

ABSTRACT
The two human neurotropic alphaherpesviruses varicella-zoster virus (VZV) and herpes simplex virus type 1 (HSV1) both establish latency in sensory ganglia. Human trigeminal ganglia are known to frequently harbor both viruses, and there is evidence to suggest the presence of both VZV and HSV1 DNA in the same neuron. We ask here whether VZV and HSV1 can exclude themselves and each other and whether they can productively infect the same cells in human neurons and human foreskin fibroblasts (HFF). Simultaneous infection (coinfection) or consecutive infection (superinfection) was assessed using cell-free HSV1 and VZV expressing fluorescent reporter proteins. Automated analysis was carried out to detect singly and dually infected cells. We demonstrate that VZV and HSV1 both display efficient superinfection exclusion (SE) in HFF, with each virus excluding either itself or the other virus. While SE also occurred in neurons, it was with much lower efficiency. Both alphaherpesviruses productively infected the same neurons, whether applied simultaneously or even consecutively, albeit at lower frequencies.
IMPORTANCE Superinfection exclusion by VZV for itself or the related neurotropic alphaherpesvirus HSV1 has been studied here for the first time. We find that while these viruses display classic SE in fibroblasts, SE is less efficient for both HSV1 and VZV in human neurons. The ability of multiple VZV strains to productively infect the same neurons has important implications in terms of recombination of both wild-type and vaccine strains in patients.
INTRODUCTION
Superinfection exclusion (SE; also called superinfection inhibition or homologous interference) is a phenomenon in which a preexisting viral infection prevents infection by another virus, whether it is of the same type, a closely related virus, or even a different virus species. Often, however, the infection by unrelated viruses is unaffected (1, 2). The phenomenon has been observed for many viruses of animals and plants (3–10). It has been used as a management tool to reduce crop losses by purposely infecting plants with mild isolates of a virus to reduce infection and losses due to more severe isolates, referred to as “cross-protection” (4, 5). The exclusion is thought to be a so-called “selfish” mechanism of the virus, as the entry of additional virus relying on the same cellular resources would decrease the progeny yield of the first virus. SE of herpesviruses was first described for herpes simplex virus type 1 (HSV1) (11). It was found that HSV1 SE results from the expression of glycoprotein D (gD), a late viral protein essential for virion entry (12), by the infected cells. After infection, gD binds a cellular receptor and renders it inactive, making subsequent infection by virions highly inefficient (11, 13, 14). In addition to inactivation, HSV-1 gD causes internalization of viral receptors during HSV1 infection, reducing their availability on the cell surface for additional virus binding (10). Studies of SE with HSV1 and other alphaherpesviruses suggested that superinfection exclusion occurs by 4 to 6 h postinfection (15). Most studies of SE by herpesviruses have evaluated subsequent infection by either the same or a very closely related virus, for example, equine herpesvirus 1 and 2 (16), the two bovine alphaherpesviruses (17), and the interaction of pseudorabies virus (14) and HSV1 (13).
To date, there has been no examination of whether the pathogen responsible for varicella and herpes zoster, varicella-zoster virus (VZV), shows the phenomenon of SE, either for itself or for the closely related pathogenic virus HSV1. This is of clinical importance, since both herpesviruses infect humans. HSV1 is a member of genus Simplexvirus and VZV of genus Varicellovirus, both in subfamily Alphaherpesvirinae (Taxonomy browser: Alphaherpesvirinae [https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode=Undef&id=10293&lvl=3&lin=f&keep=1&srchmode=1&unlock]). Both viruses produce vesicular skin lesions, with virus infecting epidermal and dermal cells of the skin. Importantly, both viruses establish latent infections in neurons of somatic sensory ganglia whose axons infiltrate the areas of skin infection (for a detailed comparison, see the recent review in reference 18). Furthermore, both VZV and HSV are widespread human pathogens, with about 60 to 90% of the population latently infected by both viruses by adulthood (19). It has been reported that both viruses can reside latently in the same trigeminal ganglion (19–22), and evidence from in situ hybridization suggests the rare presence of both HSV1 and VZV genomes in the same cells (23). However, the ability of both viruses to productively infect the same neuron has not been investigated.
In the present study, we used viruses expressing different fluorescent proteins to ask whether VZV would display SE for itself and for HSV1 and whether both viruses could productively infect the same cell simultaneously. Our experiments revealed that VZV-infected cells excluded additional virion infection by both VZV and HSV1. In addition, we showed that both viruses could replicate in the same neuron. Finally, we made the striking observation that fibroblasts displayed a much stronger SE phenomenon than neurons. These results are consistent with the possibility of individual neurons harboring both latent HSV and VZV and the coexistence of multiple VZV strains (wild type or vaccine) in the same neurons.
MATERIALS AND METHODS
Cells and viruses.
H9 (WA09; U.S. National Stem Cell Bank) human embryonic stem cells were maintained on feeder layers of mitotically inactivated human foreskin fibroblasts (HFF) in NutriStem medium (Biological Industries, Israel) with medium replacement every other day. The cells were passaged weekly at the ratio of approximately 1:30. Neonatal HFF, PA6, ARPE19 (human retinal pigment epithelium), and Vero (African green monkey kidney epithelium) cells were maintained in Dulbecco modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS), 2 mM glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin.
pOka-based VZV expressing green fluorescent protein (GFP) as a fusion protein to open reading frame 66 (ORF66) was described elsewhere (24, 25). We also exploited a new VZV, VZV-66mRFP, that was generated using the Scarless recombination system of Tischer et al. (26) with the VZV pOka bacterial artificial chromosome (BAC). Briefly, the plasmid mRFP-kan-in (a gift from Nikolaus Osterreider, Cornell University, Ithaca, NY) was PCR amplified using primers with 40-bp overhangs homologous to the preceding sequence and immediately following the ORF66-initiating ATG. The oligonucleotides were designed to place the monomeric red fluorescent protein (mRFP) gene in frame with the methionine of codon 1 of ORF66. Primer sequences are available on request. The PCR fragment was gel purified and transfected into Escherichia coli GS1783 (a gift of Gregory Smith, Northwestern University, Chicago, IL) harboring the VZV pOka BAC, and recombination induced by heating to 42°C for 15 min. BACs harboring the fragment were selected based on gain of kanamycin resistance. A second round of recombination was induced in conjunction with ISceI induction by growth on arabinose to remove the kanamycin resistance cassette. Virus resulting from MeWo cells transfected with the sequence-verified BAC expressed the mRed signal fused to the N-terminal residue of ORF66. All viruses were maintained in ARPE19 cells, as described previously (25). HSV1 expressing GFP under the promoter of the late glycoprotein C gene was described elsewhere (27). HSV1 expressing mCherry fused to VP26 was a kind gift of Prashant Desai (Johns Hopkins University, Baltimore, MD). HSV1 was propagated and titrated in Vero cells.
Infectious focus assays and multiplicity of infection (MOI) calculations.
For VZV, titration was carried as detailed previously (28). Briefly, 10-fold dilutions of the virus were inoculated onto triplicate 90% confluent ARPE19 monolayers in a 24-well plate and fluorescent foci were counted 5 days postinfection. HSV1 was titrated on Vero monolayers overlaid with 0.3% agar in medium to prevent secondary plaque formation. Fluorescent foci were counted 3 days postinfection, and the titers were verified after visualizing plaques with 0.1% crystal violet.
For the experiments described here, 2.5 × 104 HFF were seeded in each well of a 24-well plate. For neurons, the exact number of the cells in each well was variable but was estimated to be between 2,000 and 20,000 cells; this variation is due to the difference in both the size of the spheres themselves and the exact numbers of the spheres seeded in each well. The resulting MOIs for VZV were approximately 0.1 for fibroblasts and 0.1 to 1 for neurons. For HSV1, the MOI for fibroblasts was 1, while for neurons, it was estimated to have been 1 to 10. In experiments using both viruses, the MOI of HSV1 was adjusted to match that of the VZV.
Neuronal differentiation from hESC.
Neurons were differentiated from human embryonic stem cells (hESC) as described previously, where it was shown that that >90% of the cells in the cultures stained positive for the intermediate neurofilament proteins (28).
Generation of cell-free VZV for infections.
Cell-free VZV was generated as detailed previously (28). Briefly, ARPE19 cells were infected with VZV in a cell-associated manner and harvested upon the development of an extensive cytopathic effect. Cells washed in phosphate-buffered saline (PBS) were then freeze-thawed and sonicated. Following low-speed centrifugation to remove cellular debris, the supernatant (cell-free virus) and the pellet (cell debris) were aliquoted in PSGC (PBS-sucrose-glutamate-serum) buffer and stored in liquid nitrogen. The virus titer of the debris in the pellet was about 1 order of magnitude higher than the titer of the free virus in the supernatant (up to 106 versus 105 PFU/ml, respectively). Therefore, the debris fraction was used throughout this study. The pellets did not contain any live or intact cells, as tested by microscopy and by plating the debris and examining the culture for live, dividing cells.
Viral infections.
VZV sonicates were thawed, diluted 1:1 in DMEM-F12 medium (to decrease the toxic effect of serum on neurons), and allowed to adsorb to cultured cells at 37°C in a volume of 150 μl/well of a 24-well-plate. After 2 h, the inoculum was replaced with growth medium. Infection, as detected by fluorescence, was observed after 2 or 3 days. Infection by conventionally prepared cell-free HSV1 was performed as described above for VZV, and fluorescence was observed within 24 h.
Each pair of viruses was assayed for both coinfection (the application of the two viruses simultaneously) and superinfection (sequential application of the two viruses), using duplicate or triplicate wells. As a positive control, the infectiveness of each individual viral preparation by itself was assessed in parallel in every experiment. Only those experiments in which the single infection was evident within 2 days were analyzed. In superinfection experiments, the times between applications of viruses were 8 h after HSV1 introduction and 1 day after VZV introduction. Cells were checked for viability by morphology under phase-contrast microscopy before superinfections were performed. In one experiment, we verified that HSV1-infected fibroblasts were still viable when superinfected by vital propidium iodide staining. No nuclei were stained, indicating intact plasma membranes of the cells (not shown).
For experiments using inactivated VZV, virus was placed under a UV lamp for 10 min. The effectiveness of the inactivation was complete, as demonstrated by the lack of mRFP expression by cells treated with the inactivated virus at 5 days postinfection, while cells treated with the original stock displayed widespread fluorescence within the same period after exposure to the virus.
Data analysis.
In all superinfection experiments, the red-fluorescent virus was applied first. At set times after application (see Results), digital micrographs of living cultures were made using the appropriate filter sets on an Olympus IX70 inverted microscope with a 20× objective. Three to 10 fields from each well were imaged using both filter sets. For fibroblasts, random cell-containing fields were imaged. In neuronal cultures, the fields covered most of the cells on the coverslips, since most of the cells were initially plated as clumps.
The threshold was set manually for each set of images. A dilation morphological filter was then applied, and the pairs of resulting binary images were merged into the R and G channels of RGB images using the ImageJ-based image analysis program Fiji (29). The RG2B colocalization plugin (http://rsbweb.nih.gov/ij/plugins/rg2bcolocalization.html) was then used to determine which pixels contained both red and green fluorescence individually in both channels. At the magnification used in the analysis, a typical fibroblast contained about 40 by 50 pixels if round and 40 by 100 pixels if still elongated. At the same magnification, the typical dimensions of neuronal cell bodies were about 20 by 20 pixels.
The fraction of the area (pixels of each color/total pixels) of the image for each color channel and for dual fluorescence were then determined. The sum of the area fractions was defined to be 100%, and the area fractions are given as percentages of the total fluorescence. The proportions of the differently infected (and colored) cells are depicted in all figures by the height of the column colored according to the fluorescent labeling of the viruses, with yellow depicting dual fluorescence. An illustration of the counting and analysis procedure is shown in Fig. 1.
FIG 1.

Experimental procedures and analysis. (A) A graphic representation of the infection methods used in this study. Neonatal human foreskin fibroblasts or human embryonic stem cell-derived neurons were infected with cell-free preparations of red and green fluorescently labeled VZV or HSV1. Second infections were carried out either at the same time (coinfection) or after a time interval (superinfection). (B) An illustrated flowchart of the computerized method of analysis. After extensive infection was present, as determined by fluorescence, digital images of both red and green channels were made. Thresholds were set manually for fluorescent color, and the fractions of the field displaying each color and overlapping fluorescence were determined. The total amount of fluorescent pixels above the threshold was defined as 100%, and the proportions of pixels of each color are depicted in the histogram labeled “% Fluorescence.”
In order to validate this analysis procedure, red, green, and dually fluorescent cells were both counted visually and analyzed by computerized image processing in approximately 40 micrographic fields. The experiment analyzed was superinfection of HSV1 VP26-mCherry-infected cells (either fibroblasts or neurons, 20 micrographs each) with HSV1 gC-GFP. For both fibroblasts and neurons, analysis with both procedures resulted in similar results: the same percentage of dually fluorescent (yellow) cells and percentages of colocalized red and green pixels were obtained (Fig. 2). When individual colors were measured, the percentages of cells and the percentages of fluorescence were slightly less in agreement than in the dual-labeling experiments, but all the values fell within 10% of each other. Since visual counting of fluorescent cells is subject to some variability due to individual judgment and we were looking for semiquantitative phenomena rather than absolute values, we believe that our automated and potentially less biased measurement procedure provides a valid estimate for the levels of infection for fluorescent protein-tagged viruses.
FIG 2.
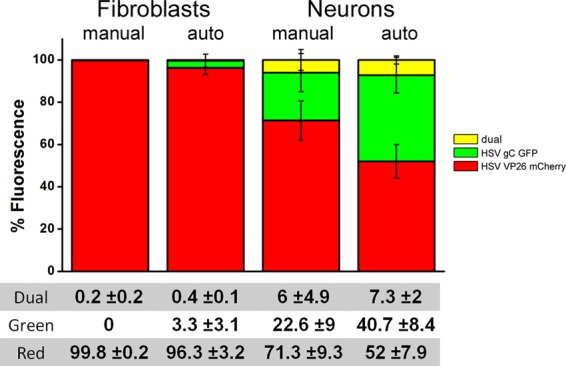
Automated analysis of fluorescence correlates with manual counting of infected cells. Approximately 40 micrographs of fibroblasts and neurons infected with red-fluorescent HSV1 and superinfected with green-fluorescent HSV1 were made with a 20× objective. The numbers of red, green, and yellow (dually fluorescent) fluorescent cells were counted (manual), and the percentage of the fields with fluorescence of each color was analyzed by the automated analysis procedure described in the text (auto). For both fibroblasts and neurons, very similar amounts of dual infection (yellow) were detected by both measurement methods. When the counts of singly infected cells and percent fluorescence of the individual channels were compared, the agreement was less good, but the values fell within ±10% of each other. The values shown are the means ± standard errors of the means (SEM).
RESULTS
Coinfection of and superinfection exclusion by human fibroblasts and neurons exposed to two differently labeled HSV1 viruses.
We first examined HSV1 infection of human foreskin fibroblasts (HFF), as both HSV1 and VZV primary infections involve infection of cutaneous fibroblasts. Preliminary experiments showed that the percentage of fluorescent pixels was a valid surrogate for the number of cells infected (Fig. 2), so we use the term “infection” to describe the proportion of pixels in micrographs displaying fluorescence.
Two recombinant HSV1 viruses, labeled with mCherry (red) or green fluorescent protein (GFP; green), were applied either simultaneously (coinfection) or sequentially (superinfection) at the same MOI (Fig. 3). When both viruses were added simultaneously (Fig. 3, left) similar amounts of infection of the fibroblasts by each virus were observed. A significant population of fibroblasts (13%) displayed dual fluorescence, indicating simultaneous productive infection when coinfected with the two HSV1 viruses. In contrast, fibroblasts infected with HSV1 VP26-mCherry very effectively excluded HSV1 gC-GFP infection when the red-fluorescent virus was applied 8 h earlier, as indicated by the much higher proportion of red-fluorescent virus infection (96% red and 3.5% green). In this superinfection experiment, virtually no dually fluorescent cells were observed.
FIG 3.
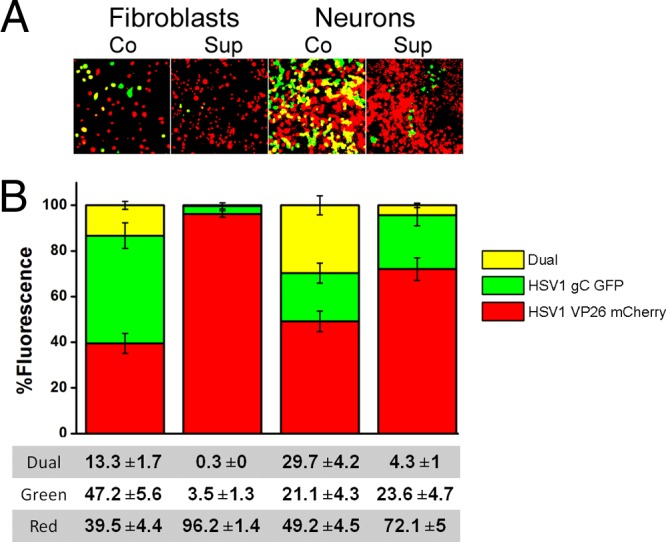
HSV1 infection of fibroblasts and neurons excludes subsequent HSV1 infection. (A) Representative micrographs of coinfection (Co) and superinfection (Sup) by fluorescent HSV1 strains after thresholds were set. (B) Graphic representation of the mean percentage of the field displaying fluorescence of each color, with yellow depicting colocalization of both colors. The values shown are the means ± standard errors of the means (SEM) of 4 independent experiments. Red-fluorescent-HSV1-infected fibroblasts and neurons were able to exclude green-fluorescent HSV1 when superinfected. The amount of double infection (yellow) was higher for neurons than for fibroblasts.
After demonstrating that fluorescently labeled HSV1 recapitulated the well described SE phenomenon for this virus when using our computerized analysis in HFF, we then repeated these experiments using neurons generated from human embryonic stem cells. When both HSV1 viruses were applied simultaneously, many neurons were observed to be singly infected. Strikingly, however, the percentage of double infection among neurons was more than double that observed when fibroblasts were coinfected. With sequential application of two HSV1 viruses, although there was clear SE, we observed that a small but significant percentage of the neurons were dually infected. The higher percentages of dually infected neurons in both the co- and superinfection experiments suggests that neurons are less efficient than fibroblasts at inducing SE for HSV.
Coinfection of and superinfection exclusion by human fibroblasts and neurons exposed to two differently labeled VZV viruses.
The ability of VZV-infected cells to exclude VZV (or HSV) has not yet been reported. In experiments studying SE for HSV1, relatively high MOIs of infecting virus are used in order to ensure that all cells are infected with the first virus. It is well known that obtaining high-titer cell-free VZV is not readily achievable from tissue cultures in research laboratories (30). Indeed, most experiments studying VZV in vitro are performed using cell-associated infection. We have recently described a method for generating relatively high titers of cell-free VZV (28). Using the cellular debris fraction, which had the higher cell-free viral titer, of ARPE19 cells infected with VZV expressing red (VZV-66mRFP) or green fluorescent (VZV-66GFP) reporters, we were not able to achieve the desired MOI of more than 1. However, as described below, the titer achieved was sufficient to give qualitative answers to whether VZV-infected cells display SE.
When both GFP- and mRFP-labeled VZV viruses were incubated simultaneously with fibroblasts, the percentage of cells infected by each virus was similar (Fig. 4A). However, when the VZV-66mRFP was applied ∼24 h before the VZV-66GFP, the GFP-labeled virus was efficiently excluded by the mRFP-labeled virus, as shown by the lower proportion of GFP fluorescence than was seen in the simultaneous infection experiments. The exclusion of the second virus applied in fibroblasts was not as effective as that observed for HSV1, probably due to the much lower MOI used in the VZV experiments.
FIG 4.
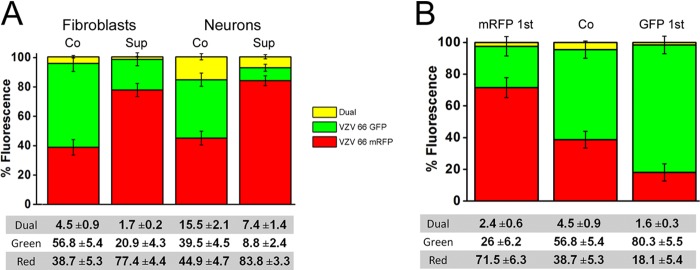
VZV infection of fibroblasts and neurons excludes subsequent VZV infection. (A) Graphic representation of the mean percentage of fields displaying fluorescence of each color, with yellow depicting colocalization of both colors. The values shown are the means ± SEM of 3 experiments. Red-fluorescent VZV excluded green-fluorescent VZV superinfection in both fibroblasts and neurons. The percentage of double-infected cells (yellow) was higher for neurons than for fibroblasts, as was observed for HSV1. VZV SE is less effective than that of HSV1, probably due to the lower MOI used in VZV infections (see the text). (B) In order to ensure there was no bias present due to higher infectivity or visibility of one of the two recombinant viruses used, a superinfection experiment of VZV on fibroblasts was carried out twice, using both possible sequences of viral application (red then green and green then red). Similar percentages of the cells were infected in both experiments, and the SE effect was clearly present regardless of the order of application of the viruses.
After determining that VZV displayed SE in fibroblasts, we performed parallel experiments using neurons. As was observed for HSV1, more dually infected neurons than fibroblasts were seen following exposure to the two viruses in both simultaneous and sequential infections. While there was exclusion of infection by a second VZV in neurons, the SE was less effective than that seen in fibroblasts.
In order to ensure that the results obtained were not dependent on different replication kinetics or attenuation of one of the recombinant viruses, we performed the sequential infection experiment in the reverse order, i.e., by exposing the fibroblasts to the GFP-labeled virus first. A similar percentage of the cells were infected by the virus that was introduced first, regardless of the label (Fig. 4B).
The observed exclusion of the virus introduced second could have been due to an effect on the infected cells by the VZV introduced first, causing, for example, virion host shutoff, which is well documented in HSV1 and has also been described for VZV (31, 32). In order to evaluate this possibility, HFF were treated with nonreplicating, UV-inactivated virus. Pretreatment of the cells with UV-inactivated VZV-66mRFP was able to reduce the infection by green virus by 85% compared to the results for parallel cultures of uninfected cells (Fig. 5). In contrast, pretreatment of the cells with debris derived from uninfected cells (mock) did not have an effect on subsequent VZV infection compared to the results for uninfected cells. Therefore, the reduction of infection by the virus introduced second is dependent upon the first viral infection.
FIG 5.
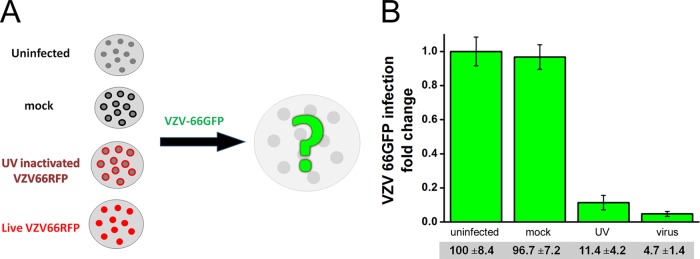
UV-inactivated VZV excludes infective VZV. (A) A schematic representation of the experimental protocol. Either live (red) or UV-treated (red outlines) VZV-66mRFP was allowed to adsorb to fibroblasts; infective VZV-66GFP was then added to the cells after 24 h (green). As a control, the same virus and concentration were used to infect either naive fibroblasts (gray) or fibroblasts inoculated with uninfected ARPE19 debris (black outlines). (B) Fold change of HFF infection by VZV-66GFP after pretreatment with UV-inactivated or infective VZV. Micrographs were captured 5 days after the infection with VZV-66GFP, and automatic analysis was carried out. The amount of VZV-66GFP infection of naive, uninfected cells was taken as 100%. The addition of UV-inactivated RFP-labeled virus prevented ∼85% of the infection when VZV-66GFP was applied after 24 h. The values shown are the means ± SEM of 2 independent experiments.
VZV- and HSV1-infected fibroblasts show mutual exclusion in superinfection experiments.
SE is often defined as the ability of identical or closely related viruses to exclude infection by one another: for example, HSV-1 glycoprotein D expression can exclude HSV1, pseudorabies virus (PrV), or bovine herpesvirus (BHV) (33). To determine whether VZV and HSV1 would each exclude infection by the other, HFF were exposed to VZV-66mRFP and HSV1 gC-GFP together (coinfection) or to the VZV before applying the HSV1 24 h later (superinfection), using approximately the same MOIs.
When the two viruses were applied simultaneously to HFF, many more cells were infected by HSV1 than by VZV (Fig. 6), due to the low level of infection achievable with cell-free VZV. Very few cells were dually infected under these experimental conditions. Sequential application of the viruses revealed that VZV-infected fibroblasts were able to successfully exclude subsequent infection by HSV1, as shown by the lower percentage of GFP fluorescence from HSV1 present in cultures previously infected with VZV. Performing a similar set of experiments with neurons revealed that neurons infected by VZV were able to prevent subsequent infection by HSV1. The SE between the viruses was less effective in neurons than in fibroblasts, with 7% of coinfected and 11% of sequentially infected neurons displaying both red and green fluorescence. These results are consistent with the results described above for HSV1-HSV1 and VZV-VZV SE and suggest that neurons can be productively coinfected, as well as superinfected, with two different alphaherpesviruses. This possibility was examined in greater detail as described below.
FIG 6.
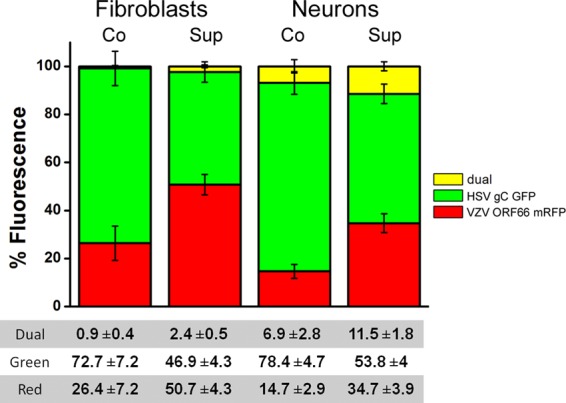
VZV infection of fibroblasts and neurons prevents subsequent HSV1 infection. Graphic representation of the mean percentage of the field displaying fluorescence of each color, with yellow depicting colocalization of both colors. When cell-free VZV-infected (red) human fibroblasts and neurons were superinfected with HSV1 (green), HSV1 infection was less extensive than when the viruses were coinfected. The percentage of double-infected cells was significantly higher for superinfected neurons than for fibroblasts (P < 0.01).
VZV and HSV1 can productively infect the same neuron.
It is well known that trigeminal ganglions from human cadavers harbor both HSV1 and VZV genomes (21). It has even been reported that transcripts of both viruses can be localized to the same neuron by in situ hybridization (23). The last experiment described above indicated that exposure of human neurons to recombinant VZV and HSV1 resulted in dually fluorescent neurons, indicating simultaneous productive infection with both viruses. To further demonstrate the possibility of HSV1 and VZV coinfection of neurons, we stained the HSV1- and VZV-infected cultures (both coinfected and superinfected) for the heavy subunit of the neurofilament protein (Fig. 7). Cells displaying red and green fluorescence were also immunopositive for neurofilament, establishing that active synthesis of proteins from both viruses can occur together in neurons. These results demonstrate that human neurons can be simultaneously infected by both alphaherpesviruses in a productive manner.
FIG 7.
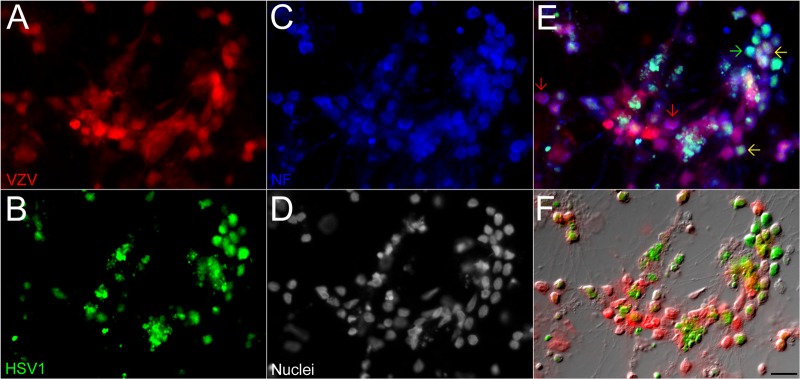
VZV and HSV1 can productively infect the same neuron. hESC-derived neurons were infected with red-fluorescent VZV and superinfected with green-fluorescent HSV1 after 1 day. Three days after infection with VZV, the culture was immunostained for a neuron-specific marker (neurofilament-H, blue). (E) Some neurons were productively infected with both viruses (yellow arrows), while others in the field were singly infected by either VZV or HSV1 (red and green arrows, respectively). (F) HSV1 and VZV fluorescence overlaid with a Nomarski optics image. NF, neurofilaments. Scale bar = 20 μm.
DISCUSSION
This study addresses for the first time the phenomenon of VZV SE. As has been shown and studied for more rapidly replicating alphaherpesviruses, as well as viruses of various families that infect animal and plants, infection of cells such as fibroblasts by VZV establishes a state that prevents the subsequent infection of the same cell by additional VZV. We also addressed SE for VZV and the related human alphaherpesvirus, HSV1, since it was feasible that both could establish infections of the same cells in vivo, and found that VZV infection effectively prevented infection of fibroblasts by HSV1. The SE caused by VZV infection was not absolute, in all likelihood due to the difficulty of obtaining efficient infections with cell-free VZV, but by comparison of sequential to simultaneous infection events, the SE phenomenon was clearly demonstrated.
It is known that HSV1 self-mediated SE is gD dependent (11–13, 34, 35). Other nonhuman viruses of the same family were also shown to exclude each other by a similar mechanism (2, 17, 33, 36). However, VZV and the related simian varicella virus (SVV) are the only known alphaherpesviruses that do not encode any obvious gD ortholog, and therefore, they must use different glycoproteins to mediate infection and SE. Studies have suggested that VZV gE in conjunction with gI may mediate both the initial receptor binding and cell-to-cell infections (37, 38). gE/gI form a heterodimeric complex, and gE is the most abundantly expressed VZV glycoprotein, with expression occurring rapidly after the initiation of infection (39); therefore, these proteins are candidates for participating in VZV SE. It is also possible that prevention of infection with a second virus is mediated through nonspecific interactions with glycosylated heparan sulfate residues on the cells, as described for HSV1 and PrV (40, 41). Indeed, a recent transcriptome study in our laboratory showed that the core protein heparan sulfate proteoglycan 2 (HSPG2) (42) is downregulated during VZV infection in human fibroblasts but not in hESC-derived neurons (A. Markus, H. Waldman BenAsher, P. R. Kinchington, and R. S. Goldstein, submitted for publication). The downregulation of this general alphaherpesvirus receptor may partially account for the exclusion of both VZV and HSV1 by VZV being more efficient in fibroblasts than in neurons. Another possible mechanism might be via receptor occupancy, which would physically interfere with virion binding to the cells. This mechanism has been reported for HSV1 and other alphaherpesviruses (11, 13, 14). A third possibility is that VZV may employ an entirely different SE mechanism, such as that recently detailed for vaccinia virus in which the early expression of surface glycoproteins in newly infected cells signals the spread of virus by promoting actin tail projections (43).
The SE we observed could also have been a consequence of the alterations in the environment of the infected cell, such as the virion host shutoff response. This response is characterized by degradation of mRNA mediated via interaction with the cellular translation initiation machinery (44, 45). The virion host shutoff response of VZV is delayed compared to that of HSV1, and VZV ORF63 was found to mediate part of the host shutoff response, in addition to the activity of the ORF41 virion host shutoff protein (32). However, our results with UV-inactivated virus indicate that even VZV that is unable to make viral proteins or replicate is still able to exclude live, infectious virus applied subsequently, at levels similar to those observed with live virus. The SE takes place only when the cells are pretreated with virus, as mock treatment with debris made from uninfected ARPE19 followed by live VZV results in the same level of infection as obtained in naive, untreated cells. Therefore, some virus-dependent mechanism, such as receptor occupancy, is likely to be responsible for the SE observed, rather than a host shutdown response.
SE was observed for both VZV and HSV1 in human neurons derived from hESC, and while each virus was able to exclude both itself and the other virus, co- and superinfection were observed to occur. Importantly, human neurons infected with one of these viruses were significantly less effective than fibroblasts at preventing superinfection by another alphaherpesvirus. The consequences of VZV infection of neurons and nonneuronal cells, including apoptosis and the ability to support latent infection, are known to differ (46, 47). The difference in SE between fibroblasts and neurons could be due to different mechanisms used by the two cell types, differences in innate immunity-related responses, or the expression of genes involved in apoptosis. Less efficient SE in neurons than in other cell types has been described for vectors derived from the Semliki Forest virus (10). The demonstration of coinfection of neurons with HSV1 and VZV, leading to apparent productive infections in the same neuron, establishes the potential for a single neuron to harbor both viruses at low frequencies in vivo, as previously suggested (23).
An important clinical aspect of the neuronal superinfection we describe here regards the potential for genetic recombination between VZV strains. There is evidence that infection by different VZV strains in individual neurons may occur in vivo in natural infections, since signs of genomic recombination between different clades of VZV have been described (48, 49). It is possible that multiple wild-type infections with VZV may occur throughout life, resulting in subclinical boosting of cellular immunity, with disease only occurring with the primary infection. It is unlikely that recombination events would occur in fibroblasts or other nonneuronal cells due to the fact that they undergo lytic cell death as a result of infection. Our demonstration of simultaneous productive infection by two VZV strains in one neuron is consistent with the possibility that that the site of viral recombination is in neurons of infected ganglia.
The varicella vaccine is routinely administered to children in a number of countries, including the United States, Canada, Germany, Greece, and Israel (50, 51). Studies examining the interaction between the wild-type and vaccine strains have been published (52–54), but there is not yet extensive information about the incidence of coinfection with both strains in the same person. The in vitro model used in the present study could be used as a tool with which to investigate recombination between wild-type and vaccine strains of VZV.
Our study only evaluated productive infections of neurons with VZV (and HSV-1). An important and clinically relevant issue is whether SE occurs within neurons that were infected at a prior time and now harbor the virus in a latent state. Another related question is whether VZV or HSV1 latently infected neurons can be “reactivated” by superinfection with one of the viruses. These questions are now being tested with the hESC-derived neuron model.
ACKNOWLEDGMENTS
This study was supported by Israel Science Foundation grant number 238/11 and a donation from the Maximillian Goode Foundation to R.S.G. and funds to P.R.K., including NIH grants NS082662 (with R.S.G.), NS064022, and EY08098, and funding from Research to Prevent Blindness, Inc., and The Eye & Ear Foundation of Pittsburgh.
Thanks as always to Chaya Morgenstern for expert technical and logistic support.
The authors declare no conflict of interest.
Footnotes
Published ahead of print 26 February 2014
REFERENCES
- 1.Folimonova SY. 2012. Superinfection exclusion is an active virus-controlled function that requires a specific viral protein. J. Virol. 86:5554–5561. 10.1128/JVI.00310-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarosinski KW. 2012. Dual infection and superinfection inhibition of epithelial skin cells by two alphaherpesviruses co-occur in the natural host. PLoS One 7:e37428. 10.1371/journal.pone.0037428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steck FT, Rubin H. 1966. The mechanism of interference between an avian leukosis virus and Rous sarcoma virus. I. Establishment of interference. Virology 29:628–641 [DOI] [PubMed] [Google Scholar]
- 4.Bratt MA, Rubin H. 1968. Specific interference among strains of Newcastle disease virus. 3. Mechanisms of interference. Virology 35:395–407 [DOI] [PubMed] [Google Scholar]
- 5.Johnston RE, Wan K, Bose HR. 1974. Homologous interference induced by Sindbis virus. J. Virol. 14:1076–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fulton RW. 1978. Superinfection by strains of tobacco streak virus. Virology 85:1–8. 10.1016/0042-6822(78)90406-3 [DOI] [PubMed] [Google Scholar]
- 7.Delwart EL, Panganiban AT. 1989. Role of reticuloendotheliosis virus envelope glycoprotein in superinfection interference. J. Virol. 63:273–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simon KO, Cardamone JJ, Jr, Whitaker-Dowling PA, Youngner JS, Widnell CC. 1990. Cellular mechanisms in the superinfection exclusion of vesicular stomatitis virus. Virology 177:375–379. 10.1016/0042-6822(90)90494-C [DOI] [PubMed] [Google Scholar]
- 9.Wildum S, Schindler M, Münch J, Kirchhoff F. 2006. Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J. Virol. 80:8047–8059. 10.1128/JVI.00252-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehrengruber MU, Goldin AL. 2007. Semliki Forest virus vectors with mutations in the nonstructural protein 2 gene permit extended superinfection of neuronal and non-neuronal cells. J. Neurovirol. 13:353–363. 10.1080/13550280701393204 [DOI] [PubMed] [Google Scholar]
- 11.Johnson RM, Spear PG. 1989. Herpes simplex virus glycoprotein D mediates interference with herpes simplex virus infection. J. Virol. 63:819–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Cohen GH, Eisenberg RJ. 2003. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J. Virol. 77:8127–8140. 10.1128/JVI.77.14.8127-8140.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campadelli-Fiume G, Qi S, Avitabile E, Foà-Tomasi L, Brandimarti R, Roizman B. 1990. Glycoprotein D of herpes simplex virus encodes a domain which precludes penetration of cells expressing the glycoprotein by superinfecting herpes simplex virus. J. Virol. 64:6070–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JS, Enquist LW, Card JP. 1999. Circuit-specific coinfection of neurons in the rat central nervous system with two pseudorabies virus recombinants. J. Virol. 73:9521–9531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Folimonova SY, Robertson CJ, Shilts T, Folimonov AS, Hilf ME, Garnsey SM, Dawson WO. 2010. Infection with strains of Citrus tristeza virus does not exclude superinfection by other strains of the virus. J. Virol. 84:1314–1325. 10.1128/JVI.02075-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dutta SK, Myrup AC, Thaker SR. 1986. In vitro interference between equine herpesvirus types 1 and 2. Am. J. Vet. Res. 47:747–750 [PubMed] [Google Scholar]
- 17.Meurens F, Schynts F, Keil GM, Muylkens B, Vanderplasschen A, Gallego P, Thiry E. 2004. Superinfection prevents recombination of the alphaherpesvirus bovine herpesvirus 1. J. Virol. 78:3872–3879. 10.1128/JVI.78.8.3872-3879.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinchington PR, Leger AJS, Guedon J-MG, Hendricks RL. 2012. Herpes simplex virus and varicella zoster virus, the house guests who never leave. Herpesviridae 3:5. 10.1186/2042-4280-3-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richter ER, Dias JK, Gilbert JE, Jr, Atherton SS. 2009. Distribution of herpes simplex virus type 1 and varicella zoster virus in ganglia of the human head and neck. J. Infect. Dis. 200:1901–1906. 10.1086/648474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoue H, Motani-Saitoh H, Sakurada K, Ikegaya H, Yajima D, Hayakawa M, Sato Y, Otsuka K, Kobayashi K, Nagasawa S, Iwase H. 2010. Detection of varicella-zoster virus DNA in 414 human trigeminal ganglia from cadavers by the polymerase chain reaction: a comparison of the detection rate of varicella-zoster virus and herpes simplex virus type 1. J. Med. Virol. 82:345–349. 10.1002/jmv.21687 [DOI] [PubMed] [Google Scholar]
- 21.Cohrs RJ, Laguardia JJ, Gilden D. 2005. Distribution of latent herpes simplex virus type-1 and varicella zoster virus DNA in human trigeminal ganglia. Virus Genes 31:223–227. 10.1007/s11262-005-1799-5 [DOI] [PubMed] [Google Scholar]
- 22.Saitoh H, Momma Y, Inoue H, Yajima D, Iwase H. 2013. Viable herpes simplex virus type 1 and varicella-zoster virus in the trigeminal ganglia of human cadavers. J. Med. Virol. 85:833–838. 10.1002/jmv.23527 [DOI] [PubMed] [Google Scholar]
- 23.Theil D, Paripovic I, Derfuss T, Herberger S, Strupp M, Arbusow V, Brandt T. 2003. Dually infected (HSV-1/VZV) single neurons in human trigeminal ganglia. Ann. Neurol. 54:678–682. 10.1002/ana.10746 [DOI] [PubMed] [Google Scholar]
- 24.Eisfeld AJ, Yee MB, Erazo A, Abendroth A, Kinchington PR. 2007. Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J. Virol. 81:9034–9049. 10.1128/JVI.00711-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markus A, Grigoryan S, Sloutskin A, Yee MB, Zhu H, Yang IH, Thakor NV, Sarid R, Kinchington PR, Goldstein RS. 2011. Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: direct demonstration of axonal infection, transport of VZV, and productive neuronal infection. J. Virol. 85:6220–6233. 10.1128/JVI.02396-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 27.Decman V, Kinchington PR, Harvey SAK, Hendricks RL. 2005. Gamma interferon can block herpes simplex virus type 1 reactivation from latency, even in the presence of late gene expression. J. Virol. 79:10339–10347. 10.1128/JVI.79.16.10339-10347.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sloutskin A, Kinchington PR, Goldstein RS. 2013. Productive vs non-productive infection by cell-free varicella zoster virus of human neurons derived from embryonic stem cells is dependent upon infectious viral dose. Virology 443:285–293. 10.1016/j.virol.2013.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harper DR, Mathieu N, Mullarkey J. 1998. High-titre, cryostable cell-free varicella zoster virus. Arch. Virol. 143:1163–1170. 10.1007/s007050050364 [DOI] [PubMed] [Google Scholar]
- 31.Kwong AD, Kruper JA, Frenkel N. 1988. Herpes simplex virus virion host shutoff function. J. Virol. 62:912–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desloges N, Rahaus M, Wolff MH. 2005. The varicella-zoster virus-mediated delayed host shutoff: open reading frame 17 has no major function, whereas immediate-early 63 protein represses heterologous gene expression. Microbes Infect. 7:1519–1529. 10.1016/j.micinf.2005.05.010 [DOI] [PubMed] [Google Scholar]
- 33.Dasika GK, Letchworth GJ., III 1999. Cellular expression of bovine herpesvirus 1 gD inhibits cell-to-cell spread of two closely related viruses without blocking their primary infection. Virology 254:24–36. 10.1006/viro.1998.9553 [DOI] [PubMed] [Google Scholar]
- 34.Campadelli-Fiume G, Arsenakis M, Farabegoli F, Roizman B. 1988. Entry of herpes simplex virus 1 in BJ cells that constitutively express viral glycoprotein D is by endocytosis and results in degradation of the virus. J. Virol. 62:159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rauch DA, Rodriguez N, Roller RJ. 2000. Mutations in herpes simplex virus glycoprotein D distinguish entry of free virus from cell-cell spread. J. Virol. 74:11437–11446. 10.1128/JVI.74.24.11437-11446.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muylkens B, Farnir F, Meurens F, Schynts F, Vanderplasschen A, Georges M, Thiry E. 2009. Coinfection with two closely related alphaherpesviruses results in a highly diversified recombination mosaic displaying negative genetic interference. J. Virol. 83:3127–3137. 10.1128/JVI.02474-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Q, Ali MA, Cohen JI. 2006. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 127:305–316. 10.1016/j.cell.2006.08.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berarducci B, Ikoma M, Stamatis S, Sommer M, Grose C, Arvin AM. 2006. Essential functions of the unique N-terminal region of the varicella-zoster virus glycoprotein E ectodomain in viral replication and in the pathogenesis of skin infection. J. Virol. 80:9481–9496. 10.1128/JVI.00533-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zerboni L, Berarducci B, Rajamani J, Jones CD, Zehnder JL, Arvin A. 2011. Varicella-zoster virus glycoprotein E is a critical determinant of virulence in the SCID mouse-human model of neuropathogenesis. J. Virol. 85:98–111. 10.1128/JVI.01902-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22. 10.1016/S0092-8674(00)80058-6 [DOI] [PubMed] [Google Scholar]
- 41.Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell. Microbiol. 6:401–410. 10.1111/j.1462-5822.2004.00389.x [DOI] [PubMed] [Google Scholar]
- 42.Lindahl U, Kjellén L. 2013. Pathophysiology of heparan sulphate: many diseases, few drugs. J. Intern. Med. 273:555–571. 10.1111/joim.12061 [DOI] [PubMed] [Google Scholar]
- 43.Doceul V, Hollinshead M, van der Linden L, Smith GL. 2010. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327:873–876. 10.1126/science.1183173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doepker RC, Hsu W-L, Saffran HA, Smiley JR. 2004. Herpes simplex virus virion host shutoff protein is stimulated by translation initiation factors eIF4B and eIF4H. J. Virol. 78:4684–4699. 10.1128/JVI.78.9.4684-4699.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saffran HA, Read GS, Smiley JR. 2010. Evidence for translational regulation by the herpes simplex virus virion host shutoff protein. J. Virol. 84:6041–6049. 10.1128/JVI.01819-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hood C, Cunningham AL, Slobedman B, Arvin AM, Sommer MH, Kinchington PR, Abendroth A. 2006. Varicella-zoster virus ORF63 inhibits apoptosis of primary human neurons. J. Virol. 80:1025–1031. 10.1128/JVI.80.2.1025-1031.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.James SF, Mahalingam R, Gilden D. 2012. Does apoptosis play a role in varicella zoster virus latency and reactivation? Viruses 4:1509–1514. 10.3390/v4091509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Breuer J. 2010. VZV molecular epidemiology. Curr. Top. Microbiol. Immunol. 342:15–42. 10.1007/82_2010_9 [DOI] [PubMed] [Google Scholar]
- 49.Chow VT, Tipples GA, Grose C. 2013. Bioinformatics of varicella-zoster virus: single nucleotide polymorphisms define clades and attenuated vaccine genotypes. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 18:351–356. 10.1016/j.meegid.2012.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flatt A, Breuer J. 2012. Varicella vaccines. Br. Med. Bull. 103:115–127. 10.1093/bmb/lds019 [DOI] [PubMed] [Google Scholar]
- 51.Megged O, Schlesinger Y. 2009. Varicella zoster infection in adults: a preventable disease. Isr. Med. Assoc. J. 11:306–307 [PubMed] [Google Scholar]
- 52.Horien C, Grose C. 2012. Neurovirulence of varicella and the live attenuated varicella vaccine virus. Semin. Pediatr. Neurol. 19:124–129. 10.1016/j.spen.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gershon AA, Chen J, Davis L, Krinsky C, Cowles R, Reichard R, Gershon M. 2012. Latency of varicella zoster virus in dorsal root, cranial, and enteric ganglia in vaccinated children. Trans. Am. Clin. Climatol. Assoc. 123:17–35 [PMC free article] [PubMed] [Google Scholar]
- 54.Pahud BA, Glaser CA, Dekker CL, Arvin AM, Schmid DS. 2011. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J. Infect. Dis. 203:316–323. 10.1093/infdis/jiq066 [DOI] [PMC free article] [PubMed] [Google Scholar]