ29 prohead RNA-connector interaction. J. Mol. Biol. 369:239–248. 10.1016/j.jmb.2007.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao F, Moll WD, Guo S, Guo P. 2005. Binding of pRNA to the N-terminal 14 amino acids of connector protein of bacteriophage ϕ29. Nucleic Acids Res. 33:2640–2649. 10.1093/nar/gki554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitamura A, Jardine PJ, Anderson DL, Grimes S, Matsuo H. 2008. Analysis of intermolecular base pair formation of prohead RNA of the phage ϕ29 DNA packaging motor using NMR spectroscopy. Nucleic Acids Res. 36:839–848. 10.1093/nar/gkm874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reid RJ, Bodley JW, Anderson D. 1994. Identification of bacteriophage ϕ29 prohead RNA domains necessary for in vitro DNA-gp3 packaging. J. Biol. Chem. 269:9084–9089 [PubMed] [Google Scholar]
- 28.Reid RJ, Zhang F, Benson S, Anderson D. 1994. Probing the structure of bacteriophage ϕ29 prohead RNA with specific mutations. J. Biol. Chem. 269:18656–18661 [PubMed] [Google Scholar]
- 29.Zhao W, Morais MC, Anderson DL, Jardine PJ, Grimes S. 2008. Role of the CCA bulge of prohead RNA of bacteriophage ϕ29 in DNA packaging. J. Mol. Biol. 383:520–528. 10.1016/j.jmb.2008.08.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Wilkinson MF. 2001. Deletion mutagenesis of large (12-kb) plasmids by a one-step PCR protocol. Biotechniques 31:722–724 [DOI] [PubMed] [Google Scholar]
- 31.Reid RJ, Bodley JW, Anderson D. 1994. Characterization of the prohead-pRNA interaction of bacteriophage ϕ29. J. Biol. Chem. 269:5157–5162 [PubMed] [Google Scholar]
- 32.Grimes S, Anderson D. 1997. The bacteriophage ϕ29 packaging proteins supercoil the DNA ends. J. Mol. Biol. 266:901–914. 10.1006/jmbi.1996.0843 [DOI] [PubMed] [Google Scholar]
- 33.Mellado RP, Moreno F, Vinuela E, Salas M, Reilly BE, Anderson DL. 1976. Genetic analysis of bacteriophage ϕ29 of Bacillus subtilis: integration and mapping of reference mutants of two collections. J. Virol. 19:495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grimes S, Anderson D. 1989. In vitro packaging of bacteriophage ϕ29 DNA restriction fragments and the role of the terminal protein gp3. J. Mol. Biol. 209:91–100. 10.1016/0022-2836(89)90172-1 [DOI] [PubMed] [Google Scholar]
- 35.Guo PX, Erickson S, Xu W, Olson N, Baker TS, Anderson D. 1991. Regulation of the phage ϕ29 prohead shape and size by the portal vertex. Virology 183:366–373. 10.1016/0042-6822(91)90149-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grimes S, Ma S, Gao J, Atz R, Jardine PJ. 2011. Role of ϕ29 connector channel loops in late-stage DNA packaging. J. Mol. Biol. 410:50–59. 10.1016/j.jmb.2011.04.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frank J, Wagenknecht T, McEwen BF, Marko M, Hsieh CE, Mannella CA. 2002. Three-dimensional imaging of biological complexity. J. Struct. Biol. 138:85–91. 10.1016/S1047-8477(02)00019-9 [DOI] [PubMed] [Google Scholar]
- 38.Morais MC, Kanamaru S, Badasso MO, Koti JS, Owen BA, McMurray CT, Anderson DL, Rossmann MG. 2003. Bacteriophage ϕ29 scaffolding protein gp7 before and after prohead assembly. Nat. Struct. Biol. 10:572–576. 10.1038/nsb939 [DOI] [PubMed] [Google Scholar]
- 39.Xiang Y, Morais MC, Battisti AJ, Grimes S, Jardine PJ, Anderson DL, Rossmann MG. 2006. Structural changes of bacteriophage ϕ29 upon DNA packaging and release. EMBO J. 25:5229–5239. 10.1038/sj.emboj.7601386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker TS, Olson NH, Fuller SD. 1999. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol. Mol. Biol. Rev. 63:862–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97. 10.1006/jsbi.1999.4174 [DOI] [PubMed] [Google Scholar]
- 42.Chang J, Weigele P, King J, Chiu W, Jiang W. 2006. Cryo-EM asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure 14:1073–1082. 10.1016/j.str.2006.05.007 [DOI] [PubMed] [Google Scholar]
- 43.Chen DH, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, Baker D, King JA, Chiu W. 2011. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc. Natl. Acad. Sci. U. S. A. 108:1355–1360. 10.1073/pnas.1015739108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo F, Liu Z, Vago F, Ren Y, Wu W, Wright ET, Serwer P, Jiang W. 2013. Visualization of uncorrelated, tandem symmetry mismatches in the internal genome packaging apparatus of bacteriophage T7. Proc. Natl. Acad. Sci. U. S. A. 110:6811–6816. 10.1073/pnas.1215563110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. 2006. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science 312:1791–1795. 10.1126/science.1127981 [DOI] [PubMed] [Google Scholar]
- 46.Tang J, Olson N, Jardine PJ, Grimes S, Anderson DL, Baker TS. 2008. DNA poised for release in bacteriophage ϕ29. Structure 16:935–943. 10.1016/j.str.2008.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harjes E, Kitamura A, Zhao W, Morais MC, Jardine PJ, Grimes S, Matsuo H. 2012. Structure of the RNA claw of the DNA packaging motor of bacteriophage ϕ29. Nucleic Acids Res. 40:9953–9963. 10.1093/nar/gks724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris S, Schroeder SJ. 2010. Nuclear magnetic resonance structure of the prohead RNA E-loop hairpin. Biochemistry 49:5989–5997. 10.1021/bi100393r [DOI] [PubMed] [Google Scholar]
- 49.Zhang H, Endrizzi JA, Shu Y, Haque F, Sauter C, Shlyakhtenko LS, Lyubchenko Y, Guo P, Chi YI. 2013. Crystal structure of 3WJ core revealing divalent ion-promoted thermostability and assembly of the ϕ29 hexameric motor pRNA. RNA 19:1226–1237. 10.1261/rna.037077.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoeprich S, Guo P. 2002. Computer modeling of three-dimensional structure of DNA-packaging RNA (pRNA) monomer, dimer, and hexamer of ϕ29 DNA packaging motor. J. Biol. Chem. 277:20794–20803. 10.1074/jbc.M112061200 [DOI] [PubMed] [Google Scholar]
- 51.Baumann RG, Mullaney J, Black LW. 2006. Portal fusion protein constraints on function in DNA packaging of bacteriophage T4. Mol. Microbiol. 61:16–32. 10.1111/j.1365-2958.2006.05203.x [DOI] [PubMed] [Google Scholar]
- 52.Hugel T, Michaelis J, Hetherington CL, Jardine PJ, Grimes S, Walter JM, Falk W, Anderson DL, Bustamante C. 2007. Experimental test of connector rotation during DNA packaging into bacteriophage ϕ29 capsids. PLoS Biol. 5:e59. 10.1371/journal.pbio.0050059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trottier M, Guo P. 1997. Approaches to determine stoichiometry of viral assembly components. J. Virol. 71:487–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chistol G, Liu S, Hetherington CL, Moffitt JR, Grimes S, Jardine PJ, Bustamante C. 2012. High degree of coordination and division of labor among subunits in a homomeric ring ATPase. Cell 151:1017–1028. 10.1016/j.cell.2012.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]