

ABSTRACT
Rates of spontaneous mutation determine viral fitness and adaptability. In RNA viruses, treatment with mutagenic nucleoside analogues selects for polymerase variants with increased fidelity, showing that viral mutation rates can be adjusted in response to imposed selective pressures. However, this type of resistance is not possible in viruses that do not encode their own polymerases, such as single-stranded DNA viruses. We previously showed that serial passaging of bacteriophage ϕX174 in the presence of the nucleoside analogue 5-fluorouracil (5-FU) favored substitutions in the lysis protein E (P. Domingo-Calap, M. Pereira-Gomez, and R. Sanjuán, J. Virol. 86:9640–9646, 2012, doi:10.1128/JVI.00613–12). Here, we found that approximately half (6/12) of the amino acid replacements in the N-terminal region of this protein led to delayed lysis, and two of these changes (V2A and D8A) also conferred partial resistance to 5-FU. By delaying lysis, the V2A and D8A substitutions allowed the virus to increase the burst size per cell in the presence of 5-FU. Furthermore, these substitutions tended to alleviate drug-induced mutagenesis by reducing the number of rounds of copying required for population growth, revealing a new mechanism of resistance. This form of mutation rate regulation may also be utilized by other viruses whose replication mode is similar to that of bacteriophage ϕX174.
IMPORTANCE Many viruses display high rates of spontaneous mutations due to defects in proofreading or postreplicative repair, allowing them to rapidly adapt to changing environments. Viral mutation rates may have been optimized to achieve high adaptability without incurring an excessive genetic load. Supporting this, RNA viruses subjected to chemical mutagenesis treatments have been shown to evolve higher-fidelity polymerases. However, many viruses cannot modulate replication fidelity because they do not encode their own polymerase. Here, we show a new mechanism for regulating viral mutation rates. We found that, under mutagenic conditions, the single-stranded bacteriophage ϕX174 evolved delayed lysis, and that this allowed the virus to increase the amount of progeny produced per cell. As a result, the viral population was amplified in fewer infection cycles, reducing the chances for mutation appearance.
INTRODUCTION
Viral mutation rates vary amply, from 10−8 to 10−4 substitutions per nucleotide per round of copying (s/n/r), and some of the biochemical and genetic determinants of this variability have been elucidated (1). All RNA virus polymerases studied, except those of coronaviruses, lack 3′exonuclease proofreading activity, making replication more error-prone than in DNA viruses (2–5). Also, single-stranded viruses tend to show higher mutation rates than double-stranded genomes (6, 7). Furthermore, in DNA viruses and, to a lesser extent, RNA viruses mutation rates correlate negatively with genome size, although the causes of this correlation remain poorly understood (8–11). In theory, mutation rates should be evolutionarily adjusted in response to several factors, including the fitness costs incurred by deleterious mutations (12–15); the energy costs of encoding replication fidelity mechanisms (15, 16); the benefits of increasing adaptability (17, 18), population size (19, 20), and structure (21); and the topology of the fitness landscape (22). The ability of viruses to modify their mutation rates has been exemplified by RNA viruses serially passaged in the presence of mutagenic nucleoside analogues. Changes in key residues of the viral polymerase led to an overall increase in replication fidelity or reduced affinity for the drug, thereby compensating for the mutagenic effect of these treatments (23–28). In principle, viruses could also modulate their mutational load through changes in life history traits, although this has not been demonstrated experimentally (29–31).
Mutation rate regulation should be more constrained in single-stranded DNA viruses which do not encode their own polymerases. Despite this, the mutation rates of the single-stranded DNA bacteriophages ϕX174 and m13 are clearly distinct from those of their host, Escherichia coli, and are highest among DNA-based genomes, averaging 10−6 s/n/r (32–34). Also, the fast molecular evolution of animal and plant single-stranded DNA viruses suggests that their mutation rates are elevated (6), although these have not been measured. The replication fidelity of bacteriophage ϕX174 is similar to that of E. coli, since the phage is replicated by the host DNA polymerase III holoenzyme (35) with apparently unaltered nucleotide selection specificity and proofreading activity (36, 37). Therefore, the elevated mutation rate of the phage probably is associated with defects in postreplicative repair (38, 39). Indeed, the 5.4-kb genome of bacteriophage ϕX174 lacks GATC sequence motifs required for methyl-directed mismatch repair (MMR) mediated by the MutHLS system (40–44). However, modification of GATC motifs across all of the viral genome is a slow process and should not provide an efficient mechanism for regulating the viral mutation rate in response to chemical mutagenesis.
In a previous work, we found that serial passaging of bacteriophage ϕX174 in the presence of 10 ng/μl 5-fluorouracil (5-FU) selected for the amino acid replacement V2A in the periplasmic N-terminal domain lysis protein E, which conferred resistance to the drug (45). Since viral lysis proteins have not been implicated previously in nucleoside analogue resistance, we sought to explore the role played by this region of protein E in determining the lytic properties of the phage, 5-FU resistance, and the viral mutation rate. Site-directed mutagenesis analysis revealed that single-amino-acid replacements in the N terminus of the E protein frequently led to delayed lysis, and two of these substitutions (V2A and D8A) also conferred 5-FU resistance. In the presence of 5-FU, the V2A and D8A substitutions significantly increased the per-cell viral burst size and reduced the estimated viral mutation rate by approximately 2-fold.
We propose a simple model whereby the increased burst size afforded by delayed lysis allows the virus to reduce the number of infection cycles required for population expansion. As a result, the number of replication cycles during which mutations can accumulate becomes smaller, alleviating the mutational load induced by 5-FU and contributing to drug resistance. This demonstrates that mutation rate regulation can be mediated by proteins not directly involved in replication. Other viruses with replication modes similar to those of bacteriophage ϕX174 also may adjust their mutation rates by modifying lysis time and burst size.
MATERIALS AND METHODS
Bacteriophage and cells.
The E. coli C strain IJ1862 was obtained from James J. Bull. The gro87 mutant was provided by Bentley A. Fane. Our laboratory strain of bacteriophage ϕX174 has been described previously and was adapted to IJ1862 cells by long-term serial passaging (46).
Site-directed mutagenesis.
The ϕX174 double-stranded DNA replicative form was purified from infected cultures using a standard miniprep kit (Qiagen), and 500 pg of this DNA served as the template for PCR-based mutagenesis using Phusion high-fidelity DNA polymerase (New England BioLabs) and a pair of contiguous, divergent, 5′-phosphorylated primers, of which the reverse primer carried the desired nucleotide substitution. PCR products were circularized with a Quick Ligation kit (New England BioLabs) and used for transfecting competent IJ1862 cells by the heat shock method. A single plaque was picked, resuspended in LB, and stored at −70°C. The presence of the substitution was confirmed by Sanger sequencing.
Lysis time.
By following methods of a previous work (47), we inoculated exponentially growing IJ1862 cells (0.5 ml) at a multiplicity of infection (MOI) of 3 PFU per cell to achieve a rapid infection of the entire culture, which was incubated at 37°C and 650 rpm in a Thermomixer shaker (Eppendorf). Lysis was assessed by measuring the optical density at 600 nm (OD600) of the culture at different time points and fitting the following logistic model to the data by nonlinear least-squares regression: OD600(t) = ODmin + (ODmax − ODmin)/[1 + (t/L)h], where t is time postinoculation (min), L is the lysis time (defined as the time required to lyse 50% of the cells), h is the maximal slope of the lysis curve (Hill coefficient), and ODmax and ODmin are the initial and final optical densities, respectively. Each virus was assayed at least in triplicate.
Growth curves.
Exponentially growing IJ1862 cultures (0.5 ml; approximately 108 cells/ml) were infected with 104 PFU and incubated as described above. Samples were collected at different time points, cells were removed by centrifugation, and the supernatants were aliquoted, stored at −70°C, and titrated. A logistic growth model of the form Nt = K/(1 + ec − rt) was fit to the data, where r is the population growth rate, K is the maximal viral titer, and c sets the initial conditions. Each virus was assayed in triplicate.
Burst size.
Exponentially growing cells (0.5 ml; 108 cells/ml) were inoculated at an MOI of 0.5 PFU/ml, and after adsorption, cultures were diluted 104-fold to impede subsequent spread to other cells, as previously described (47). Samples from this initial time point were titrated to determine the total number of free particles and infected cells (Nfree + NC). Another aliquot of the same time point was treated with 10% chloroform to lyse cells and then was titrated. Since intracellular viral particles were not mature at this time point, they did not yield plaques in chloroform-treated samples, allowing us to determine Nfree alone. The difference in titer between these two plating assays was used to calculate NC. Infected cultures were incubated for approximately twice the lysis time (50 min for the WT and 72 min for the V2A and D8A mutants in the absence of 5-FU; 1.5 h for all viruses in the presence of 5-FU) to allow completion of the infection cycle and titrated (without chloroform) to determine the final number of free PFU (N1). The burst size (B) was then calculated as B = (N1 − Nfree)/Nc. Six independent replicates were performed for each virus and condition. For assays performed with 5-FU, one replicate of each of the WT, V2A, and D8A viruses was removed because the number of infected cells was too small (NC < 10).
Intracellular virion accumulation and eclipse time.
Exponentially growing cells (0.5 ml; 108 cells/ml) were inoculated and diluted 104-fold after adsorption. Samples were taken at different time points until shortly before lysis, treated with chloroform to lyse cells, and titrated to determine the number of mature phage particles. We used the following equation to model the data: Nt = N0e−aHt + B/(1 + ec − RT). The first term describes the loss of titer due to adsorption of N0 initial mature particles to host cells (H) at rate a. The second term is a logistic intracellular growth model with growth rate R, maximal per-cell yield B (burst size), and a parameter, c, that sets initial conditions. The model was fit by nonlinear least-squares regression using experimentally determined N0, H, and B values to infer R, c, and a. These values were used to calculate the eclipse time, E, defined as the time from inoculation to the maturation of the first phage particle. Since at t = E we have Nt = N0, the eclipse time can be obtained as E = [c − ln (B/N0)]/R.
Luria-Delbrück fluctuation tests.
Each Luria-Delbrück fluctuation test consisted of 24 independent IJ1862 cultures (0.5 ml) inoculated with the indicated initial number (N0) of PFU and incubated (37°C, 650 rpm) to allow viral growth. The final number of PFU, N1, was determined by titrating 6/24 random cultures. To score mutants, the culture was centrifuged to remove IJ1862 cells and 0.4 ml was plated without dilution on the restrictive gro87 strain, a mutant of the rep gene (48, 49) in which only mutants with certain mutations in the N-terminal end of protein A can form plaques. Since the total plating volume was 4 ml, mutant scoring was done in the presence of 1/10 of the 5-FU dose used during the growth phase, which could affect plating efficiency. We estimated the rate (m) at which gro87-resistant mutants appeared using the null-class method, which is based on counting the proportion of cultures showing zero versus at least one mutant. Since mutation is a rare and random event, the number of mutations per culture should follow a Poisson distribution with parameter λ = m(N1 − N0), such that the expected probability of no mutants in a culture is P0 = e−m(N1 − N0). Mutation rates were then transformed to substitutions per nucleotide per round of copying (s/n/r) using μ = 3m/Ts (1), where Ts = 7 is the total number of different substitutions leading to this phenotype under our laboratory conditions (32). Fluctuation tests were performed in two experimental blocks, and the results were analyzed using a two-way analysis of variance (ANOVA) in which the block was treated as a random factor and the virus type as a fixed factor. Data were log transformed for statistical analysis. Fluctuation tests carried out with and without 5-FU were analyzed separately.
RESULTS
Substitutions in the N-terminal region of protein E can delay lysis and confer partial resistance to 5-FU.
We used site-directed mutagenesis to individually modify residues 2 to 9 of the N-terminal region of the lysis protein E by introducing single-nucleotide substitutions that would not alter the overlapping reading frame of the scaffolding protein D (Fig. 1). Of the 16 mutants assayed, 12 yielded plaques that could be successfully amplified, the other being probably lethal or highly deleterious. Serial optical measurements of infected cultures provided strong evidence for delayed lysis in 6/12 cases (Table 1), whereas the other six mutations changed lysis time only slightly in either direction. We then performed a preliminary test of the ability of these six mutants to grow in the presence of 10 ng/μl 5-FU by determining the fold increase in titer after 3 h of incubation (RFU). This suggested that viruses carrying substitutions V2A (RFU = 21.0 ± 7.1) and D8A (RFU = 6.3 ± 1.6) had greater abilities to grow in the presence of 5-FU than the nonmutated WT virus (RFU = 3.7 ± 0.9). To confirm this, we performed growth curves of the WT, V2A, and D8A viruses in the presence/absence of 5-FU, and lysis times were also determined in the presence of the drug (Fig. 2A to D). Treatment with 5-FU delayed lysis considerably, but the V2A and D8A mutants again showed significantly delayed lysis compared with the WT, although differences were small (3 to 6 min) (Table 2). Both substitutions increased the population growth rate (r) in the presence of 5-FU but were costly in the absence of the drug (Table 2). The V2A and WT viruses showed similar maximal viral titers (K) in the presence of 5-FU, indicating that r is the main fitness component determining 5-FU resistance. We also found that the D8A mutant showed strongly reduced K, although the interpretation of this parameter is less clear because it reflects exhaustion of susceptible cells, which in turn can be determined by the ability of the virus to infect the culture rapidly or to continue replicating as the cell population approaches the stationary phase.
FIG 1.
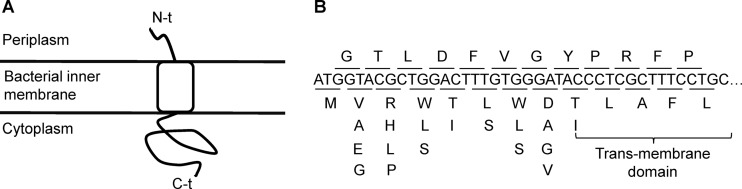
Single-amino-acid replacements introduced in the N-terminal region of the lysis protein. (A) Scheme of the cytoplasmic, transmembrane, and periplasmic domains of the lysis protein E. All substitutions were introduced in the periplasmic domain. (B) Nucleotide sequence of the region studied, showing the two overlapping reading frames (top, gene D; bottom, gene E) and the amino acid changes introduced.
TABLE 1.
Lysis time of N-terminal mutants of the lysis protein E
| Genomea | Protein E | Lysis timec (min) |
|---|---|---|
| WT | WT | 24.5 ± 0.2 |
| T572C | V2A | 36.2 ± 0.6*** |
| T572A | V2E | 27.8 ± 0.2* |
| T572G | V2G | NDb |
| G575A | R3H | 24.4 ± 1.0 |
| G575C | R3P | 38.0 ± 0.2** |
| G575T | R3L | 23.6 ± 0.3 |
| G578C | W4S | ND |
| G578T | W4L | 27.6 ± 0.4* |
| C581T | T5I | 23.2 ± 0.1* |
| T584C | L6S | 23.0 ± 0.6* |
| G587T | W7L | 35.9 ± 1.1** |
| G587C | W7S | 36.3 ± 0.8*** |
| A590T | D8V | ND |
| A590C | D8A | 38.5 ± 1.0*** |
| A590G | D8G | ND |
| C593T | T9I | 33.8 ± 1.0** |
Numbering is according to that of GenBank accession number GQ153915.
ND, lethal or highly deleterious mutant, based on titration.
Asterisks indicate values significantly different from those for the WT (*, P < 0.05; **, P < 0.01; ***, P < 0.001, as determined by the t test).
FIG 2.

Effect of the V2A and D8A substitutions on lytic and growth properties of the virus. Standard growth curves (A and B), lysis assays (C and D), and intracellular growth assays (E and F) are shown for the WT (black circles), V2A (white squares), and D8A (white triangles) viruses in the absence (A, C, and E) and in the presence (B, D, and F) of 10 ng/μl 5-FU. Error bars in panels A to D indicate the standard errors of the means (SEM) and are not shown in panels E and F for clarity.
TABLE 2.
Characteristics for the WT, V2A, and D8A viruses in the presence/absence of 5-FU
| Virus and treatment | r (min−1) | K (PFU, ×10−8) | L (min) | B (PFU/cell) | aHa (ml/min) | R (min−1) | E (min) | Predicted rb | Predicted rc |
|---|---|---|---|---|---|---|---|---|---|
| None | |||||||||
| WT | 0.130 ± 0.002 | 9.21 ± 1.12 | 24.5 ± 0.2 | 25.4 ± 4.4 | 0.524 ± 0.062 | 0.877 ± 0.027 | 13.6 ± 0.2 | 0.132 | 0.123 |
| V2Ad | 0.089 ± 0.002*** | 5.01 ± 0.50* | 36.2 ± 0.6*** | 37.2 ± 7.5 | 0.287 ± 0.028* | 0.540 ± 0.049** | 19.3 ± 0.3*** | 0.099 | 0.092 |
| D8Ad | 0.090 ± 0.005** | 0.160 ± 0.010*** | 38.5 ± 1.0*** | 18.2 ± 1.5 | 0.111 ± 0.021** | 0.400 ± 0.148 | 20.9 ± 0.9** | 0.075 | 0.064 |
| 5-FU | |||||||||
| WT | 0.021 ± 0.001 | 0.059 ± 0.010 | 43.0 ± 0.8 | 1.73 ± 0.19 | 0.379 ± 0.052 | 0.348 ± 0.057 | 20.1 ± 0.2e | 0.013 | 0.012 |
| V2Ad | 0.029 ± 0.002* | 0.061 ± 0.003 | 46.3 ± 0.7* | 2.47 ± 0.19* | 0.279 ± 0.181 | 0.283 ± 0.113 | 33.0e | 0.020 | 0.018 |
| D8Ad | 0.030 ± 0.001** | 0.015 ± 0.005* | 49.0 ± 1.3** | 3.01 ± 0.24** | ND | ND | ND | 0.022 | 0.019f |
For a cell density of H = 108 cells/ml.
Predicted from r = lnB/L.
Predicted from r = aH(Be−Lr − 1).
Asterisks indicate significant differences from WT values by t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Calculated from two (WT) and one (V2A) of the three replicates due to insufficient viral growth in the other replicates.
Using aH = 0.111.
Substitutions V2A and D8A increase the viral burst size in the presence of 5-FU.
We hypothesized that by delaying lysis, the V2A and D8A substitutions provide additional time for mature viral particles to accumulate inside cells and may increase the per-cell viral progeny (burst size). Thus, we measured the burst size of each virus in the presence/absence of 5-FU by determining the amount of infectious viral particles released from a known number of infected cells. In the absence of 5-FU, the V2A mutant showed greater burst size than the WT, whereas the D8A mutant had lower burst size; these differences were not statistically significant (Table 2). The lower burst size of the D8A mutant may be explained by its less efficient lysis, as judged by minimal optical density values (Fig. 2C). In any case, these results indicate that, in the absence of 5-FU, the payoffs of retarding lysis are small or absent. Addition of 10 ng/μl 5-FU strongly reduced the burst size, showing that the cytotoxic effects of the drug had a major impact on the infection. However, under these conditions both the V2A and D8A viruses showed significantly higher burst size than the WT (Table 2), indicating that delayed lysis allowed these mutants to produce more progeny than the WT in cells treated with 5-FU.
V2A and D8A retard the intracellular accumulation of mature viral particles.
We characterized the intracellular accumulation of mature viral particles within the time frame of a single infection cycle by lysing cells prematurely with chloroform (Fig. 2E and F). In the first 10 min following inoculation, the number of mature particles declined as a result of viral adsorption, increased exponentially as newly synthesized virions accumulated inside infected cells, and finally stagnated 5 to 10 min before lysis in the absence of 5-FU and 10 to 20 min before lysis in the presence of 5-FU. From these data, we inferred the viral adsorption rate (a) and the eclipse time (defined as the time span from inoculation to phage maturation, E). In the absence of 5-FU, substitutions V2A and D8A retarded the intracellular accumulation of mature particles mainly due to a longer eclipse time (Table 2); on the other hand, particle accumulation stagnated later (approximately 26 min postinoculation) than for the WT (approximately 18 min postinoculation) (Fig. 2E). Mutants V2A and D8A also showed retarded yet prolonged particle accumulation in the presence of 5-FU, although inference of eclipse times was complicated by the low and fluctuating viral titers (Fig. 2F).
The changes in lysis time and burst size produced by V2A and D8A explain 5-FU resistance.
A simple expression describing viral growth when cells are not a limiting resource is Ng = N0Bg, where N0 is the initial population size and g is the number of viral generations (i.e., infection cycles). This can be rewritten as Ng = N0erLg, because the lysis time (L) here is equivalent to the generation time; thus, r = lnB/L. The growth rates predicted by this model using the experimentally determined B and L values correlated well with the observed rates (Pearson r = 0.986, P < 0.001) and showed that the changes in burst size and lysis time produced by V2A and D8A should be deleterious in the absence of 5-FU but beneficial in the presence of the drug (Fig. 3). A more detailed growth model is given by r = aH(Be−Lr − 1) (47). This equation was solved numerically given the values for parameters a, H, B, and L. The predicted growth rates were similar to those obtained with the simpler model (Table 2), also correlated with the observed values (Pearson r = 0.979, P < 0.001), and confirmed that the FU-dependent fitness effects of substitutions V2A and D8A could be explained by their effects on lysis time and burst size.
FIG 3.

Expected effect of changes in lysis time and burst size on viral growth rate. The growth rates of the WT (black circles), V2A (white squares), and D8A (white triangles) viruses were predicted based on their experimentally determined lysis times and burst sizes (x axis) and compared to measured growth rates (y axis) in the presence and in the absence of 10 ng/μl 5-FU.
V2A and D8A alleviate the mutagenic effect of 5-FU.
Since the V2A and D8A substitutions conferred resistance to 5-FU, we sought to test whether they contributed to compensating for the mutagenic effects of this drug. We used the Luria-Delbrück fluctuation test to infer viral mutation rates in the presence/absence of 5-FU based on the appearance of mutants capable of growing in a nonpermissive host strain (gro87). The WT showed an average spontaneous mutation rate of μ = (0.86 ± 0.10) × 10−6 s/n/r, consistent with previous work (32, 34), and the V2A and D8A substitutions had no significant effect on this rate in the absence of 5-FU (P = 0.399 by two-way ANOVA). In contrast, in cells treated with 5-FU, there were differences among the mutation rates of the WT, V2A, and D8A viruses (P = 0.004). The V2A substitution had a significant antimutagenic effect, whereas the effect of D8A was smaller and did not reach statistical significance (Table 3). However, mutation rate differences were probably underestimated in these assays, because 5-FU reduced plating efficiency during mutant scoring by approximately 2-fold for the WT but had no such effect on the V2A and D8A viruses.
TABLE 3.
Mutation rate estimates from fluctuation tests in the presence/absence of 10 ng/μl 5-FU
| Virus and treatment | No. of testsa | N0b (PFU) | N1c (PFU, ×10−5) | P0d | μe (s/n/r, ×106) |
|---|---|---|---|---|---|
| None | |||||
| WT | 4 | 110, 432, 178, 323f | 5.2, 3.6, 2.3, 13.1 | 0.33, 0.46, 0.58, 0.17 | 0.864 ± 0.096 |
| V2Ag | 3 | 192, 382, 468 | 3.6, 1.8, 1.4 | 0.38, 0.67, 0.58 | 1.26 ± 0.19 |
| D8Ag | 5 | 743, 397, 198, 305, 397 | 9.1, 5.0, 6.2, 3.2, 6.6 | 0.17, 0.67, 0.58, 0.79, 0.63 | 0.436 ± 0.102 |
| 5-FU | |||||
| WT | 6 | 213, 90, 157, 375, 343, 462 | 0.042, 0.092, 0.087, 0.15,0.23, 0.084 | 0.83, 0.67, 0.71, 0.17, 0.25, 0.54 | 27.9 ± 5.4 |
| V2Ag | 3 | 235, 140, 252 | 0.13, 0.15, 0.13 | 0.75, 0.75, 0.79 | 8.73 ± 0.59** |
| D8Ag | 3 | 687, 407, 368 | 0.025, 0.099, 0.092 | 0.96, 0.58, 0.75 | 16.2 ± 4.2 |
Number of independent fluctuation tests performed.
Initial number of PFU per culture.
Final number of PFU per culture.
Fraction of 24 cultures showing no gro87-resistant plaques.
For each test, the mutation rate was calculated as μ = [−lnP0/(N1 − N0)] × (3/Ts), where Ts = 7.
Fluctuation tests were performed in two experimental blocks (differentiated by underlining).
Asterisks indicate significant differences in mutation rates in a t test against the WT (**, P < 0.01).
The antimutagenic effects of V2A and D8A are explained by differences in burst size.
Considering the results described above, we hypothesized that the greater burst size conferred by the V2A and D8A substitutions allow the virus to reduce the number of viral generations required for infecting a given number of cells and, as a result, to reduce the rate at which mutations accumulate. To test this, we used the growth model described above, Ng = N0Bg. For the average N0 (261 PFU) and N1 (6.03 × 105 PFU) values from fluctuation tests performed with the WT in the absence of 5-FU, the calculated number of viral generations was g = ln(N1/N0)/lnB = 2.39. For the same level of population expansion, the V2A and D8A substitutions should modify the number of generations by 10% or less (g = 2.14 and g = 2.67, respectively). In contrast, in fluctuation tests performed with 5-FU, the calculated number of WT generations was 6.81 (using N0 = 240 PFU and N1 = 3.68 × 105 PFU); the numbers were 4.13 and 3.39 for the V2A and D8A mutants, which represent 1.7- and 2.0-fold reductions, respectively. In general, the number of generations required to infect a given number of host cells (H) is g = 1 + lnH/lnB. Based on the B estimates, the V2A and D8A substitutions should have a small effect on the rate of mutation accumulation in the absence of 5-FU, whereas they should significantly reduce these rates in the presence of the drug (Fig. 4).
FIG 4.

Predicted number of viral generations required to infect a given number of host cells for the WT (black circles), V2A (white squares), and D8A (white triangles) viruses with and without 5-FU. For an infection starting with 1 PFU, generations were calculated as g = 1 + lnH/lnB, where H is the number of infected cells and B is the burst size.
DISCUSSION
We have identified two single-amino-acid replacements (V2A and D8A) in the lysis protein E of bacteriophage ϕX174 that modify lysis time and burst size, confer partial resistance to the nucleoside analogue 5-FU, and tend to alleviate the mutagenic effect of the drug. The N terminus of the E protein has been implicated previously in determining lysis times (50). Interestingly, the V2A substitution was found to promote lysis in an alanine mutagenesis scan, but these assays were based on expressing the E protein from a plasmid under the control of the T7 promoter and may not reproduce the expression levels of the phage (50). The concentration of E protein should be critical for lysis, since a threshold amount of the protein should be needed for effectively inhibiting cell wall synthesis and triggering lysis. For instance, it was found that a substitution in the adjacent residue (R3H) increased the intracellular concentration of the protein, probably by increasing the translation rate of the mRNA, and that this allowed the phage to lyse cells even in the absence of SlyD, which normally contributes to lysis by binding and stabilizing the E protein (51).
Together with these previous findings, our results show that the N-terminal region of the E protein can alter viral fitness by modifying lysis time. Delayed lysis has the expected payoff of increasing the burst size; on the other hand, it retards the infection of new cells. Therefore, viral fitness should be maximized for some intermediate, optimal lysis time (47, 52, 53). It has been suggested that this optimum can be calculated as a function of the eclipse time (E) and the maximal population growth rate (r̂) as L̂ = E + 1/r̂ (54). This assumes that mature viral particles accumulate inside cells linearly at rate R during the posteclipse phase of the infection until they are released by lysis, such that the burst size is B = R(L − E), but our results indicate that intracellular mature particle accumulation was exponential and plateaued before lysis (notice that this does not preclude linear accumulation of viral genomes). Therefore, optimal lysis times cannot be derived from these expressions. However, regardless of the intracellular growth model assumed, our data show that the burst size and lysis time of the WT are fitter than those of the V2A and D8A mutants in the absence of 5-FU, whereas the latter are fitter in the presence of 5-FU. It has been pointed out previously that optimal lysis times should depend on cell physiology (52, 55). 5-FU induces severe metabolic changes in the cell, including altered replication, transcription, and translation; thymidine deprivation; DNA strand breaks; and abnormal cell wall synthesis, which can lead to lysis (56, 57). Taking this into account, there are at least two explanations for why the V2A and D8A substitutions increase viral fitness in the presence of 5-FU. First, the drug should slow down phage replication and morphogenesis; therefore, delayed lysis may provide the extra time needed to produce enough mature viral particles. Second, since both 5-FU and the E protein trigger lysis through defects in cell wall synthesis, the drug may lower the threshold concentration of E protein required for lysis, and the V2A and D8A mutant proteins may have higher threshold concentrations than the WT, compensating for the effects of 5-FU.
Our results suggest that, in addition to modifying the lytic properties of the phage, substitutions in the N terminus of the E protein can change the dynamics of intracellular virus accumulation, and the D8A substitution even appeared to alter viral adsorption. These effects were unexpected, because the lysis protein E is not part of the virion and is expressed during the late phase of the infection. However, delayed production of mature particles was also observed in another mutant of gene E synonymous with the overlapping reading frame D (58). Since the cell's gene expression patterns and physiology are altered as the infection progresses, a possible consequence of delayed lysis is that virion morphogenesis takes place in an unusual cellular environment, potentially leading to virions with modified physical properties. It is also possible that, despite being synonymous with the scaffolding protein reading frame, the V2A and D8A substitutions produce disfavored codons, reducing the translation rate of protein D and negatively impacting virion maturation.
The observation that V2A (and, to a lesser extent, D8A) modified the estimated viral mutation rate was also unexpected, because the lysis protein does not participate in replication. The ϕX174 genome is copied following a rolling-circle mechanism in which most or all progeny molecules are synthesized from a single template (1, 58, 59). Hence, replication conforms to a stamping machine model in which there is essentially one replication event per cell (i.e., two rounds of copying, one for each polarity of the genome) regardless of burst size. In contrast, greater burst size should allow the virus to reduce the number of viral generations required to infect a population of susceptible cells and, as a result, the overall number of rounds of copying. Therefore, the lysis protein may act as a regulator of the mutation rate by inducing changes in lysis time and burst size. Since the null-class method used here for mutation rate estimation scores the probability of no mutations after N1 − N0 new progeny have been produced, it should not be sensitive to the number of generations elapsed (60, 61). However, the observed titers should be substantially lower than the actual number of viral particles produced because plating is not fully efficient, making the method sensitive to the actual number of mutants present in each culture and to the number of generations elapsed.
The V2A substitution was originally isolated from lines serially passaged in the presence of 10 ng/μl 5-FU, and the primary selective pressure favoring this mutation was probably related to the strong physiological changes produced by the drug. In fact, our results showed that V2A and D8A directly increased the viral growth rate in the presence of 10 ng/μl 5-FU. However, these substitutions should also allow the virus to alleviate the mutational load produced by 5-FU over the long term. Based on their effects on burst size, V2 and D8A should reduce the rate of mutation accumulation in the population to approximately one-half. This is similar to the effect of some nucleoside analog resistance mutations previously reported in RNA viruses. For instance, serial passaging of poliovirus in the presence of the nucleoside analogue ribavirin selected for an amino acid replacement in the viral polymerase (G64S) that increased overall replication fidelity by 3-fold (27). Similarly, Chikungunya virus treated with the same 5-FU dose used here evolved the C483Y replacement near the conserved active site of the nsP4 replicase, reducing the genetic variability of the virus in cell culture by slightly less than 2-fold, as determined by molecular clone sequencing (25).
Selection for delayed lysis in harsh and mutagenic environments and its modulatory effect on viral mutation may apply to other viruses. As opposed to cellular semiconservative replication, viruses follow a variety of replication mechanisms which, in many cases, incorporate a stamping machine-like feature where each template is copied multiple times to produce progeny genomes. As a consequence, an increase in burst size should come about with a reduction of the number of infection cycles required for viral population expansion, diminishing the accumulation of deleterious mutations.
ACKNOWLEDGMENTS
We thank Silvia Torres and Pablo Hernández for technical assistance.
This work was supported by grants from the Spanish Ministerio de Economía y Competitividad (BFU2011-25271) and the European Research Council (ERC-2011-StG-281191-VIRMUT) to R.S. and by a Ph.D. fellowship from the Spanish Ministerio de Educación to M.P.-G.
Footnotes
Published ahead of print 19 February 2014
REFERENCES
- 1.Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R. 2010. Viral mutation rates. J. Virol. 84:9733–9748. 10.1128/JVI.00694-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denison MR, Graham RL, Donaldson EF, Eckerle LD, Baric RS. 2011. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 8:270–279. 10.4161/rna.8.2.15013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menéndez-Arias L. 2009. Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses 1:1137–1165. 10.3390/v1031137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts JD, Bebenek K, Kunkel TA. 1988. The accuracy of reverse transcriptase from HIV-1. Science 242:1171–1173. 10.1126/science.2460925 [DOI] [PubMed] [Google Scholar]
- 5.Steinhauer DA, Domingo E, Holland JJ. 1992. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 122:281–288. 10.1016/0378-1119(92)90216-C [DOI] [PubMed] [Google Scholar]
- 6.Duffy S, Shackelton LA, Holmes EC. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9:267–276. 10.1038/nrg2323 [DOI] [PubMed] [Google Scholar]
- 7.Sanjuán R. 2012. From molecular genetics to phylodynamics: evolutionary relevance of mutation rates across viruses. PLoS Pathog. 8:e1002685. 10.1371/journal.ppat.1002685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradwell K, Combe M, Domingo-Calap P, Sanjuán R. 2013. Correlation between mutation rate and genome size in riboviruses: mutation rate of bacteriophage Qβ. Genetics 195:243–251. 10.1534/genetics.113.154963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drake JW, Charlesworth B, Charlesworth D, Crow JF. 1998. Rates of spontaneous mutation. Genetics 148:1667–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drake JW, Holland JJ. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 96:13910–13913. 10.1073/pnas.96.24.13910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch M. 2010. Evolution of the mutation rate. Trends Genet. 26:345–352. 10.1016/j.tig.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.André JB, Godelle B. 2006. The evolution of mutation rate in finite asexual populations. Genetics 172:611–626. 10.1534/genetics.105.046680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson T. 1999. The approach to mutation-selection balance in an infinite asexual population, and the evolution of mutation rates. Proc. R. Soc. Lond. B Biol. Sci. 266:2389–2397. 10.1098/rspb.1999.0936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimura M. 1967. On the evolutionary adjustment of spontaneous mutation rates. Genet. Res. 9:23–34. 10.1017/S0016672300010284 [DOI] [Google Scholar]
- 15.Sniegowski PD, Gerrish PJ, Johnson T, Shaver A. 2000. The evolution of mutation rates: separating causes from consequences. Bioessays 22:1057–1066. [DOI] [PubMed] [Google Scholar]
- 16.Dawson KJ. 1998. Evolutionarily stable mutation rates. J. Theor. Biol. 194:143–157. 10.1006/jtbi.1998.0752 [DOI] [PubMed] [Google Scholar]
- 17.Johnson T, Barton NH. 2002. The effect of deleterious alleles on adaptation in asexual populations. Genetics 162:395–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orr HA. 2000. The rate of adaptation in asexuals. Genetics 155:961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynch M. 2011. The lower bound to the evolution of mutation rates. Genome Biol. Evol. 3:1107–1118. 10.1093/gbe/evr066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M. 2012. Drift-barrier hypothesis and mutation-rate evolution. Proc. Natl. Acad. Sci. U. S. A. 109:18488–18492. 10.1073/pnas.1216223109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang X, Mu B, Huang Z, Zhang M, Wang X, Tao S. 2010. Impacts of mutation effects and population size on mutation rate in asexual populations: a simulation study. BMC Evol. Biol. 10:298. 10.1186/1471-2148-10-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clune J, Misevic D, Ofria C, Lenski RE, Elena SF, Sanjuán R. 2008. Natural selection fails to optimize mutation rates for long-term adaptation on rugged fitness landscapes. PLoS Comput. Biol. 4:e1000187. 10.1371/journal.pcbi.1000187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agudo R, Ferrer-Orta C, Arias A, de la Higuera I, Perales C, Pérez-Luque R, Verdaguer N, Domingo E. 2010. A multi-step process of viral adaptation to a mutagenic nucleoside analogue by modulation of transition types leads to extinction-escape. PLoS Pathog. 6:e1001072. 10.1371/journal.ppat.1001072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arias A, Arnold JJ, Sierra M, Smidansky ED, Domingo E, Cameron CE. 2008. Determinants of RNA-dependent RNA polymerase (in)fidelity revealed by kinetic analysis of the polymerase encoded by a foot-and-mouth disease virus mutant with reduced sensitivity to ribavirin. J. Virol. 82:12346–12355. 10.1128/JVI.01297-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coffey LL, Beeharry Y, Bordería AV, Blanc H, Vignuzzi M. 2011. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl. Acad. Sci. U. S. A. 108:16038–16043. 10.1073/pnas.1111650108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graci JD, Gnadig NF, Galarraga JE, Castro C, Vignuzzi M, Cameron CE. 2012. Mutational robustness of an RNA virus influences sensitivity to lethal mutagenesis. J. Virol. 86:2869–2873. 10.1128/JVI.05712-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfeiffer JK, Kirkegaard K. 2003. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. U. S. A. 100:7289–7294. 10.1073/pnas.1232294100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sierra M, Airaksinen A, González-López C, Agudo R, Arias A, Domingo E. 2007. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: implications for error catastrophe. J. Virol. 81:2012–2024. 10.1128/JVI.01606-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sardanyés J, Elena SF. 2011. Quasispecies spatial models for RNA viruses with different replication modes and infection strategies. PLoS One 6:e24884. 10.1371/journal.pone.0024884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sardanyés J, Solé RV, Elena SF. 2009. Replication mode and landscape topology differentially affect RNA virus mutational load and robustness. J. Virol. 83:12579–12589. 10.1128/JVI.00767-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thebaud G, Chadoeuf J, Morelli MJ, McCauley JW, Haydon DT. 2010. The relationship between mutation frequency and replication strategy in positive-sense single-stranded RNA viruses. Proc. Biol. Sci. 277:809–817. 10.1098/rspb.2009.1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuevas JM, Duffy S, Sanjuán R. 2009. Point mutation rate of bacteriophage ϕX174. Genetics 183:747–749. 10.1534/genetics.109.106005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kunkel TA. 1985. The mutational specificity of DNA polymerase-beta during in vitro DNA synthesis. Production of frameshift, base substitution, and deletion mutations. J. Biol. Chem. 260:5787–5796 [PubMed] [Google Scholar]
- 34.Raney JL, Delongchamp RR, Valentine CR. 2004. Spontaneous mutant frequency and mutation spectrum for gene A of ϕX174 grown in E. coli. Environ. Mol. Mutagen. 44:119–127. 10.1002/em.20041 [DOI] [PubMed] [Google Scholar]
- 35.Wickner S, Hurwitz J. 1974. Conversion of ϕX174 viral DNA to double-stranded form by purified Escherichia coli proteins. Proc. Natl. Acad. Sci. U. S. A. 71:4120–4124. 10.1073/pnas.71.10.4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fersht AR. 1979. Fidelity of replication of phage ϕX174 DNA by DNA polymerase III holoenzyme: spontaneous mutation by misincorporation. Proc. Natl. Acad. Sci. U. S. A. 76:4946–4950. 10.1073/pnas.76.10.4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fersht AR, Knill-Jones JW. 1981. DNA polymerase accuracy and spontaneous mutation rates: frequencies of purine.purine, purine.pyrimidine, and pyrimidine.pyrimidine mismatches during DNA replication. Proc. Natl. Acad. Sci. U. S. A. 78:4251–4255. 10.1073/pnas.78.7.4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuevas JM, Pereira-Gomez M, Sanjuán R. 2011. Mutation rate of bacteriophage PhiX174 modified through changes in GATC sequence context. Infect. Genet. Evol. 11:1820–1822. 10.1016/j.meegid.2011.08.024 [DOI] [PubMed] [Google Scholar]
- 39.Laengle-Rouault F, Maenhaut-Michel G, Radman M. 1986. GATC sequence and mismatch repair in Escherichia coli. EMBO J. 5:2009–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deschavanne P, Radman M. 1991. Counterselection of GATC sequences in enterobacteriophages by the components of the methyl-directed mismatch repair system. J. Mol. Evol. 33:125–132. 10.1007/BF02193626 [DOI] [PubMed] [Google Scholar]
- 41.Friedberg EC. 2003. DNA damage and repair. Nature 421:436–440. 10.1038/nature01408 [DOI] [PubMed] [Google Scholar]
- 42.Marti TM, Kunz C, Fleck O. 2002. DNA mismatch repair and mutation avoidance pathways. J. Cell. Physiol. 191:28–41. 10.1002/jcp.10077 [DOI] [PubMed] [Google Scholar]
- 43.McClelland M. 1984. Selection against dam methylation sites in the genomes of DNA of enterobacteriophages. J. Mol. Evol. 21:317–322 [DOI] [PubMed] [Google Scholar]
- 44.Modrich P, Lahue R. 1996. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65:101–133. 10.1146/annurev.bi.65.070196.000533 [DOI] [PubMed] [Google Scholar]
- 45.Domingo-Calap P, Pereira-Gomez M, Sanjuán R. 2012. Nucleoside analogue mutagenesis of a single-stranded DNA virus: evolution and resistance. J. Virol. 86:9640–9646. 10.1128/JVI.00613-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Domingo-Calap P, Cuevas JM, Sanjuán R. 2009. The fitness effects of random mutations in single-stranded DNA and RNA bacteriophages. PLoS Genet. 5:e1000742. 10.1371/journal.pgen.1000742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heineman RH, Bull JJ. 2007. Testing optimality with experimental evolution: lysis time in a bacteriophage. Evolution 61:1695–1709. 10.1111/j.1558-5646.2007.00132.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ekechukwu MC, Oberste DJ, Fane BA. 1995. Host and ϕX174 mutations affecting the morphogenesis or stabilization of the 50S complex, a single-stranded DNA synthesizing intermediate. Genetics 140:1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tessman ES, Peterson PK. 1976. Bacterial rep- mutations that block development of small DNA bacteriophages late in infection. J. Virol. 20:400–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka S, Clemons WM., Jr 2012. Minimal requirements for inhibition of MraY by lysis protein E from bacteriophage ϕX174. Mol. Microbiol. 85:975–985. 10.1111/j.1365-2958.2012.08153.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernhardt TG, Roof WD, Young R. 2002. The Escherichia coli FKBP-type PPIase SlyD is required for the stabilization of the E lysis protein of bacteriophage ϕX174. Mol. Microbiol. 45:99–108. 10.1046/j.1365-2958.2002.02984.x [DOI] [PubMed] [Google Scholar]
- 52.Bull JJ, Pfennig DW, Wang IN. 2004. Genetic details, optimization and phage life histories. Trends Ecol. Evol. 19:76–82. 10.1016/j.tree.2003.10.008 [DOI] [PubMed] [Google Scholar]
- 53.Chantranupong L, Heineman RH. 2012. A common, non-optimal phenotypic endpoint in experimental adaptations of bacteriophage lysis time. BMC Evol. Biol. 12:37. 10.1186/1471-2148-12-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bull JJ. 2006. Optimality models of phage life history and parallels in disease evolution. J. Theor. Biol. 241:928–938. 10.1016/j.jtbi.2006.01.027 [DOI] [PubMed] [Google Scholar]
- 55.Wang I-N, Dykhuizen DE, Slobodkin LB. 1996. The evolution of phage lysis timing. Evol. Ecol. 10:545–558. 10.1007/BF01237884 [DOI] [Google Scholar]
- 56.Adelberg EA, Coughlin CA. 1956. Bacterial mutation induced by thymine starvation. Nature 178:531–532. 10.1038/178531a0 [DOI] [PubMed] [Google Scholar]
- 57.Drake JW, Greening EO. 1970. Suppression of chemical mutagenesis in bacteriophage T4 by genetically modified DNA polymerases. Proc. Natl. Acad. Sci. U. S. A. 66:823–829. 10.1073/pnas.66.3.823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gillam S, Astell CR, Jahnke P, Hutchison CA, III, Smith M. 1984. Construction and properties of a ribosome-binding site mutation in gene E of ϕX174 bacteriophage. J. Virol. 52:892–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hutchison CA, III, Sinsheimer RL. 1966. The process of infection with bacteriophage phi-X174. X. Mutations in a phi-X Lysis gene. J. Mol. Biol. 18:429–447 [DOI] [PubMed] [Google Scholar]
- 60.Rosche WA, Foster PL. 2000. Determining mutation rates in bacterial populations. Methods 20:4–17. 10.1006/meth.1999.0901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng Q. 1999. Progress of a half century in the study of the Luria-Delbruck distribution. Math. Biosci. 162:1–32. 10.1016/S0025-5564(99)00045-0 [DOI] [PubMed] [Google Scholar]