

ABSTRACT
Tegument proteins pp150 and pUL96 function at a late step in cytomegalovirus (CMV) maturation. Here, we show that pp150 interacts directly with pUL96; however, the N-terminal region of pp150 and the C-terminal region of pUL96, which are critical for these proteins to function, are not required for this interaction. Moreover, the largely dispensable C-terminal region of pp150 is critical for pp150-pUL96 interaction. To further study the role of pUL96, several point and clustered mutations were engineered into the CMV Towne bacterial artificial chromosome (Towne-BAC) genome, replacing the conserved negatively charged C-terminal residues of pUL96. Although individual point mutations (E122A, D124A, and D125A) reduced virus growth slightly, the clustered mutations of 122EVDDAV127 significantly reduced virus growth, produced small syncytial plaque phenotypes, and impacted a late stage of virus maturation. When the UL96 C-terminal alanine conversion mutant (B6-BAC) virus was serially passaged in cell culture, it gained a plaque size comparable to that of Towne-BAC, displayed an altered restriction fragment length pattern, and replicated with increased growth kinetics. Whole-genome sequencing of this passaged virus (UL96P10) and the similarly passaged Towne-BAC virus revealed major differences only in the RNA4.9 and UL96 regions. When one of the mutations in the UL96 coding region was engineered into the B6-BAC virus, it significantly increased the plaque size and rescued the virus growth rate. Thus, accumulation of compensatory mutations only in UL96 in this revertant and the specific involvement of functionally dispensable regions of pp150 in the pUL96-pp150 interaction point toward a role for pUL96 in virus maturation that does not depend upon pp150.
IMPORTANCE Human cytomegalovirus causes significant medical problems in newborns, as well as in people with low immunity. In this study, we investigated the functions of two essential virus proteins, pp150 and pUL96, and determined the impact of their mutual interaction on virus replication. These studies provide valuable information that is critical for the development of targeted antiviral therapies.
INTRODUCTION
The proteinaceous layer between the capsid and the envelope in a herpesvirus virion, known as the tegument, is analogous to the matrix layer found in some RNA viruses. Tegument proteins function in at least four different ways; they (i) are delivered into host cells upon virus entry as prepackaged transactivation factors that can promote infection, e.g., cytomegalovirus (CMV) pp71 (1, 2); (ii) are critical for the acquisition of the virus envelope and act as a bridge between the nucleocapsids and the envelope layer (3, 4); (iii) provide capsid stability during virus maturation and trafficking, e.g., CMV pp150 and pUL96 (5, 6); and (iv) help in the trafficking of virus in the cell by engaging the host cytoskeleton, e.g., herpes simplex virus (HSV) UL36 (7–9). Herpesvirus tegument has been thought to be amorphous in nature (3, 4, 10), although a more ordered structure has been assigned to the inner tegument layer, which is in close proximity to the nucleocapsid (11–13). In CMV, this inner tegument includes pp150, the product of the UL32 gene that binds to the capsids directly (13, 14) and plays a major role in maintaining the integrity of capsids during maturation (5, 6, 15). More than 20 tegument proteins are packaged in a CMV virion (10), but very few functional interactions among these proteins have been reported. Moreover, it is hypothesized that intact capsids are required for the visualization of capsid-tegument interactions in CMV, a proposition strengthened by the detection of pp150 binding to intact capsids but not with individual capsid proteins (14).
Studies of bacteriophages, which have a capsid structure very similar to that of herpesviruses, have shown that the viral genome is packaged in an energy-driven process to almost liquid crystalline density and thus the interior walls of the capsids sustain significant positive pressure from inside (16). A changing environment during the translocation of nucleocapsids from the cell nucleus through the cytoplasm to the outside of cell can compromise the protective capabilities of the capsid shell in the absence of any stabilizing factors. Herpesvirus tegument proteins include important structural components that serve this capsid-stabilizing function (3–6). The size, shape, and structure of betaherpesvirus capsids are very similar to those of alphaherpesvirus capsids (11, 12, 17). Evidently, CMV DNA is packed at a significantly higher pressure than HSV DNA because the CMV genome is approximately 1.5 times as large as that of HSV despite their similar capsid sizes (18). Thus, CMV may need additional accessory factors to stabilize the nucleocapsids. The CMV pp150 and pUL96 tegument proteins (19, 20) are critical for virus growth in cell culture (21, 22) but lack obvious functional or sequence homologs in either alpha- or gammaherpesviruses (4), indicating that these proteins have important betaherpesvirus-specific functions. Both of these proteins are essential for efficient virus maturation (4–6), and they impact the accumulation of intact enveloped virus in the cytoplasmic virus assembly compartment (AC) (5, 6). pp150 binds to nucleocapsids through its amino-terminal region (14), and two small conserved regions (CR-1 and-2) located in this region determine the role of pp150 in virus maturation (5). The interactions of pUL96 with capsids have not yet been established. Deletion of the UL96 gene from the virus, insertion of stop codons at different positions in this gene, or conditional degradation of the pUL96 protein renders the virus unable to replicate (6). The defects in UL96 mutant viruses are reflected at a late stage of the virus life cycle, despite the fact that pUL96 is an early protein (6). The phenotype of these viruses is comparable to but less severe than that of a pp150 mutant virus (5). During wild-type infection, pp150 and pUL96 colocalize in infected cells. Also, cells show increased accumulation of pp150 in the nucleus following infection with a UL96 mutant virus (6). We hypothesized that pUL96 could either enhance pp150-capsid association or act as a chaperone to assist in the relocation of pp150-associated nucleocapsids from the nucleus to the cytoplasm.
In this study, we explored the interaction between pp150 and pUL96 and its impact on virus maturation. While full-length pp150 and pUL96 interacted with each other, mutation of amino acid residues in both pp150 and pUL96 that determine the functions of these two proteins in virus maturation did not abolish this interaction. Specific negatively charged amino acids within the C-terminal region of the protein were found to be critical for pUL96 function. When a replication-deficient UL96 mutant virus was passaged in fibroblasts, it accumulated compensatory mutations in UL96 but not in any other gene. These data indicate that although pUL96 and pp150 are required at similar steps in the virus life cycle and manifest similar mutant phenotypes, their functions during virus maturation may not be dependent on each other.
MATERIALS AND METHODS
Sequences and alignments.
The human CMV (Merlin) UL96 amino acid sequence (YP_081543) was aligned with the amino acid sequences of pUL96 homologs from rhesus CMV (RhCMV; YP_068222), chimpanzee CMV (CCMV; AAM00734), human herpesvirus 6 (HHV6; CAA58360), and HHV7 (YP_073808). The alignments were done in VectorNTI (ver 11; Invitrogen Corporation, Carlsbad, CA) with the ClustalW algorithm. Predictprotein 2.0 (23) and Protein Investigator (ver. 3.0.2) (http://intro.bio.umb.edu/PI/) were used to predict the protein folding and secondary structure of pUL96 and its variants. Cloning of green fluorescent protein (GFP) and bacterial artificial chromosome (BAC) sequences into the CMV Towne strain (American Type Culture Collection) genome has been reported previously (21). For sequencing of the viral genomes, viral DNA was purified from pelleted virions from the cleared cell culture medium with the DNeasy Tissue kit (Qiagen, Inc.) and the resultant double-stranded DNA was quantified with a Qubit fluorometric assay (Invitrogen). Multiplexed paired-end libraries (2 × 150 bp) were prepared with the Nextera XT DNA Sample Preparation kit (Illumina). Whole-genome sequencing was done with an Illumina MiSeq. CLC Genomics Workbench v6.0 software (CLCbio) was used for quality trimming of the reads and de novo assembly. An average of 20.5 million paired-end reads per virus were collected. The fastq files were imported into CLC Genomics Workbench 6.0. The reads were trimmed for quality with a limit of 0.05 (P of error, equivalent to Q13) and a maximum of two ambiguities, and reads of <15 bp were discarded. The de novo assembly used a word size of 45, a bubble size of 90, and a minimum contig length of 1,000; paired distances were autodetected; and reads were mapped back onto contigs. All other parameters were default. The number of scaffolds, scaffold N50, and total sequence length, respectively, for each virus were as follows: Towne-BAC, 4, 49 kb, and 232 kb; UL96P10, 4, 49 kb, and 232 kb. Each scaffold from each virus had an average coverage of >7,000. Gaps in the genome sequence were filled by PCR and DNA sequencing of resultant amplicons. Open reading frames were identified by NCBI-BLAST search and annotated in Sequin 12.30.
Cells.
Primary human foreskin-derived fibroblasts (HF) or 293T cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen Corporation, Carlsbad, CA) containing 4.5 g/ml glucose, 10% fetal bovine serum (S1245OH; Atlanta Biologicals, Lawrenceville, GA), 1 mM sodium pyruvate, 2 mM l-glutamine, and 100 U/ml penicillin-streptomycin (Cellgro, Manassas, VA) at 37°C with 5% CO2. HF between passages 5 and 15 were used for transfections and infections. The cell culture medium was changed (for transfections) or additional medium was added (for infections) every other day.
BAC mutagenesis and recombinant viruses.
BAC recombineering protocols (24) were followed to mutate regions of UL96 with a KanSacB cassette as reported earlier (6). From the selected BAC clone (δUL96BAC), additional clones were derived following the introduction of a PCR fragment containing wild-type UL96 (rescued BAC [R-BAC]), E122A (B1-BAC), D124A (B2-BAC), D125A (B3-BAC), a 122EVDDAV127-to-KRKKRR conversion (B4-BAC), a 122EVDDAV127-to-DEEEDE conversion (B5-BAC), or a 122EVDDAV127-to-AAAAAA conversion (B6-BAC) and subsequent screening for the loss of kanamycin resistance, as well as sucrose sensitivity. Insertion of the desired mutation, as well as the absence of any undesired changes in the genome, was confirmed by restriction fragment length pattern (RFLP) analysis and further by DNA sequencing of the UL96 region of these viruses. At 10 days posttransfection (dpt), supernatants from HF cultures transfected with replication-competent BACs were harvested for recovery of recombinant viruses. Single-step growth curves were obtained by infecting confluent HF at a multiplicity of infection [MOI] of 3.0 in triplicate and harvesting cells plus supernatants at designated time points postinfection. Each sample was sonicated (Sonicator 3000; Misonix Incorporated, Farmingdale, NY) on ice for 30 s before freezing or direct titration. The samples were diluted 10- to 100,000-fold for titration. At 10 days postinfection (dpi), cells were fixed in methanol, stained with Giemsa stain (Sigma-Aldrich, St. Louis, MO), and allowed to dry before enumeration of PFU under an optical microscope. The R-BAC construct does not differ in its properties from Towne-BAC, as demonstrated earlier (6). For passaging of the mutant viruses, BAC-transfected cells from the six-well tissue culture dishes were split (1:2) at 10 dpt into new six-well tissue culture dishes and incubated with a change of medium every alternate day. The mutant virus was allowed to grow, and its growth was monitored by enhanced GFP (eGFP) fluorescence. When eGFP was spread to the entire monolayer, the cells were split (1:2) into a T75 flask. Growth of virus was monitored in a similar manner, and the cells were then split into a T175 flask maintaining the approximately 1:2 split ratio. Virus was harvested when the cytopathic effect reached 100% and then passaged 10 times in T175 flasks before virus stocks were prepared and the growth rates of viruses were compared as described above.
Antibodies and IB analyses.
293T cells were used for plasmid transfections. Cells were harvested at 48 h posttransfection for analysis by immunoprecipitation (IP) and immunoblotting (IB). Anti-c-Myc affinity gel (Sigma-Aldrich, St. Louis, MO) was used for IP. Anti-FLAG M2 antibody (Agilent Technologies, Santa Clara, CA) was used as the primary antibody, and peroxidase-labeled horse anti-mouse IgG (Vector Laboratories, Burlingame, CA) was used as the secondary antibody for IB analyses. Blots were detected with ECL Western blotting detection reagents (GE Healthcare, Buckinghamshire, United Kingdom).
Microscopy.
Samples for transmission electron microscopy (TEM) were prepared by transfecting HF with B4-BAC or Towne-BAC bacmids and passaging the HF once at 10 dpt and fixing them at 3 days postseeding. TEM of UL96P10, UL96ATG, or Towne-BAC virus-infected HF was performed by infecting HF at an MOI of 3.0 and fixing the cells at 5 dpi. In each case, cells were fixed at the endpoint in 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) for 2 h at room temperature. Cells were then washed with the same buffer and postfixed with buffered 1.0% osmium tetroxide at room temperature for 1 h. Following several washes with 0.1 M cacodylate buffer, cells were dehydrated with ethanol, infiltrated, and embedded in Eponate 12 resin (Ted Pella Inc., Redding, CA). Cell culture plates were cracked with a hammer to release the resin after it had solidified, and ultrathin sections (60 to 70 nm) of monolayer cells were cut and counterstained with uranyl acetate and lead citrate. Examination of ultrathin sections by TEM was carried out on a Hitachi H-7500 operated at 75 kV, and images were captured with a Gatan BioScan charge-coupled device camera (Gatan Inc., Pleasanton, CA). The images were acquired and analyzed with the DigitalMicrograph software (Gatan Inc.).
Nucleotide sequence accession numbers.
The whole-genome sequences of the UL96P10 virus and the Towne-BAC passaged virus have been deposited in the GenBank database and assigned accession numbers KF493876 and KF493877, respectively (25, 26).
RESULTS
pUL96 interacts with pp150.
C-terminal truncations of pUL96 arrest virus growth similarly to that of a UL96-null virus, indicating the functional importance of the UL96 C-terminal region (6). We generated UL96 expression clones that were either full length (UL96FL) or truncated by the conserved C-terminal region (UL961-101) (Fig. 1A). For pp150, several expression clones that either truncate or mutate this protein (Fig. 1B) have been characterized earlier (15). pp150 mutations located in the N-terminal capsid-binding region of the protein (pON2781, pON2783, and pON2784) (Fig. 1B) affect virus growth to various degrees (5, 15); however, the C-terminal mutant protein (pON2782) functions similarly to the wild-type protein (15). Interactions between Myc epitope-tagged pp150 and Flag epitope-tagged pUL96 clones were investigated by IP and IB (Fig. 1C and D). pp150 interacted with full-length, as well as C-terminally truncated, pUL96 (Fig. 1C). In a different experiment, pUL96 interacted with all of the N-terminally mutated forms of pp150; however, a C-terminal mutation significantly reduced pp150-pUL96 interaction (Fig. 1D). Thus, interaction between pp150 and pUL96 is determined by the C terminus of pp150 and the N terminus of pUL96. Surprisingly, the N-terminal region of pp150 and the C-terminal region of pUL96, which are critical for these two proteins to function, are not essential for their direct interaction. Moreover, the C-terminal region of pp150, which is largely dispensable for function (15), appears to be important for pp150-pUL96 interaction. These data suggest independent roles of pp150 and pUL96 in virus maturation despite their direct interaction.
FIG 1.
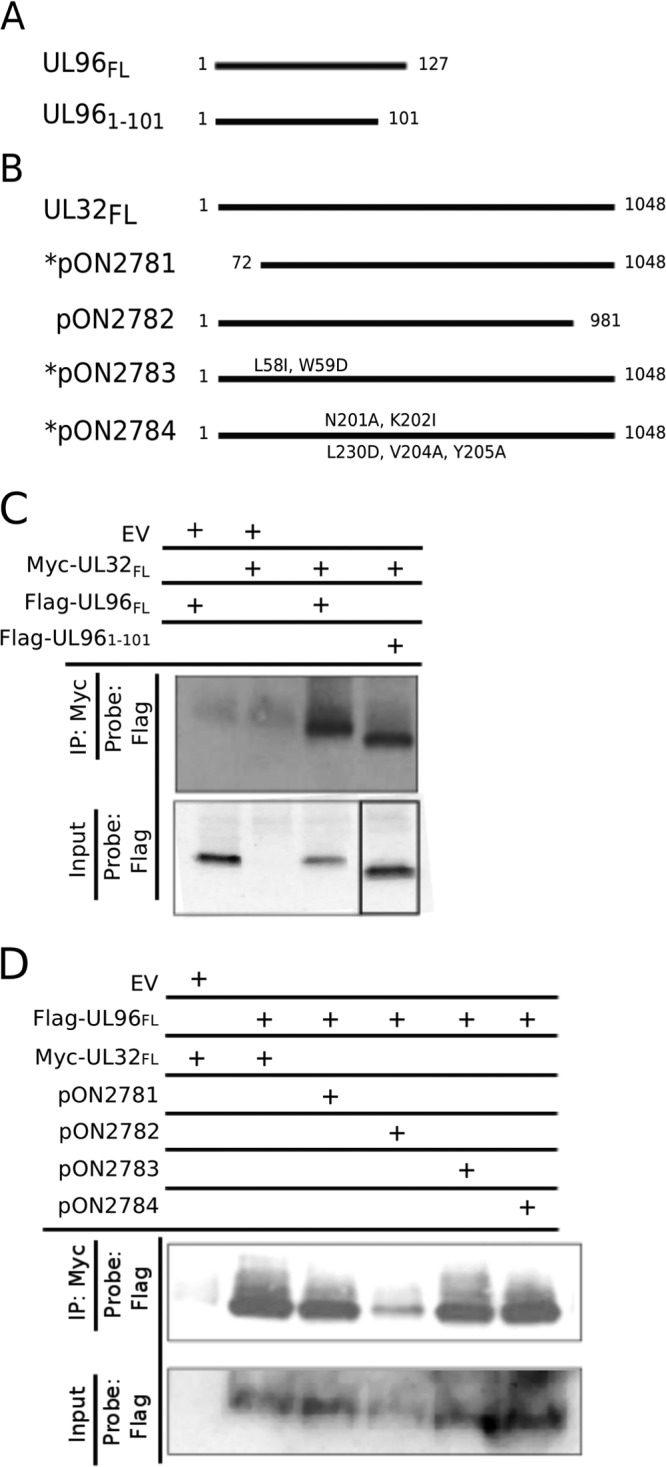
pp150 and pUL96 directly interact with each other; the N-terminal region of pUL96 and the C-terminal region of pp150 determine this interaction. Shown are line diagrams representing the Flag epitope-tagged UL96 expression plasmids (A) and Myc epitope-tagged UL32 expression plasmids used in this study (B). N-terminal pp150 mutants are denoted with an asterisk. (C) Full-length pp150 (UL32FL) interacts with full-length pUL96 (UL96FL), as well as the C-terminal truncated pUL96 (UL961-101), but not with the empty vector (EV) in an IP-IB assay after expression in 293T cells. (D) UL96FL interacts with UL32FL, as well as with the N-terminal mutants of pp150 (pON2781, pON2783, and pON2784), but only slightly with the C-terminal mutant of pp150 (pON2782) in an IP-IB assay after expression in 293T cells. The empty vector (EV) was used as a control.
Conserved UL96 C-terminal residues are critical for virus maturation.
A cluster of negatively charged amino acids in the pUL96 C-terminal region is highly conserved among betaherpesviruses (Fig. 2). A virus truncated by this region of pUL96 is arrested for growth in cell culture (6). To map the amino acids that determine pUL96 function in virus maturation, we engineered several point and clustered mutations replacing these amino acids in Towne-BAC by genetic recombineering (5, 6, 24) (Fig. 2 and 3). Individual amino acids E122, D124, and D125 were converted to alanine residues (B1-, B2-, and B3-BAC, respectively) (Fig. 3A). On the basis of the protein folding prediction of pUL96 and the degree of change in predicted protein folding upon the introduction of these mutations (Fig. 4), the entire amino acid cluster (122EVDDAV127) was converted to positively charged residues (KRKKRR [B4-BAC]), alternative negatively charged residues (DEEEDE [B5-BAC]), or neutral alanines (AAAAAA [B6-BAC]) (Fig. 3A and B). While converting the 122EVDDAV127 cluster to positively charged or neutral residues makes the UL96 C terminus more compact, changing the positions of negative charges makes it more linear (Fig. 4). Resulting mutant BACs were transfected into HF and analyzed for growth by monitoring the eGFP+ plaque size (Fig. 3B) and single-step growth curves of harvested viruses (Fig. 3C). Individual point mutations (E122A, D124A, and D125A) reduced the virus plaque size (Fig. 3B) but did not inhibit the production of infectious virus (Fig. 3C). While the B1, B2, and B3 viruses grew more slowly than Towne-BAC, all of these point mutant viruses had approximately equivalent final virus yields (Fig. 3C). In contrast, all three-cluster mutant viruses (B4, B5, and B6) showed impaired growth in fibroblasts, produced small syncytial plaque phenotypes (Fig. 3B), and failed to release infectious virus into the supernatant of transfected cells. In these mutant infections, pUL96 expression was not impaired (data not shown).
FIG 2.

pUL96 is a conserved betaherpesvirus protein critical for CMV maturation. Shown is an amino acid sequence alignment of pUL96 homologs from human CMV (HCMV; YP_081543), RhCMV (YP_068222), CCMV (AAM00734), HHV6 (CAA58360), and HHV7 (YP_073808). Identical amino acids (black) and similar amino acids (gray) are highlighted. The locations of the mutations introduced into the C-terminal region of pUL96 in this study are depicted. The last six amino acids of HCMV_UL96 are termed the EVDDAV cluster.
FIG 3.

Generation of UL96 mutant viruses by recombineering of the Towne-BAC genome. (A) Schematic of the UL95-UL97 region in Towne-BAC (top line) with other bacmid constructs shown below. δUL96BAC is depicted with a KanSacB insert replacing UL96. In B1-BAC, glutamate 122; in B2-BAC, aspartate 124; and in B3-BAC, aspartate 125 have been replaced with an alanine residue. In B4-, B5-, and B6-BAC, a negatively charged cluster of pUL96 C-terminal residues (EVDDAV) has been replaced with positively charged residues KRKKRR, the negative charge alteration DEEEDE, or an alanine conversion of all six residues, respectively. R-BAC, constructed from δUL96BAC, is depicted with the repaired UL96. (B) Typical eGFP+ plaque phenotypes of indicated BAC constructs at 7 dpt in HF. Original magnification, ×400. (C) Single-step growth curves of Towne-BAC and UL96 mutant viruses. HF were infected (MOI of 3.0), the total viruses (cells plus medium) were harvested at the indicated times postinfection, and their titers were determined on HF monolayers. The viral titers represent the averages of triplicate experiments, and the standard errors are within the symbols. The datum point at 0 dpi indicates the input virus dose.
FIG 4.

Protein Investigator (ver. 3.0.2)-based prediction of folding of the C-terminal 20 amino acids of wild type (WT) pUL96 and predicted changes in protein folding as a result of the clustered mutations (B4, B5, and B6) that were introduced in this study. The above program folds the protein on the basis of the interactions between the side chains only and does not model secondary or quaternary structure. The circles are colored to indicate the relative hydrophobicities of the side chains. The most hydrophilic amino acids are white, the most hydrophobic amino acids are black, and gray amino acids are intermediate. Amino acids with positively charged side chains have a small plus sign on their symbol; those with negatively charged side chains have a small minus sign.
To study the stage of viral infection impacted by clustered mutations in pUL96, we performed TEM of infected HF that were passaged once after transfections (Fig. 5). This single passaging was done to eliminate the possible secondary effects of transfection on cell morphology before TEM. Towne-BAC-infected cells manifested a kidney-shaped nucleus and a large single juxtanuclear virus AC in the cytoplasm, where mature and immature virions, as well as dense bodies (27–29), accumulated (Fig. 5A). B4-BAC-transfected fibroblasts (Fig. 5B) had multiple nuclei that were round or oval, and these nuclei surrounded a central cytoplasmic region where mature and immature virions, morphologically abnormal particles, and dense bodies accumulated. Less than 25% of the virus particles observed in the AC of B4-BAC-infected cells appeared regular (Fig. 5F, 6, and 7B), compared to the almost 80% of the particles in the AC of Towne-BAC-infected cells that appeared intact and regularly shaped (Fig. 5E, 6, and 7B). However, nuclear stages and the proportions of A, B, and C capsids (Fig. 5C and D) were comparable in both infections. Thus, pUL96 cluster mutations affect a late stage of virus maturation in the cytoplasm of infected cells, similar to the phenotype of a UL96 deletion virus described earlier (6).
FIG 5.

Transmission electron microscopy (TEM) of infected fibroblasts. HF transfected with Towne-BAC (A, C, E) or B4-BAC (B, D, F) were passaged once at 10 dpt and fixed for TEM at 3 days postseeding. The infected cell (A, B), the nucleus (C, D), and the cytoplasm (E, F) are shown. Bars, 5 μm (A, B) or 1 μm (C to F).
FIG 6.

Criteria for the enumeration of CMV particle types in the cytoplasm of infected cells.
FIG 7.
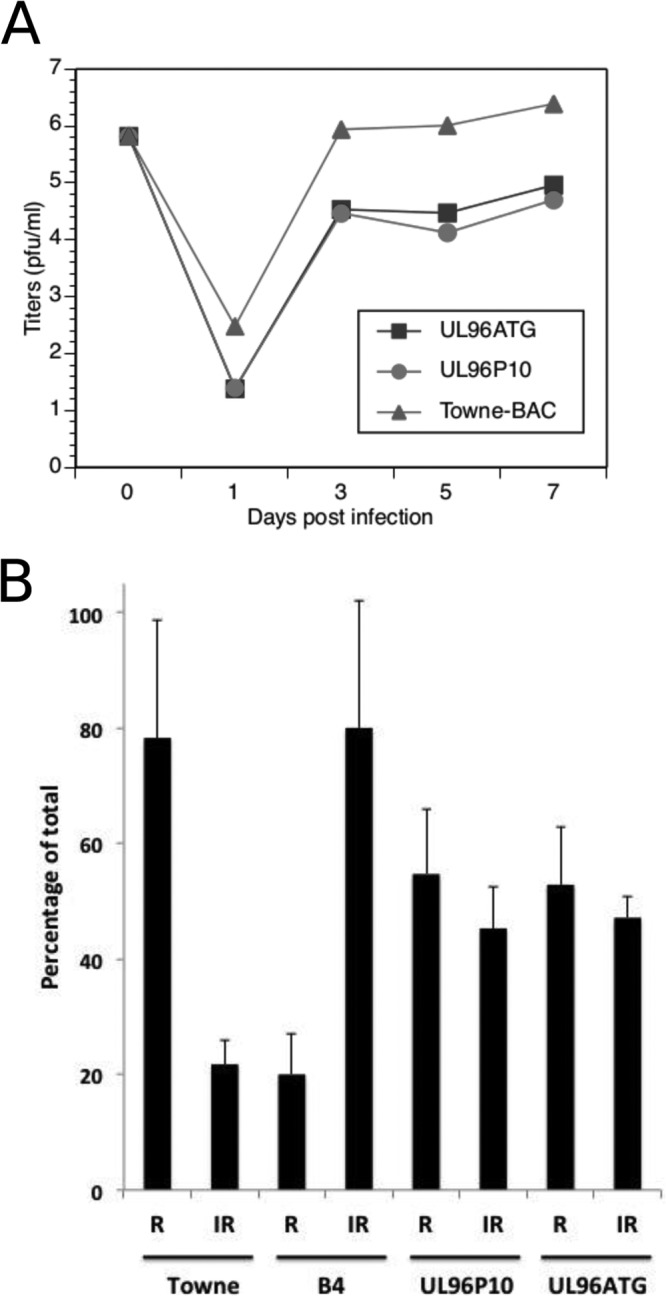
Reengineering of the UL96ATG (171819 C-to-T) mutation in the B6-BAC genome leads to recovery of virus growth and an increase in regularly shaped virus particles in the cytoplasm of infected cells. (A) Single-step growth curves of Towne-BAC, UL96P10, and UL96ATG viruses. (B) Percentages of particles that appeared irregular (IR) versus regular (R) (Fig. 6) in the cytoplasm of cells infected with the indicated viruses at a late stage of infection on the basis of average counts from five individual infected cells. Printed images of sections of electron micrographs (Fig. 5 and 9) were used for visual counting by two study-blind volunteers who were supplied with the criteria listed in Fig. 6. Tegument-filled dark particles (dense bodies) were excluded from counts. Approximately 150 particles were counted per sample.
Virus containing a UL96 C-terminal cluster mutation gains replication competency on repeated passaging.
One of the UL96 cluster mutants (B6-BAC) was allowed to accumulate compensatory mutations by repeated passage on fibroblasts. This was accomplished by passaging the BAC-transfected cells repeatedly into new tissue culture flasks until a 100% cytopathic effect was observed. Infectious virus was then harvested and passaged in tissue culture a total of 10 times. Genomic DNA harvested from this passaged virus (UL96P10) showed an RFLP different from that of the parent B6-BAC or Towne-BAC virus (Fig. 8). This UL96P10 virus replicated with a higher growth rate (Fig. 7A) than the nonreplicating parent B6-BAC virus. To identify the changes in the genome that accounted for a changed RFLP and the gain of replication competency in B6-BAC, we performed whole-genome sequencing of UL96P10 and a similarly passaged Towne-BAC virus on the Illumina platform. No major insertions or deletions were observed in the passaged Towne-BAC (KF493877) and UL96P10 (KF493876) genomes. However, several nucleotide differences between the Towne-BAC and UL96P10 viruses were noticed (Table 1). Most of these differences were located in the nonessential or noncoding regions of the genome, which are known to accumulate mutations during passaging (30). These changes did not overlap the known microRNA and noncoding RNAs that are present in the HCMV genome (31–33). When the UL96P10 and Towne-BAC viruses were compared, most noticeable were two point mutations in the UL96 region of the genome at positions 171699 and 171819 (Table 1).
FIG 8.
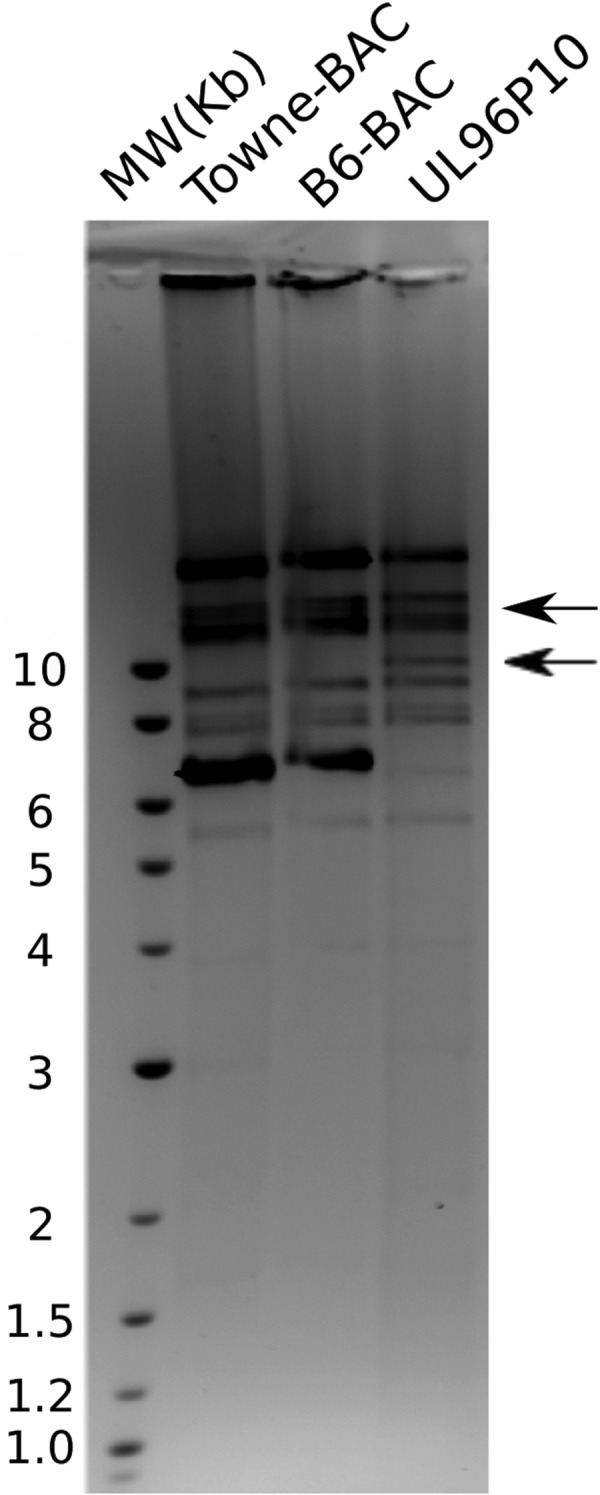
The passaged UL96P10 virus displays a restriction profile different from that of the parental B6-BAC or Towne-BAC virus. HindIII digests of viral DNA harvested from Towne-BAC, B6-BAC, and UL96P10 virus-infected fibroblasts were run on a 1% agarose gel and stained with ethidium bromide. The shift in band migration at ∼10 and ∼12 kb in the UL96P10 virus points toward genomic changes occurring in this virus during repeated passaging. Lane MW contains molecular size standards whose sizes are shown on the left in kilobases.
TABLE 1.
Summary of nucleotide differences between the Towne-BAC passaged virus (KF493877) and the UL96P10 virus (KF493876) revealed by whole-genome sequencing and subsequent comparison of nucleotide sequencesa
| Reference location(s) | Type of change | Description | Gene region | Protein coding or noncodingb |
|---|---|---|---|---|
| 27623 | Insertion | G | Upstream of TRS1 | NC |
| 124412 | Deletion | G | Upstream of RNA4.9 | NC |
| 171699 | Conversion | A→G | UL96 | C |
| 171819 | Conversion | C→T | UL96 | C |
| 171900 | Conversion | A→G | Upstream of UL97 | NC |
| 211946, 211947 | Insertion | GT | Downstream of UL145 | NC |
| 232634 | Conversion | A→G | Downstream of eGFP | NC |
| 124724 | Conversion | C→A | RNA 4.9 | NC |
| 124728 | Insertion | C | RNA 4.9 | NC |
Virus DNA was purified from pelleted virions from the cleared cell culture medium. The Nextera XT DNA Sample Preparation kit (Illumina) was used to prepare the multiplexed paired-end libraries (2 × 150 bp) before sequencing with an Illumina MiSeq. Genomic regions and open reading frames were identified by BLAST searching on the NCBI server.
C, coding; NC, noncoding.
Compensatory mutations in UL96 are responsible for the gain of replication competency in the UL96 C-terminal cluster mutant.
To study the potential impact of UL96 mutations discovered in UL96P10, we engineered the C-to-T conversion (position 171819, Table 1) in the B6-BAC mutant background in Towne-BAC by recombineering as described above. This particular mutation was chosen for its proximity to the original mutations that we engineered. The resultant UL96ATG BAC produced infectious virus, unlike the parent B6-BAC, and grew with kinetics comparable to those of the UL96P10 virus (Fig. 7A). Ultrastructural analysis of fibroblasts infected with the UL96ATG virus revealed a proportion of regularly shaped virus particles in the cytoplasm of infected cells that was comparable to that of the UL96P10 virus infections, although it did not quite reach the level of Towne-BAC virus (Fig. 7B and 9). This correlates well with the differences in final yield between the UL96P10 and UL96ATG viruses and the Towne-BAC virus (Fig. 7A). In summary, a mutation discovered in the UL96 coding region of the UL96P10 virus accounts for the gain of replication competency in this virus.
FIG 9.

Impact of reengineering of the UL96ATG (171819 C-to-T) mutation in the B6-BAC genome on the infected-cell phenotype and virus morphology. Shown are representative transmission electron micrographs of HF infected with Towne-BAC (A, D, G, J), UL96P10 (B, E, H, K), or UL96ATG (C, F, I, L) virus and processed for imaging at 5 dpi. The images show the whole infected cell (A, B, C), the cytoplasmic AC (D, E, F), the nucleus (Nuc) (G, H, I), or the cytoplasm (Cyt) (J, K, L). Bars, 5 μm (B), 2 μm (A, C, E), 1 μm (D, F), or 0.2 μm (G, H, I, J, K, L).
DISCUSSION
Tegument proteins in herpesviruses are known to interact among themselves, as well as with the capsid and envelope components of the virion and also with a variety of host proteins (14, 34–50). These interactions have important consequences in terms of virus tegumentation, envelopment, cellular trafficking, virion stability, and initiation of virus infection. In the present study, we show that CMV tegument proteins pp150 and pUL96 directly interact with each other and that the N-terminal region of pUL96 and the C-terminal region of pp150 determine this interaction (Fig. 1). These two proteins are conserved in betaherpesviruses and are known to be critical for virus maturation (5, 6, 15). Earlier studies suggested that the phenotype of a UL96 mutant virus may depend on pp150 based on the similar stage of virus replication block in UL96 mutant, UL32 mutant, and UL96 UL32 dual mutant virus-infected cells (6). Also, the accumulation of pp150 in the nuclei of UL96 mutant virus-infected cells suggested that pUL96 either enhances pp150-capsid association or functions as a chaperone to assist in the translocation of pp150-labeled nucleocapsids from the nucleus to the cytoplasm. To test the possibility of a direct interaction between pp150 and pUL96, we used IP and IB. As expected, the two proteins interacted with each other, strengthening the possibility of a functional interaction. However, a domain analysis of interaction revealed that the UL32 N-terminal and UL96 C-terminal regions are not required for this interaction. This came as a surprise because these regions determine the function of these two proteins in virus maturation (5, 6, 15). Thus, it appears that the roles of pp150 and pUL96 in virus maturation do not depend on their mutual interaction; however, it is possible that pp150 and pUL96 rely on an alternative mechanism to function when a direct interaction between these two proteins is abolished, explaining the dispensable nature of the C-terminal region of pp150 (15).
We then focused on the role of pUL96 in virus maturation by mapping the residues that are critical for function. Earlier, we demonstrated that the UL96 C terminus is important for virus maturation (6). An alignment of the pUL96 amino acid sequence reveals a stretch of negatively charged conserved amino acids at the C terminus of the protein (Fig. 2). These residues are conserved among primate CMVs, and similar negatively charged residues are present at the C termini of HHV6 and HHV7. We hypothesized that pUL96 might use these amino acids for interaction with a positively charged region of another viral or host protein; thus, exploring the phenotype of viruses containing mutations in these residues could lead to a better understanding of the mechanism of pUL96 function. Mutation of these aspartate and glutamate residues in pUL96 (122EVDDAV127) led to the arrest of virus replication at the stage of virus maturation. Ultrastructural analysis showed that these mutations affect nucleocapsid stability in the cytoplasm of infected cells, which is reminiscent of the phenotype of a UL96 null virus (6). We took advantage of the capability of the 122EVDDAV127 mutant viruses to grow to some extent in cell culture and subjected them to repeated passages. This way, we intentionally allowed the accumulation of compensatory mutations in the CMV genome. One possible outcome of this approach would be the accumulation of compensatory mutations in a virus protein that critically interacts with pUL96. In that case, it would be possible to define the interactions between pUL96 and the other candidate protein. One of the clustered mutants (B6-BAC) gained replication competency in 10 passages (UL96P10) and showed an altered RFLP on analysis of the genome. Interestingly, the B6-BAC virus was able to overcome the block at the multinucleate cell (syncytium) stage. Glycoprotein H (gH), gL, and gB are the main fusogenic proteins in CMV and are often implicated in syncytium formation (51). We hypothesized that the UL96P10 virus may be able to overcome this block by accumulating compensatory mutations in gH-gL or gB. Also, if pUL96-pp150 interaction had a role in virus maturation, we would expect to see compensatory changes in pp150. Hence, we sequenced the complete genomes of the UL96P10 virus and a similarly passaged Towne-BAC virus. On comparing these genomes, we discovered differences in the RNA4.9 and UL96 regions (Table 1); however, the gH, gL, gB, and pp150 regions were identical in all three viruses. Instead, two nonsynonymous mutations in UL96 were identified as being unique to the UL96P10 virus (Table 1). Moreover, when the C-to-T conversion mutation (position 171819) was reengineered into B6-BAC, it restored the virus growth rate to the UL96P10 level and increased the stability of virus particles in the cytoplasm (Fig. 7 and 9). Thus, results from this study show the accumulation of compensatory mutations only in the UL96 gene in a revertant virus. This, combined with the involvement of a functionally dispensable region of pp150 in the pUL96-pp150 interaction, points toward a role for pUL96 in virus maturation that does not depend upon pp150; however, the possibility remains that alternative pathways are used by pp150 and pUL96 to function when intact forms of both proteins, instead of the mutated forms, are present during infection.
ACKNOWLEDGMENTS
We are thankful to Hong Yi at the Robert P. Apkarian Integrated Electron Microscopy Core at Emory University for help with electron microscopy. Virus whole-genome sequencing was performed at the UMMC Molecular and Genomics Facility. Xiao Luo prepared the viral genome library, Ashley Robinson assembled the contigs, and Molly Tyner annotated the genome sequences. Dongmei Yang and Dirk Dittmer at The University of North Carolina at Chapel Hill helped with the analysis of genomic data. Madeline Aylward helped with cell culture in the Tandon lab.
Public Health Service grant (RO1 AI20211 [principal investigator, E. S. Mocarski]) and an Intramural Research Support Program award from the University of Mississippi Medical Center to Ritesh Tandon supported this research.
Footnotes
Published ahead of print 5 February 2014
REFERENCES
- 1.Penkert RR, Kalejta RF. 2012. Tale of a tegument transactivator: the past, present and future of human CMV pp71. Future Virol. 7:855–869. 10.2217/fvl.12.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Homer EG, Rinaldi A, Nicholl MJ, Preston CM. 1999. Activation of herpesvirus gene expression by the human cytomegalovirus protein pp71. J. Virol. 73:8512–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Britt W. 2007. CMV maturation and egress, p 311–323 In Arvin AM, Mocarski ES, Moore P, Whitley R, Yamanishi K, Campadelli-Fiume G, Roizman B. (ed), Human herpesviruses: biology, therapy and immunoprophylaxis. Cambridge Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 4.Mocarski ES, Jr, Shenk T, Pass RF. 2006. Cytomegaloviruses, p 2701–2772 In Knipe DM, Howley PM. (ed), Fields virology, 5th edition Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 5.Tandon R, Mocarski ES. 2008. Control of cytoplasmic maturation events by cytomegalovirus tegument protein pp150. J. Virol. 82:9433–9444. 10.1128/JVI.00533-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tandon R, Mocarski ES. 2011. Cytomegalovirus pUL96 is critical for the stability of pp150-associated nucleocapsids. J. Virol. 85:7129–7141. 10.1128/JVI.02549-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shanda SK, Wilson DW. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 82:7388–7394. 10.1128/JVI.00225-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolfstein A, Nagel CH, Radtke K, Dohner K, Allan VJ, Sodeik B. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227–237. 10.1111/j.1600-0854.2005.00379.x [DOI] [PubMed] [Google Scholar]
- 9.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201–209. 10.1128/JVI.80.1.201-209.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson W. 2008. Structure and formation of the cytomegalovirus virion. Curr. Top. Microbiol. Immunol. 325:187–204. 10.1007/978-3-540-77349-8_11 [DOI] [PubMed] [Google Scholar]
- 11.Trus BL, Gibson W, Cheng N, Steven AC. 1999. Capsid structure of simian cytomegalovirus from cryoelectron microscopy: evidence for tegument attachment sites. J. Virol. 73:2181–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen DH, Jiang H, Lee M, Liu F, Zhou ZH. 1999. Three-dimensional visualization of tegument/capsid interactions in the intact human cytomegalovirus. Virology 260:10–16. 10.1006/viro.1999.9791 [DOI] [PubMed] [Google Scholar]
- 13.Yu X, Shah S, Lee M, Dai W, Lo P, Britt W, Zhu H, Liu F, Zhou ZH. 2011. Biochemical and structural characterization of the capsid-bound tegument proteins of human cytomegalovirus. J. Struct. Biol. 174:451–460. 10.1016/j.jsb.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baxter MK, Gibson W. 2001. Cytomegalovirus basic phosphoprotein (pUL32) binds to capsids in vitro through its amino one-third. J. Virol. 75:6865–6873. 10.1128/JVI.75.15.6865-6873.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.AuCoin DP, Smith GB, Meiering CD, Mocarski ES. 2006. Betaherpesvirus-conserved cytomegalovirus tegument protein ppUL32 (pp150) controls cytoplasmic events during virion maturation. J. Virol. 80:8199–8210. 10.1128/JVI.00457-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DL, Bustamante C. 2001. The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature 413:748–752. 10.1038/35099581 [DOI] [PubMed] [Google Scholar]
- 17.Homa F, Huffman J, Toropova K, Lopez H, Makhov A, Conway J. 2013. Structure of the pseudorabies virus capsid: comparison with herpes simplex virus type 1 and differential binding of essential minor proteins. J. Mol. Biol. 425:3415–3428. 10.1016/j.jmb.2013.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhella D, Rixon FJ, Dargan DJ. 2000. Cryomicroscopy of human cytomegalovirus virions reveals more densely packed genomic DNA than in herpes simplex virus type 1. J. Mol. Biol. 295:155–161. 10.1006/jmbi.1999.3344 [DOI] [PubMed] [Google Scholar]
- 19.Gibson W. 1981. Structural and nonstructural proteins of strain Colburn cytomegalovirus. Virology 111:516–537. 10.1016/0042-6822(81)90354-8 [DOI] [PubMed] [Google Scholar]
- 20.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966. 10.1128/JVI.78.20.10960-10966.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228. 10.1073/pnas.2334032100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401. 10.1073/pnas.1635160100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rost B, Yachdav G, Liu J. 2004. The PredictProtein server. Nucleic Acids Res. 32:W321–W326. 10.1093/nar/gkh377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomason LC, Court DL, Bubunenko M, Costantino N, Wilson H, Datta S, Oppenheim A. 2007. Recombineering: genetic engineering in bacteria using homologous recombination. Curr. Protoc. Mol. Biol. Chapter 1:Unit 1.16. 10.1002/0471142727.mb0116s78 [DOI] [PubMed] [Google Scholar]
- 25.Brechtel T, Tyner M, Tandon R. 2013. Complete genome sequence of a cytomegalovirus Towne-BAC (bacterial artificial chromosome) isolate maintained in Escherichia coli for 10 years and then serially passaged in human fibroblasts. Genome Announc. 1:e00693–13. 10.1128/genomeA.00693-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brechtel TM, Tyner M, Tandon R. 2013. Complete genome sequence of a UL96 mutant cytomegalovirus Towne-BAC (bacterial artificial chromosome) isolate passaged in fibroblasts to allow accumulation of compensatory mutations. Genome Announc. 1:e00901–13. 10.1128/genomeA.00901-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarov I, Abady I. 1975. The morphogenesis of human cytomegalovirus. Isolation and polypeptide characterization of cytomegalovirions and dense bodies. Virology 66:464–473 [DOI] [PubMed] [Google Scholar]
- 28.Fiala M, Honess RW, Heiner DC, Heine JW, Jr, Murnane J, Wallace R, Guze LB. 1976. Cytomegalovirus proteins. I. Polypeptides of virions and dense bodies. J. Virol. 19:243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stinski MF. 1976. Human cytomegalovirus: glycoproteins associated with virions and dense bodies. J. Virol. 19:594–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang A, Ren L, Abenes G, Hai R. 2009. Genome sequence divergences and functional variations in human cytomegalovirus strains. FEMS Immunol. Med. Microbiol. 55:23–33. 10.1111/j.1574-695X.2008.00489.x [DOI] [PubMed] [Google Scholar]
- 31.Dunn W, Trang P, Zhong Q, Yang E, van Belle C, Liu F. 2005. Human cytomegalovirus expresses novel microRNAs during productive viral infection. Cell. Microbiol. 7:1684–1695. 10.1111/j.1462-5822.2005.00598.x [DOI] [PubMed] [Google Scholar]
- 32.Meshesha MK, Veksler-Lublinsky I, Isakov O, Reichenstein I, Shomron N, Kedem K, Ziv-Ukelson M, Bentwich Z, Avni YS. 2012. The microRNA transcriptome of human cytomegalovirus (HCMV). Open Virol. J. 6:38–48. 10.2174/1874357901206010038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatherer D, Seirafian S, Cunningham C, Holton M, Dargan DJ, Baluchova K, Hector RD, Galbraith J, Herzyk P, Wilkinson GW, Davison AJ. 2011. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. U. S. A. 108:19755–19760. 10.1073/pnas.1115861108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maringer K, Stylianou J, Elliott G. 2012. A network of protein interactions around the herpes simplex virus tegument protein VP22. J. Virol. 86:12971–12982. 10.1128/JVI.01913-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Omar OS, Simmons AJ, Andre NM, Wilson DW, Gross ST. 2013. Pseudorabies virus and herpes simplex virus type 1 utilize different tegument-glycoprotein interactions to mediate the process of envelopment. Intervirology 56:50–54. 10.1159/000339467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354. 10.1016/j.virol.2008.05.035 [DOI] [PubMed] [Google Scholar]
- 37.Coller KE, Lee JI, Ueda A, Smith GA. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 81:11790–11797. 10.1128/JVI.01113-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566–9571. 10.1128/JVI.79.15.9566-9571.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klupp BG, Fuchs W, Granzow H, Nixdorf R, Mettenleiter TC. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76:3065–3071. 10.1128/JVI.76.6.3065-3071.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye GJ, Vaughan KT, Vallee RB, Roizman B. 2000. The herpes simplex virus 1 U(L)34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 74:1355–1363. 10.1128/JVI.74.3.1355-1363.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips SL, Cygnar D, Thomas A, Bresnahan WA. 2012. Interaction between the human cytomegalovirus tegument proteins UL94 and UL99 is essential for virus replication. J. Virol. 86:9995–10005. 10.1128/JVI.01078-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cygnar D, Hagemeier S, Kronemann D, Bresnahan WA. 2012. The cellular protein SPT6 is required for efficient replication of human cytomegalovirus. J. Virol. 86:2011–2020. 10.1128/JVI.06776-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips SL, Bresnahan WA. 2011. Identification of binary interactions between human cytomegalovirus virion proteins. J. Virol. 85:440–447. 10.1128/JVI.01551-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Indran SV, Ballestas ME, Britt WJ. 2010. Bicaudal D1-dependent trafficking of human cytomegalovirus tegument protein pp150 in virus-infected cells. J. Virol. 84:3162–3177. 10.1128/JVI.01776-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chevillotte M, Landwehr S, Linta L, Frascaroli G, Luske A, Buser C, Mertens T, von Einem J. 2009. Major tegument protein pp65 of human cytomegalovirus is required for the incorporation of pUL69 and pUL97 into the virus particle and for viral growth in macrophages. J. Virol. 83:2480–2490. 10.1128/JVI.01818-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamil JP, Coen DM. 2007. Human cytomegalovirus protein kinase UL97 forms a complex with the tegument phosphoprotein pp65. J. Virol. 81:10659–10668. 10.1128/JVI.00497-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schierling K, Stamminger T, Mertens T, Winkler M. 2004. Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J. Virol. 78:9512–9523. 10.1128/JVI.78.17.9512-9523.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hofmann H, Sindre H, Stamminger T. 2002. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 76:5769–5783. 10.1128/JVI.76.11.5769-5783.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ogawa-Goto K, Irie S, Omori A, Miura Y, Katano H, Hasegawa H, Kurata T, Sata T, Arao Y. 2002. An endoplasmic reticulum protein, p180, is highly expressed in human cytomegalovirus-permissive cells and interacts with the tegument protein encoded by UL48. J. Virol. 76:2350–2362. 10.1128/jvi.76.5.2350-2362.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol. 20:392–401. 10.1016/j.tim.2012.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kinzler ER, Compton T. 2005. Characterization of human cytomegalovirus glycoprotein-induced cell-cell fusion. J. Virol. 79:7827–7837. 10.1128/JVI.79.12.7827-7837.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]