

ABSTRACT
N-acetyl- and N-glycolylneuraminic acids (Sia) and α2β1 integrin are frequently used by rotaviruses as cellular receptors through recognition by virion spike protein VP4. The VP4 subunit VP8*, derived from Wa rotavirus, binds the internal N-acetylneuraminic acid on ganglioside GM1. Wa infection is increased by enhanced internal Sia access following terminal Sia removal from main glycan chains with sialidase. The GM1 ligand cholera toxin B (CTB) reduces Wa infectivity. Here, we found sialidase treatment increased cellular GM1 availability and the infectivity of several other human (including RV-3) and animal rotaviruses, typically rendering them susceptible to methyl α-d-N-acetylneuraminide treatment, but did not alter α2β1 usage. CTB reduced the infectivity of these viruses. Aceramido-GM1 inhibited Wa and RV-3 infectivity in untreated and sialidase-treated cells, and GM1 supplementation increased their infectivity, demonstrating the importance of GM1 for infection. Wa recognition of α2β1 and internal Sia were at least partially independent. Rotavirus usage of GM1 was mapped to VP4 using virus reassortants, and RV-3 VP8* bound aceramido-GM1 by saturation transfer difference nuclear magnetic resonance (STD NMR). Most rotaviruses recognizing terminal Sia did not use GM1, including RRV. RRV VP8* interacted minimally with aceramido-GM1 by STD NMR. Unusually, TFR-41 rotavirus infectivity depended upon terminal Sia and GM1. Competition of CTB, Sia, and/or aceramido-GM1 with cell binding by VP8* from representative rotaviruses showed that rotavirus Sia and GM1 preferences resulted from VP8*-cell binding. Our major finding is that infection by human rotaviruses of commonly occurring VP4 serotypes involves VP8* binding to cell surface GM1 glycan, typically including the internal N-acetylneuraminic acid.
IMPORTANCE Rotaviruses, the major cause of severe infantile gastroenteritis, recognize cell surface receptors through virus spike protein VP4. Several animal rotaviruses are known to bind sialic acids at the termini of main carbohydrate chains. Conversely, only a single human rotavirus is known to bind sialic acid. Interestingly, VP4 of this rotavirus bound to sialic acid that forms a branch on the main carbohydrate chain of the GM1 ganglioside. Here, we use several techniques to demonstrate that other human rotaviruses exhibit similar GM1 usage properties. Furthermore, binding by VP4 to cell surface GM1, involving branched sialic acid recognition, is shown to facilitate infection. In contrast, most animal rotaviruses that bind terminal sialic acids did not utilize GM1 for VP4 cell binding or infection. These studies support a significant role for GM1 in mediating host cell invasion by human rotaviruses.
INTRODUCTION
Rotaviruses are a leading cause of severe infantile gastroenteritis through their infection of intestinal epithelial cells. The outer layer of the infectious particle is comprised of VP4 spikes projecting through a VP7 shell (1–4). VP4 mediates rotavirus-cell attachment, and VP7 and VP4 determine rotavirus G and P serotype specificities, respectively (5, 6). Trypsin cleavage of VP4 into VP8* and VP5* subunits activates virion infectivity but is not required for virus attachment to host cell carbohydrate receptors (5, 7). Two VP8* lectin-like domains located at the activated spike tip can mediate attachment to cell surface sialylglycoconjugates containing the N-acylneuraminic acids N-acetylneuraminic acid and N-glycolylneuraminic acid (sialic acids; Sia) (1, 3). VP5* subunits within the spike body expose a recognition sequence for the α2β1 integrin (3, 8, 9). During rotavirus-cell attachment and entry, activated VP4 undergoes a major conformational change that facilitates membrane disruption by VP5* hydrophobic domains (9, 10). VP4 may define rotavirus internalization, which commonly but not universally appears to involve clathrin-mediated endocytosis (11–13).
VP4 recognition of Sia, gangliosides, and/or α2β1 are important for cell binding and infection (8, 14–20). Histoblood group antigens bound by VP8* also are implicated as cellular receptors for certain rotaviruses (21, 22). Additional proposed entry factors include αxβ2 and αvβ3 integrins, heat shock cognate protein 70, and lipid membrane microdomains sensitive to cholesterol depletion (8, 15, 23–26). Naturally occurring mammalian rotaviruses exhibit various abilities to utilize Sia, gangliosides, and integrins (7, 14, 23, 27–31). Rotavirus usage of α2β1 and terminal Sia each segregate independently with P serotype (15, 32). However, how α2β1 usage relates to Sia and ganglioside recognition is unknown. The α2β1 integrin itself contains substantial amounts of N-acylneuraminic acid (33).
Terminal Sia dependence (sialidase sensitivity) has been demonstrated for several animal rotaviruses by infectivity blockade with the N-acylneuraminic acid-based glycosides methyl α-d-N-acetylneuraminide (Neu5Acα2Me) and methyl α-d-N-glycolylneuraminide (Neu5Gcα2Me), as well as with cellular treatment with bacterial sialidases (31, 34–38). These sialidases efficiently remove only terminal and unbranched N-acylneuraminic acids from host cells in culture, including from gangliosides GD1a and GM3 (39). Ganglioside GM1 has internal (branched) N-acylneuraminic acid and is resistant to these sialidases (39, 40).
Many rotaviruses were originally reported to show unchanged infectivity in sialidase-treated cells and have been termed sialidase insensitive or sialidase resistant (29, 41). However, sialidase treatment increases the infectivity of sialidase-insensitive human rotaviruses Wa and DS-1 (31, 34). Apart from elucidation of receptor specificity, the biological relevance of this infectivity enhancement lies in the potential for bacterial sialidases present in the gut to remove terminal Sia from epithelial cells and increase rotavirus infection. In contrast to the sialidase-sensitive, simian rotavirus RRV, Wa infection is inhibited by cellular treatment with the cholera toxin B subunit (CTB), a GM1 glycan ligand, implicating GM1 in Wa infectivity (7, 31, 42). Wa infectivity in sialidase-treated cells is substantially reduced by Neu5Acα2Me, indicating a role for internal N-acetylneuraminic acid residues such as those present in GM1 (31). This role was further defined by saturation transfer difference nuclear magnetic resonance (STD NMR) experiments that demonstrated Wa VP8* binding to aceramido-GM1 (a-GM1), involving the internal N-acetylneuraminic acid of a-GM1 (31). Although reduced by cellular CTB treatment, Wa infectivity in untreated cells is not inhibited by Neu5Acα2Me. This may be because Neu5Acα2Me can only compete with VP8* binding to GM1 via the internal Sia, whereas CTB has an extensive a-GM1 binding footprint [the I3-sialosylgangliotriaose moiety, Galβ1,3GalNAcβ1,4(Siaα2,3)Galβ1], and high (sub-μM) GM1 affinity (43, 44). The possibility also exists that Wa preferentially uses alternative receptors on untreated cells, such as the α2β1 integrin, in a manner that is unaffected by Neu5Acα2Me competition. It is currently unknown if sialidase-insensitive rotaviruses other than Wa also share these GM1 recognition properties.
Here, we establish the role of GM1 in cell binding by VP8* and rotavirus infectivity in relation to virus usage of α2β1 and Sia. Sialidase treatment was found to enhance cellular GM1 surface exposure and the infectivity of several rotaviruses, rendering most of them susceptible to Neu5Acα2Me treatment, but it did not alter the extent of rotavirus usage of α2β1. CTB and/or a-GM1 competition reduced the infectivity of these viruses, indicating their GM1 dependence. Wa recognition of α2β1 and internal Sia were at least partially independent. Rotavirus GM1 usage was mapped to VP4 using rotavirus reassortants. STD NMR spectra revealed that recombinant VP8* of RV-3 rotavirus recognized a-GM1, whereas RRV VP8* showed only weak STD NMR effects for a-GM1, suggesting a low affinity for the ligand. Analysis of recombinant VP8*-cell binding by flow cytometry showed that this binding determines the Sia recognition properties of sialidase-sensitive bovine (NCDV) and porcine (CRW-8) rotaviruses, as well as sialidase-insensitive human (Wa, RV-3) rotaviruses. These data provide evidence that several human rotaviruses utilize GM1 glycans during infection.
MATERIALS AND METHODS
Viruses and cells.
The origins, cultivation in MA104 cells, and infectivity assay of human rotavirus strains Wa (G1P1A[8]), RV-5 (G2P1B[4]), RV-3 (G3P2A[6]), and S12/85 (G3P2A[6]); monkey rotavirus RRV (G3P5B[3]); bovine rotaviruses NCDV (G6P6[1]) and UK (G6P7[5]); and porcine rotaviruses CRW-8 (G3P9[7]) and TFR-41 (G5P9[7]) have been described previously (15, 45–48). Reassortant rotaviruses 12-1 and 28-1 were produced and characterized previously (49). 12-1 contains VP2, VP7, and NSP5/6 from RRV, with the remaining genes coming from UK, and 28-1 contains VP3, VP4, and NSP5/6 genes from RRV, with the remaining genes coming from UK.
Enzymes, reagents, and antibodies.
Working solutions of sialidase from Vibrio cholerae (Sigma) were prepared in Dulbecco's modification of Eagle's medium containing 2 mM l-glutamine (Sigma-Aldrich), 20 mM HEPES (Roche), and antibiotics (DMEM), which was brought to pH 5.5 with HCl. Neu5Acα2Me and Neu5Gcα2Me were synthesized in house by methods adapted from those previously described (50, 51) and dissolved in DMEM at neutral pH. Gangliosides GM1 and GM3 (from bovine milk) were obtained from Avanti, and their aceramido (glycan) forms, a-GM1 and a-GM3, were purchased from Elicityl. Neutralizing monoclonal antibodies directed to Wa VP4 (1A10), RV-5 VP4 (RV-5:2), RV-3 and CRW-8 VP7 (RV-3:1), and RRV VP4 (2G4) were produced as mouse ascites fluids as previously described (18, 52, 53). Monoclonal antibody H7/D2 was produced as ascites fluid using gradient-purified UK rotavirus as the mouse immunogen and shown to be directed to VP7 and to neutralize UK infectivity, as previously described (54, 55). These antibodies showed reciprocal neutralization titers of ≥3 × 104 against the rotavirus strains of homologous serotype (18, 52, 53) and were used at dilutions 10-fold higher than their endpoint titers with each rotavirus strain. Monoclonal antibodies AK7 (directed to the α2 subunit of the α2β1 integrin) and MOPC21 (isotype control) were obtained and used as described before (18, 56). The B subunit of the Vibrio cholerae toxin (CTB; Sigma) was diluted in DMEM as before (31). Fluorescein isothiocyanate isomer I (FITC) and CTB conjugated to FITC (FITC-CTB) were purchased from Sigma and diluted as described above.
Production of recombinant rotavirus VP8* proteins.
The RRV, CRW-8, and NCDV VP8* cores (amino acids [aa] 64 to 224 of VP4) were expressed as glutathione S-transferase (GST) fusion proteins from cloned VP4 gene cDNA as previously reported (37, 57, 58). The wild-type CRW-8 VP8* containing Pro rather than Ser at position 157 was produced (38). The Wa VP8* core (aa 64 to 223) was expressed as described before (59). RV-3 VP8*64-223 was cloned, expressed, and purified as described for the Wa VP8* core. In brief, cDNA produced from viral double-stranded RNA (dsRNA) was used as the template for PCR to produce the VP8* gene fragment, which was cloned into the pGEX-4T-1 vector. DNA sequencing indicated that the predicted amino acid sequence of pGEX-RV-3-VP8* was identical to the most recent database entry for RV-3 VP4 (GenBank accession no. FJ998273). Our sequence and FJ998273 both differ from the original published sequence (GenBank accession no. U16299) in showing serine rather than proline at amino acid position 71 (48, 60). The proteins were soluble, and the same batches proved suitable for structural studies (7, 31, 61).
Detection of surface GM1 on adherent cells by fluorimetry.
Washed, confluent MA104 cell monolayers in 96-well trays were treated with sialidase or DMEM at pH 5.5 as described above, cooled to 4°C, washed, and reacted with equimolar amounts of CTB-FITC or unconjugated FITC at 4°C for 1 h. Fluorescence by washed cell monolayers was detected in a FLUOStar Omega microplate reader (BMG Labtech). CTB-FITC fluorescence was adjusted to compensate for background levels of FITC fluorescence, which comprised <15% of the CTB-FITC reading.
Assays for inhibition of virus infection.
The assays for inhibition of virus infection were adapted from previously described methods used in related studies (15, 16, 31, 34, 48). Rotavirus infectivity was activated with porcine pancreatic trypsin (Sigma) at 20 μg/ml (human strains) or 10 μg/ml (animal strains) for 20 min at 37°C. Neu5Acα2Me, Neu5Gcα2Me, a-GM1, or a-GM3 (10 mM), rotavirus-neutralizing antibody, or diluent (DMEM) was incubated with trypsin-activated rotavirus for 1 h at 37°C. Confluent MA104 cell monolayers containing 8 × 104 cells were treated with sialidase at 0.52 U/ml or maintained in DMEM at pH 5.5 (untreated cells) for 1 h at 37°C. Incubated virus-ligand mixtures and treated cells were separately cooled to 4°C. At this temperature, virus binds but cannot enter cells, and N-acylneuraminic acid regeneration on cells is prevented. Sialidase was removed from cells and replaced with the cold virus-ligand mixture. Sialidase-treated and untreated cells were inoculated with virus or virus-ligand mixtures at the optimum multiplicity of infection (determined as fluorescent cell-forming units [FCFU]/cell) of 0.02 at 4°C for 1 h. Following inoculum removal and washing as described above, infected cells were incubated for 15 h at 37°C in 95% (vol/vol) air with 5% (vol/vol) CO2. Virus titers, in FCFU per ml, were determined in acetone-fixed cell monolayers by indirect immunofluorescence as described before (50). For CTB competition assays, prior to virus inoculation and infectivity assay as described above, cells were treated for 1 h at 4°C with DMEM, CTB at 1.0 μg/ml or 0.1 μg/ml, or 1.0 μg/ml CTB that had been heat inactivated at 100°C for 10 min.
Ganglioside supplementation of cells.
The ganglioside supplementation method has been described previously (62). Briefly, MA104 cells were grown to 70% confluence in 96-well trays and incubated with DMEM supplemented with 10% (vol/vol) fetal bovine serum (DMEM-FBS) containing 3 μM GM1 or GM3 for 17 h or with DMEM-FBS alone. Medium containing the gangliosides was removed, and the cells were washed with additional medium to remove any unincorporated gangliosides. The cells were then infected with Wa or RV-3, and infectious titers were determined by indirect immunofluorescent staining as described above.
STD NMR spectroscopy of a-GM1 interactions with rotavirus VP8*.
STD NMR spectra were acquired with a Bruker 600 MHz Avance spectrometer at 288 K using a conventional 1H/13C/15N gradient cryoprobe. a-GM1 ligand (2.4 mM) in complex with 24 μM RV-3 VP8*, Wa VP8* or RRV VP8* as a GST fusion protein was prepared in 250 μl deuterated 20 mM phosphate buffer containing 70 mM NaCl, pD 7.1, to give a final ligand/protein ratio of 100:1. The protein was saturated with a cascade of 40 Gaussian-shaped pulses with a duration of 50 ms each at −0.1 ppm, resulting in a total saturation time of ∼2 s. The off-resonance frequency was set to 33 ppm, and a total of 4,096 scans were acquired. A WATERGATE sequence was used to suppress the residual deuterium protium oxide signal, and a spinlock filter with a strength of 5 kHz and duration of 10 ms was applied to suppress the protein background. As a control experiment, 2.4 mM a-GM1 in complex with 24 μM GST protein in the same phosphate buffer was measured using an identical NMR setup.
Flow-cytometric analysis of surface GM1 levels and VP8* binding.
Cells from confluent MA104 cell monolayers were placed in suspension by brief treatment with trypsin-EDTA or mechanical scraping, washed, resuspended in DMEM containing 10% (vol/vol) fetal bovine serum, and rested for 30 min at 37°C. Trypsin-EDTA was used, unless otherwise stated, to optimize single-cell production. Cells passed through a 40-μm filter were quantitated for viability in a hemocytometer by trypan blue exclusion and seeded into 96-well V-bottom trays (Nunc, Denmark) at 8 × 105 cells/well in medium containing 1.0% (vol/vol) fetal bovine serum. The following reactions were performed at 4°C. Cell surface GM1 was analyzed by staining with equimolar amounts of FITC-CTB or FITC for 45 min. Cell binding by GST-VP8* from Wa, RV-3, RRV, UK, CRW-8, and NCDV rotaviruses was assayed as previously described (30, 63). GST-VP8* at 5 to 600 μg/ml was reacted with cells for 45 min. Although GST protein equimolar to the GST-VP8* initially was reacted with the cells as a control, this was not routinely performed, as rabbit serum showed similar results (see below). Cell-bound protein was detected with rabbit antiserum to GST diluted 1:500. Similarly diluted, normal rabbit serum served as a negative control. Both of these sera showed undetectable antibodies to rotavirus (enzyme immunoassay and neutralization titers of <1:100). Bound rabbit antibodies were detected with FITC-conjugated goat anti-rabbit IgG (Invitrogen). For sialidase studies, cells seeded as described above were resuspended in DMEM at pH 5.5 in the presence or absence of 0.52 U sialidase and incubated for 1 h at 37°C. After cooling at 4°C for 15 min, cells were washed, reacted with GST-VP8*, and stained as described above. N-acylneuraminic acid blockade was conducted by mixing VP8* with Neu5Acα2Me, Neu5Gcα2Me, or DMEM for 1 h at 4°C prior to VP8* addition to cells. CTB competition was measured by cellular treatment with 1.0 μg/ml CTB or heat-inactivated CTB for 1 h at 4°C prior to GST-VP8* addition. Stained single cells were analyzed by flow cytometry as before (30). The level of FITC-CTB or GST-VP8* bound to cells relative to the negative control was expressed as the relative linear median fluorescence intensity (RLMFI), with an RLMFI value of ≥1.20 considered to indicate cell binding (30). The results provided are representative of those obtained from at least two independent experiments, each with 2 to 3 replicates.
Statistical analysis.
Student's t test or analysis of variance (ANOVA) was used, with significance set at the 95% level. For all assays, data represent the means of triplicate samples from at least two independent experiments, and error bars on graphs indicate the standard deviations (SD).
RESULTS
Sialidase treatment increased surface GM1 detection on cell monolayers.
Sialidase-treated cells support increased Wa infection, which is susceptible to Neu5Acα2Me competition (31). To determine if this related to altered GM1 glycan levels, CTB-FITC binding to sialidase-treated or control MA104 cell monolayers was assayed. Sialidase treatment increased bound CTB-FITC fluorescence (means ± SD) from 8,920 ± 2,470 U to 21,900 ± 6,190 U (2.5-fold) at 2 μg/ml and from 45,100 ± 7,490 U to 64,700 ± 10,600 U (1.4-fold) at 5 μg/ml of input (P = 0.0098 and P = 0.0019, respectively; data not shown). This increase in GM1 detection on sialidase-treated cells presumably resulted from the enzymatic removal of terminal N-acetylneuraminic acids from gangliosides such as GM3 and GD1a, as has been reported previously for other cell types (64).
Usage of internal N-acetylneuraminic acids and α2β1 integrin by Wa rotavirus was at least partially additive.
Wa uses α2β1 and recognizes the α2 subunit I domain (15, 28, 65). The abilities of Neu5Acα2Me (Fig. 1A), antibody to α2β1, and their combination to inhibit Wa infection were determined in untreated and sialidase-treated MA104 cells (Fig. 1B). As before (15, 31), anti-α2 antibody AK7 reduced Wa infectivity in untreated cells, whereas Wa incubation with Neu5Acα2Me had no effect. Consistent with this, combined treatment with anti-α2 antibody and Neu5Acα2Me reduced Wa infectivity by 42% ± 7%, an effect similar to anti-α2 alone. Wa infectivity increased >2.5-fold in sialidase-treated cells and was significantly reduced by Neu5Acα2Me (P < 0.0001), as found previously (31). Anti-α2 antibody reduced Wa infection after sialidase treatment (P < 0.0001). This reduction was proportionally similar to that in untreated cells, although it represented a greater reduction in virus titer (Fig. 1B). Combined treatment of Wa with anti-α2 antibody and Neu5Acα2Me produced a significantly greater infectivity reduction in sialidase-treated cells (51% ± 3%; P < 0.0001) than either antibody or Neu5Acα2Me alone (Fig. 1B). Thus, the extent of Wa usage of α2β1 is maintained in sialidase-treated cells, and this property is at least partially independent of Neu5Acα2Me inhibition.
FIG 1.

Effects of Neu5Acα2Me and antibody to α2β1 integrin on human rotavirus infection of untreated and sialidase-treated MA104 cells. (A) Chemical structures of monomeric N-acetylneuraminic acid and its corresponding methyl glycoside. (B to E) Rotavirus reacted with 10 mM Neu5Acα2Me or diluent (control) was adsorbed to cells treated with sialidase (0.52 U/ml) and/or α2β1 antibody (20 μg/ml). Infected cells were enumerated at 16 h postinfection following indirect immunofluorescent staining. Infectious virus titers are expressed as a percentage of the titers produced in control untreated cells. These control infectious titers, expressed as mean FCFU/ml ± SD, were 7.7 × 103 ± 1.3 × 103 (B), 1.2 × 104 ± 0.1 × 104 (C), 8.8 × 103 ± 0.7 × 103 (D), and 1.6 × 104 ± 0.3 × 104 (E). Positive-control neutralizing monoclonal antibodies (Ab) were 1A10 (Wa), RV-5:2 (RV-5), and RV-3:1 (RV-3; S12/85). Cells treated with isotype control MOPC21 at 20 μg/ml produced virus titers that were indistinguishable from those of untreated cells.
Several human rotaviruses showed enhanced infectivity, susceptibility to Neu5Acα2Me treatment, and maintenance of extent of α2β1 usage in sialidase-treated cells.
RV-5, but not RV-3 or S12/85, uses α2β1 (15). As expected, anti-α2 antibody reduced RV-5 but not RV-3 infectivity in untreated cells (Fig. 1C and D). Neu5Acα2Me did not affect these three viruses, either alone or in combination with anti-α2 (Fig. 1C, D, and E). Sialidase treatment increased RV-5, RV-3, and S12/85 infectivity (P < 0.0001) (Fig. 1C, D, and E), but it did not convert RV-3 to α2β1 dependence. This finding for RV-5 is consistent with the increase reported for DS-1, another G2P1B[4] rotavirus (34, 42). Neu5Acα2Me treatment reduced the elevated RV-5 and RV-3 titers (P < 0.0001), but interestingly, it did not affect S12/85 (Fig. 1E) (P = 0.38). Like Wa, the reduction in RV-5 infectivity due to anti-α2 was similar before (41% ± 8%) and after (31% ± 12%) sialidase treatment (Fig. 1C). Unlike Wa, the effect of anti-α2 and Neu5Acα2Me combined on RV-5 infection in sialidase-treated cells could not be analyzed due to our inability to sufficiently increase RV-5 infectivity.
Overall, the infectivity of human rotaviruses was increased after terminal N-acetylneuraminic acid removal, and typically they became sensitive to Neu5Acα2Me competition (Table 1). Taken together with the raised GM1 level on these sialidase-treated cells described above, these data suggest that cellular sialidase treatment increased the infectivity of these viruses through elevated GM1 availability. After terminal Sia removal, the extent of α2β1 usage was maintained, and α2β1 usage was least partly independent of Neu5Acα2Me sensitivity.
TABLE 1.
Summary of α2β1, GM1, and N-acetylneuraminic acid usage for infection by human rotaviruses
| Virus strain | α2β1 usagea | Untreated cell findings |
Infectivity in sialidase-treated cells |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Proposed internalization pathwayc | CTB inhibition of given propertyb (% at 1 μg/ml) |
Infectivity reduced by Neu5Acα2Me | Infectivity reduced by a-GM1e (% at 10 mM) | Increased | Reduced by Neu5Acα2Me | Reduced by a-GM1e (% at 10 mM) | |||
| VP8* bindinge | Infectivity | ||||||||
| RV-5 | + | CE | ND | 29 | −e | ND | +e | +e | ND |
| Wa | + | CE | 21 | 48 | −d | 57 | +d | +d | 35 |
| RV-3 | − | ND | 39 | 41 | −e | 49 | +e | +e | 43 |
| S12/85 | − | ND | ND | 48 | −e | ND | +e | −e | ND |
Shown in references 8, 15, 18, 32, and 35. The extent of α2β1 usage is not affected by sialidase treatment of cells, as shown in this study.
Shown in this study and reference 31 for Wa. GM1 usage also was shown by STD NMR detection of a-GM1 binding by Wa VP8* (31) and RV-3 VP8* (this study). CE, clathrin-mediated endocytosis; ND, not determined.
Data are from references 11 to 13. DS-1 uses CE (12). RV-5 and DS-1 share the G2P1B serotype, differing in VP8* sequence at amino acid positions 82, 89, and 160, so RV-5 also may enter cells by CE.
Shown in this study and reference 31.
Shown in this study.
Relative usage of Sia and α2β1 integrin by animal rotaviruses.
RRV virions and expressed RRV VP5* bind the α2 subunit I domain, and binding is abolished by mutation of the VP5* integrin recognition sequence or the I domain (15, 28, 30). CRW-8 infection requires terminal N-acylneuraminic acid on the glycan main chain (Fig. 2A) but not α2β1 (15, 16, 31). As before (29, 31, 36, 37), RRV (Fig. 2B) and CRW-8 (Fig. 2C) infectivity was substantially reduced by Neu5Acα2Me blockade or prior sialidase treatment. In untreated cells, anti-α2 antibody inhibited RRV infection by 36% ± 2% (Fig. 2B) (P = <0.0001) but not CRW-8 infection (Fig. 2C) (P = 0.49), as before (15). After sialidase treatment, anti-α2 inhibited RRV infection by 29% ± 8% (Fig. 2B) (P < 0.0001) but had no effect on CRW-8 (Fig. 2C) (P = 0.14). Neu5Acα2Me treatment of RRV and CRW-8 also did not alter their interactions with sialidase-treated cells (P > 0.05). These data confirmed that terminal Sia on glycan main chains are important receptors for these rotaviruses. Additionally, α2β1 integrin usage by RRV occurred to a similar extent during infection of untreated and sialidase-treated cells, and CRW-8 infection after sialidase treatment remained independent of α2β1. The infectivity of porcine rotavirus TFR-41 was highly sialidase sensitive (Fig. 2D), as previously reported (66). Unusually, however, TFR-41 infectivity in untreated cells was only slightly reduced by Neu5Acα2Me (20% ± 7%; P = 0.01) and was unaffected by Neu5Gcα2Me (P = 0.34). Neu5Acα2Me and Neu5Gcα2Me did not alter TFR-41 infection in sialidase-treated cells.
FIG 2.

Effects of Neu5Acα2Me, antibody to α2β1 integrin, and Neu5Gcα2Me on animal rotavirus infection of untreated and sialidase-treated MA104 cells. (A) Chemical structures of monomeric N-acylneuraminic acids and their corresponding methyl glycosides. (B to F) Rotavirus reacted with 10 mM Neu5Acα2Me, 10 mM Neu5Gcα2Me, or diluent (control) was adsorbed to cells treated with sialidase (0.52 U/ml) and/or α2β1 antibody (20 μg/ml). Infected cells were enumerated at 16 h postinfection following indirect immunofluorescent staining. Rotavirus infectious titers are expressed as a percentage of the titers produced in control untreated cells. These control infectious titers, expressed as mean FCFU/ml ± SD, were 1.2 × 104 ± 0.2 × 104 (B), 9.0 × 103 ± 0.5 × 103 (C), 1.1 × 104 ± 0.1 × 104 (D), 2.6 × 104 ± 0.1 × 104 (E), and 1.2 × 104 ± 0.2 × 104 (F). Positive control neutralizing monoclonal antibodies (Ab) were 2G4 (RRV), RV-3:1 (CRW-8) and H7/D2 (UK). No positive-control antibody was available for TFR-41 or NCDV. Cells treated with isotype control antibody MOPC21 produced virus titers that were indistinguishable from the titers in untreated cells.
NCDV infection of MA104 cells is reduced by Neu5Acα2Me and Neu5Gcα2Me (38), as shown in Fig. 2E. Sialidase treatment reduced NCDV infectivity by 69% ± 2.8% (Fig. 2E), consistent with a previous report (27). Neu5Gcα2Me (P = 0.001) but not Neu5Acα2Me (P = 0.06) further inhibited NCDV infection in sialidase-treated cells (Fig. 2E). Infection by bovine UK rotavirus in untreated cells was reduced by Neu5Gcα2Me treatment (Fig. 2F). In sialidase-treated cells, UK infectivity increased by 187% ± 13%, and Neu5Acα2Me reduced this titer by 29% ± 6% (Fig. 2F) (P < 0.0001). Neu5Gcα2Me did not significantly alter UK infection of sialidase-treated cells (P = 0.28). These data showed that UK may use terminal N-glycolylneuraminic acid and also accessed N-acetylneuraminic acid for infection once terminal Sia were removed. The extent of α2β1 usage by animal rotaviruses was maintained irrespective of the effect on infectivity of terminal Sia removal and N-acetylneuraminic acid blockade (Table 2).
TABLE 2.
Summary of α2β1, GM1, and N-acylneuraminic acid usage for cell binding and/or infection by sialidase-sensitive (animal) rotaviruses
| Virus strain | α2β1 usagea | Untreated cells |
Sialidase-treated cells |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Infectivity reduced by CTBb | Proposed internalization pathwayc | Reduced by Neu5Acα2Me (% at 10 mM) |
Reduced by Neu5Gcα2Me (% at 10 mM) |
Infectivity reduced by a-GM1d | Effect of sialidase on given property |
Infectivity reduced by Neu5Acα2Me | Infectivity reduced by Neu5Gcα2Med | |||||
| VP8* boundd | Infectivity | VP8* boundd | Infect | VP8* boundd | Infectivity | |||||||
| TFR-41 | − | + | CE | ND | 19d | ND | 0d | ND | ND | Reducedd | −d | − |
| CRW-8 | − | − | ND | 41 | 46e | 43 | 66e | ND | ND | Reducede,f | −f | ND |
| RRV | + | − | Non-CE | ND | 60g | ND | 40e | − | ND | Reducedh | −d | ND |
| NCDV | + | − | ND | 34 | 29e | 41 | 41e | ND | Reduced | Reducedi | −d | + |
Shown in references 8, 15, 18, 32, and 35. In this study, the extent of α2β1 usage was shown not to be affected by sialidase treatment of cells.
Shown by CTB infectivity inhibition in this study for UK, TFR-41, and NCDV and in references 7 and 31 for CRW-8 and RRV. STD NMR analysis showed minimal RRV VP8* binding to a-GM1 (this study).
Data are from references 11 and 13. CE, clathrin-mediated endocytosis; Non-CE, endocytosis independent of clathrin and caveolin; ND, not determined.
Shown in this study.
Shown in this study and reference 38.
Shown in this study and reference 31.
The infectivity of several rotaviruses was reduced by GM1 ganglioside competition by CTB, which was related to VP4.
To determine if GM1 plays a role in infection by rotaviruses other than Wa, the effect of CTB on their infectivity was evaluated (Fig. 3). CTB shows high affinity and specificity for GM1, recognizing the I3-sialosylgangliotriaose moiety, Galβ1,3GalNAcβ1,4(Siaα2,3) Galβ1 (43, 44). In preliminary studies, Wa infectivity was inhibited to the same extent (48% ± 3%) by CTB at 10 μg/ml as at 1 μg/ml (Fig. 3A and data not shown). Treatment with 1 μg/ml CTB also reduced the infectivity of human rotaviruses RV-5, RV-3, and S12/85 by 29% ± 3%, 41% ± 3%, and 48% ± 6%, respectively (P < 0.0001) (Fig. 3A). For several viruses, this infectivity reduction was CTB dose dependent. As shown in Fig. 2, the infectivity of RRV, CRW-8, NCDV, and TFR-41 is sialidase sensitive, while UK infectivity is sialidase insensitive (66). Infection by RRV and CRW-8 is unaffected by CTB treatment (7, 31, 38). NCDV infectivity was tested here and found to be similarly unaffected by CTB (Fig. 3B). Notably, TFR-41 and UK infectivity was reduced by 33% ± 4% and 31% ± 3%, respectively, with 1 μg/ml CTB (P < 0.0001) (Fig. 3B). Interestingly, 0.1 μg/ml CTB also inhibited these rotaviruses by 30% ± 8% and 28% ± 5%, respectively (P < 0.0001) (Fig. 3B), in contrast to RV-3 and Wa, where 0.1 μg/ml CTB reduced infectivity by 9% ± 6% (P > 0.05) and 17% ± 7% (P = 0.04), respectively (Fig. 3A). This suggests that GM1 binding by TFR-41 and UK is weaker than that of RV-3 and Wa. Overall, GM1 usage was common for both human and animal rotaviruses, although it was more frequent among the human strains used here. The detected association of GM1 usage with sialidase insensitivity was not absolute, as sialidase-sensitive TFR-41 also utilized GM1.
FIG 3.

Effects of CTB, a-GM1 treatment, and GM1 or GM3 supplementation on rotavirus infectivity in MA104 cells and mapping UK rotavirus dependence on GM1 to VP4. (A) CTB treatment inhibited infection by human rotaviruses RV-5, RV-3, S12/85, and Wa. (B) CTB inhibition of the infectivity of NCDV, TFR-41, UK, and RRV rotaviruses and reassortant rotaviruses 12-1 and 28-1. Heat-inactivated CTB (Inact.) was included as a negative control, as described before (31). (C) Effects of exposure to 10 mM a-GM1 or a-GM3 on the infectivity of Wa and RV-3 in untreated and sialidase-treated cells. (D) Effects of cell supplementation with 3 μM GM1 or GM3 on Wa and RV-3 infectivity. Rotavirus infectious titers are expressed as a percentage of the titers produced in control untreated cells. These control infectious titers (mean FCFU/ml ± SD) were 3.0 × 104 ± 0.1 × 104 (RV-5), 2.0 × 104 ± 0.1 × 104 (RV-3), 1.8 × 104 ± 0.3 × 104 (S12/85), 2.4 × 104 ± 0.1 × 104 (Wa), 2.6 × 104 ± 0.1 × 104 (NCDV), 2.6 × 104 ± 0.2 × 104 (TFR-41), 1.1 × 104 ± 0.1 × 104 (UK), 1.2 × 104 ± 0.2 × 104 (RRV), 2.7 × 104 ± 0.1 × 104 (12-1), and 7.9 × 103 ± 0.8 × 103 (28-1).
As before, RRV infectivity was unaffected by CTB treatment (Fig. 3B). The infectivity of reassortant rotavirus 12-1 (containing UK VP4 and RRV VP7) was reduced by 47% ± 2% at 1 μg/ml CTB (P < 0.0001), whereas that of 28-1 (containing RRV VP4 and UK VP7) was unaffected (P = 0.45) (Fig. 3B). CTB at 0.1 μg/ml had little effect on 12-1 infectivity, suggesting that the extent or strength of GM1 binding by UK VP4 was changed when its genetic background was altered. As only VP4 and VP7 are relevant to rotavirus cell binding and entry, the parental source of the other genes is not important for these experiments, and the modified effect of CTB on 12-1 infectivity probably relates to the presence of RRV VP7. Overall, CTB sensitivity was mapped to the UK rotavirus gene encoding VP4, providing evidence that VP4 of UK rotavirus, containing the VP8* subunit, determines GM1 usage.
The infectivity of GM1-using rotaviruses but not RRV was reduced by competition with a-GM1.
In order to confirm rotavirus usage of the GM1 glycan for infection, the ability of the GM1 pentasaccharide, a-GM1, to inhibit rotavirus infection was determined. Importantly, 10 mM a-GM1 treatment substantially reduced the infectivity of Wa and RV-3 in both untreated and sialidase-treated cells (P < 0.05 by ANOVA) (Fig. 3C). The respective infectivity reductions in untreated cells of 57% and 49% were of comparable magnitude (P > 0.05), as were the respective infectivity reductions in sialidase-treated cells, 35% and 43% (P > 0.05). The magnitude of the infectivity reductions by a-GM1 did not differ between untreated and sialidase-treated cells (P > 0.05). In contrast, 10 mM a-GM3 treatment did not affect the infectivity of Wa and RV-3 in untreated or sialidase-treated cells (P > 0.05) (Fig. 3C). Overall, these data show that the GM1 glycan specifically blocks infection by Wa and RV-3 in both untreated and sialidase-treated cells.
RRV VP8* binds Neu5Acα2Me, Neu5Gcα2Me, and terminal Sia on the main chain of a-GM3 (7, 38, 67, 68). Antibody to a-GM3 inhibits RRV infection, whereas CTB blockade of GM1 does not (7) (Fig. 3B). To confirm the lack of RRV dependence on GM1, the ability of a-GM1 to inhibit RRV infection was assessed. Treatment with 10 mM a-GM1 did not affect RRV infectivity, which was 100% ± 3% of the infectivity in untreated cells (P = 0.80) (data not shown). This provides additional evidence that RRV does not recognize the GM1 glycan for infectivity and further demonstrates the specificity of the a-GM1 inhibition of infection by Wa and RV-3.
Incorporation of exogenous GM1 into cell membranes increased the infectivity of Wa and RV-3.
We showed above that sialidase treatment increased MA104 cell surface GM1 expression, and a-GM1 reduced the infectivity of Wa and RV-3 in both untreated and sialidase-treated cells. Therefore, we asked whether the incorporation of additional GM1 onto untreated MA104 cells would increase Wa and RV-3 infection. After preincubation with GM1, GM3, or medium, cells were infected with Wa or RV-3. GM1 addition to the cells before infection increased infectious titers of Wa and RV-3 by at least 1.5-fold over their titers in control cells (P < 0.001) (Fig. 3D). In contrast, GM3 supplementation did not affect Wa and RV-3 infectivity (P > 0.05). These results confirm that Wa and RV-3 specifically interact with GM1 and further demonstrate the importance of this interaction for infection.
STD NMR spectroscopy showed RV-3 VP8* interaction with a-GM1 but only weak a-GM1 signals for RRV VP8*.
The capacity for recombinant VP8* of RRV, Wa, and RV-3 to recognize a-GM1 was investigated by STD NMR spectroscopy in a manner we have previously reported (31). The 1H NMR control spectrum of a-GM1 is depicted in Fig. 4A. In the STD NMR spectrum, the height of the STD proton signal is proportional to the distance of the proton to the protein surface. Strong STD NMR signals imply extensive interactions of the ligand and the protein surface, while weak STD NMR signals suggest that the proton is more solvent exposed. Only weak STD NMR signals could be observed for a-GM1 in complex with RRV GST-VP8*, predominantly through its Neu5Ac moiety, suggesting limited, if any, interaction of the GalNAc residue (Fig. 4B). A low STD NMR signal intensity can be a direct consequence of low-affinity binding, and this result is consistent with the inability of CTB (7) (Fig. 3B) and a-GM1 to inhibit RRV infection. GST-fused Wa VP8* bound a-GM1 (Fig. 4C) in a manner identical to the one we previously reported for untagged Wa VP8* (31), with both the GalNAc and Neu5Ac moieties being equally important in the binding event, suggesting that the sialic acid is not the only residue that is important for protein recognition. Thus, as expected, a more extensive GM1 glycan engagement through both the Neu5Ac and GalNAc moieties was observed for VP8* of Wa than for that of RRV. Consequently, the GST tag did not affect the interaction between the glycan and VP8*. RV-3 VP8* also bound a-GM1 in a manner similar to that of Wa VP8*, suggesting a similar binding mode, as expected for another sialidase-insensitive human rotavirus strain (Fig. 4D). To further ensure that the fusion protein, GST, did not participate in ganglioside recognition, a control experiment was performed that clearly showed no recognition of a-GM1 by the GST (Fig. 4E). Thus, the binding epitopes on a-GM1 for the VP8* of Wa and RV-3 include both the Neu5Ac and GalNAc residues, whereas the RRV VP8* epitope encompasses only the Neu5Ac moiety (Fig. 4F).
FIG 4.

NMR spectra of a-GM1. (A) Control 1H NMR spectrum of a-GM1. STD NMR spectra of a-GM1 in the presence of RRV GST-VP8* (B), Wa GST-VP8* (C), RV-3 VP8* (D), and with GST alone (E) are shown. In all panels, the signals of the two N-acetamido groups' methyl protons at approximately 1.70 ppm are depicted separately on the right at a uniform magnification of ×2.5. All spectra were acquired in deuterated phosphate buffer at 600 MHz and 280 K. (F) Structure of a-GM1 with the binding epitopes of VP8* encircled. The Wa and RV-3 VP8* epitopes include both the Neu5Ac and GalNAc residues (left), whereas the RRV VP8* epitope encompasses only the Neu5Ac moiety (right).
Cell binding by NCDV and CRW-8 VP8* showed the same terminal Sia dependence as virion infectivity.
The ability of recombinant VP8* of RRV, NCDV, and CRW-8 to bind MA104 cells was determined by flow cytometry using cells placed into suspension with trypsin-EDTA. Over the VP8* concentration range of 5 μg/ml to 150 μg/ml, cell binding by RRV VP8* was saturated at 5 μg/ml with an RLMFI (means ± SD) of 1.8 ± 0.1 (Fig. 5A). Neu5Acα2Me and Neu5Gcα2Me inhibit MA104 cell infection by NCDV and CRW-8 (38). CRW-8 VP8* binds Neu5Acα2Me and Neu5Gcα2Me, as well as N-acetylneuraminic acids on a-GD1a and a-GM3 but not on a-GM1 (31, 38, 68). Here, NCDV VP8* showed dose-dependent increases in binding between 5 μg/ml (RLMFI of 1.8) and 300 μg/ml (RLMFI of 2.5) (Fig. 5B). NCDV binding increased further at 600 μg/ml, showing a mean RLMFI of 4.5 (Fig. 5C and D). CRW-8 VP8* binding was high level and dose dependent, approaching saturation at 150 μg/ml to 300 μg/ml (RLMFI of 38.4 and 47.6, respectively), with an RLMFI of 10.0 at 5 μg/ml (Fig. 5E, F, and G). The effects of Sia competition and sialidase treatment on VP8*-cell binding were analyzed. NCDV VP8* treatment with Neu5Acα2Me or Neu5Gcα2Me dose dependently reduced the RLMFI by up to 34% and 41%, respectively (Fig. 5C and D). CRW-8 VP8* binding blockade data obtained with low (5 μg/ml) and near-saturating (150 μg/ml) VP8* were equivalent, so only 5 μg/ml CRW-8 VP8* data are illustrated (Fig. 5F and G). CRW-8 VP8* treatment with Neu5Acα2Me or Neu5Gcα2Me dose dependently reduced the RLMFI by up to 41% and 43%, respectively. Sialidase treatment inhibited NCDV VP8* binding by 86% (Fig. 5H), comparable to the 69% reduction in NCDV infectivity (Fig. 2E). Substantial concordance in the degrees of blockade of VP8* binding and homologous rotavirus infectivity (reported in reference 38) was evident (Table 2). In particular, after Neu5Gcα2Me blockade, VP8*-cell binding and infectivity correlated for NCDV (Pearson r = 0.99; P = 0.005) and CRW-8 (r = 0.98; P = 0.016). Thus, the Neu5Acα2Me, Neu5Gcα2Me, and sialidase effects on VP8* binding reflected those seen for virus infectivity. This verified our flow-cytometric assay for VP8*-cell binding and indicated that virion VP8* is sufficient for the cell binding and infectivity that is mediated through terminal cellular N-acylneuraminic acids.
FIG 5.

Studies of Sia competition and sialidase sensitivity of cell binding by animal rotavirus VP8*. Cell binding by recombinant VP8* of RRV (A) and NCDV (B) at concentrations (μg/ml) of 5, 50, and 150 to cells placed into suspension using trypsin-EDTA treatment. NCDV GST-VP8* also was tested at 300 μg/ml. The histograms for NCDV VP8* at 150 μg/ml and 50 μg/ml fell close to and between those for 300 μg/ml and 5 μg/ml and are not shown for the sake of clarity. Cell binding by NCDV VP8* at 600 μg/ml (C and D) and CRW-8 VP8* at concentrations (μg/ml) of 5, 50, 150 and 300 (E) also are illustrated. Reduced cell binding by NCDV VP8* (C and D) at 600 μg/ml and CRW-8 VP8* at 5 μg/ml (F and G) following Neu5Acα2Me (C and F) and Neu5Gcα2Me (D and G) treatment is shown. (H) Cellular sialidase treatment reduced binding by NCDV VP8* (600 μg/ml). Reductions in GST-VP8* binding in panels C to G were analyzed for Neu5Acα2Me and Neu5Gcα2Me at concentrations (mM) of 1.25, 2.5, 5.0, and 10. Histograms for NCDV VP8* treated with 1.25 mM Neu5Gcα2Me were indistinguishable from those for untreated NCDV. Data for CRW-8 VP8* incubated with 2.5 mM Neu5Acα2Me or 2.5 mM Neu5Gcα2Me were indistinguishable from those of CRW-8 reacted with the respective Sia at 1.25 mM.
VP8* of Wa and RV-3 bound to cell surface GM1.
Flow cytometry requires suspended single cells, which can show reduced surface GM1 availability due to increased internalization (69). Confirming this, surface GM1 levels on single MA104 cells were reduced by 4.2-fold after suspension with trypsin-EDTA (RLMFI of 35) compared to suspension by scraping (RLMFI of 144) (Fig. 6A). In parallel with this reduced GM1 surface exposure, levels of cell binding by VP8* of Wa and RV-3 to cells placed into suspension with trypsin-EDTA were relatively low, with saturation evident at 5 μg/ml and a positive RLMFI (means ± SD) of 1.6 ± 0.2 and 1.7 ± 0.1, respectively, over the VP8* concentration range of 5 μg/ml to 300 μg/ml (Fig. 6B and C). From the histogram fluorescence intensity profiles, the mean fluorescence intensity of cell binding by VP8* of Wa (Fig. 6D) and RV-3 (Fig. 6E) was reduced by 22% and 39%, respectively, following CTB treatment of MA104 cells placed into suspension with trypsin-EDTA. A replicate experiment gave similar results. In contrast, cellular CTB treatment did not alter CRW-8 VP8* binding (Fig. 6F), as expected from its lack of effect on CRW-8 infectivity and the inability of CRW-8 VP8* to bind a-GM1 in STD NMR studies (31). These data indicated that VP8* of Wa and RV-3 specifically bound GM1 glycan on the cell surface. As described in Table 1, these VP8*-cell binding reductions were commensurate with the Wa and RV-3 infectivity titer reductions also seen in these CTB-treated cells (Fig. 3A) and the extent of Wa and RV-3 infectivity inhibition by a-GM1 (Fig. 3C).
FIG 6.
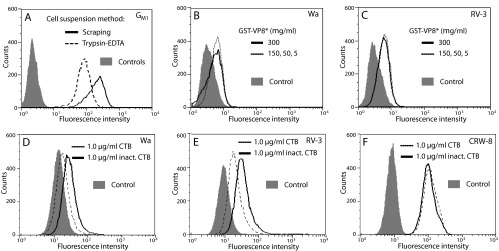
Flow-cytometric analysis of MA104 cell surface GM1 availability and effect of CTB on cell binding by VP8* of Wa, RV-3, and CRW-8. (A) Trypsin-EDTA treatment reduced cell surface GM1 availability. Histograms indicate the GM1 surface exposure, determined by incubation with FITC-CTB, on cells that were placed into suspension by trypsin-EDTA or scraping and rested for 30 min. Each cell preparation was reacted with unconjugated FITC for the controls, which each showed similar histograms. (B and C) Cell binding by recombinant VP8* of Wa (B) and RV-3 (C). Wa and RV-3 GST-VP8* proteins were tested for binding at concentrations (μg/ml) of 5, 50, 150, and 300 to cells placed into suspension using trypsin-EDTA treatment. Effect of cellular treatment with CTB or inactivated (inact.) CTB at 1 μg/ml on cell binding by VP8* of Wa (D), RV-3 (E), and CRW-8 (F). Wa, RV-3, or CRW-8 GST-VP8* protein was added at 300 μg/ml to cells placed into suspension with trypsin-EDTA. (D to F) The histogram of each VP8* bound to untreated cells was indistinguishable from that of the corresponding VP8* bound to cells treated with inactivated CTB.
DISCUSSION
It is demonstrated here that human rotaviruses of the most common serotypes recognize GM1 ganglioside during cellular infection, together with particular animal strains, namely, the UK bovine and TFR-41 porcine rotaviruses. GM1 glycan usage by rotaviruses was demonstrated through reduced infectivity following competition with CTB and/or a-GM1. Our new results showing increased infectivity of Wa and RV-3 rotaviruses after cell supplementation with exogenous GM1 show the importance of GM1 usage for infection by these human rotaviruses. Our finding that an Sia-containing saccharide (a-GM1) inhibits infection by sialidase-insensitive rotaviruses in normal (not sialidase-treated) cells also is novel. The infectivity reduction after a-GM1 but not N-acetylneuraminic acid treatment in normal cells indicates the importance of glycan moieties other than that of Neu5Ac in rotavirus recognition of a-GM1. VP8* of RV-3, like that of Wa, was shown here to bind a-GM1 by STD NMR, involving both the Neu5Ac and GalNAc moieties. Thus, it is expected that both of these residues are involved in virion recognition of GM1. The CTB inhibition of cell binding by VP8* of Wa and RV-3 also supports this view. Overall, we have now shown that VP8* from these rotaviruses binds GM1 glycan on the host cell surface to facilitate infectivity. In sialidase-treated cells, which expressed increased GM1 levels and supported enhanced infectivity of these sialidase-insensitive rotaviruses, the extent of a-GM1 usage for infection was maintained and was associated with de novo susceptibility of infection to competition by N-acetylneuraminic acid (summarized in Table 1). These findings suggest that the increased GM1 levels in sialidase-treated cells, perhaps combined with reduced GD1a and GM3 (64, 70), facilitated virion access to GM1 and the sensitivity of detection of N-acetylneuraminic acid infectivity blockade.
Most rotaviruses tested utilized either GM1 (Wa, RV-5, RV-3, S12/85, and UK) or terminal Sia (RRV, CRW-8, and NCDV). The inability of radiolabeled NCDV to bind pure GM1 supports these findings (71). For CRW-8 and NCDV, the effects of N-acetyl- and N-glycolylneuraminic acid competition and/or sialidase on VP8* cell binding recapitulated the effects of these treatments on their infectivity (38). Thus, VP8* is sufficient for infectious virus binding to terminal Sia. We found that RRV infectivity is unaffected by treatment with CTB (7) (Fig. 3B) or a-GM1 (Fig. 3C). This is entirely consistent with our STD NMR studies, where RRV VP8* interacted only weakly with GM1, predominantly through the Neu5Ac moiety and not the GalNAc residue. Another study has detected GM1 inhibition of RRV infection (72). However, this may relate to the presence of the ceramide moiety, which was absent from our a-GM1, rather than the GM1 glycan itself. Via this ceramide, GM1 and gangliosides generally would be expected to insert into the cellular plasma membrane under the experimental conditions used (43, 73, 74). This cellular GM1 incorporation might indirectly influence RRV recognition of its cellular receptors and/or reduce RRV cell entry by altering membrane lipid microdomains (lipid rafts), as GM1 partitions with lipid rafts.
Interestingly, TFR-41 alone utilized both terminal Sia and GM1, and its infectivity showed the least dependence on terminal Sia of the sialidase-dependent viruses (Table 2). This is consistent with TFR-41 usage of GM1, which lacks terminal Sia. Our findings that GM1 is used by UK and TFR-41 extend the recent report that suppression of ganglioside synthesis inhibits the infectivity of these rotaviruses (72).
As CRW-8 and TFR-41 share VP4 serotype specificity, their various abilities to use GM1 and terminal Sia are intriguing. The particular virus passages we studied differ in VP8* amino acid sequence at positions 88, 92, 93, 114, 116, and 157 (V. T. Dang, G. Holloway, and B. S. Coulson, unpublished data), implicating one or more of these changes in the altered glycan binding. Residues at positions 88, 114, and 116 are located within neutralization epitopes (75). The CRW-8 passage 31 virus used for these studies has Ser157 (38), whereas TFR-41 has Pro157. Our group showed previously that replacement of Ser157 in CRW-8 VP8* with Pro157 facilitates significantly stronger binding to N-acylneuraminic acids without altering the N-acylneuraminic acid specificity of VP8* binding (38). Pro157 in TFR-41 VP8* may play a role in modulating Sia binding to influence usage of either or both terminal Sia and GM1.
The VP8* binding sites for terminal Sia, GM3 glycan, and the type A histoblood group antigen overlap substantially, but the VP8* binding site for GM1 is unknown (21, 68). GM1 is the sole cellular receptor identified for RV-3 to date (15, 27, 48). The lack of Neu5Acα2Me blockade of S12/85 infectivity in sialidase-treated cells suggests that this virus has a reduced dependence on the internal Sia for GM1 recognition. Interestingly, the S12/85 and RV-3 VP8* amino acid sequences differ at residues 71, 106, 133, and 215 only, so one or more of these amino acid changes may be responsible for the altered GM1 recognition. The difference at position 133 has been associated with the ability of S12/85 to cause symptomatic disease in older children and the asymptomatic infection of infants by RV-3 (76). Given their shared origins with Wa-like viruses and RV-3, respectively, it is expected that the Rotarix and RV-3 vaccine viruses also recognize the internal N-acetylneuraminic acid of GM1 and depend to a degree on GM1 for infectivity.
The independence of GM1 and internal Sia recognition by rotaviruses from their α2β1 usage (Tables 1 and 2) extends previous data showing a lack of segregation between rotavirus usage of terminal Sia and α2β1 (15). Furthermore, Wa recognition of α2β1 and internal N-acetylneuraminic acid was at least partially additive. However, it remains possible that internal and/or terminal Sia on α2β1 also act as rotavirus receptors, as previously proposed (15). Sialidase treatment of α3β1 and α5β1 integrins removes terminal Sia from both α and β subunits and substantially affects natural ligand binding (77). As the extent of α2β1 integrin α2 subunit I domain recognition by rotaviruses was unaltered by sialidase treatment, it is unlikely that removal of terminal Sia from glycan main chains affected infectivity through altered binding of the integrin recognition sequence in VP5* to the α2 I domain. The α2β1 amino acids required for rotavirus and type I collagen binding only partially overlap (28), so any effect of sialidase on α2β1 function would not necessarily be expected to alter rotavirus binding.
Rotavirus recognition of α2β1 does not correlate with proposed rotavirus internalization routes (Tables 1 and 2) (11). However, α2β1 recognition might affect the entry process for some rotaviruses, such as the laboratory-derived Nar 3 variant of RRV that no longer depends on terminal Sia for infection (12). Rotavirus usage of GM1 shows a possible association with internalization by clathrin-mediated endocytosis (Tables 1 and 2). MA104 cell entry by Wa, DS-1, UK, and TFR-41 is blocked by treatments that inhibit clathrin-mediated endocytosis and alter endosomal pH, suggesting that they utilize a pathway that includes clathrin-mediated endocytosis (11, 12). UK VP4 is sufficient to direct rotavirus internalization by this pathway (12). DS-1 and RV-5 rotaviruses differ in only 3 amino acids in VP8*, so it is possible that RV-5 also uses this route. In contrast, these treatments do not affect RRV internalization, which occurs by an endocytic route that may be independent of clathrin and caveola (11, 13). The Nar 3 variant of RRV is considered to utilize a clathrin-dependent pathway, possibly due to a single VP8* amino acid change from RRV (Lys187Arg) (12). Interestingly, an amino acid sequence difference at this position between RRV (Lys187) and CRW-8 (Gly187) is a major determinant dictating the preference of CRW-8 for N-glycolyl- over N-acetylneuraminic acid, with the bulkier Lys187 in RRV impeding such a preference, although it permits N-glycolylneuraminic acid recognition (38). As RRV VP8* amino acid 187 is associated with both Sia specificity and internalization pathway preference, these virus properties may be linked.
Two lectin-like VP8* domains are considered to be presented on each virion spike (3). We hypothesize that rotavirus VP8* binding to GM1 assists infection by facilitating the lateral movement of VP8* monomers initially bound to terminal Sia or other receptors. During VP4 conformational change, it has been proposed that the connection between the cell membrane-bound heads of the two VP8* monomers with VP5* is lost, with the VP8* head most likely moving apart while remaining tethered to the spike foot to allow access of the VP5* hydrophobic domains to the cell membrane for penetration (3). Possibly, GM3 and GD1a binding by VP8* of sialidase-sensitive rotaviruses also enhances lateral movement of VP8* and/or plays a role in directing virus internalization. GM1- and GM3-enriched membrane domains are segregated from each other and differ in size and association with cell membrane glycoproteins and kinases (78, 79). Further studies are necessary to determine precisely how interactions between particular ganglioside receptors and VP8* mediate rotavirus-cell attachment and participate in rotavirus internalization.
ACKNOWLEDGMENTS
We thank Milton Kiefel for helpful suggestions and synthesis of Neu5Acα2Me, Harry Greenberg for provision of reassortant rotaviruses 12-1 and 28-1, and Con Sonza for the H7/D2 antibody.
The financial support of the National Health and Medical Research Council of Australia (ID509006 and ID628319 to B.S.C. and ID597439 to M.V.I., H.B., and B.S.C.) and the Australian Research Council (DP1094393 and DP0774383 to M.V. I., H.B., and B.S.C.) is gratefully acknowledged.
B.S.C. is a Senior Research Fellow of the NHMRC (ID628319).
Footnotes
Published ahead of print 5 February 2014
This study is dedicated to the memory of Ian H. Holmes, whose conviction that sialic acids would prove to be important in cell attachment or entry for many rotaviruses helped provide the impetus for this work.
REFERENCES
- 1.Li Z, Baker ML, Jiang W, Estes MK, Prasad BV. 2009. Rotavirus architecture at subnanometer resolution. J. Virol. 83:1754–1766. 10.1128/JVI.01855-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prasad BV, Wang GJ, Clerx JP, Chiu W. 1988. Three-dimensional structure of rotavirus. J. Mol. Biol. 199:269–275. 10.1016/0022-2836(88)90313-0 [DOI] [PubMed] [Google Scholar]
- 3.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC. 2011. Atomic model of an infectious rotavirus particle. EMBO J. 30:408–416. 10.1038/emboj.2010.322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeager M, Dryden KA, Olson NH, Greenberg HB, Baker TS. 1990. Three-dimensional structure of rhesus rotavirus by cryoelectron microscopy and image reconstruction. J. Cell Biol. 110:2133–2144. 10.1083/jcb.110.6.2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Estes MK, Cohen J. 1989. Rotavirus gene structure and function. Microbiol. Rev. 53:410–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorziglia M, Larralde G, Kapikian AZ, Chanock RM. 1990. Antigenic relationships among human rotaviruses as determined by outer capsid protein VP4. Proc. Natl. Acad. Sci. U. S. A. 87:7155–7159. 10.1073/pnas.87.18.7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haselhorst T, Fiebig T, Dyason JC, Fleming FE, Blanchard H, Coulson BS, von Itzstein M. 2011. Recognition of the GM3 ganglioside glycan by Rhesus rotavirus particles. Angew. Chem. Int. Ed. Engl. 50:1055–1058. 10.1002/anie.201004116 [DOI] [PubMed] [Google Scholar]
- 8.Coulson BS, Londrigan SL, Lee DJ. 1997. Rotavirus contains integrin ligand sequences and a disintegrin-like domain that are implicated in virus entry into cells. Proc. Natl. Acad. Sci. U. S. A. 94:5389–5394. 10.1073/pnas.94.10.5389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dormitzer PR, Nason EB, Prasad BV, Harrison SC. 2004. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature 430:1053–1058. 10.1038/nature02836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim IS, Trask SD, Babyonyshev M, Dormitzer PR, Harrison SC. 2010. Effect of mutations in VP5 hydrophobic loops on rotavirus cell entry. J. Virol. 84:6200–6207. 10.1128/JVI.02461-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutierrez M, Isa P, Sanchez-San Martin C, Perez-Vargas J, Espinosa R, Arias CF, Lopez S. 2010. Different rotavirus strains enter MA104 cells through different endocytic pathways: the role of clathrin-mediated endocytosis. J. Virol. 84:9161–9169. 10.1128/JVI.00731-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diaz-Salinas MA, Romero P, Espinosa R, Hoshino Y, Lopez S, Arias CF. 2013. The spike protein VP4 defines the endocytic pathway used by rotavirus to enter MA104 cells. J. Virol. 87:1658–1663. 10.1128/JVI.02086-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolf M, Vo PT, Greenberg HB. 2011. Rhesus rotavirus entry into a polarized epithelium is endocytosis dependent and involves sequential VP4 conformational changes. J. Virol. 85:2492–2503. 10.1128/JVI.02082-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rolsma MD, Kuhlenschmidt TB, Gelberg HB, Kuhlenschmidt MS. 1998. Structure and function of a ganglioside receptor for porcine rotavirus. J. Virol. 72:9079–9091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham KL, Halasz P, Tan Y, Hewish MJ, Takada Y, Mackow ER, Robinson MK, Coulson BS. 2003. Integrin-using rotaviruses bind α2β1 integrin α2 I domain via VP4 DGE sequence and recognize αXβ2 and αVβ3 by using VP7 during cell entry. J. Virol. 77:9969–9978. 10.1128/JVI.77.18.9969-9978.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graham KL, Zeng W, Takada Y, Jackson DC, Coulson BS. 2004. Effects on rotavirus cell binding and infection of monomeric and polymeric peptides containing α2β1 and αxβ2 integrin ligand sequences. J. Virol. 78:11786–11797. 10.1128/JVI.78.21.11786-11797.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halasz P, Holloway G, Turner SJ, Coulson BS. 2008. Rotavirus replication in intestinal cells differentially regulates integrin expression by a phosphatidylinositol 3-kinase-dependent pathway, resulting in increased cell adhesion and virus yield. J. Virol. 82:148–160. 10.1128/JVI.01980-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Londrigan SL, Graham KL, Takada Y, Halasz P, Coulson BS. 2003. Monkey rotavirus binding to α2β1 integrin requires the α2 I domain and is facilitated by the homologous β1 subunit. J. Virol. 77:9486–9501. 10.1128/JVI.77.17.9486-9501.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Londrigan SL, Hewish MJ, Thomson MJ, Sanders GM, Mustafa H, Coulson BS. 2000. Growth of rotaviruses in continuous human and monkey cell lines that vary in their expression of integrins. J. Gen. Virol. 81:2203–2213 [DOI] [PubMed] [Google Scholar]
- 20.Ciarlet M, Crawford SE, Estes MK. 2001. Differential infection of polarized epithelial cell lines by sialic acid-dependent and sialic acid-independent rotavirus strains. J. Virol. 75:11834–11850. 10.1128/JVI.75.23.11834-11850.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu L, Crawford SE, Czako R, Cortes-Penfield NW, Smith DF, Le Pendu J, Estes MK, Prasad BV. 2012. Cell attachment protein VP8* of a human rotavirus specifically interacts with A-type histo-blood group antigen. Nature 485:256–259. 10.1038/nature10996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang P, Xia M, Tan M, Zhong W, Wei C, Wang L, Morrow A, Jiang X. 2012. Spike protein VP8* of human rotavirus recognizes histo-blood group antigens in a type-specific manner. J. Virol. 86:4833–4843. 10.1128/JVI.05507-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guerrero CA, Mendez E, Zarate S, Isa P, Lopez S, Arias CF. 2000. Integrin αvβ3 mediates rotavirus cell entry. Proc. Natl. Acad. Sci. U. S. A. 97:14644–14649. 10.1073/pnas.250299897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerrero CA, Zarate S, Corkidi G, Lopez S, Arias CF. 2000. Biochemical characterization of rotavirus receptors in MA104 cells. J. Virol. 74:9362–9371. 10.1128/JVI.74.20.9362-9371.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gualtero DF, Guzman F, Acosta O, Guerrero CA. 2007. Amino acid domains 280-297 of VP6 and 531-554 of VP4 are implicated in heat shock cognate protein hsc70-mediated rotavirus infection. Arch. Virol. 152:2183–2196. 10.1007/s00705-007-1055-5 [DOI] [PubMed] [Google Scholar]
- 26.Perez-Vargas J, Romero P, Lopez S, Arias CF. 2006. The peptide-binding and ATPase domains of recombinant hsc70 are required to interact with rotavirus and reduce its infectivity. J. Virol. 80:3322–3331. 10.1128/JVI.80.7.3322-3331.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciarlet M, Estes MK. 1999. Human and most animal rotavirus strains do not require the presence of sialic acid on the cell surface for efficient infectivity. J. Gen. Virol. 80:943–948 [DOI] [PubMed] [Google Scholar]
- 28.Fleming FE, Graham KL, Takada Y, Coulson BS. 2011. Determinants of the specificity of rotavirus interactions with the α2β1 integrin. J. Biol. Chem. 286:6165–6174. 10.1074/jbc.M110.142992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukudome K, Yoshie O, Konno T. 1989. Comparison of human, simian, and bovine rotaviruses for requirement of sialic acid in hemagglutination and cell adsorption. Virology 172:196–205. 10.1016/0042-6822(89)90121-9 [DOI] [PubMed] [Google Scholar]
- 30.Graham KL, Takada Y, Coulson BS. 2006. Rotavirus spike protein VP5* binds α2β1 integrin on the cell surface and competes with virus for cell binding and infectivity. J. Gen. Virol. 87:1275–1283. 10.1099/vir.0.81580-0 [DOI] [PubMed] [Google Scholar]
- 31.Haselhorst T, Fleming FE, Dyason JC, Hartnell RD, Yu X, Holloway G, Santegoets K, Kiefel MJ, Blanchard H, Coulson BS, von Itzstein M. 2009. Sialic acid dependence in rotavirus host cell invasion. Nat. Chem. Biol. 5:91–93. 10.1038/nchembio.134 [DOI] [PubMed] [Google Scholar]
- 32.Ciarlet M, Crawford SE, Cheng E, Blutt SE, Rice DA, Bergelson JM, Estes MK. 2002. VLA-2 (α2β1) integrin promotes rotavirus entry into cells but is not necessary for rotavirus attachment. J. Virol. 76:1109–1123. 10.1128/JVI.76.3.1109-1123.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hemler ME, Jacobson JG, Strominger JL. 1985. Biochemical characterization of VLA-1 and VLA-2. Cell surface heterodimers on activated T cells. J. Biol. Chem. 260:15246–15252 [PubMed] [Google Scholar]
- 34.Ludert JE, Feng N, Yu JH, Broome RL, Hoshino Y, Greenberg HB. 1996. Genetic mapping indicates that VP4 is the rotavirus cell attachment protein in vitro and in vivo. J. Virol. 70:487–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zarate S, Espinosa R, Romero P, Guerrero CA, Arias CF, Lopez S. 2000. Integrin α2β1 mediates the cell attachment of the rotavirus neuraminidase-resistant variant nar3. Virology 278:50–54. 10.1006/viro.2000.0660 [DOI] [PubMed] [Google Scholar]
- 36.Mendez E, Arias CF, Lopez S. 1993. Binding to sialic acids is not an essential step for the entry of animal rotaviruses to epithelial cells in culture. J. Virol. 67:5253–5259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haselhorst T, Blanchard H, Frank M, Kraschnefski MJ, Kiefel MJ, Szyczew AJ, Dyason JC, Fleming F, Holloway G, Coulson BS, von Itzstein M. 2007. STD NMR spectroscopy and molecular modeling investigation of the binding of N-acetylneuraminic acid derivatives to rhesus rotavirus VP8* core. Glycobiology 17:68–81. 10.1093/glycob/cwl051 [DOI] [PubMed] [Google Scholar]
- 38.Yu X, Dang VT, Fleming FE, von Itzstein M, Coulson BS, Blanchard H. 2012. Structural basis of rotavirus strain preference towards N-acetyl- or N-glycolylneuraminic acid-containing receptors. J. Virol. 86:13456–13466. 10.1128/JVI.06975-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saito M, Sugano K, Nagai Y. 1979. Action of Arthrobacter ureafaciens sialidase on sialoglycolipid substrates. Mode of action and highly specific recognition of the oligosaccharide moiety of ganglioside GM1. J. Biol. Chem. 254:7845–7854 [PubMed] [Google Scholar]
- 40.Venerando B, Cestaro B, Fiorilli A, Ghidoni R, Preti A, Tettamanti G. 1982. Kinetics of Vibrio cholerae sialidase action on gangliosidic substrates at different supramolecular-organizational levels. Biochem. J. 203:735–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Isa P, Arias CF, Lopez S. 2006. Role of sialic acids in rotavirus infection. Glycoconj. J. 23:27–37. 10.1007/s10719-006-5435-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mendez E, Lopez S, Cuadras MA, Romero P, Arias CF. 1999. Entry of rotaviruses is a multistep process. Virology 263:450–459. 10.1006/viro.1999.9976 [DOI] [PubMed] [Google Scholar]
- 43.Nagafuku M, Okuyama K, Onimaru Y, Suzuki A, Odagiri Y, Yamashita T, Iwasaki K, Fujiwara M, Takayanagi M, Ohno I, Inokuchi J. 2012. CD4 and CD8 T cells require different membrane gangliosides for activation. Proc. Natl. Acad. Sci. U. S. A. 109:E336–342. 10.1073/pnas.1114965109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Angstrom J, Backstrom M, Berntsson A, Karlsson N, Holmgren J, Karlsson KA, Lebens M, Teneberg S. 2000. Novel carbohydrate binding site recognizing blood group A and B determinants in a hybrid of cholera toxin and Escherichia coli heat-labile enterotoxin B-subunits. J. Biol. Chem. 275:3231–3238. 10.1074/jbc.275.5.3231 [DOI] [PubMed] [Google Scholar]
- 45.Woode GN, Bridger JC, Hall G, Dennis MJ. 1974. The isolation of a reovirus-like agent associated with diarrhoea in colostrum-deprived calves in Great Britain. Res. Vet. Sci. 16:102–105 [PubMed] [Google Scholar]
- 46.Beisner B, Kool D, Marich A, Holmes IH. 1998. Characterisation of G serotype dependent non-antibody inhibitors of rotavirus in normal mouse serum. Arch. Virol. 143:1277–1294. 10.1007/s007050050375 [DOI] [PubMed] [Google Scholar]
- 47.Coulson BS. 1993. Typing of human rotavirus VP4 by an enzyme immunoassay using monoclonal antibodies. J. Clin. Microbiol. 31:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirkwood CD, Bishop RF, Coulson BS. 1998. Attachment and growth of human rotaviruses RV-3 and S12/85 in Caco-2 cells depend on VP4. J. Virol. 72:9348–9352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalica AR, Flores J, Greenberg HB. 1983. Identification of the rotaviral gene that codes for hemagglutination and protease-enhanced plaque formation. Virology 125:194–205. 10.1016/0042-6822(83)90073-9 [DOI] [PubMed] [Google Scholar]
- 50.Kiefel MJ, Beisner B, Bennett S, Holmes IH, von Itzstein M. 1996. Synthesis and biological evaluation of N-acetylneuraminic acid-based rotavirus inhibitors. J. Med. Chem. 39:1314–1320. 10.1021/jm950611f [DOI] [PubMed] [Google Scholar]
- 51.Sherman AA, Yudina ON, Shashkov AS, Menshov VM, Nifantiev NE. 2002. Preparative route to N-glycolylneuraminic acid phenyl 2-thioglycoside donor and synthesis of Neu5Gc-α-(2→3′)-lactosamine 3-aminopropyl glycoside. Carbohydr. Res. 337:451–457. 10.1016/S0008-6215(02)00003-4 [DOI] [PubMed] [Google Scholar]
- 52.Padilla-Noriega L, Dunn SJ, Lopez S, Greenberg HB, Arias CF. 1995. Identification of two independent neutralization domains on the VP4 trypsin cleavage products VP5* and VP8* of human rotavirus ST3. Virology 206:148–154. 10.1016/S0042-6822(95)80029-8 [DOI] [PubMed] [Google Scholar]
- 53.Coulson BS, Tursi JM, McAdam WJ, Bishop RF. 1986. Derivation of neutralizing monoclonal antibodies to human rotaviruses and evidence that an immunodominant neutralization site is shared between serotypes 1 and 3. Virology 154:302–312. 10.1016/0042-6822(86)90456-3 [DOI] [PubMed] [Google Scholar]
- 54.Sonza S, Breschkin AM, Holmes IH. 1983. Derivation of neutralizing monoclonal antibodies against rotavirus. J. Virol. 45:1143–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sonza S, Breschkin AM, Holmes IH. 1984. The major surface glycoprotein of simian rotavirus (SA11) contains distinct epitopes. Virology 134:318–327. 10.1016/0042-6822(84)90300-3 [DOI] [PubMed] [Google Scholar]
- 56.Hewish MJ, Takada Y, Coulson BS. 2000. Integrins α2β1 and α4β1 can mediate SA11 rotavirus attachment and entry into cells. J. Virol. 74:228–236. 10.1128/JVI.74.1.228-236.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scott SA, Holloway G, Coulson BS, Szyczew AJ, Kiefel MJ, von Itzstein M, Blanchard H. 2005. Crystallization and preliminary X-ray diffraction analysis of the sialic acid-binding domain (VP8*) of porcine rotavirus strain CRW-8. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61:617–620. 10.1107/S1744309105013849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu X, Guillon A, Szyczew AJ, Kiefel MJ, Coulson BS, von Itzstein M, Blanchard H. 2008. Crystallization and preliminary X-ray diffraction analysis of the carbohydrate-recognizing domain (VP8*) of bovine rotavirus strain NCDV. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64:509–511. 10.1107/S1744309108011949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kraschnefski MJ, Scott SA, Holloway G, Coulson BS, von Itzstein M, Blanchard H. 2005. Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of the VP8* carbohydrate-binding protein of the human rotavirus strain Wa. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61:989–993. 10.1107/S1744309105032999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rippinger CM, Patton JT, McDonald SM. 2010. Complete genome sequence analysis of candidate human rotavirus vaccine strains RV-3 and 116E. Virology 405:201–213. 10.1016/j.virol.2010.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blanchard H, Yu X, Coulson BS, von Itzstein M. 2007. Insight into host cell carbohydrate-recognition by human and porcine rotavirus from crystal structures of the virion spike associated carbohydrate-binding domain (VP8*). J. Mol. Biol. 367:1215–1226. 10.1016/j.jmb.2007.01.028 [DOI] [PubMed] [Google Scholar]
- 62.Low JA, Magnuson B, Tsai B, Imperiale MJ. 2006. Identification of gangliosides GD1b and GT1b as receptors for BK virus. J. Virol. 80:1361–1366. 10.1128/JVI.80.3.1361-1366.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warner S, Hartley CA, Stevenson RA, Ficorilli N, Varrasso A, Studdert MJ, Crabb BS. 2001. Evidence that Equine rhinitis A virus VP1 is a target of neutralizing antibodies and participates directly in receptor binding. J. Virol. 75:9274–9281. 10.1128/JVI.75.19.9274-9281.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller-Podraza H, Bradley RM, Fishman PH. 1982. Biosynthesis and localization of gangliosides in cultured cells. Biochemistry 21:3260–3265. 10.1021/bi00257a002 [DOI] [PubMed] [Google Scholar]
- 65.Fleming FE, Graham KL, Taniguchi K, Takada Y, Coulson BS. 2007. Rotavirus-neutralizing antibodies inhibit virus binding to integrins α2β1 and α4β1. Arch. Virol. 152:1087–1101. 10.1007/s00705-007-0937-x [DOI] [PubMed] [Google Scholar]
- 66.Ciarlet M, Ludert JE, Iturriza-Gomara M, Liprandi F, Gray JJ, Desselberger U, Estes MK. 2002. Initial interaction of rotavirus strains with N-acetylneuraminic (sialic) acid residues on the cell surface correlates with VP4 genotype, not species of origin. J. Virol. 76:4087–4095. 10.1128/JVI.76.8.4087-4095.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dormitzer PR, Sun ZY, Blixt O, Paulson JC, Wagner G, Harrison SC. 2002. Specificity and affinity of sialic acid binding by the rhesus rotavirus VP8* core. J. Virol. 76:10512–10517. 10.1128/JVI.76.20.10512-10517.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu X, Coulson BS, Fleming FE, Dyason JC, von Itzstein M, Blanchard H. 2011. Novel structural insights into rotavirus recognition of ganglioside glycan receptors. J. Mol. Biol. 413:929–939. 10.1016/j.jmb.2011.09.005 [DOI] [PubMed] [Google Scholar]
- 69.del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RG, Schwartz MA. 2004. Integrins regulate Rac targeting by internalization of membrane domains. Science 303:839–842. 10.1126/science.1092571 [DOI] [PubMed] [Google Scholar]
- 70.Guo CT, Nakagomi O, Mochizuki M, Ishida H, Kiso M, Ohta Y, Suzuki T, Miyamoto D, Hidari KI, Suzuki Y. 1999. Ganglioside GM(1a) on the cell surface is involved in the infection by human rotavirus KUN and MO strains. J. Biochem. 126:683–688. 10.1093/oxfordjournals.jbchem.a022503 [DOI] [PubMed] [Google Scholar]
- 71.Delorme C, Brussow H, Sidoti J, Roche N, Karlsson K-A, Neeser J-R, Teneberg S. 2001. Glycosphingolipid binding specifities of rotavirus: identification of a sialic acid-binding epitope. J. Virol. 75:2276–2287. 10.1128/JVI.75.5.2276-2287.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martinez MA, Lopez S, Arias CF, Isa P. 2012. Gangliosides have a functional role during rotavirus cell entry. J. Virol. 87:1115–1122. 10.1128/JVI.01964-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Magaldi TG, Buch MH, Murata H, Erickson KD, Neu U, Garcea RL, Peden K, Stehle T, DiMaio D. 2012. Mutations in the GM1 binding site of simian virus 40 VP1 alter receptor usage and cell tropism. J. Virol. 86:7028–7042. 10.1128/JVI.00371-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chinnapen DJ, Hsieh WT, te Welscher YM, Saslowsky DE, Kaoutzani L, Brandsma E, D'Auria L, Park H, Wagner JS, Drake KR, Kang M, Benjamin T, Ullman MD, Costello CE, Kenworthy AK, Baumgart T, Massol RH, Lencer WI. 2012. Lipid sorting by ceramide structure from plasma membrane to ER for the cholera toxin receptor ganglioside GM1. Dev. Cell 23:573–586. 10.1016/j.devcel.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dormitzer PR, Sun ZY, Wagner G, Harrison SC. 2002. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 21:885–897. 10.1093/emboj/21.5.885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kirkwood CD, Coulson BS, Bishop RF. 1996. G3P2 rotaviruses causing diarrhoeal disease in neonates differ in VP4, VP7 and NSP4 sequence from G3P2 strains causing asymptomatic neonatal infection. Arch. Virol. 141:1661–1676. 10.1007/BF01718290 [DOI] [PubMed] [Google Scholar]
- 77.Pan D, Song Y. 2010. Role of altered sialylation of the I-like domain of β1 integrin in the binding of fibronectin to β1 integrin: thermodynamics and conformational analyses. Biophys. J. 99:208–217. 10.1016/j.bpj.2010.03.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fujita A, Cheng J, Hirakawa M, Furukawa K, Kusunoki S, Fujimoto T. 2007. Gangliosides GM1 and GM3 in the living cell membrane form clusters susceptible to cholesterol depletion and chilling. Mol. Biol. Cell 18:2112–2122. 10.1091/mbc.E07-01-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frisz JF, Lou K, Klitzing HA, Hanafin WP, Lizunov V, Wilson RL, Carpenter KJ, Kim R, Hutcheon ID, Zimmerberg J, Weber PK, Kraft ML. 2013. Direct chemical evidence for sphingolipid domains in the plasma membranes of fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 110:E613–E622. 10.1073/pnas.1216585110 [DOI] [PMC free article] [PubMed] [Google Scholar]