

ABSTRACT
The extent to which HIV-1 clade B strains exhibit population-specific adaptations to host HLA alleles remains incompletely known, in part due to incomplete characterization of HLA-associated HIV-1 polymorphisms (HLA-APs) in different global populations. Moreover, it remains unknown to what extent the same HLA alleles may drive significantly different escape pathways across populations. As the Japanese population exhibits distinctive HLA class I allele distributions, comparative analysis of HLA-APs between HIV-1 clade B-infected Japanese and non-Asian cohorts could shed light on these questions. However, HLA-APs remain incompletely mapped in Japan. In a cohort of 430 treatment-naive Japanese with chronic HIV-1 clade B infection, we identified 284 HLA-APs in Gag, Pol, and Nef using phylogenetically corrected methods. The number of HLA-associated substitutions in Pol, notably those restricted by HLA-B*52:01, was weakly inversely correlated with the plasma viral load (pVL), suggesting that the transmission and persistence of B*52:01-driven Pol mutations could modulate the pVL. Differential selection of HLA-APs between HLA subtype members, including those differing only with respect to substitutions outside the peptide-binding groove, was observed, meriting further investigation as to their mechanisms of selection. Notably, two-thirds of HLA-APs identified in Japan had not been reported in previous studies of predominantly Caucasian cohorts and were attributable to HLA alleles unique to, or enriched in, Japan. We also identified 71 cases where the same HLA allele drove significantly different escape pathways in Japan versus predominantly Caucasian cohorts. Our results underscore the distinct global evolution of HIV-1 clade B as a result of host population-specific cellular immune pressures.
IMPORTANCE Cytotoxic T lymphocyte (CTL) escape mutations in HIV-1 are broadly predictable based on the HLA class I alleles expressed by the host. Because HLA allele distributions differ among worldwide populations, the pattern and diversity of HLA-associated escape mutations are likely to be somewhat distinct to each race and region. HLA-associated polymorphisms (HLA-APs) in HIV-1 have previously been identified at the population level in European, North American, Australian, and African cohorts; however, large-scale analyses of HIV-1 clade B-specific HLA-APs in Asians are lacking. Differential intraclade HIV-1 adaptation to global populations can be investigated via comparative analyses of HLA-associated polymorphisms across ethnic groups, but such studies are rare. Here, we identify HLA-APs in a large Japanese HIV-1 clade B cohort using phylogenetically informed methods and observe that the majority of them had not been previously characterized in predominantly Caucasian populations. The results highlight HIV's unique adaptation to cellular immune pressures imposed by different global populations.
INTRODUCTION
HIV cytotoxic T lymphocyte (CTL) escape occurs in a manner that is highly reproducible in the context of the HLA class I alleles expressed by the host (1–8). By extension, HIV sequences circulating in a given host population exhibit polymorphisms that reflect the HLA allele distribution of that population (9). Because HLA class I allele distributions differ among racial and ethnic groups worldwide (10), the pattern and diversity of HLA-associated escape mutations are also likely to be somewhat distinct to each race and region. Numerous population-based studies identifying HLA-associated polymorphisms (HLA-APs) have been conducted in European, North American, Australian, and African cohorts (2, 6, 8). However, fewer have been undertaken in Asian cohorts, where HIV-1 prevalence is also substantial (11). Since Asian populations differ in their HLA allele distributions from the cohorts previously studied, it is important to identify and analyze HLA-APs to achieve a better understanding of HIV-1 pathogenesis in Asia and to inform future HIV vaccine design efforts targeted to these populations. The Japanese epidemic is unique in Asia. While clades A/E and C predominate in many Asian countries (12–14), the Japanese HIV-1 epidemic comprises 80% clade B infections (12). As such, the analysis of Japanese cohorts also provides the opportunity to undertake comparative analyses of HLA-APs between Asian and non-Asian populations infected with HIV clade B.
Previous studies have investigated differential HLA-driven HIV evolution across human populations. For example, a study of HLA-specific adaptations in HIV Pol in a Mexican cohort identified “unique” HLA-APs in this population that were not present in an international cohort from Canada, the United States, and Australia, even though both cohorts harbored HIV clade B (15). Most of the unique Mexican HLA-APs were restricted by HLA alleles particular to this population (e.g., HLA-B*39) but that were underrepresented or absent in the international cohort (15). This study, therefore, illustrates population-specific HIV adaptation in its most intuitive manifestation, i.e., where distinctive HLA-associated polymorphisms are observed in a population due to the presence (or comparatively high frequency) of an HLA allele in that population compared to another.
What remains unknown, however, is the extent to which the same HLA allele may drive divergent escape pathways in different human populations. Two critical features are required to address this question. First, the identification of HLA-APs must be undertaken at the HLA subtype level. This is because the majority (>60%) of HLA-associated polymorphisms are best defined at the subtype level (16), even for closely related HLA subtype members that present the same or similar peptide epitopes (16, 17, 18, 19). Comparative studies undertaken at allele level (two-digit) resolution cannot disentangle whether population-specific HLA-APs are attributable to differential HLA subtype distributions between cohorts or whether they are “true” cases where the same HLA subtype drives different escape pathways across populations. Indeed, a study investigating >500 Americans with chronic HIV-1 clade B infection observed distinct patterns of HLA-APs among white, black, and Hispanic individuals that were likely attributable to the differential distribution of closely related HLA subtypes among these groups (18) rather than true differential escape. The present study is therefore undertaken at subtype level resolution. Second, the identification of population-specific escape pathways driven by the same HLA allele requires a method to do so. Here, we adapt phylogenetically corrected statistical methods originally developed to assess differential escape among related HLA subtypes (17) and apply them to investigate differential escape across host populations.
The present study is divided into two parts, each with a specific major objective. Our first objective was to identify and characterize HLA-APs in HIV-1 Gag, Pol, and Nef proteins in a cohort of 430 chronically clade B-infected Japanese individuals using phylogenetically informed approaches (20) and to investigate their associations with clinical parameters (CD4+ T cell count [CD4 count] and plasma viral load [pVL]). Importantly, HLA genotyping (and thus HLA-AP identification) was undertaken at subtype level resolution, allowing us to analyze the effects of genetic differences among closely related HLA subtypes on the selection of HLA-APs in the Japanese cohort as part of this objective. Our second major objective was to perform a comparative analysis of HLA-APs identified in Japan and those identified in a large international (Canada/United States/Australia) cohort of antiretroviral-naive, chronically clade B-infected, predominantly Caucasian individuals. As expected, a substantial proportion of Japanese HLA-APs were restricted by alleles unique to (or highly enriched in) Japan compared to the non-Asian cohort. Notably, we also observed numerous cases where the same HLA allele drove significantly different—sometimes opposing—escape pathways in these two populations. Our results highlight HIV's unique adaptation to cellular immune pressures imposed by different global populations.
MATERIALS AND METHODS
Ethics statement.
This study was approved as part of the study of immunological and virological analysis in HIV-1 infection (number 540) by the ethics committee for epidemiology and general study in the Faculty of Life Science of Kumamoto University and the National Center for Global Health and Medicine (NCGHM). All studied individuals were adults. Written informed consent was obtained from all studied individuals according to the Declaration of Helsinki.
Subjects.
Four-hundred thirty treatment-naive Japanese individuals with chronic HIV-1 clade B infection were enrolled in the NCGHM from 2008 to 2011. The HLA alleles of these individuals were determined at the four-digit level by a probe-based sequence-specific oligonucleotide (SSO) typing method (HLA Laboratory, Kyoto, Japan). The median CD4 count and pVL at the first visit to the NCGHM were 321 cells/μl (interquartile range [IQR], 190 to 440 cells/μl) and 25,000 copies/ml (IQR, 6,800 to 98,000 copies/ml), respectively.
HLA-associated polymorphisms derived from the International HIV Adaptation Collaborative (IHAC) cohort, comprising 1,888 treatment-naive individuals with chronic clade B infection from Canada, the United States, and Western Australia (16), identified by identical methods, were used for comparison. The IHAC cohort comprises predominantly Caucasian individuals, with Asians making up less than 5% of the total. The median CD4 count in the IHAC cohort was 260 cells/μl (IQR, 110 to 418 cells/μl).
RT-PCR and sequencing of plasma HIV RNA.
HIV-1 RNA was extracted from plasma samples using either a QIAamp MinElute virus spin kit (Qiagen, Valencia, CA) or an EZ1 Virus Mini Kit v2.0 (Qiagen, Valencia, CA). Reverse transcription (RT) was performed using random hexamers with the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA). HIV-1 Gag, Pol, and Nef genes were amplified from cDNA by nested PCR using Taq DNA polymerase (Promega, Fitchburg, WI) and 10 primer pairs that were designed based on the clade B strain. For subjects with a viral load below 1,000 copies/ml, RT-PCR was performed with region-specific primers using the SuperScript III One-Step RT-PCR System with Platinum Taq kit (Invitrogen, Carlsbad, CA). The 1st-round PCR product was then used in the 2nd-round PCR amplification using Taq DNA polymerase (Promega, Fitchburg, WI) and the 10 primer pairs. The 2nd-round PCR product was purified by using the ExoSap-It reagent containing exonuclease I and alkaline phosphatase (GE Healthcare, Buckinghamshire, United Kingdom). Gag, Pol, and Nef sequences were determined by using the BigDye Terminator v3.1 cycle-sequencing kit (Applied Biosystems, Carlsbad, CA) and an ABI 3500 genetic analyzer (Applied Biosystems, Carlsbad, CA). Sequencing reactions were performed in both the 5′ and 3′ directions to yield a minimum of bidirectional coverage of all regions. The sequence data were then aligned by using SeqScape software (Applied Biosystems, Carlsbad, CA) based on the HXB2 reference sequence (K03455).
Identification of HLA-associated polymorphisms.
HLA-APs can be identified in large cross-sectional linked data sets of host (HLA) and HIV genotypes using statistical-association approaches that identify viral polymorphisms significantly over- or underrepresented in individuals harboring a specific HLA class I allele (1, 2, 4, 16–18, 21). HLA-APs that are overrepresented in individuals harboring the relevant HLA are commonly referred to as “adapted” forms, while those underrepresented in individuals harboring the relevant HLA are referred to as “nonadapted” forms (2, 18). As such, “nonadapted” and “adapted” forms can be conceptualized to represent the “immunologically susceptible” and “escape mutant” forms, respectively, for the specific HLA allele in question at that HIV codon position. Statistical association approaches for the identification of HLA-APs also correct for the confounding influences of viral phylogeny, HIV codon covariation, and linkage disequilibrium (LD) between HLA class I alleles (2, 16, 17, 21).
Associations between HLA class I alleles and HIV-1 amino acid polymorphisms in the Japanese and IHAC data sets were identified using a published phylogenetically corrected logistical-regression model that corrects for HLA LD, HIV phylogeny, and HIV codon covariation as potential confounders (17, 20). Briefly, maximum-likelihood phylogenetic trees were constructed using Gag, Pol, and Nef sequences (one tree per gene), and a model of conditional adaptation was inferred for each observed amino acid at each codon. Amino acids are assumed to evolve independently along the phylogeny to the tree tips (representing the present host). In each host, HLA-mediated selection and HIV amino acid covariation are directly modeled using weighted logistical regression, in which the individual's HLA repertoire and covarying HIV amino acids are used as binary predictors and the bias is determined by the possible transmitted sequences as inferred from the phylogeny (17). To identify which factors (HLA and/or HIV covariation) contribute to selection pressure, we employ a forward-selection procedure where the most significant association is iteratively added to the model, with P values computed using the likelihood ratio test. We performed post hoc filtering of the resulting HLA-associated-polymorphism list, restricting our output to instances in which at least 10 individuals carried the allele or polymorphism and at least 10 individuals did not carry the allele or polymorphism. Multiple tests were accounted for using q values, the P value analog of the false-discovery rate (FDR) (22). The FDR is the expected proportion of false positives among results deemed significant at a given threshold; for example, at a q value of <0.2, we expect 20% of identified associations to be false positives. In the analyses identifying HLA-APs, a significance threshold of a q value of <0.2 was employed.
Statistical analysis.
Correlations between the total number of HLA-associated substitutions in each individual and clinical parameters (pVL and CD4 count) were performed using Spearman's correlation. To determine the total number of HLA-associated substitutions within a given HIV-1 sequence, we first identified all HIV-1 sites within that sequence known to be associated with any HLA allele. The specific residue at each site was counted as “HLA associated” if it matched any HLA-associated adapted form or any residue other than a nonadapted form identified at that position. The HLA alleles expressed by the individual were not considered (unless specifically stated); rather, our goal was to enumerate the HLA-APs associated with any HLA allele in each viral sequence. In analyses where host HLA alleles were not considered, HIV sites harboring residues that simultaneously represented a nonadapted and an adapted form associated with different HLA alleles were excluded from consideration.
Detection of differential escape between closely related HLA alleles and between cohorts.
Two types of differential escape were investigated. First, we investigated differential escape between closely related HLA class I alleles, defined here as (four-digit) HLA subtype members belonging to the same (two-digit) allele group in the Japanese cohort. Specifically, seven HLA allele groups (A*02, A*26, B*15, B*40, C*03, C*08, and C*14) for which a minimum of two subtype members were represented in the Japanese cohort were investigated. For example, the HLA-A*02 allele group featured subtypes A*02:01, A*02:06, and A*02:07, while the A*26 allele group featured subtypes A*26:01 and A*26:03. For each allele group, we took the union of all HLA-APs identified for all subtype members of the group. Then, in a pairwise manner, we compared their strengths of selection between all HLA subtype members using a previously described phylogenetically corrected interaction test (17). In this analysis, thresholds of a P value of <0.05 and a q value of <0.2 were used to define significance.
Second, we investigated differential HLA-driven escape pathways between Japanese and IHAC cohorts. As outlined in the introduction, HLA-APs identified in human populations differ to some extent due to the presence (or enrichment) of certain HLA alleles in one population versus another. However, in this analysis, we were specifically interested in identifying cases where the same HLA allele drove significantly different escape pathways in the two cohorts. To do this, we took the union of all HLA-APs identified in the Japan and IHAC cohorts that were restricted by HLA subtypes observed a minimum of 10 times in both cohorts. We then compared the strength of selection of each HLA-AP in a pairwise manner between cohorts. The statistical methods used to investigate differential escape between the Japanese and IHAC cohorts are similar to those used to investigate differential escape between HLA subtype members (17), with some modifications, as follows. Briefly, a phylogenetically corrected logistical-regression model was constructed using a single HLA allele as a predictor. Using a likelihood ratio test, we then compared this model to a more expressive one that included an additional interaction term that was 1 if the individual expressed the HLA allele and was in the IHAC cohort or 0 otherwise. In this way, we could obtain a P value, testing the hypothesis that selection is the same in both cohorts (the null hypothesis) or whether selection differs across cohorts (alternative hypothesis). In contrast to the HLA-AP analyses described thus far, the present one does not feature corrections for HLA LD or HIV codon covariation and therefore yields odds ratios of association and P values that differ slightly from the original cohort-specific values. In the intercohort differential-escape analysis, significance was defined as a P value of <0.01 and a q value of <0.05.
Nucleotide sequence accession numbers.
The accession numbers for the sequences determined in this study are AB873205 to AB873601 (Gag), AB873908 to AB874270 (Pol), and AB873602 to AB873907 (Nef).
RESULTS
Identification of HLA-associated polymorphisms in chronically HIV-1 clade B-infected Japanese individuals.
The first objective of our study was to identify and characterize HLA-APs in Japan, a unique population in terms of its HLA class I distribution and predominantly HIV clade B epidemic. Toward this end, we analyzed linked HIV-HLA genotypes from 430 antiretroviral-therapy-naive Japanese individuals chronically infected with HIV-1 clade B. A total of 78 unique HLA class I alleles, defined at subtype level (four-digit) resolution, were observed in our cohort (see Fig. S1 in the supplemental material) at frequencies consistent with those in the published literature (23). Of these, 37 (including 9 HLA-A, 17 HLA-B, and 11 HLA-C alleles) were observed in at least 10 individuals and thus were included in the statistical analysis of HLA-APs (see Materials and Methods). Amplification and sequencing of HIV-1 Gag, Pol without the transframe (TF) protein, and Nef was successful for 397 (92.3%), 363 (84.4%), and 306 (71.2%) individuals, respectively. As described in Materials and Methods, HLA-APs within these three genes were identified using a phylogenetically corrected logistical-regression model that corrects for the confounding effects of viral phylogeny, HIV-1 codon covariation, and linkage disequilibrium between host HLA class I alleles (16, 17, 20). A false-discovery rate (q value) approach was employed to address multiple tests.
At a threshold of a q value of <0.2, a total of 284 HLA-APs, comprising 143 adapted and 141 nonadapted associations, were identified in Gag (n = 94 associations), Pol (n = 86 associations), and Nef (n = 104 associations) (Fig. 1; see Table S1 in the supplemental material). HLA-APs were more frequently detected in Nef (occurring at 45 of 206 codons [21.8%]) compared to Gag (51 of 500 codons [10.2%]) or Pol (51 of 947 codons [5.1%]). Although HLA class I allele frequencies in Japan are somewhat distinct globally, the distribution of HLA-APs across HIV-1 proteins was consistent with that reported in previous studies of other populations infected with clade B or C (1, 2, 6, 7, 16). Broken down by HLA locus, the numbers of HLA-A-, HLA-B-, and HLA-C-associated polymorphisms were 78, 140, and 66, respectively, numbers that were also consistent with previous reports from Caucasian and African cohorts that HLA-B alleles restrict more associations than HLA-A or HLA-C alleles (1, 6, 18).
FIG 1.

Escape map of HLA-APs for Gag, Pol, and Nef in the Japanese cohort. The escape maps indicate the locations, specific residues, and HLA restrictions of HLA-APs (all q < 0.2). The global HIV-1 clade B consensus amino acid sequence is used as a reference. The shaded vertical bars separate blocks of 10 amino acids. Adapted amino acids (those significantly overrepresented in the presence of a given HLA allele) are red. Nonadapted amino acids (those significantly underrepresented in the presence of a given HLA allele) are blue. Polymorphisms associated with the same HLA allele that occur in proximity to one another are grouped together in yellow boxes. A list of all HLA-APs is provided in Table S1 in the supplemental material.
Correlation between the total number of HLA-associated substitutions and clinical parameters in Japanese individuals.
We next wished to investigate the relationship between the presence of HLA-associated substitutions in each gene and the patient HIV-1 pVL and CD4 count in the Japanese cohort. As described in Materials and Methods, substitutions within a given HIV-1 sequence were counted as HLA associated if they had been identified as being associated with any HLA class I allele in our study, regardless of the HLA alleles expressed by the patient. For example, Gag-9S is an HLA-B*15:01-associated nonadapted polymorphism (Fig. 1; see Table S1 in the supplemental material); as such, any amino acid other than S at codon 9 was counted as an HLA-associated substitution. Similarly, Gag-123G is an HLA-C*01:02-associated adapted polymorphism (but no specific nonadapted forms, restricted by C*01:02 or others, were identified at this position); as such, any sequence harboring G at codon 123 was counted as having an HLA-associated substitution at this site.
A weak yet statistically significant inverse correlation was observed between pVL and the total number of HLA-associated substitutions in Pol (Spearman's R = −0.11; P = 0.04) (Fig. 2A). However, no such correlations were observed for Gag (Spearman's R = −0.056; P = 0.3) or Nef (Spearman's R = −0.029; P = 0.6) (Fig. 2A). Moreover, no significant correlations were observed between the total number of HLA-associated substitutions in any HIV protein and the CD4 count (Fig. 2A). Though the overall association is weak, the results raise the intriguing hypothesis that selection of certain HLA-driven substitutions in Pol could modulate the pVL in the Japanese population.
FIG 2.

Correlations between HLA-associated substitutions in Gag, Pol, and Nef and viral load or CD4 count. The total number of HLA-associated substitutions in each subject's Gag, Pol, and Nef sequence was determined (see Materials and Methods). (A) Correlation between the number of HLA-associated substitutions in Gag, Pol, or Nef and pVL or CD4 count. (B) Correlation between pVL and the number of HLA-associated substitutions in Pol, with HLA-B*52:01-associated substitutions excluded. (C) Correlation between pVL and the number of HLA-B*52:01-associated substitutions in Pol (all patients). (D) Correlation between the number of HLA-B*52:01-associated substitutions in Pol in HLA-B*52:01-positive individuals (left) and HLA-B*52:01-negative individuals (right). Analyses were performed using Spearman's correlation. Linear regression lines are included in the plots.
We next wondered whether the observed correlation between Pol polymorphisms and lower pVL could be attributed to polymorphisms restricted by HLA alleles that are protective in Japanese populations. HLA-B*67:01 and the HLA-B*52:01-HLA-C*12:02 haplotype are examples of such protective alleles (24). As such, we investigated whether they could play a role in the observed pVL correlation. No HLA-B*67:01-associated substitution was identified in Pol, whereas four HLA-B*52:01-associated and one HLA-C*12:02-associated substitutions were detected in the protein (see Table S1 in the supplemental material). Exclusion of the single HLA-C*12:02-associated substitution from analysis did not affect the relationship between the number of HLA-associated substitutions in Pol and pVL (data not shown). In contrast, exclusion of the four HLA-B*52:01-associated Pol substitutions substantially weakened the overall relationship between the number of HLA-associated Pol substitutions and pVL (Spearman's R = −0.057; P = 0.3) (Fig. 2B). Similarly, specific consideration of only HLA-B*52:01-associated Pol substitutions revealed a highly significant inverse correlation with pVL (Spearman's R = −0.18; P = 0.0007) (Fig. 2C) that represented the strongest such relationship detected in Pol for common HLA alleles observed in our cohort (see Fig. S2 in the supplemental material). We therefore reasoned that B*52:01-restricted substitutions were likely to be critical mediators of the observed pVL effect.
Finally, stratification of B*52:01-associated Pol substitutions by host B*52:01 expression revealed that the inverse correlation with pVL remained strongly detectable in HLA-B*52:01− individuals (Spearman's R = −0.18; P = 0.003), but not in HLA-B*52:01+ individuals (Spearman's R = 0.026; P = 0.8) (Fig. 2D). We interpret our observations as suggesting that HLA-B*52:01-restricted Pol substitutions possess fitness costs that manifest themselves in terms of lower pVL upon transmission to, and persistence in, HLA-B*52:01− individuals. In contrast, no such pVL effects are detectable in B*52:01+ individuals, likely because the fitness costs of these substitutions are outweighed by the advantages conferred by immune escape.
Differential escape between HLA subtypes in Japanese individuals.
Our final goal in characterizing HLA-APs in Japan was to investigate the extent of differential escape between closely related HLA subtypes. In particular, we hypothesized that HLA subtype members differing with respect to the amino acids located within in the peptide-binding groove of the HLA molecule may differ with respect to the nature (or binding affinity) of the specific HIV epitopes presented (25–28), and therefore, that they may exhibit differential escape pathways. In contrast, we hypothesized that HLA subtype members that differ with respect to amino acids located outside the peptide-binding groove may be more likely to present the same epitopes (29–31) and therefore will generally exhibit less evidence for differential escape between them. Of the 284 HLA-APs identified in our cohort, 128 were restricted by HLA allele groups (A*02, A*26, B*15, B*40, C*03, C*08, and C*14) containing two or more subtype members (see Table S1 in the supplemental material). For five of these allele groups (A*02, A*26, B*15, B*40, and C*08), subtype members differed by substitutions within the peptide-binding groove (see Fig. S3 in the supplemental material), supporting their potential as candidates for differential HLA-AP selection. In contrast, members of the C*03 and C*14 subtypes differed by substitutions outside the peptide-binding groove (see Fig. S3 in the supplemental material), suggesting that their epitope repertoires (and thus escape pathways) would be more similar to one another.
We began by simply comparing HLA-APs identified in the context of the different HLA subtypes. As expected, viral polymorphisms associated with HLA subtype members differing within their peptide-binding grooves appeared to be quite specific to each HLA subtype (see Fig. S3A to D and F in the supplemental material). Surprisingly, however, viral polymorphisms associated with HLA subtype members differing only with respect to amino acids located outside their peptide-binding grooves also appeared to be quite specific to each HLA subtype (see Fig. S3E and G in the supplemental material). For example, HLA-C*03:03 and C*03:04, which differ only by substitutions at position 91 that have no contact with the groove (29–31), were associated with a total of 11 HLA-APs, none of which appeared to be shared (see Fig. S2E in the supplemental material). Similarly, HLA-C*14:02 and C*14:03, which differ only by a substitution at position 21 located outside the floor of the peptide-binding groove (see Fig. S2G in the supplemental material), shared only 10 of the 24 HLA-APs identified between them.
However, qualitative comparisons of HLA-APs meeting a specific significance threshold, such as those described above, are not statistically robust (since individual associations may fail to meet the threshold and thus not be detected, or variations in allele frequency may limit the power to detect associations). Thus, to explicitly investigate whether the above-mentioned examples represent statistically significant instances of differential escape between subtype members, we applied a phylogenetically corrected interaction test to compare their strengths of selection between subtypes (17). For each HLA allele group, we took the union of all HLA-APs identified for all subtype members and compared their strengths of selection between all subtype members in a pairwise manner. Representative examples of our results are shown in Fig. 3. For example, HLA-A*26:01 and -A*26:03 differ with respect to substitutions at amino acids 74, 76, and 77, located within the peptide-binding groove of the HLA molecule (see Fig. S3B in the supplemental material). A total of 10 HLA-APs, located at 8 HIV codons, were originally identified as associated with either HLA-A*26:01 or -A*26:03 (see Fig. S3B in the supplemental material). Although qualitatively, all 10 HLA-APs appear to be differentially selected by HLA-A*26:01 or -A*26:03 (see Fig. S3B in the supplemental material), the phylogenetically corrected interaction test revealed only 3 of them (located at Pol residues 276 and 551 and Nef residue 85) to be significantly differentially selected in terms of their natural logarithms of the odds ratios (lnORs) of association (P < 0.05; q < 0.2) (Fig. 3A). Surprisingly, significant differential escape was also observed between subtype members that differed only with respect to substitutions outside their peptide-binding grooves: 3 of 9 (33.3%) sites restricted by HLA-C*03 allele group members and 5 of 14 (35.7%) sites restricted by C*14 allele group members similarly exhibited statistically significant evidence of differential selection (Fig. 3B and C).
FIG 3.

Polymorphic positions in HLA class I molecules and differential escape between pairs of HLA subtypes. In each ribbon diagram depicting the HLA-peptide-binding groove, the locations of residues differing among subtype members of the HLA-A*26 (A), HLA-C*03 (B), and HLA-C*14 (C) allele groups are highlighted in red and labeled with their locations and amino acids. HLA-AP comparisons between subtype members are shown in the corresponding plot below. The horizontal bars represent the lnORs, with colors indicating the restricting allele. Infinite lnORs are set to values of ±4. Boldface type indicates HLA-APs whose strengths of selection are statistically significantly different between the two subtype members (P < 0.05; q < 0.2). a.a., amino acid.
To determine whether the extent of differential escape between HLA subtype members varied between HLA allele groups that differed with respect to substitutions within or outside the binding groove, we asked whether the extent of differential escape between subtype members of the former group (comprising A*02, A*26, B*15, B*40, and C*08) differed from those of the latter group (comprising HLA-C*03 and C*14). Overall, we found no significant differences in the proportions of differential escape between them (34.8% for HLA-C*03/C*14 subtypes compared to 36.8% for subtypes of all other HLA alleles; P = 0.5) (see Table S2 in the supplemental material). This intriguing result suggests that variations outside the HLA binding groove may contribute as much to differential escape as variations within the binding groove.
Comparison of HLA-APs between Japanese and non-Asian individuals chronically infected with HIV-1 clade B.
Our second objective was to investigate HLA-APs identified in Japan versus those previously identified in non-Asian cohorts infected with HIV clade B. The comparison cohort in this analysis was the IHAC cohort, comprising 1,888 antiretroviral-naive individuals with chronic clade B infection in Canada, the United States, and Australia (in which <5% of cohort participants were Asian) (16).
HLA-APs differ to some extent between human populations due to the presence (or enrichment) of certain HLA alleles in one population versus another. Indeed, HLA allele frequencies differed markedly between the Japan and IHAC cohorts (see Fig. S1 in the supplemental material). As such, we began with a qualitative comparison of HLA-APs between them, starting with a simple positional analysis. In the Japanese cohort, HLA-APs were observed at a total of 147 codon positions in Gag, Pol, and Nef (Fig. 4). Of these, 117 (79.6%) were also associated with at least one HLA allele in the IHAC cohort. In contrast, the remaining 30 positions (including 16, 7, and 7 in Gag, Pol, and Nef, respectively) that harbored HLA associations in Japan were not associated with any HLA alleles in the IHAC cohort (Fig. 4). That 30/147 (20.4%) HIV codons exhibited evidence of HLA-driven selection in Japan but not in the IHAC cohort already strongly suggests that HIV is evolving under population-specific selection pressures in Japan compared to other regions.
FIG 4.
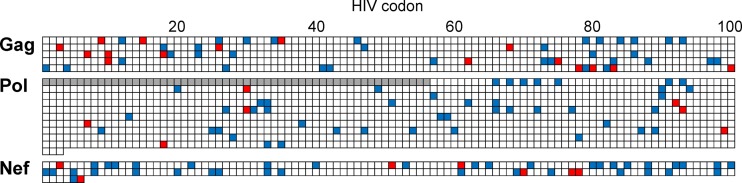
Locations of HLA-associated sites common to HIV-1 clade B-infected Japanese and Caucasian cohorts and those unique to Japan. The locations of all HLA-APs in Gag (500 codons), Pol (1,003 codons), and Nef (206 codons) are illustrated. The residues in the Pol TF protein were not analyzed in the IHAC cohort and are thus excluded (gray bar). The blue squares identify codons that harbored at least one HLA-AP in both Japanese and IHAC cohorts. The red squares indicate codons that harbored HLA-APs in Japan but that were not associated with any HLA alleles in the IHAC cohort.
Next, we compared HLA-APs over HIV position and specific HLA restriction. Of the 284 HLA-APs identified in Japan, 188 (66.2%) were not reported in the IHAC cohort. As expected, a substantial portion of these (46 of 188 [24.5%]) were associated with 8 HLA subtypes (A*26:03, B*40:06, B*54:01, B*55:02, B*59:01, B*67:01, C*08:03, and C*14:03) common in Japan but essentially absent (<1% frequency) in the IHAC cohort. Others were likely attributable to alleles observed at much higher frequencies in Japan than in the IHAC cohort: for example, an additional 27.1% were associated with HLA alleles present in both cohorts but whose frequencies were at least 4-fold higher in Japan than in the IHAC cohort. Overall, the results suggest that HLA-APs identified in Japan are quite distinctive, in large part reflecting the unique HLA allele distribution in the Japanese population.
We also wished to investigate the existence of differential HLA-associated escape pathways between the two populations that are not attributable to HLA frequency differences between them—in other words, cases where the same HLA subtypes drive significantly different escape pathways in the Japan and IHAC cohorts. This required the application of statistical tests (see Materials and Methods and below). Specifically, we first identified a list of 551 HLA-APs in HIV Gag, Pol, and Nef, which represented the union of all HLA-APs identified in either the Japan or IHAC cohort for which both the viral polymorphism and the restricting HLA allele were observed in a minimum of 10 individuals per cohort (not shown). The latter criterion was employed in order to achieve some minimal statistical power to compare the strengths of individual associations between cohorts. It is important to emphasize that these criteria would by definition exclude HLA alleles (and/or viral polymorphisms) present in one cohort but essentially absent in the other (as we would have no power, and in fact no rationale, to test whether their strengths of selection were statistically significantly different between cohorts).
For each HLA-AP, we calculated its lnOR of association in each cohort—a measure that can be interpreted as an estimate of the strength of selection exerted by the HLA allele on that particular HIV codon in that cohort. We then applied a phylogenetically corrected interaction test (17) to assess whether these lnORs of selection were significantly different in the Japanese versus the IHAC cohort. In these analyses, statistical significance was defined as a P value of <0.01 and a q value of <0.05.
Overall, 71 of 551 (12.8%) HLA-APs originally identified in either the Japan or IHAC cohort exhibited significantly different strengths of selection between the two populations (Fig. 5; see Table S3 in the supplemental material). The HLA-B*44:03-associated 125H substitution in Nef serves as an example of how to interpret these data. The lnOR of this association is 1.73 in Japan (with a cohort-specific P value of 3.26 × 10−6) versus 0.42 for the IHAC cohort (with a cohort-specific P value of 0.36). Both lnORs are positive, indicating that 125H is positively associated with B*44:03 in both cohorts, but the higher lnOR in Japan indicates that the strength of selection of Nef-125H by B*44:03 is greater in Japan than in the IHAC cohort (indeed, the cohort-specific P values reveal that this association is significant in Japan but not in the IHAC cohort). Finally, the P and q values for the intercohort comparison (P = 1.02 × 10−6 and q = 1.19 × 10−4) (see Table S3 in the supplemental material) confirm that the strength of selection of Nef-125H by B*44:03 is significantly greater in Japan than in the IHAC cohort. Importantly, this difference is not simply attributable to intercohort differences in B*44:03 frequencies (which are comparable between populations [see Fig. S1 in the supplemental material]).
FIG 5.

HLA-APs displaying significantly different strengths of selection between Japanese and IHAC cohorts. A phylogenetically corrected interaction test was used to compare the lnOR of selection of HLA-APs in the Japanese cohort versus the IHAC cohort. Comparisons with a P value of <0.01 and a q value of <0.05 are shown. The bars represent the lnORs. Infinite lnORs are set to values of ±4. Boldface type indicates HLA-APs that display diametrically opposed directions of selection between the cohorts (defined here as lnORs of association that were positive in one cohort but negative in the other, where the cohort-specific P values were <0.05 in both cases). A complete list of all comparisons with a P value of <0.05 is available in Table S3 in the supplemental material.
In addition to the HLA-B*44:03-associated 125H polymorphism in Nef, we identified 21 other HLA-APs whose strengths of selection were significantly greater in Japan than in the IHAC cohort, yielding a total of 22 (out of 71 [31.0%]) HLA-APs in this category. Conversely, 39 of 71 (54.9%) differentially selected HLA-APs exhibited strengths of selection that were greater in the IHAC cohort than in Japan. The HLA-A*26:01-associated 889S substitution in Pol serves as an example. The lnOR of this association is −0.18 in Japan (with a cohort-specific P value of 0.3) versus −1.17 for the IHAC cohort (with a cohort-specific P value of 7.92 × 10−9). Both lnORs are negative, indicating that 889S is negatively associated with A*26:01 in both cohorts, but the more negative value for the IHAC cohort indicates that this association is stronger in the IHAC cohort than in Japan. Finally, the P and q values for the intercohort comparison (P = 1.15 × 10−4 and q = 4.48 × 10−3 [see Table S3 in the supplemental material]) confirm that the strength of the negative association between Pol-889S by A*26:01 is significantly greater in the IHAC cohort than in Japan.
Strikingly, the remaining 10 (out of 71 [14.1%]) differentially selected HLA-APs displayed diametrically opposed directions of selection between the cohorts (defined here as lnORs of association that were positive in one cohort but negative in the other, where the cohort-specific P values were <0.05 in both cases) (Fig. 5). The HLA-B*44:03-associated 120F substitution in Nef serves as an example. The lnOR of this association is 1.44 in Japan (with a cohort-specific P value of 2.03 × 10−4), indicating that HLA-B*44:03 is significantly positively associated with 120F in Japan. In contrast, the lnOR of this association is −0.69 in the IHAC cohort (with a cohort-specific P value of 9.50 × 10−3), indicating that HLA-B*44:03 is significantly negatively associated with 120F in IHAC. The P and q values for the intercohort comparison (P = 2.15 × 10−8 and q = 3.75 × 10−6 [see Table S3 in the supplemental material]) confirm that the opposing directions of selection of Nef-120F by B*44:03 between the Japanese and IHAC cohorts is a statistically significant observation.
Of interest, the 71 HLA-APs identified as being under significantly different selection in the Japan and IHAC cohorts were differentially distributed across HLA loci and HIV proteins (Fig. 6A and B). Specifically, HLA-A-associated polymorphisms that were significantly differentially selected across cohorts were most abundant in Gag, followed by Pol and Nef, whereas differentially selected HLA-B-associated and HLA-C-associated polymorphisms were most numerous in Nef, followed by Pol and Gag. Taken together, the results support the existence of HLA class I alleles that drive significantly different HIV escape pathways in global populations infected with the same viral clade. The uneven distribution of the locations of these differentially selected polymorphisms across HLA loci and HIV regions raises the intriguing hypothesis that Gag and Pol/Nef may differentially evolve under selection pressures dominated by HLA-A versus HLA-B/C allele-restricted immune responses, respectively.
FIG 6.

HLA-APs identified as being under differential strengths of selection in Japanese and IHAC cohorts. At a P value of <0.01 and a q value of <0.05, a total of 71 HLA-APs were identified as being under significantly different strengths of selection in the Japanese and IHAC cohorts. (A) Restricting HLA alleles and their HIV-1 protein locations. (B) Numbers of differentially selected HLA-APs, broken down by HLA locus and HIV-1 protein.
DISCUSSION
The present study comprised two major objectives, both of which are novel in terms of populations studied and/or analytical methods used. First, we characterized HLA-APs in HIV-1 clade B Gag, Pol, and Nef and their relationship with clinical parameters in a large Japanese cohort. Second, we compared HLA-APs in Japanese versus non-Asian populations infected with HIV clade B to identify population-specific differences in their selection. In particular, we wished to identify HLA-APs that are unique to Japan by virtue of the distinctive HLA distribution in this population, as well as cases where the same HLA allele drives divergent escape pathways in Japan versus non-Asian populations.
This study is the first to identify HLA-APs in HIV-1's structural and functional genes in Japanese populations. Only one previous study investigated HLA-APs in HIV-1 clade B-infected Asians (11): the study comprised 231 Chinese individuals infected during a narrow-source outbreak and identified 141 HLA-associated polymorphisms at two-digit resolution. Our study differs from the previous study with respect to the cohort size, HLA genetics of the host population, HLA-typing resolution, and type of epidemic. Using phylogenetically informed approaches, we identified 284 HLA-APs within HIV-1 Gag, Pol, and Nef in our cohort, supporting a strong influence of population-specific, HLA-driven immune pressures in shaping HIV-1 evolution in Japan. In contrast to a previous study undertaken in a predominantly Caucasian population that observed approximately one-half of the total number of Gag HLA-APs to be located within or flanking reported CTL epitopes (3), the majority of HLA-APs identified in the present study were not located near reported CTL epitopes. This discrepancy may be due to the limited number of Asian-specific HLA-restricted CTL epitopes identified to date, underscoring the need for further epitope discovery in these populations.
This study revealed differential frequencies of HLA-APs across HIV genes in the Japanese population. Consistent with previous studies of HLA-APs in HIV clade B (2, 16, 18), HLA-APs were more frequently detected in Nef than in Gag and Pol. Also consistent with previous observations in Caucasian, African, Chinese, and Mexican populations (1, 6, 11, 15, 18), the number of HLA-B-associated polymorphisms in our cohort was higher than that of HLA-A- or HLA-C-associated polymorphisms, further supporting a dominant role of HLA-B in HIV evolution (32). An interesting feature of the Japanese population is that approximately 70% of individuals carry HLA-A*24:02 (23). Despite sufficient statistical power to detect HLA-A*24:02-associated polymorphisms in our cohort, we identified only 9 of them, 6 of which were located in epitopes identified by our group (33–35). A possible explanation for the relatively low number of A*24:02-associated polymorphisms in Japan is that they have accumulated over time in circulating sequences so that they are no longer significantly enriched among persons expressing HLA-A*24:02. Further analysis of mutations selected by HLA-A*24:02-restricted CTLs should clarify the mechanism whereby high-frequency HLA alleles influence the formation of HIV-1 polymorphisms.
Protective HLA alleles, such as HLA-B*57, -B*58, and -B*27, select Gag mutations affecting viral replication in Caucasians and Africans (36–41) that may also provide some clinical benefit if they are transmitted to hosts lacking these alleles (42, 43). HLA-B*57, -B*58, and -B*27 are not present at appreciable frequencies in Japan (23). It is therefore perhaps unsurprising that no correlations between HLA-associated substitutions in Gag and HIV clinical parameters were observed in our cohort. In contrast, we observed a weak but significant inverse correlation between the frequency of HLA-APs in Pol and the plasma viral load, which appeared to be driven by polymorphisms selected by HLA-B*52:01, an allele identified as protective in Japan (24). Upon further stratification by HLA-B*52:01 expression, the inverse correlation between VL and the total number of B*52:01-associated Pol substitutions was maintained in HLA-B*52:01− but not in HLA-B*52:01+ individuals. Taken together, these findings suggest that transmitted B*52:01-associated polymorphisms could reduce viral fitness in a dose-dependent manner, though further studies will be required to assess this. In addition, these substitutions were not located within or near known B*52:01-restricted epitopes. Thus, further research will be required to identify these epitopes and elucidate their mechanisms of escape.
Many previous studies of HLA-APs were performed at two-digit HLA resolution (1–4, 6). Here, we performed HLA genotyping at four-digit resolution, which allowed us to investigate differential escape between closely related HLA subtypes in the Japanese cohort. Nearly one-half of the HLA-APs identified in Japan were restricted by HLA allele groups containing two or more subtype members (A*02, A*26, B*15, B*40, C*03, C*08, and C*14). For five of these groups (A*02, A*26, B*15, B*40, and C*08), subtype members differed by substitutions within the peptide-binding groove, while for the remaining two groups (HLA-C*03 and -C*14), subtype members differed by substitutions located outside the peptide-binding groove. Reasoning that amino acid differences located within the peptide-binding groove could modulate the nature or presentation of CTL epitopes, we hypothesized that the former group would generally exhibit distinct HLA-APs between subtype members, while the latter would generally exhibit similar or identical HLA-APs. However, we were surprised to observe substantial evidence for differential HLA-AP selection between closely related HLA subtypes regardless of whether they differed in sequence within or outside the peptide-binding groove. Significantly differential HLA-AP selection was observed at 3 of 9 HLA-C*03-associated sites and 5 of 14 HLA-C*14-associated sites (Fig. 3), proportions that were not significantly lower than the frequency of differential selection between subtypes that differed in their peptide-binding grooves.
This observation raised several hypotheses. HLA polymorphic sites outside the peptide-binding groove may indirectly influence the binding groove conformation, thus altering HLA-peptide interactions and/or T cell recognition. Another possibility is selection by NK cells, as KIR may recognize sites outside the peptide-binding groove. Indeed, KIR3DL1 binds to the loop including position 91 of HLA-B*57:01 (44). However, it is not clear whether KIR2DLs, which are receptors for HLA-C, can bind to the loop outside the peptide-binding groove of HLA-C molecules. A recent study showed that HLA-C antigens are expressed at different levels on the cell surface, even among HLA-C subtypes (45). This study also observed a strong positive correlation between the HLA-C expression level and the strength of HLA-C-mediated selection pressure conferred on HIV. Differential expression levels of these HLA-C subtype members in Japanese populations thus provide another potential explanation for this observation for future follow-up.
Our second objective was to investigate differential HLA-APs between Japanese and non-Asian cohorts infected with HIV clade B. Here, the IHAC cohort (comprising clade B-infected Canadians, Americans, and Australians) was used as a comparison group (16). HLA-APs identified in human populations differ to some extent due to population-specific HLA distributions, yielding population-specific HLA-APs driven by HLA alleles present in one population but not another (15). Indeed, two-thirds of the HLA-APs identified in Japan had not previously been identified due to the presence of the restricting HLA alleles in Japan but their absence (or far lower prevalence) in the IHAC cohort.
What remains unknown however, is the extent to which the same HLA allele may drive significantly different escape pathways in different human populations. To this end, we applied novel phylogenetically corrected statistical approaches to assess the extent to which HLA-APs identified in either Japan or the IHAC cohort restricted by HLA alleles present in both populations exhibited significantly different strengths of selection. Of the 551 HLA-APs investigated, 71 (12.9%) were significantly differentially selected in Japan versus the IHAC cohort at a stringent statistical threshold of a q value of <0.05. Of these 71, 31% exhibited significantly greater strengths of selection in Japan than in the IHAC cohort, whereas 55% exhibited greater strengths of selection in the IHAC cohort than in Japan. Surprisingly, the remaining 14% displayed diametrically opposed selection pathways in the two cohorts (where an HIV polymorphism represented the adapted form associated with a given allele in one cohort but the nonadapted form associated with the same allele in the other cohort). It is important to emphasize that these significantly different pathways of HLA-AP selection are not simply attributable to differences in HLA frequency between the cohorts.
We feel that these are intriguing observations that merit further study. Nevertheless, we propose the following potential interpretations. First, these differences could be explained by functional differences in HIV-1-specific T cells elicited between the Japanese and Caucasian cohorts, possibly as a result of differences in host genetics (for example, in the genes that encode the T-cell receptor and/or modulate its expression). Such differences may influence the structure of the T-cell receptor(s) and thus the quality, quantity, and/or makeup of the HIV-1-specific T cell repertoire, thus influencing the specific escape mutations selected in the context of peptide-bound HLA. Further analysis of HIV-1-specific T cells driving the selection of these mutants in both cohorts is therefore warranted. It is also important to note that the intercohort HLA-AP comparisons, unlike previous analyses, did not correct for HLA LD or HIV codon covariation. Although both the Japan and IHAC cohorts feature HIV clade B infections, intraclade differences in the viral backbone could also influence differential escape via epistatic effects. In-depth analyses of intercohort differences in HIV codon covariation relationships are therefore also warranted. Intercohort differences in HLA LD are another possible contributor. Finally, the HLA-APs differentially selected between cohorts appeared to be unevenly distributed by HLA locus; while HLA-A-associated polymorphisms exhibiting differential selection between cohorts were more abundant in Gag than in other proteins, HLA-B- and HLA-C-associated polymorphisms exhibiting differential selection between cohorts tended to be more abundant in Nef. This suggests that intercohort differential HLA-APs across HIV proteins may be arising as a result of cellular immune pressures exerted by distinct HLA class I loci, though this also requires further study.
Nevertheless, the present study confirms the existence of population-specific HIV-1 adaptations that are attributable to the unique HLA allele distributions of the population (15). We additionally provide evidence of population-specific HIV adaptation to HLA-restricted immune responses that cannot be explained by differential HLA frequencies alone, cases where the same HLA allele drives significantly different, sometimes opposing, escape pathways in different host populations. Taken together, the results support differential HIV-1 adaptation to human populations worldwide that might be driven by multiple host and viral mechanisms.
Supplementary Material
ACKNOWLEDGMENTS
We thank Madoka Koyanagi and Rie Maruyama for the collection of samples from patients, Mari Hasegawa and Sayaka Nagata for technical assistance, Sachiko Sakai for her secretarial assistance, and Kyle Cobarrubias for assistance in constructing Fig. 1.
T.C. is a JSPS Research Fellow. A.Q.L. is the recipient of a Frederick Banting and Charles Best Masters award from the Canadian Institutes for Health Research (CIHR). Z.L.B. is the recipient of a New Investigator Award from the CIHR and a Scholar Award from the Michael Smith Foundation for Health Research (MSFHR). This research was supported by the Global COE program Global Education and Research Center Aiming at the Control of AIDS, launched as a project commissioned by the Ministry of Education, Science, Sports, and Culture, Japan, and by Grants-in-Aid for AIDS Research from the Ministry of Health, Labor, and Welfare, Japan.
We have no financial conflicts of interest.
Footnotes
Published ahead of print 12 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00147-14.
REFERENCES
- 1.Brumme ZL, Brumme CJ, Heckerman D, Korber BT, Daniels M, Carlson J, Kadie C, Bhattacharya T, Chui C, Szinger J, Mo T, Hogg RS, Montaner JS, Frahm N, Brander C, Walker BD, Harrigan PR. 2007. Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog. 3:e94. 10.1371/journal.ppat.0030094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, Swenson LC, Tao I, Szeto S, Rosato P, Sela J, Kadie CM, Frahm N, Brander C, Haas DW, Riddler SA, Haubrich R, Walker BD, Harrigan PR, Heckerman D, Mallal S. 2009. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 4:e6687. 10.1371/journal.pone.0006687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brumme ZL, Tao I, Szeto S, Brumme CJ, Carlson JM, Chan D, Kadie C, Frahm N, Brander C, Walker B, Heckerman D, Harrigan PR. 2008. Human leukocyte antigen-specific polymorphisms in HIV-1 Gag and their association with viral load in chronic untreated infection. AIDS 22:1277–1286. 10.1097/QAD.0b013e3283021a8c [DOI] [PubMed] [Google Scholar]
- 4.Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439–1443. 10.1126/science.1069660 [DOI] [PubMed] [Google Scholar]
- 5.Rolland M, Carlson JM, Manocheewa S, Swain JV, Lanxon-Cookson E, Deng W, Rousseau CM, Raugi DN, Learn GH, Maust BS, Coovadia H, Ndung'u T, Goulder PJ, Walker BD, Brander C, Heckerman DE, Mullins JI. 2010. Amino-acid co-variation in HIV-1 Gag subtype C: HLA-mediated selection pressure and compensatory dynamics. PLoS One 5:e12463. 10.1371/journal.pone.0012463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rousseau CM, Daniels MG, Carlson JM, Kadie C, Crawford H, Prendergast A, Matthews P, Payne R, Rolland M, Raugi DN, Maust BS, Learn GH, Nickle DC, Coovadia H, Ndung'u T, Frahm N, Brander C, Walker BD, Goulder PJ, Bhattacharya T, Heckerman DE, Korber BT, Mullins JI. 2008. HLA class I-driven evolution of human immunodeficiency virus type 1 subtype c proteome: immune escape and viral load. J. Virol. 82:6434–6446. 10.1128/JVI.02455-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang YE, Li B, Carlson JM, Streeck H, Gladden AD, Goodman R, Schneidewind A, Power KA, Toth I, Frahm N, Alter G, Brander C, Carrington M, Walker BD, Altfeld M, Heckerman D, Allen TM. 2009. Protective HLA class I alleles that restrict acute-phase CD8+ T-cell responses are associated with viral escape mutations located in highly conserved regions of human immunodeficiency virus type 1. J. Virol. 83:1845–1855. 10.1128/JVI.01061-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang KH, Goedhals D, Carlson JM, Brockman MA, Mishra S, Brumme ZL, Hickling S, Tang CS, Miura T, Seebregts C, Heckerman D, Ndung'u T, Walker B, Klenerman P, Steyn D, Goulder P, Phillips R, Bloemfontein-Oxford Collaborative Group. van Vuuren C, Frater J. 2011. Progression to AIDS in South Africa is associated with both reverting and compensatory viral mutations. PLoS One 6:e19018. 10.1371/journal.pone.0019018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, Kuse N, Oka S, Duda A, Prendergast A, Crawford H, Leslie A, Brumme Z, Brumme C, Allen T, Brander C, Kaslow R, Tang J, Hunter E, Allen S, Mulenga J, Branch S, Roach T, John M, Mallal S, Ogwu A, Shapiro R, Prado JG, Fidler S, Weber J, Pybus OG, Klenerman P, Ndung'u T, Phillips R, Heckerman D, Harrigan PR, Walker BD, Takiguchi M, Goulder P. 2009. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458:641–645. 10.1038/nature07746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solberg OD, Mack SJ, Lancaster AK, Single RM, Tsai Y, Sanchez-Mazas A, Thomson G. 2008. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: a meta-analytic review of 497 population studies. Hum. Immunol. 69:443–464. 10.1016/j.humimm.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong T, Zhang Y, Xu KY, Yan H, James I, Peng Y, Blais ME, Gaudieri S, Chen X, Lun W, Wu H, Qu WY, Rostron T, Li N, Mao Y, Mallal S, Xu X, McMichael A, John M, Rowland-Jones SL. 2011. Extensive HLA-driven viral diversity following a narrow-source HIV-1 outbreak in rural China. Blood 118:98–106. 10.1182/blood-2010-06-291963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lau KA, Wang B, Saksena NK. 2007. Emerging trends of HIV epidemiology in Asia. AIDS Rev. 9:218–229 [PubMed] [Google Scholar]
- 13.Rodrigo C, Rajapakse S. 2009. Current status of HIV/AIDS in South Asia. J. Glob. Infect. Dis. 1:93–101. 10.4103/0974-777X.56249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruxrungtham K, Brown T, Phanuphak P. 2004. HIV/AIDS in Asia. Lancet 364:69–82. 10.1016/S0140-6736(04)16593-8 [DOI] [PubMed] [Google Scholar]
- 15.Avila-Rios S, Ormsby CE, Carlson JM, Valenzuela-Ponce H, Blanco-Heredia J, Garrido-Rodriguez D, Garcia-Morales C, Heckerman D, Brumme ZL, Mallal S, John M, Espinosa E, Reyes-Teran G. 2009. Unique features of HLA-mediated HIV evolution in a Mexican cohort: a comparative study. Retrovirology 6:72. 10.1186/1742-4690-6-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, Chui CK, Cotton LA, Knapp DJ, Riddler SA, Haubrich R, Nelson G, Pfeifer N, Deziel CE, Heckerman D, Apps R, Carrington M, Mallal S, Harrigan PR, John M, Brumme ZL, International HIVAC 2012. Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J. Virol. 86:13202–13216. 10.1128/JVI.01998-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson JM, Listgarten J, Pfeifer N, Tan V, Kadie C, Walker BD, Ndung'u T, Shapiro R, Frater J, Brumme ZL, Goulder PJ, Heckerman D. 2012. Widespread impact of HLA restriction on immune control and escape pathways of HIV-1. J. Virol. 86:5230–5243. 10.1128/JVI.06728-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.John M, Heckerman D, James I, Park LP, Carlson JM, Chopra A, Gaudieri S, Nolan D, Haas DW, Riddler SA, Haubrich R, Mallal S. 2010. Adaptive interactions between HLA and HIV-1: highly divergent selection imposed by HLA class I molecules with common supertype motifs. J. Immunol. 184:4368–4377. 10.4049/jimmunol.0903745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leslie A, Price DA, Mkhize P, Bishop K, Rathod A, Day C, Crawford H, Honeyborne I, Asher TE, Luzzi G, Edwards A, Rousseau CM, Mullins JI, Tudor-Williams G, Novelli V, Brander C, Douek DC, Kiepiela P, Walker BD, Goulder PJ. 2006. Differential selection pressure exerted on HIV by CTL targeting identical epitopes but restricted by distinct HLA alleles from the same HLA supertype. J. Immunol. 177:4699–4708 [DOI] [PubMed] [Google Scholar]
- 20.Carlson JM, Brumme ZL, Rousseau CM, Brumme CJ, Matthews P, Kadie C, Mullins JI, Walker BD, Harrigan PR, Goulder PJ, Heckerman D. 2008. Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLoS Comput. Biol. 4:e1000225. 10.1371/journal.pcbi.1000225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlson J, Kadie C, Mallal S, Heckerman D. 2007. Leveraging hierarchical population structure in discrete association studies. PLoS One 2:e591. 10.1371/journal.pone.0000591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U. S. A. 100:9440–9445. 10.1073/pnas.1530509100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itoh Y, Mizuki N, Shimada T, Azuma F, Itakura M, Kashiwase K, Kikkawa E, Kulski JK, Satake M, Inoko H. 2005. High-throughput DNA typing of HLA-A, -B, -C, and -DRB1 loci by a PCR-SSOP-Luminex method in the Japanese population. Immunogenetics 57:717–729. 10.1007/s00251-005-0048-3 [DOI] [PubMed] [Google Scholar]
- 24.Naruto T, Gatanaga H, Nelson G, Sakai K, Carrington M, Oka S, Takiguchi M. 2012. HLA class I-mediated control of HIV-1 in the Japanese population, in which the protective HLA-B*57 and HLA-B*27 alleles are absent. J. Virol. 86:10870–10872. 10.1128/JVI.00689-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sette A, Sidney J. 1999. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics 50:201–212. 10.1007/s002510050594 [DOI] [PubMed] [Google Scholar]
- 26.Reche PA, Reinherz EL. 2007. Definition of MHC supertypes through clustering of MHC peptide-binding repertoires. Methods Mol. Biol. 409:163–173. 10.1007/978-1-60327-118-9_11 [DOI] [PubMed] [Google Scholar]
- 27.Sidney J, Southwood S, Sette A. 2005. Classification of A1- and A24-supertype molecules by analysis of their MHC-peptide binding repertoires. Immunogenetics 57:393–408. 10.1007/s00251-005-0004-2 [DOI] [PubMed] [Google Scholar]
- 28.Sidney J, Peters B, Frahm N, Brander C, Sette A. 2008. HLA class I supertypes: a revised and updated classification. BMC Immunol. 9:1. 10.1186/1471-2172-9-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kostyu DD, Hannick LI, Traweek JL, Ghanayem M, Heilpern D, Dawson DV. 1997. HLA class I polymorphism: structure and function and still questions. Hum. Immunol. 57:1–18. 10.1016/S0198-8859(97)00175-4 [DOI] [PubMed] [Google Scholar]
- 30.Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. 2000. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature 405:537–543. 10.1038/35014520 [DOI] [PubMed] [Google Scholar]
- 31.Fan QR, Wiley DC. 1999. Structure of human histocompatibility leukocyte antigen (HLA)-Cw4, a ligand for the KIR2D natural killer cell inhibitory receptor. J. Exp. Med. 190:113–123. 10.1084/jem.190.1.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L, Zimbwa P, Moore S, Allen T, Brander C, Addo MM, Altfeld M, James I, Mallal S, Bunce M, Barber LD, Szinger J, Day C, Klenerman P, Mullins J, Korber B, Coovadia HM, Walker BD, Goulder PJ. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432:769–775. 10.1038/nature03113 [DOI] [PubMed] [Google Scholar]
- 33.Fujiwara M, Tanuma J, Koizumi H, Kawashima Y, Honda K, Mastuoka-Aizawa S, Dohki S, Oka S, Takiguchi M. 2008. Different abilities of escape mutant-specific cytotoxic T cells to suppress replication of escape mutant and wild-type human immunodeficiency virus type 1 in new hosts. J. Virol. 82:138–147. 10.1128/JVI.01452-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koizumi H, Iwatani T, Tanuma J, Fujiwara M, Izumi T, Oka S, Takiguchi M. 2009. Escape mutation selected by Gag28-36-specific cytotoxic T cells in HLA-A*2402-positive HIV-1-infected donors. Microbes Infect. 11:198–204. 10.1016/j.micinf.2008.11.005 [DOI] [PubMed] [Google Scholar]
- 35.Ikeda-Moore Y, Tomiyama H, Miwa K, Oka S, Iwamoto A, Kaneko Y, Takiguchi M. 1997. Identification and characterization of multiple HLA-A24-restricted HIV-1 CTL epitopes: strong epitopes are derived from V regions of HIV-1. J. Immunol. 159:6242–6252 [PubMed] [Google Scholar]
- 36.Altfeld M, Kalife ET, Qi Y, Streeck H, Lichterfeld M, Johnston MN, Burgett N, Swartz ME, Yang A, Alter G, Yu XG, Meier A, Rockstroh JK, Allen TM, Jessen H, Rosenberg ES, Carrington M, Walker BD. 2006. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T cell response against HIV-1. PLoS Med. 3:e403. 10.1371/journal.pmed.0030403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, Desouza I, Ryvkin F, Derdeyn CA, Allen S, Hunter E, Mulenga J, Goepfert PA, Walker BD, Allen TM. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 81:12608–12618. 10.1128/JVI.01369-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao X, Bashirova A, Iversen AK, Phair J, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, Altfeld M, O'Brien SJ, Carrington M. 2005. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat. Med. 11:1290–1292. 10.1038/nm1333 [DOI] [PubMed] [Google Scholar]
- 39.Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, Conlon C, Workman C, Shaunak S, Olson K, Goulder P, Brander C, Ogg G, Sullivan JS, Dyer W, Jones I, McMichael AJ, Rowland-Jones S, Phillips RE. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375–386. 10.1084/jem.193.3.375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282–289. 10.1038/nm992 [DOI] [PubMed] [Google Scholar]
- 41.Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, Rinaldo CR, Craggs SL, Allgaier RL, Power KA, Kuntzen T, Tung CS, LaBute MX, Mueller SM, Harrer T, McMichael AJ, Goulder PJ, Aiken C, Brander C, Kelleher AD, Allen TM. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 81:12382–12393. 10.1128/JVI.01543-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chopera DR, Woodman Z, Mlisana K, Mlotshwa M, Martin DP, Seoighe C, Treurnicht F, de Rosa DA, Hide W, Karim SA, Gray CM, Williamson C, CAPRISA 002 Study Team 2008. Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog. 4:e1000033. 10.1371/journal.ppat.1000033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson JM, Derdeyn CA, Tang J, Kaslow RA, Bansal A, Yusim K, Heckerman D, Mulenga J, Allen S, Goulder PJ, Hunter E. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 205:1009–1017. 10.1084/jem.20072457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vivian JP, Duncan RC, Berry R, O'Connor GM, Reid HH, Beddoe T, Gras S, Saunders PM, Olshina MA, Widjaja JM, Harpur CM, Lin J, Maloveste SM, Price DA, Lafont BA, McVicar DW, Clements CS, Brooks AG, Rossjohn J. 2011. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature 479:401–405. 10.1038/nature10517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Apps R, Qi Y, Carlson JM, Chen H, Gao X, Thomas R, Yuki Y, Del Prete GQ, Goulder P, Brumme ZL, Brumme CJ, John M, Mallal S, Nelson G, Bosch R, Heckerman D, Stein JL, Soderberg KA, Moody MA, Denny TN, Zeng X, Fang J, Moffett A, Lifson JD, Goedert JJ, Buchbinder S, Kirk GD, Fellay J, McLaren P, Deeks SG, Pereyra F, Walker B, Michael NL, Weintrob A, Wolinsky S, Liao W, Carrington M. 2013. Influence of HLA-C expression level on HIV control. Science 340:87–91. 10.1126/science.1232685 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.