

ABSTRACT
During dengue virus infection of host cells, intracellular membranes are rearranged into distinct subcellular structures such as double-membrane vesicles, convoluted membranes, and tubular structures. Recent electron tomographic studies have provided a detailed three-dimensional architecture of the double-membrane vesicles, representing the sites of dengue virus replication, but temporal and spatial evidence linking membrane morphogenesis with viral RNA synthesis is lacking. Integrating techniques in electron tomography and molecular virology, we defined an early period in virus-infected mosquito cells during which the formation of a virus-modified membrane structure, the double-membrane vesicle, is proportional to the rate of viral RNA synthesis. Convoluted membranes were absent in dengue virus-infected C6/36 cells. Electron tomographic reconstructions elucidated a high-resolution view of the replication complexes inside vesicles and allowed us to identify distinct pathways of particle formation. Hence, our findings extend the structural details of dengue virus replication within mosquito cells and highlight their differences from mammalian cells.
IMPORTANCE Dengue virus induces several distinct intracellular membrane structures within the endoplasmic reticulum of mammalian cells. These structures, including double-membrane vesicles and convoluted membranes, are linked, respectively, with viral replication and viral protein processing. However, dengue virus cycles between two disparate animal groups with differing physiologies: mammals and mosquitoes. Using techniques in electron microscopy, we examined the differences between intracellular structures induced by dengue virus in mosquito cells. Additionally, we utilized techniques in molecular virology to temporally link events in virus replication to the formation of these dengue virus-induced membrane structures.
INTRODUCTION
Dengue virus (DENV) is a flavivirus, within the Flaviviridae family. There are four distinct serotypes, referred to as DENV-1, -2, -3, and -4. DENV is an enveloped virus with an 11-kb positive-sense RNA genome encoding a polyprotein which is co- and posttranslationally processed. Three structural proteins (C, prM, and E) constitute the virus particle, and the seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) function in viral RNA replication (1). DENV causes one of the most aggressive arthropod-borne viral diseases, with approximately 100 to 350 million cases annually. Of these, approximately 500,000 patients are admitted to hospitals with a more severe form of the disease, referred to as dengue hemorrhagic fever and/or dengue shock syndrome (2).
During infection, the DENV RNA is translated into a single polyprotein associated with the endoplasmic reticulum (ER) membrane, and cellular and viral proteases cleave the polyprotein, generating the individual proteins required for subsequent viral RNA synthesis and virion assembly. Following cleavage, the viral proteins remain associated with the ER membrane either on the cytoplasmic side or in the ER lumen. The three structural proteins and the replicase proteins, NS1, NS2A, NS2B, NS4A, and NS4B, are all integrated into the ER membrane. The C protein will engage with newly synthesized RNA on the cytoplasmic side of the ER and form the capsid-RNA complex. Together with the lipid bilayer of the ER, the transmembrane prM and E proteins residing within the ER lumen form an envelope that will enclose the capsid-RNA complex, generating immature virus particles that bud into the ER. NS1 is involved in virus replication, as it has been shown to reside within the viral replicase complex (3). However, a major portion of the NS1 protein is localized within the ER lumen, and thus it is unclear how it interacts with other components of the replication complex. While the function of NS2A is not known, NS2B is a cofactor for the viral protease NS3 and is involved in viral polyprotein processing. Additionally, NS3 has a helicase activity that presumably unwinds the RNA template during viral RNA synthesis, which is carried out by the RNA-dependent RNA polymerase (RdRp), NS5. NS4A and NS4B are integral membrane proteins. NS4A, in concert with other viral and cellular proteins, is believed to provide a scaffold for the formation of the replication complex (4–6). NS4B is considered a negative modulator for helicase activity (7, 8). Both NS4A and NS4B have also been implicated in driving the reorganization of cellular membranes observed in virus-infected cells (5, 6).
Numerous positive-strand RNA viruses have been shown to induce subcellular membrane alterations to promote their replication. These include rubiviruses (9–11), nodaviruses (12), picornaviruses (13, 14), arteriviruses (15), coronaviruses (16, 17), alphaviruses (18–20), and flaviviruses (3, 21–31). Combined immunoelectron microscopy (IEM) and electron tomography (ET) studies have provided significant insight into the architecture of flavivirus-induced membrane structures associated with functional replication complexes within mammalian cells (22, 28, 31). A complex set of DENV-modified structures, including double-membrane vesicles (Ve) enclosed within membrane packets (Vp), convoluted membranes (CM), and tubular structures (T), is observed, covered by a single continuous ER membrane (28). Ve are comprised of viral RNA, replication proteins, and probably cellular proteins and form the replication complex where RNA synthesis occurs (28, 32–34). CM are likely to be the sites for viral protein translation and processing, as shown with Kunjin virus-infected Vero cells (3). IEM analyses on Huh-7 cells have also suggested that the CM consists of a pool of nonstructural proteins and lipids involved in DENV replication (28).
The morphological characteristics of virus-induced structures have been well addressed for mammalian cell systems. However, little is known about these structures within cells of the insect vector. Although the morphological structures induced by DENV within mosquito cells had been examined by transmission electron microscopy (TEM) (23, 24), the limitation of conventional chemical fixation for sample visualization presents obstacles in obtaining sufficient morphological detail. Here we exploited the advantage of high-pressure freezing (HPF) and freeze-substitution (FS) techniques to improve the preservation of virally modified structures within DENV-infected C6/36 cells. We applied a combination of molecular virology and electron microscopy techniques (immunoelectron microscopy [IEM] and electron tomography [ET]) to obtain a three-dimensional (3D) perspective of DENV replication, assembly, and budding within cells derived from the insect vector.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
C6/36 cells, a mosquito cell line (ATCC), were maintained in minimum essential medium (MEM) supplemented with 2 mM l-glutamine, 0.1 mM nonessential amino acids (Gibco, Grand Island, NY), and heat-inactivated 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO) at 30°C in the presence of 5% CO2. Huh-7 cells were seeded in Dulbecco's modified eagle medium (DMEM) (Gibco, Grand Island, NY) supplemented with heat-inactivated 10% FBS at 37°C with 5% CO2. MEM and DMEM containing 2% FBS were used to maintain DENV-infected C6/36 cells and Huh-7 cells, respectively. DENV serotype 2, strain 16681 (kindly provided by Richard Kinney, CDC, Fort Collins, CO), was amplified in C6/36 cells.
Antibodies against DENV proteins (C, prM, E, NS1, NS3, and NS5) were kindly provided by James Strauss (Division of Biology, California Institute of Technology, Pasadena, CA). An anti-double-stranded RNA (anti-dsRNA) monoclonal antibody was obtained from English & Scientific Consulting (Szirak, Hungary). An antibromodeoxyuridine (anti-BrdU) monoclonal antibody and a tetramethyl rhodamine isocyanate (TRITC)-conjugated goat anti-mouse secondary antibody were purchased from Sigma-Aldrich (St. Louis, MO) and Abcam (Cambridge, MA), respectively.
Time course experiments.
C6/36 cells, seeded in 35-mm culture dishes, were washed twice and infected with virus at a multiplicity of infection (MOI) of 5 for 2 h at room temperature. Infected cells were washed extensively with MEM and incubated further in MEM supplemented with 2% FBS at 30°C in the presence of 5% CO2. At the indicated time points (0, 4, 8, 12, 14, 16, 20, 24, 36, and 48 h) after washing/or adding of the maintenance medium, infected cells and culture media were harvested for the measurement of total cell number, intracellular viral RNA, and infectious cell-associated and extracellular viruses. A subset of infected cells was also processed for transmission electron microscopy analysis.
qRT-PCR.
Measurement of intracellular viral RNA was performed using quantitative real-time reverse transcription-PCR (qRT-PCR) (35). Infected cells were lysed with TRIzol reagent according to the manufacturer's protocol (Invitrogen, Carlsbad, CA), and total RNA was recovered by chloroform precipitation. qRT-PCR was performed using a TaqMan probe amplification system (Invitrogen, Carlsbad, CA). As a standard reference for measuring viral RNA quantity, 10-fold serial dilutions of a known concentration of full-length in vitro-transcribed DENV-2 RNA covering the range of 8.67 × 103 to 8.67 × 108 copies/ml was used along with the samples. A standard curve was then used to calculate the viral RNA concentration in the samples, and the data were expressed as RNA copies/total cells and RNA copies/cell.
Focus immunoassay titration.
Focus immunoassay titration was used to determine the titer of infectious DENV-2 as described previously (36) with the following modifications. Briefly, C6/36 cells seeded in 96-well plates were infected with equal volumes of serial 10-fold dilutions of virus samples. Virus adsorption was carried out at 30°C for 2 h, following which the cells were overlaid with 1.5% (vol/vol) carboxymethyl cellulose (Sigma, St. Louis, MO) in MEM medium containing 4% (vol/vol) FBS. Foci of infected cells were visualized by successively employing 4G2, an anti-E primary antibody, followed by alkaline phosphatase-conjugated goat anti-mouse IgG(H+L) secondary antibody (Sigma, St. Louis, MO) and a chromogenic substrate mixture (Sigma, St. Louis, MO). The virus titer was expressed as focus-forming units (FFU)/ml.
Immunofluorescence microscopy and BrUTP labeling of viral RNA.
DENV-infected C6/36 cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) at room temperature for 10 min and then permeabilized with 2% Triton X-100 in PBS. Cell monolayers were reacted with each of the primary antibodies specific for C, prM, E, NS1, NS3, NS5, and dsRNA at 37°C for 1 h. Cells were washed 3 times with PBS and reacted with a goat anti-mouse TRITC-conjugated secondary antibody. Following extensive washing with PBS, nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole), and the cells were mounted on glass slides and examined at a magnification of ×60 using an IX81 inverted fluorescence microscope (Olympus, Tokyo, Japan).
For the detection of the replicative form of the viral RNA, C6/36 cells were infected with DENV-2 for 12 h, washed 3 times with PBS, and starved in FBS-free MEM for 1 h. Cellular protein synthesis was then inhibited with a final concentration of 5 μg/ml actinomycin D prior to treatment with 10 mM BrUTP (Sigma, St. Louis, MO) for an additional 12 h at 30°C. The cells were fixed, and the presence of replicative-form RNA was detected by fluorescence microscopy employing an anti-BrdU primary antibody (Sigma, St. Louis, MO).
Sample preparation for conventional transmission electron microscopy.
C6/36 or Huh-7 cells seeded on Thermanox coverslips were infected with DENV-2 at a multiplicity of infection of 5. At 48 h after virus adsorption, cells were fixed in 2% paraformaldehyde and 1.5% glutaraldehyde in 0.1 M cacodylate buffer for a minimum of 30 min at room temperature. Cells were washed 3 times with 0.1 M cacodylate buffer and then reacted with 2% osmium and 1.5% potassium ferricyanide in distilled water (dH2O) for 1 h. After washing with deionized H2O, cells were stained with 2% uranyl acetate for 90 min, dehydrated gradually in a series of ethanol concentrations (10%, 30%, 50%, 70%, 90%, and absolute ethanol), and then infiltrated in ethanol-Epon 812 (Electron Microscopy Sciences, Hatfield, PA) mixtures at 3:1, 2:2, 1:3, and 1:9 prior to embedding in 100% Epon 812 at 50°C for 48 h. Thin sections of resin-embedded cells were prepared using an ultramicrotome (Leica, Bannockburn, IL), placed on Formvar-carbon coated copper grids, and stained with 2% uranyl acetate followed by Sato's lead. The sections were then examined using a CM-10 transmission electron microscope (Philips) at 80 kV. Approximately 50 cell profiles (18) were randomly observed for the presence of the virus-induced structures, and the numbers of Vp and Ve per 10 cell profiles were determined at different time points following virus adsorption.
High-pressure freezing and freeze-substitution.
DENV-infected cells on sapphire discs or cell pellets harvested at 48 h after virus adsorption were placed into the EM PACT2 membrane carrier shortly before they were subjected to rapid freezing using the EM PACT2 high-pressure freezer (Leica, Bannockburn, IL). The frozen cells were then subjected to freeze-substitution using the AFS2 automatic freeze-substitution system (Leica, Bannockburn, IL). Briefly, frozen cells were stained in 1% tannic acid in acetone at −90°C for 8 h. The cells were washed 3 times with cold acetone and stained with a mixture of 0.5% osmium and 2% uranyl acetate in acetone for 8 h at −90°C. The temperature was then raised in increments of 5°C/h to −20°C, where it was held for 8 h, and then warmed up to 0°C at the same rate. At 0°C, cells were washed 3 times with cold acetone. Samples were then moved to room temperature to start the infiltration process with the series of acetone-Epon 812 or acetone-Durcupan (Sigma-Aldrich, St. Louis, MO) mixtures at 3:1, 1:1, 1:3, and 1:9 before final embedding in either 100% Epon 812 or Durcupan at 50°C for 48 h. The 250-nm-thick sections were prepared and placed on glow-discharged Formvar-carbon coated copper slot grids. The sections were poststained with 2% uranyl acetate followed by Sato's lead. Gold beads (10 nm) were applied on both sides of the grid for use as fiducial markers.
For immunoelectron microscopy, the cells were subjected to HPF and FS in 2% uranyl acetate at −90°C for 8 h and then warmed to −30°C in increments of 5°C/h. The temperature was held at −30°C for another 8 h before it was raised to 0°C for further dehydration in ethanol. Cells were then subjected to infiltration using a series of HM20 (Electron Microscopy Sciences, Hatfield, PA) and ethanol mixtures before the final embedding with HM20 at −20°C for 48 h. Resin-embedded DENV-infected C6/36 cell sections were incubated with 1% bovine serum albumin (BSA) in PBS for 15 min to block nonspecific binding and reacted with monoclonal anti-NS1, -NS3, and -NS5 antibodies as well as the anti-dsRNA antibody at room temperature for 18 h. For the detection of E protein by immunoelectron microscopy, infected cells were subjected to FS in a mixture of 80% (wt/wt) epoxy embedding medium in acetone using the same parameters as described above before embedding with Durcupan. The sections were then stained with anti-E antibody 4G2. After incubation with various primary antibodies, sections were washed 3 times on a drop of PBS and incubated with a goat anti-mouse IgG antibody that was conjugated with 10-nm gold particles (EMS, Hatfield, PA) at room temperature for 1 h. The washing step was repeated 3 times with a drop of PBS and 2 times with a drop of deionized H2O. The sections were treated with 2% uranyl acetate, air dried, and examined under the CM-10 electron microscope.
Electron tomography.
Resin-embedded blocks of DENV-infected C6/36 cells were cut to a thickness of 250 nm and placed on Formvar-carbon coated slot grids. Sections were poststained with 2% uranyl acetate followed by Sato's lead. Ten-nanometer gold particles were applied on both sides of the sections to serve as a fiducial marker. Grids were placed in the Autoloader sample holder on a Titan Krios electron microscope (FEI, Eindhoven, Netherlands) operated at 300 kV. Digital images were recorded as a single-axis tilt series across a range of −60° to 60° in 1° increments using a 4k Ultrascan 950 (Gatan, Warrendale, PA). Images were acquired at a defocus level of −0.2 μm. Tomograms were reconstructed using the IMOD software package (version 4.3.7) (37). The three-dimensional surface models were generated by manually tracing the areas of interest from unfiltered tomograms followed by smoothing labels and gap filling.
RESULTS
Measurement of DENV replication parameters within mosquito cells.
In order to establish the most suitable time point for EM analysis of DENV-infected C6/36 mosquito cells, a single-step kinetic study was employed to measure DENV infection parameters. C6/36 cells seeded in 35-mm tissue culture dishes were infected with DENV-2 strain 16681 at an MOI of 5. After 2 h of adsorption at room temperature, the infected cells were extensively washed with FBS-free culture medium and then maintained in 2% FBS-containing minimal essential medium. At the indicated time points, the infected cells were harvested and lysed for the measurement of intracellular viral RNA by qRT-PCR. Newly synthesized intracellular viral RNA was first detected at 8 h postinfection (p.i.) and increased until it reached a plateau producing 9.5 × 1010 RNA copies/ml at 24 h p.i. (Fig. 1A). Based on the focus immunoassay titration that was performed to determine the infectious, cell-associated virus, the progeny virus particles became detectable only at 12 h, indicating that there was a 4-h delay in virus assembly compared to RNA replication (Fig. 1B, open circles). The cell-associated virus detected during the exponential period, however, increased simultaneously with the RNA levels and peaked at the same time point. A similar result was observed for the infectious extracellular virus during the exponential period (Fig. 1B, closed circles), with a slight increase in virus release detected after 36 h p.i. Immunofluorescence analysis was also employed using a DENV anti-NS3 antibody to measure infection. While the peak of RNA synthesis would be the most appropriate time point for investigating the functional correlation of DENV-induced structures with RNA replication, the cells at this stage were not all infected, which would have complicated the IEM and ET analyses. Therefore, the 48-h time point (when all cells were infected) was chosen to observe different stages of the infection.
FIG 1.
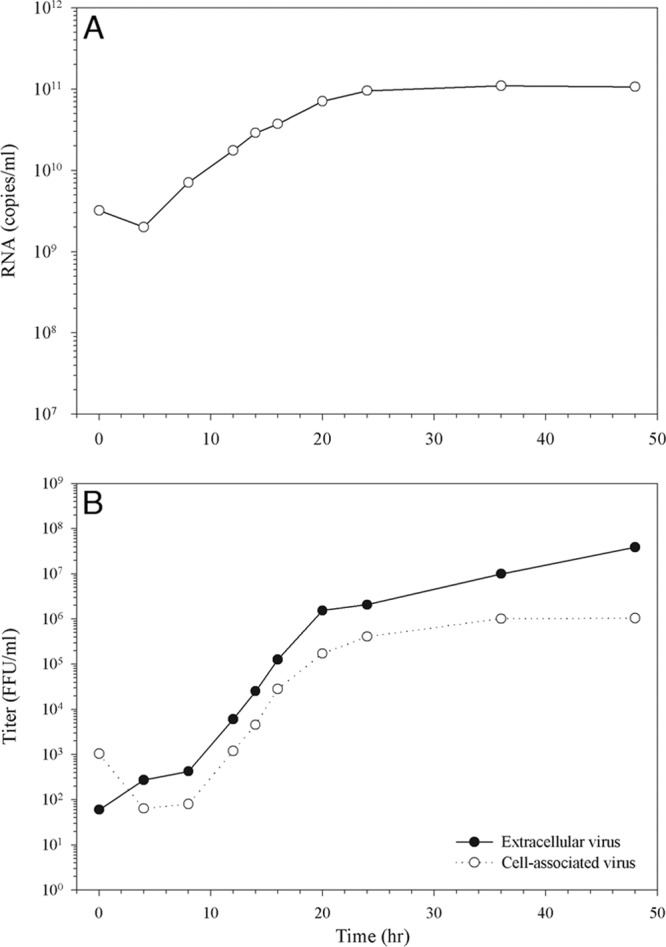
Time course of virus replication. C6/36 mosquito cells were infected with DENV at an MOI of 5. At the indicated time points infected cells were harvested. (A) Viral RNA was extracted from the cells and quantified by qRT-PCR using a DENV-specific TaqMan probe. (B) The cell-associated virus titers (open circles) in infected cell lysates or the culture supernatant of infected cells harvested at each time point were used to determine the extracellular virus titer (closed circles) using a focus immunoassay titration method.
DENV-induced membrane structure alterations occur within C6/36 cells.
Chemically fixed mock-infected and DENV-infected C6/36 cells at 48 h p.i. were examined under the transmission electron microscope to delineate structures that were induced by DENV infection. Compared with mock-infected C6/36 cells, DENV-infected cells exhibited a complex set of virus-induced subcellular structures, including large membrane-bound Vp, which enclosed multiple round Ve and, in some instances, a number of virus particles (Vi) (Fig. 2A). A proportion of Ve contained irregular thread-like electron-dense material previously identified in other flaviviruses and single-stranded, positive-sense RNA viruses as the replication complex (RC) based on the presence of double-stranded RNA (15, 22). In addition, tubular structures (T) were observed in the vicinity of Vp (Fig. 2A). To improve the visualization of these structures, DENV-infected C6/36 cells were prepared by HPF for TEM. Strikingly, compact areas of electron-dense material were observed in the internal part of some Ve (Fig. 2B), suggesting that the thread-like material detected with chemically fixed sections might represent larger structures that were distorted during the chemical fixation process. With both chemically fixed and HPF-derived sections, only Vp, Ve, and T were identified in DENV-infected C6/36 cells, and CM was not observed. In contrast, four morphologically distinct structures, Vp, Ve, T, and CM, were observed in our parallel study on DENV-infected Huh-7 cells (Fig. 2C and D), in agreement with previous studies (22, 28).
FIG 2.
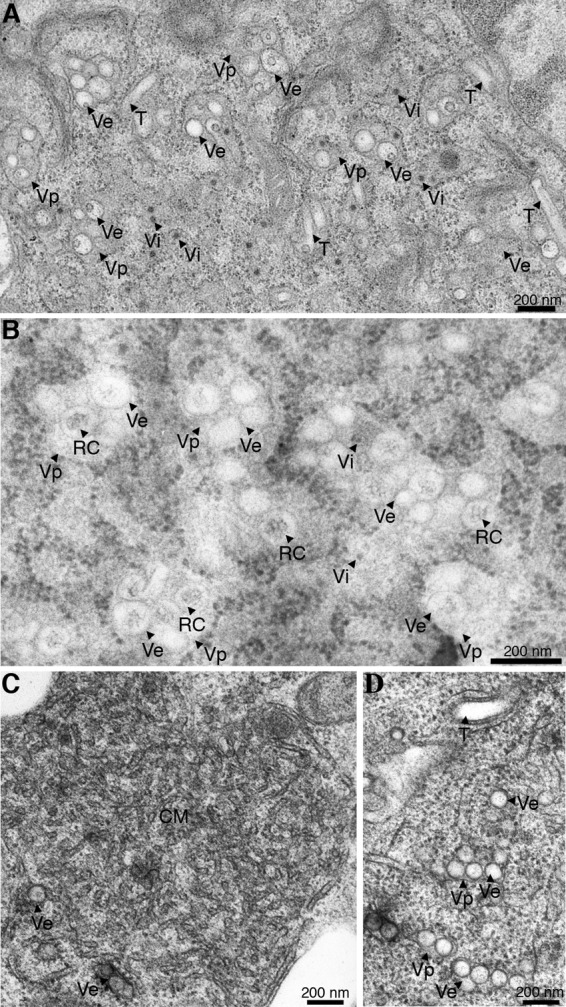
DENV-induced structural rearrangements within C6/36 cells. Thin-section TEM images of dengue virus-infected resin-embedded C6/36 cells are shown. (A) DENV-infected C6/36 cells at 48 h of infection were chemically fixed with 2% paraformaldehyde and 1.5% glutaraldehyde for a minimum of 30 min at room temperature. Fixed cells were stained with 2% osmium followed by 2% aqueous uranyl acetate for 90 min. The cells were then dehydrated gradually with a series of ethanol-resin mixtures and processed for embedding with Epon 812. DENV-infected C6/36 cells showed three types of virus-induced structures: the large Vp containing Ve within and the T. The dark, small, electron-dense spheres corresponded to the morphology and size of virions (Vi). (B) DENV-infected cells were processed for high-pressure freezing and freeze-substitution in 0.5% osmium in acetone. The resin-embedded sections were stained with uranyl acetate followed by Sato's lead citrate and examined under a CM-10 transmission electron microscope (Philips). In addition to the structures observed by chemical fixation in panel A, more-detailed structures were obtained by using high-pressure freezing of cell preparations. The thread-like electron material inside the vesicles was clearly observed by this fixation method. These structures represent the RC. (C and D) Comparison of the different structures that were induced during DENV replication within C6/36 cells and Huh-7 cells. TEM micrographs of DENV-infected Huh-7 cells are shown. Infected cells were chemically fixed before processing for dehydration, infiltration, and embedding as described in Materials and Methods. The 90-nm thin sections were stained with uranyl acetate and Sato's lead citrate before examination under a CM-10 electron microscope at 80 kV. High magnification of TEM micrographs revealed CM and Ve (C) and Vp, Ve, and T (D) induced by DENV within Huh-7 cells. The CM represent morphological differences between structures induced in Huh-7 cells and C6/36 cells during DENV infection.
Temporal changes in membrane rearrangements induced by DENV infection.
A time course study was then performed to visualize how DENV-induced membrane rearrangements progress during infection. At various time points after DENV-2 infection at an MOI of 5, C6/36 cells were chemically fixed, and 50 cell profiles were observed at random for each time point. At 0 and 4 h p.i., there was no distinct difference in the intracellular membrane architecture between mock-infected and DENV-infected cells. Isolated Vp containing one or two Ve were first observed at 8 h p.i. (Fig. 3). At 12 and 16 h p.i. single Vp were found to enclose a few more Ve. The number of Ve inside the Vp increased after 20 h of infection (Fig. 3). Virus particles were detected inside and around Vp starting at 20 h p.i., whereas tubular structures became detectable at 36 h p.i., when Vp occupied a large fraction of the cytoplasm of infected cells (Fig. 3). A quantitative analysis, which was performed by manually counting Vp and Ve found in 10 cells from a single slice at each time point, revealed a time-dependent accumulation of Vp and Ve between 20 and 48 h p.i. (Fig. 4A). When the average number of Ve (per cell) was plotted against the intracellular viral RNA content (per cell), a linear relationship between the Ve content and intracellular viral RNA content was observed between 8 and 24 h p.i. (Fig. 4C). This relationship did not extend into the 36- and 48-h time points due to a reduction of the intracellular viral RNA content (per cell) after 24 h p.i. (Fig. 4B). This result suggested that during the early period of dengue virus infection, formation of new Ve structures is coupled with an increase in viral RNA replication. After this period, limitation of essential substances required for RNA replication or, more likely, the formation and subsequent release of viral particles may contribute to a depletion of the intracellular viral RNA pool, resulting in the loss of linearity between these two parameters.
FIG 3.
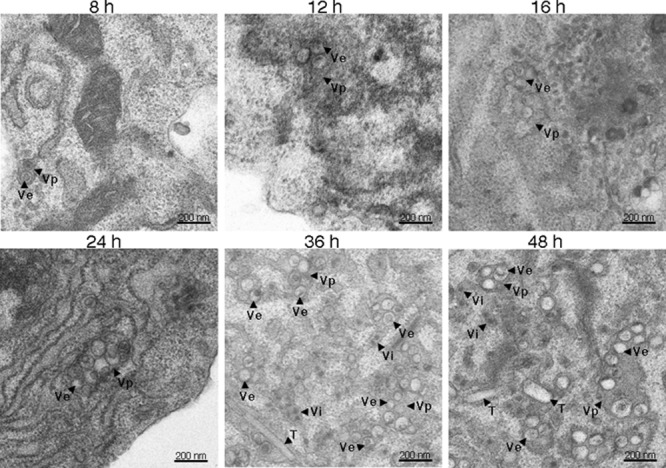
Progression of DENV-induced ultrastructure alterations. Electron micrographs representing chemically fixed DENV-infected cells at 8 h (A), 12 h (B), 16 h (C), 24 h (D), 36 h (E), and 48 h (F) after infection are shown. A minimum of 50 cells was examined for morphological changes that occur during DENV infection of C6/36 cells. An image of each representative area is shown. The Vp and Ve structures were first observed at 8 h p.i. (A). Minimal expansion in the numbers of these structures was observed at 12 p.i. (B) and 16 h p.i. (C). A significant expansion of Vp and Ve formation was observed at 24 h p.i. (D). The T were visualized only at 36 h p.i. (E), with the presence of Vp and Ve structures occupying a significant volume of infected cells. Scale bars, 200 nm.
FIG 4.
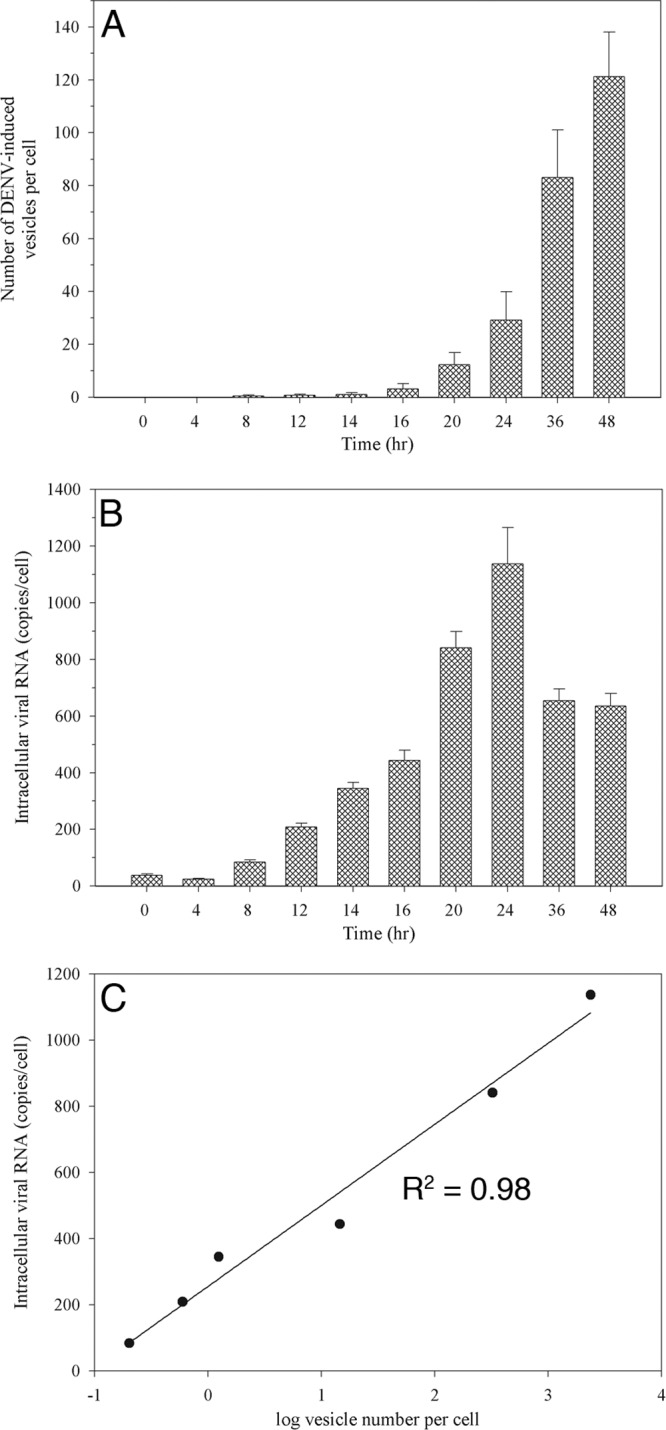
Quantitative analysis of vesicle formation during the course of DENV infection. C6/36 cells infected with DENV at 8, 12, 14, 16, 20, 24, 36, and 48 h after infection were chemically fixed and processed for TEM. Ten different cell profiles were taken randomly using a CM-10 electron microscope for measurement of Vp and Ve numbers per cell. (A) The average number of Ve per cell at each time point was plotted. The 48-h time point showed the maximum number of Ve. (B) Intracellular viral RNA content per cell. Peak RNA levels were detected at 24 h p.i. (C) The graph shows a linear correlation of log vesicle number versus total intracellular viral RNA levels. The Ve number per cell was converted to a log and subjected to linear regression analysis with total intracellular RNA copies per cell. The r2 and P values were 0.98 and 0.0002, respectively, and indicated the correlative multiplication of Ve formation to the viral RNA that was being produced per cell during the period of 8 to 24 h postinfection.
Distribution of dengue viral proteins and newly synthesized viral RNA in the cell.
To analyze the localization of viral proteins within the DENV-induced structures, immunofluorescence microscopy was performed on DENV-infected C6/36 cells. At 48 h p.i., infected cells were fixed and reacted with antibodies specific for the following viral proteins: C, prM, E, NS1, NS3, and NS5. These antibodies displayed distinct staining patterns in DENV-infected cells compared to mock-infected cells. C, prM, NS1, and NS3 were distributed in the perinuclear region, whereas NS5 was observed predominantly in the nucleus (Fig. 5A). The C protein displayed small speckled patterns of staining (Fig. 5A, C), whereas the prM protein showed a more diffuse pattern (Fig. 5A, prM). This is different from observations in mammalian cells, where the C protein localized predominantly to the nucleus (38). The localization of the E protein was similar to the pattern observed for prM (data not shown). Interestingly, NS1 showed the same pattern of staining as C (Fig. 5A, NS1). NS3 was observed as a large diffuse signal in the perinuclear region and the cytoplasm of infected cells (Fig. 5A, NS3). Under the same conditions, an anti-dsRNA antibody, which recognized the replicative intermediate form of the RNA, revealed a coarse speckled pattern at the perinuclear region and throughout the cytoplasm of infected cells (Fig. 5A, dsRNA). To further delineate the distribution of viral RNA, newly synthesized RNA was metabolically labeled with BrUTP in the presence of actinomycin D, and the BrUTP-labeled RNA was stained with an anti-BrUTP antibody. In these samples, the speckled pattern of staining was also observed at the perinuclear region and cytoplasm of infected cells (Fig. 5B). Therefore, the pattern of staining observed for the dsRNA antibody represented the area where newly synthesized viral RNA was localized.
FIG 5.

Localization of DENV proteins and newly synthesized RNA in infected cells. (A) Mock- and DENV-infected C6/36 cells at 48 h p.i. were fixed with 3.7% formaldehyde in PBS for 10 min at room temperature. The fixed cells were then permeabilized with 2% Triton X-100 in PBS for 10 min. Cells were washed 5 times with PBS before being probed with antibodies specific to DENV C protein, prM, NS1, NS3, NS5, and dsRNA. The signal was developed using a TRITC-conjugated anti-mouse secondary antibody (red, left panels). The cell nucleus was stained with DAPI (blue, middle panels). The merged images are shown in the right panels. (B) Mock- and DENV-infected cells at 12 h p.i. were treated with 10 μg/ml of actinomycin D and starved with low-glucose DMEM at 30°C for 1 h. The cells were then labeled with 10 mM BrUTP using Lipofectamine 2000. After 6 h of labeling, the cells were fixed and permeabilized as described above. Fixed cells were incubated with a anti-BrdU antibody followed by a goat anti-mouse TRITC-conjugated secondary antibody to detect newly synthesized viral RNA (red, left panels). Cell nuclei were stained with DAPI (blue, middle panels). Merged images are shown in the right panels.
Immunoelectron microscopy reveals DENV replication sites.
Immunoelectron microscopy was performed with the same set of antibodies to localize viral proteins on the DENV-induced membrane structures. NS1 proteins displayed a tendency to accumulate on the Ve membrane (Fig. 6A), whereas NS3 proteins were more distributed in all areas of Ve (Fig. 6B). NS5 was also associated with the vesicle structures but localized predominantly in the nuclei of infected cells (Fig. 6C and E), as expected from the immunofluorescence staining. Specific labeling of the vesicle structures by the dsRNA antibody was also observed (Fig. 6D). In contrast, the E protein was not associated with the DENV-induced structures but appeared to be colocalized with virus particles (Fig. 6F). Combining the observation that NS1, NS3, NS5, and dsRNA were localized on the Ve, the results indicated that these vesicle structures function as the site for viral RNA replication.
FIG 6.

Immunoelectron microscopy of viral replication proteins and dsRNA in vesicle structures. C6/36 cells infected with DENV were high-pressure frozen at 48 h postinfection. The cells were processed for freeze-substitution, dehydration, and embedding as described in Materials and Methods. The 90-nm thick sections were placed on gold-coated grids for antibody labeling. The resin-embedded cell sections were reacted with antibodies specific to DENV proteins NS1, NS3, and NS5 and dsRNA. The specific labeling signal of the deposited small gold particles is observed in black. (A and B) Anti-NS1 (A) and anti-NS3 (B) antibodies showed labeling in the cytoplasm of infected cells, where gold particles deposited specifically to the Ve. (C and E) In contrast, anti-NS5 labeling was observed mostly in the nuclei of infected cells, while only a few gold particles was associated with the Ve. (D) Specific labeling of dsRNA was observed on the Ve. (F) The sections were stained with anti-E antibody. No labeling signal of this viral protein was detected on Ve structures. Association of viral nonstructural proteins with the Ve indicated that these structures are involved in viral RNA synthesis. N, nucleus. Black arrows depict the gold labeling signal.
Virus replication complex structures.
Electron tomographic analysis of 250-nm sections of DENV-infected C6/36 cells revealed large sac-like Vp with double-layer membranes in the cytoplasm of infected cells (Fig. 7). These structures were not observed from 2D micrographs of chemically fixed DENV-infected C6/36 cells, which showed a single-layer membrane surrounding the Vp. Decoration of ribosomes implied that Vp were rough-ER-derived virally modified structures (Fig. 7 and 8; see Movies S1 and S2 in the supplemental material). We observed evidence of membrane connections between Vp and membrane sacs enclosing a number of virus particles (Fig. 8A and B; see Movies S1 and S2 in the supplemental material), suggesting functional association of these two structures, possibly linking RNA replication to virus particle budding. In addition, spherical coreless particles comprised of double-layer membranes were observed within the Vp and ER membranes adjacent to virus particles (Fig. 7; see Movie S1 in the supplemental material). These structures were similar in size to virus particles and are presumably the large form of subviral particles devoid of a viral genome. These observations imply that formation of infectious dengue virions and formation of subviral particles take place in the same location.
FIG 7.

Electron tomography of DENV-infected C6/36 cells. An overview of reconstructed tomograms of DENV-infected C6/36 cells at 48 h p.i. is shown. Infected cells were processed for high-pressure freezing and freeze-substitution in 0.5% osmium and 2% uranyl acetate. The set of tilt tomograms obtained from −60°C to 60°C with 1°C increments were collected at 300 kV. The tomograms were processed for reconstruction using the IMOD software package. Tomogram slices showed a number of Vp as double-layered membranes that enclosed Ve. The RC was clearly observed inside the Ve structures with different structural features. Electron-dense spheres smaller than virus particles decorated the Vp membrane and represented ribosomes. See Movie S1 in the supplemental material for the reconstruction tomogram and 3D rendering.
FIG 8.
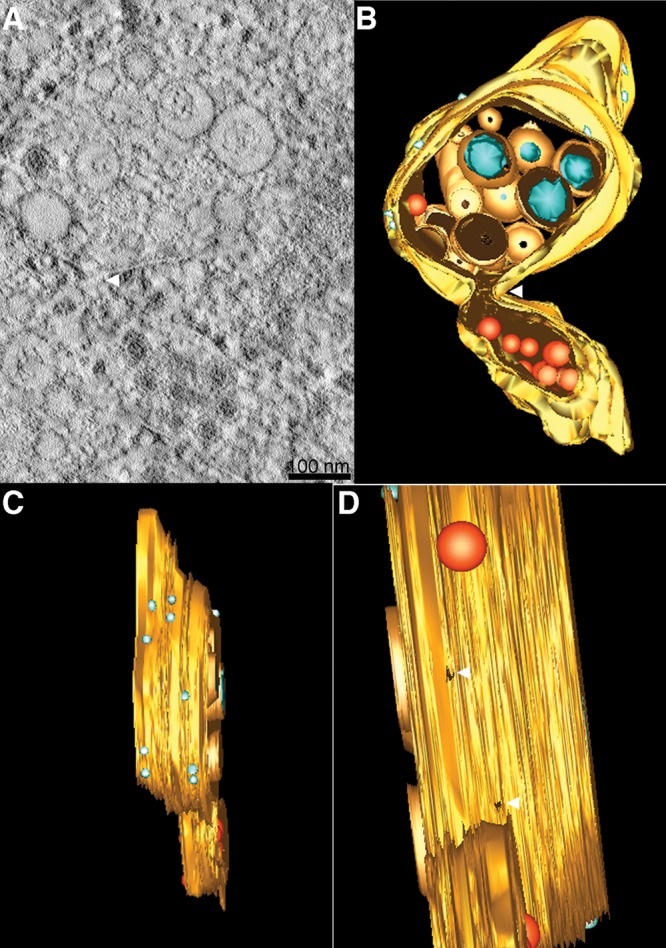
ET reveals a connection between Vp and virus particles on ER membranes. (A) Close inspection of an ET slice shows connection between Vp membrane- and ER membrane-containing virus particles. (B) A 3D surface-rendered model shows the cluster of Vp (yellow), Ve (orange), RC (blue), ribosome (light blue), and virus particles and subviral particles (red). The model depicts the connection between the Vp membrane and the membrane enclosing the virus particles. (C) A side view of the 3D model demonstrates the decoration of ribosomes on the Vp membrane. (D) An alternative side view of the 3D model shows the pores in the Vp membrane. See Movie S2 in the supplemental material for the reconstruction tomogram and 3D rendering.
The Ve are approximately 80 to 120 nm in diameter, are composed of single- or double-layer membranes, and are enclosed within the Vp (Fig. 7; see Movie S1 in the supplemental material). In our study, the most remarkable result was the observation of the RC structures within the Ve (Fig. 7; see Movie S1 in the supplemental material). These structures were positioned close to one side of the inner Ve membrane, while the other side was left as an empty space (see Movie S1 in the supplemental material). The unoccupied space may be reserved for structural transitions of the replication complex during the RNA replication process. Different features of the RC observed in each Ve throughout the reconstructed volume may reflect the state of RNA synthesis (see Movie S1 in the supplemental material). Close inspection revealed neck-liked structures of Ve located at the edge of the Vp forming the Vp pore (Fig. 8D and 9A; see Movie S2 in the supplemental material). This demonstrates that the ER membrane curves inward, forming the Ve within the lumen of Vp. This membrane architecture allows exposure of the environment within the Ve to the cytoplasmic space. Neck-like structures were also found opening into smaller budding vesicles through the connection of the two vesicle membranes (Fig. 9B and C), suggesting a cooperative function of these two structures. It possibly represents the junction where viral RNA replication and virus assembly occur. The 3D model of a single Ve revealed the orientation of the replication complex pointing straight forward toward the pore, which is juxtaposed close to the site of virus budding from another Vp (Fig. 9D and E; see Movie S3 in the supplemental material).
FIG 9.
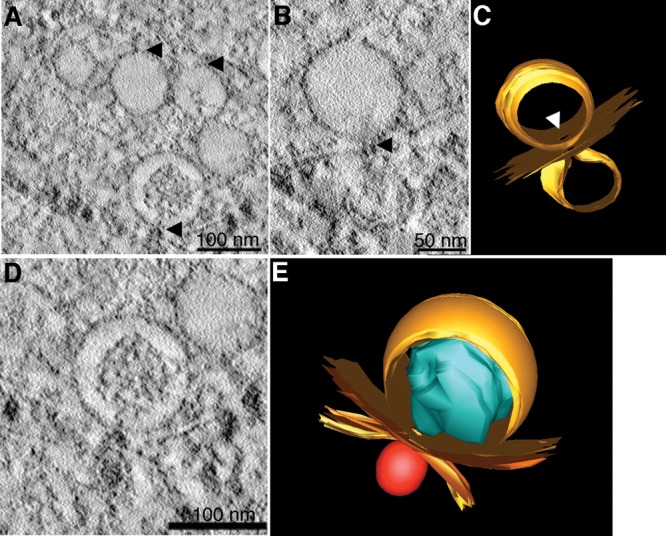
Virus-induced vesicles contain pores open to the cytoplasm. Electron tomography of DENV-infected C6/36 cells shows Vp pores. (A) The images suggest that the neck of the Ve gives rise to the Vp pores. (B) Membrane connection of Ve pores and budding vesicles. (C and E) The 3D surface-rendered model of DENV-infected C6/36 cells. (C) Structures of Ve membrane (top orange) and budding vesicles (bottom orange) demonstrate the position of a pore and the connection between the two membranes vesicles (derived from panel B). (D) Close association of two different Vp membranes containing the Ve and virus particles. (E) The 3D model shows close association between two independent Vp membranes (yellow) that contain Ve (orange) and a virus particle (red) (derived from panel D). The RC structure (blue) is oriented in the direction of the Ve pore. See Movie S3 in the supplemental material for the reconstruction tomogram and 3D rendering.
Tubular structures were also observed in our reconstructed tomograms (data not shown). These structures have two membrane layers and were located around the area of Vp and Ve. It is unclear whether these structures are surrounded with one ER membrane and connected to other virally modified structures. As previously mentioned, the CM structures were not observed in our tomography studies of DENV-infected C6/36 cells.
DENV budding at the Vp membrane.
Our electron tomography studies detected virus budding events within (or into) the Vp and also revealed virus particles and subviral particles located close to the Vp structures. While immunoelectron microscopy was attempted with anti-prM, -C, and -E antibodies, the binding of the antibodies were too weak to detect a signal in our studies. The data suggest that once the viral RNA-capsid protein complex is formed within the cytoplasm, it is encapsidated by the Vp membrane containing E and prM proteins. Initially, the membrane curves to form the neck of the budding vesicle (Fig. 10B and C, right particle, and D, left particle) and then forms the neck (Fig. 10D right particle, and C and B, left particle) to help drive the fission of the membrane of the virus particle into the Vp lumen (Fig. 10E, right particle, and A, left particle). The connection between viral membrane and Vp membrane implies that the virus particle is budding into the Vp (Fig. 10F). Hence, these data support the hypothesis that Vp is the site for virus replication and budding.
FIG 10.

Electron tomography captures virus budding. Different Z slices of a reconstructed tomogram of DENV-infected C6/36 cells show two nascent virus particles at different stages of budding. (A to E) Different Z slices of the tomogram provide a three-dimensional perspective of these two nascent particles. Black arrows indicate the necks of the nascent particles. Scale bars represent 50 nm. (F) Three-dimensional model of budding particles shown in panels A to E. The particle on the lower left has completed budding from the ER membrane, whereas the particle on the right has a narrow neck connecting the viral and ER membranes.
DISCUSSION
TEM and ET were used to visualize membrane rearrangements induced by DENV within insect cells. In the ET studies we were able to resolve the morphological appearance of virus-induced structures by taking advantage of HPF and FS techniques to improve the quality of sample preservation. This approach provided us with the ability to achieve a higher resolution during imaging than with conventional chemical fixation methods (21, 23, 24, 39). Consistent with previous findings (21, 23, 24, 27), three types of virally modified structures were observed: Vp, Ve, and T, which are also known as the “vesiculotubular structures” or “smooth membrane structures” (SMS). However, we were not able to identify paracrystalline arrays and crystalloid aggregates that were previously observed in DENV-infected cells (24). An important observation in this study is the absence of CM within mosquito cells. These structures have been detected exclusively within flavivirus-infected mammalian cells (22, 25, 28, 29) and coronavirus infected-mammalian cells (16, 17). This suggests some level of similarity among positive-strand RNA viruses that modify the structure of organelles during their replication. The purpose of CM structures during viral replication has yet to be identified. Localization of nonstructural proteins to the CM, as shown by IEM analysis, suggests a role in viral protein translation and processing (3). Alternatively, it may serve as the pool of viral proteins and lipids involved in RNA replication (28). As for insect cells, formation of Vp, Ve, and T without CM suggests that DENV accomplishes RNA replication using species-specific modification of host membranes. This is likely controlled by fundamental differences in lipid and protein homeostasis required for establishing the membrane rearrangements of mammalian and insect cells. Considering that insects are cholesterol auxotrophs, as they lack several enzymes in the cholesterol biosynthesis pathway (40), it is possible that cholesterol may be a key component of CM structures and thus they do not form in insect cells. It has been shown that West Nile virus redistributes cholesterol and cholesterol-generating proteins to virally modified structures within mammalian cells (41). In C6/36 cells, the cellular homeostasis of phospholipid species is perturbed during DENV replication (42). These results suggest that specific host lipid metabolites may contribute differently to virus-induced membrane reorganization. It is necessary to elucidate how the virus usurps host lipid metabolites to generate the distinct virus-induced structures found in mammalian and insect cells.
The tubular structure is another feature induced by DENV within mosquito cells. Consistent with previous reports, our TEM analysis showed a low number of T during 48 h of infection (23, 24). The generation of these structures has no apparent correlation with viral replication, and they were detectable only after the multiple rounds of DENV replication (36 h postinfection). This phenomenon was also observed in other positive-strand RNA virus infections (17). A large number of T were observed following chronic infection (7 to 14 days) of DENV in C6/36 cells (24). Electron tomography revealed that these structures are part of one continuous ER membrane covering Vp and Ve (28). However, the functional contribution of this virally modified structure has not yet been addressed. In the case of enterovirus infection of host cells, structural transformations have been observed from single-membrane tubules into double-membrane vesicles, concomitant with the time of infection (14). This transformation may reflect the progression to later stages of the replication cycle, possibly encapsidation (13).
Previous time course TEM studies of DENV-infected cells revealed a correlation between virus production and intracellular membrane rearrangements (24). Our studies provide a direct correlative comparison between functional membrane remodeling and RNA synthesis during DENV replication. We monitored membrane reorganization every 4 h from 0 to 24 h (one cycle of virus replication) and every 12 h from 24 to 48 h after infection along with quantification of virus replication. The appearances of Vp, Ve, T, and virus particles were observed at different time points during the infection and correlated well with the kinetics of RNA synthesis, viral particle assembly, and virus egress. Vp and Ve were first formed at 8 h after infection, when the rate of newly synthesized viral RNA started to increase. This observation implies that there is an association between membrane remodeling and RNA synthesis and that there is a linear correlation between Ve number and RNA copies during 8 to 24 h postinfection. However, this correlation did not apply after 24 h, where saturation of Ve formation was observed and RNA synthesis leveled off. We suspect that the observed reduction in the rate of viral RNA synthesis is primarily due to the increased rate of genome packaging and virus release at the later time points, thus preventing a measurable accumulation of the RNA in the cytoplasm. It could also result from the depletion of the RNA building blocks supplied by the host, a supply that is almost usurped by the virus during the log phase of replication.
The virus-modified structures formed within the cytoplasm of infected cells clearly show the rearrangement of the ER membrane into Vp containing Ve. Arrays of ribosomes decorated on Vp membranes support our conclusion that Vp are derived from the rough ER and further support the hypothesis that Vp may function in viral protein translation. RNA replication occurs on Ve membranes as demonstrated with IEM, where viral proteins NS1, NS3, and NS5 and dsRNA localized to these structures. Our conclusions are consistent with previous studies in a number of mammalian cell lines infected with DENV or Kunjin virus (22, 23, 25, 28, 29). Unlike the case for other flaviviruses that show distribution of NS5 within the cytoplasm (43–45), the NS5 protein from DENV is located primarily within the nucleus despite the fact that RNA replication occurs within the cytoplasm. We have observed this in both mammalian and insect cells. This suggests that during the replication process, DENV does not need the majority of NS5 in the cytoplasm to efficiently replicate within both insect and mammalian host cells. Our IEM result also showed less NS5 localized to the Ve.
Resolution of prominent RC structures within the Ve provides evidence for the hypothesis that Ve are sites for viral RNA replication. It has been shown for flaviviruses and other positive-stranded RNA viruses that dsRNA accumulates within these structures (15, 23). The RC structures observed in our study appear to be more compact and larger, as they fill most of the space within the Ve, than previous structures described for Kunjin virus-infected cells (22). Gillespie et al. observed cellular structures using the chemical fixation method and suggested that this could have affected the preservation of these structures. Since we are able to preserve a more native state of the subcellular structures using HPF and FS, the structures visualized should represent more accurate models of RCs among flaviviruses. The heterogeneity of RC structures detected in our tomograms may represent the different dynamic states of RNA replication events. The next step is to investigate the structural details of the RC and the viral and host components within these structures.
For RNA synthesis to occur within the lumen of the Ve, a pore is required to transport the building blocks from the cytoplasm. This pore allows the release of newly synthesized viral RNA into the cytoplasm for further encapsidation (22, 28, 46). From the ET results, we suggest that Ve pores are created by negative membrane curvature to allow invagination of double membranes forming Ve within the Vp. In studies of other positive-stranded RNA viruses, open pores were not observed in the Ve (13–15). In those studies, there is still some controversy about how the transportation of building blocks and newly synthesized RNA would occur to support efficient RNA replication. In the case of flaviviruses, the characteristic membrane topology of viral proteins could contribute to the observed membrane curvature. All flaviviral proteins are associated with membranes. NS1 and parts of NS4A/4B are localized to the luminal side of the ER, whereas NS2A, NS3, and NS5 are located on the cytoplasmic side of the ER. The membrane-anchored C protein is also on the cytoplasmic side of the ER. The transmembrane proteins E and prM are located in the ER lumen. Additionally, the altered lipid composition in DENV-infected cells may assist in driving the required membrane curvature. Presumably with the assistance of these membrane-associated viral proteins, host proteins, and host lipids, the Ve structures are formed through the invagination of the Vp membrane into the Vp lumen. This invagination event encloses the cytoplasm containing NS1, NS3, and NS5 (our observations) and other nonstructural proteins required for RNA synthesis, such as NS4A (5, 6) and possibly some cellular proteins. The C, prM, and E proteins are excluded (28). While RNA replication occurs within the Ve lumen, the Ve pore functions to supply (from the cytoplasm) the building blocks necessary for continued RNA replication within the Ve lumen. Following synthesis, the RNA must form a complex with the transmembrane C protein integrated on the ER membrane outside the Ve. Again, the Ve pore functions to secrete this genome RNA for capture by the C protein. The capsid protein-RNA complex is enveloped by the ER membrane containing E and prM proteins and buds into the ER lumen. From our tomograms, we directly visualized virally modified structures that are linked to budding. We observed the connection of the Ve pore to the neck of a budding vesicle and the arrangement of RC structures that correlates with the direction of the opened pore, with virus particles opposite to the Ve pore and inside the Vp. The most striking finding in our study is evidence of virus budding, which has not been observed in other studies on DENV.
Interestingly, we also observed coreless subviral particles within the Vp, with a membrane about the same size as that found in virus particles. Since they became apparent only by electron tomography, we were unable to demonstrate the presence of the E protein on these particles by IEM. However, the clear double-membrane features present within the Vp or ER lumen most likely represent subviral particles. This would suggest that assembly of both infectious virions and subviral particles occurs within the Vp of insect cells.
In conclusion, we directly visualized the subcellular membrane reorganization induced by DENV within mosquito cells by using advanced high-pressure freezing and freeze-substitution and electron tomography techniques. In combination with IEM and other molecular virology methods, these data provide insight into the link between DENV RNA replication, assembly, and budding and the process of virus-induced membrane remodeling. Future research needs to focus on characterization of the components of these virally modified structures in terms of metabolites, lipids, and host and viral proteins that are recruited for membrane formation and to interrogate the biological functions of such components in DENV replication.
Supplementary Material
ACKNOWLEDGMENTS
We thank Debra Sherman for assistance with sample preparations for electron microscopy and Chia-Ping Huang for sectioning the resin-embedded C6/36 cells and Huh-7 cells. We also thank Nopporn Sittissombut for critical reading of the manuscript and Vitara Pungpapong for helpful discussions on data analysis. We also acknowledge the excellent clerical assistance of Anita Robinson.
This work was supported by NIH grants R21 AI083984 and AI076331 to R.J.K. from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print 12 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00118-14.
REFERENCES
- 1.Lindenbach BD, Rice CM. 2001. The viruses and their replication: Flaviviridae, p 991–1041 In Fields BN, Knipe DN, Howley PM. (ed), Fields virology, 4th ed. Lippincott-Raven, New York, NY [Google Scholar]
- 2.Gubler DJ. 2002. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 10:100–103. 10.1016/S0966-842X(01)02288-0 [DOI] [PubMed] [Google Scholar]
- 3.Mackenzie JM, Jones MK, Young PR. 1996. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 220:232–240. 10.1006/viro.1996.0307 [DOI] [PubMed] [Google Scholar]
- 4.Mclean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. 2011. Flavivirus NSA-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 286:22147–22159. 10.1074/jbc.M110.192500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenshclager R. 2007. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 282:8873–8882. 10.1074/jbc.M609919200 [DOI] [PubMed] [Google Scholar]
- 6.Roosendaal J, Westaway EG, Khromykh A, Mackenzie JM. 2006. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 80:4623–4632. 10.1128/JVI.80.9.4623-4632.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller S, Sparacio S, Bartenschalager R. 2006. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J. Biol. Chem. 281:8854–8863. 10.1074/jbc.M512697200 [DOI] [PubMed] [Google Scholar]
- 8.Umareddy I, Chao A, Sampath A, Gu F, Vasudevan SG. 2006. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J. Gen. Virol. 87:2605–2614. 10.1099/vir.0.81844-0 [DOI] [PubMed] [Google Scholar]
- 9.Magliano D, Marshall JA, Bowden DS, Vardaxis N, Meanger J, Lee JY. 1998. Rubella virus replication complexes are virus-modified lysosomes. Virology 240:57–63. 10.1006/viro.1997.8906 [DOI] [PubMed] [Google Scholar]
- 10.Lee JY, Marshall JA, Bowden DS. 1992. Replication complexes associated with the morphogenesis of rubella virus. Arch. Virol. 122:95–106. 10.1007/BF01321120 [DOI] [PubMed] [Google Scholar]
- 11.Lee JY, Marshall JA, Bowden DS. 1994. Characterization of rubella virus replication complexes using antibodies to double-stranded RNA. Virology 200:307–312. 10.1006/viro.1994.1192 [DOI] [PubMed] [Google Scholar]
- 12.Miller DJ, Schwartz MD, Ahlquist P. 2001. Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J. Virol. 75:11664–11676. 10.1128/JVI.75.23.11664-11676.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. 2012. Complex dynamic development of poliovirus membrane replication complexes. J. Virol. 86:302–312. 10.1128/JVI.05937-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Limpens RW, van der Schaar HM, Kumar D, Koster AJ, Snijder EJ, van Kuppeveld FJ, Barcena M. 2011. The transformation of enterovirus replication structures: a three-dimensional study of single-and double-membrane compartments. mBio 2:e00166–11. 10.1128/mBio.00166-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knoops K, Barcena M, Limpens RW, Koster AJ, Mommaas AM, Snijder EJ. 2012. Ultrastructural characterization of arterivirus replication structures: reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 86:2474–2487. 10.1128/JVI.06677-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knoops K, Kikkert M, Van den Worm S, Zevenhoven-Dobbe J, Van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. 2008. SARS-coronavirus replication is supported by a reticulovasicular network of modified endoplasmic reticulum. PLoS Biol. 6:1957–1974. 10.1371/journal.pbio.0060226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ulasli M, Verheije MH, de Haan CA, Reggiori F. 2010. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 12:844–861. 10.1111/j.1462-5822.2010.01437.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Froshauer S, Kartenbeck J, Helenius A. 1988. Alphavirus RNA replicase is located on the cytoplasmic surface of endosmoes and lysosomes. J. Cell Biol. 107:2075–2086. 10.1083/jcb.107.6.2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kujala P, Ahola T, Ehsani N, Auvinen P, Vihinen H, Kaariainen L. 1999. Intracellular distribution of rubella virus nonstructural protein P150. J. Virol. 73:7805–7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kujala P, Ikaheimonen A, Ehsani N, Vihinen H, Auvinen P, Kaariainen L. 2001. Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol. 75:3873–3884. 10.1128/JVI.75.8.3873-3884.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barth OM. 1992. Replication of dengue viruses in mosquito cell cultures—a model from ultrastructural observations. Mem. Inst. Oswaldo Cruz 87:565–574. 10.1590/S0074-02761992000400017 [DOI] [PubMed] [Google Scholar]
- 22.Gillespie LK, Hoenen A, Morgan G, Mackenzie JM. 2010. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 84:10438–10447. 10.1128/JVI.00986-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grief C, Galler R, Cortes LM, Barth OM. 1997. Intracellular localisation of dengue-2 RNA in mosquito cell culture using electron microscopy in situ hybridization. Arch. Virol. 142:2347–2357. 10.1007/s007050050247 [DOI] [PubMed] [Google Scholar]
- 24.Ko KK, Igarashi A, Fukai K. 1979. Electron microscopic observation on Aedes albopictus cells infected with dengue viruses. Arch. Virol. 62:41–52. 10.1007/BF01314902 [DOI] [PubMed] [Google Scholar]
- 25.Mackenzie JM, Jones MK, Westaway EG. 1999. Markers for trans-Golgi membranes and the intermediate compartment localize to induced membranes with distinct replication functions in flaviviruses-infected cells. J. Virol. 73:9555–9567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng ML. 1987. Ultrastructural studies of Kunjin virus-infected Aedes albopictus cells. J. Gen. Virol. 68:577–582. 10.1099/0022-1317-68-2-577 [DOI] [PubMed] [Google Scholar]
- 27.Ng ML, Yeong FM, Tan SH. 1994. Cryosubstitution technique reveals new morphology of flavivirus-induced structures. J. Virol. Methods 49:305–314. 10.1016/0166-0934(94)90145-7 [DOI] [PubMed] [Google Scholar]
- 28.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CKE, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell 5:365–375. 10.1016/j.chom.2009.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. 1997. Ultrastructural of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 71:6650–6661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westaway EG, Khromykh AA, Mackenzie JM. 1999. Nascent flavivirus RNA colocalized in situ with double-stranded RNA in stable replication complexes. Virology 258:108–117. 10.1006/viro.1999.9683 [DOI] [PubMed] [Google Scholar]
- 31.Offerdahl DK, Dorward DW, Hansen BT, Bloom ME. 2012. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS One 7:e47912. 10.1371/journal.pone.0047912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackenzie J. 2005. Wrapping things up about virus RNA replication. Traffic. 6:967–977. 10.1111/j.1600-0854.2005.00339.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6:363–374. 10.1038/nrmicro1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salonen A, Ahola T, Kaariainen L. 2005. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 285:139–173. 10.1007/3-540-26764-6_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laue T, Emmerich P, Schmitz H. 1999. Detection of dengue virus RNA in patients after primary or secondary dengue infection by using the TaqMan automated amplification system. J. Clin. Microbiol. 37:2543–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keelapang P, Sriburi R, Supasa S, Panyadee N, Songjaeng A, Jairungsri A, Putthkhunt C, Kasinrerk W, Malasit P, Sittisombut N. 2004. Alterations of pr-M cleavage and virus export in pr-M junction chimeric dengue viruses. J. Virol. 78:2367–2381. 10.1128/JVI.78.5.2367-2381.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kremer JM, Mastronarde DN, Mcitosh JR. 1996. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116:71–76. 10.1006/jsbi.1996.0013 [DOI] [PubMed] [Google Scholar]
- 38.Sangiambut S, Keelapang P, Aaskov J, Puttikhunt C, Kasinrerk W, Malasit P, Sittisombut N. 2008. Multiple regions in dengue virus capsid protein contribute to nuclear localization during virus infection. J. Gen. Virol. 89:1254–1264. 10.1099/vir.0.83264-0 [DOI] [PubMed] [Google Scholar]
- 39.Sosinsky GE, Crum J, Jones YZ, Lanman J, Smarr B, Terada M, Martone ME, Deerinck TJ, Johnson JE, Ellisman MH. 2008. The combination of chemical fixation procedures with high pressure freezing and freeze substitution preserves highly labile tissue ultrastructure for electron tomography applications. J. Struct. Biol. 161:359–371. 10.1016/j.jsb.2007.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rawson RB. 2003. The SREBP pathway—insights from insigs and insects. Nat. Rev. Mol. Cell Biol. 8:631–640. 10.1038/nrm1174 [DOI] [PubMed] [Google Scholar]
- 41.Mackenzie JM, Khromykh AA, Parton RG. 2007. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2:229–239. 10.1016/j.chom.2007.09.003 [DOI] [PubMed] [Google Scholar]
- 42.Perera R, Riley C, Isaac G, Hopf-Jannasch AS, Moore RJ, Weitz KW, Pasa-Tolic L, Metz TO, Adamec J, Kuhn RJ. 2012. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 8:e1002584. 10.1371/journal.ppat.1002584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackenzie JM, Kenney MT, Westaway EG. 2007. West Nile virus strain Kunjin NS5 polymerase is a phosphoprotein localized at the cytoplasmic site of viral RNA synthesis. J. Gen. Virol. 88:1163–1168. 10.1099/vir.0.82552-0 [DOI] [PubMed] [Google Scholar]
- 44.Werme K, Wigerius M, Johansson M. 2008. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell. Microbiol. 10:696–712. 10.1111/j.1462-5822.2007.01076.x [DOI] [PubMed] [Google Scholar]
- 45.Yang SH, Liu ML, Tien CF, Chou SJ, Chang RY. 2009. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) interaction with 3′ ends of Japanese encephalitis virus RNA and colocalization with the viral NS5 protein. J. Biomed. Sci. 16:40. 10.1186/1423-0127-16-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kopek BG, Perkins G, Miller DJ, Ellisman MH, Ahlquist P. 2007. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 5:e220. 10.1371/journal.pbio.0050220 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.