

ABSTRACT
Members of the genus Parvovirus are small, nonenveloped single-stranded DNA viruses that are nonpathogenic in humans but have potential utility as cancer therapeutics. Because the innate immune response to parvoviruses has received relatively little attention, we compared the response to parvoviruses to that of several other types of viruses in human cells. In normal human glia, fibroblasts, or melanocytes, vesicular stomatitis virus evoked robust beta interferon (IFN-β) responses. Cytomegalovirus, pseudorabies virus, and Sindbis virus all evoked a 2-log-unit or greater upregulation of IFN-β in glia; in contrast, LuIII and MVMp parvoviruses did not evoke a detectable IFN-β or interferon-stimulated gene (ISG; MX1, oligoadenylate synthetase [OAS], IFIT-1) response in the same cell types. The lack of response raised the question of whether parvoviral infection can be attenuated by IFN; interestingly, we found that IFN did not decrease parvovirus (MVMp, LuIII, and H-1) infectivity in normal human glia, fibroblasts, or melanocytes. The same was true in human cancers, including glioma, sarcoma, and melanoma. Similarly, IFN failed to attenuate transduction by the dependovirus vector adeno-associated virus type 2. Progeny production of parvoviruses was also unimpaired by IFN in both glioma and melanoma, whereas vesicular stomatitis virus replication was blocked. Sarcoma cells with upregulated IFN signaling that show high levels of resistance to other viruses showed strong infection by LuIII. Unlike many other oncolytic viruses, we found no evidence that impairment of innate immunity in cancer cells plays a role in the oncoselectivity of parvoviruses in human cells. Parvoviral resistance to the effects of IFN in cancer cells may constitute an advantage in the virotherapy of some tumors.
IMPORTANCE Understanding the interactions between oncolytic viruses and the innate immune system will facilitate employing these viruses as therapeutic agents in cancer patients. The cancer-selective nature of some oncolytic viruses is based on the impaired innate immunity of many cancer cells. The parvoviruses H-1, LuIII, and MVM target cancer cells; however, their relationship with the innate immune system is relatively uncharacterized. Surprisingly, we found that these parvoviruses do not evoke an interferon response in normal human fibroblasts, glia, or melanocytes. Furthermore, unlike most other types of virus, we found that parvovirus infectivity is unaffected by interferon treatment of human normal or tumor cells. Finally, parvoviral replication was unimpaired by interferon in four human tumor types, including those with residual interferon functionality. We conclude that deficits in the interferon antiviral response of cancer cells do not contribute to parvoviral oncoselectivity in human cells. The interferon-resistant phenotype of parvoviruses may give them an advantage over interferon-sensitive oncolytic viruses in tumors showing residual interferon functionality.
INTRODUCTION
Viruses within the genus Parvovirus (e.g., MVMp, LuIII, H-1) are nonenveloped, have a small (diameter, approximately 26 nm) icosahedral capsid, and contain a single-stranded DNA genome with telomeric hairpins (1). After binding to a sialoglycoprotein receptor(s) and subsequent endocytosis, these viruses deploy a tethered phospholipase domain of the capsid polypeptide via a pore within the capsid shell; this enables virion exit from the endosome into the cytoplasm (2). From there, a small subset of internalized virions translocates to the nucleus by mechanisms that require both microtubules (3) and the proteasome (4). Once in the nucleus, the uncoated genome waits for the cell to spontaneously enter S phase, at which point a double-stranded form of the genome that is competent to serve as a template for transcription is generated (5). The early promoter (P4) then drives expression of nonstructural (NS) proteins NS1 and NS2; NS1 transactivates the late viral promoter, driving capsid gene expression. Packaging of single-stranded genomes into intact empty capsids occurs in the nucleus, and progeny are released by exocytosis or cell lysis (1). This viral life cycle presents several potential opportunities for detection by the innate immune system.
The innate immune system recognizes moieties associated with pathogens, also known as pathogen-associated molecular patterns (PAMPs), by virtue of cognate pattern recognition receptors (PRRs) distributed throughout different regions of the cell (6). Stimulation of these receptors typically leads to secretion of type I interferons (alpha interferon [IFN-α] and IFN-β), which stimulate the type I IFN receptor (IFNAR), leading to the upregulation of a large number of interferon-stimulated genes (ISGs), many of which have direct antiviral activity (6). Innate immune detection and inhibition of parvoviruses are topics that have received relatively little attention; however, as understanding of the innate immune system has increased and as the potential utility of parvoviruses as cancer therapeutics has become increasingly supported by recent studies, the relationship of parvoviruses to the innate immune system in human cells merits greater study.
MVMp, H-1, and LuIII parvoviruses and derivatives thereof are of interest for their potential utility as cancer therapeutics both as replication-competent viruses and as replication-incompetent transgene-delivering vectors (7). Oncosuppressive efficacy with these three parvoviruses has been demonstrated in diverse in vivo models of tumors, including glioma (8, 9), pancreatic cancer (10, 11), and lymphoma (12); a clinical trial is under way for H-1 therapy of glioblastoma (ClinicalTrials.gov trial NCT01301430), and we recently reported that LuIII may hold even greater promise than H-1 for treatment of glioma in humans (8). These viruses are not associated with any pathogenicity in humans (13); in fact, their growth and toxicity are innately more selective for a variety of oncogenically transformed human cells than for their normal precursors (14). The mechanisms of this oncoselectivity have been partly elucidated and include a Ras-responsive early viral promoter (P4) (15, 16). An additional possible mechanism, which is important to the oncoselectivity of certain other oncolytic viruses (17), relates to defects in innate immunity, often present in cancer cells (18–20), which may selectively disinhibit parvoviral replication in tumor cells. This possibility has found some support in murine models of transformation but has not yet found support in human models; two separate groups have demonstrated that murine embryonic fibroblasts (MEFs), but not transformed murine cells, can mount a type I IFN response to parvoviral infection (21, 22). Among human cells, however, peripheral blood mononuclear cells (PBMCs) alone have been demonstrated to produce an IFN response to parvovirus infection (23).
Vesicular stomatitis virus (VSV), a negative-stranded RNA virus, is sensitive to the effects of interferon, which VSV effectively triggers in most normal human cells. In transformed human cells, VSV often fails to trigger this response or produces a response that is ineffective against infection (24). This results in the selective growth of VSV in most transformed human tumors compared to in their untransformed normal cell counterparts of the same tissue type (25–28). Other oncolytic viruses whose oncoselectivity depends, at least in part, on disruption of innate immunity in transformed cells include Newcastle disease virus (NDV) and myxoma virus (29).
To examine the ability of parvoviruses to stimulate the innate immune system, we infected normal human glial cells, melanocytes, and fibroblasts with MVMp, H-1, and LuIII; we detected no upregulation of IFN-β or ISG expression in any of these cells; in contrast, normal glia mounted a strong IFN response against nonrelated cytomegalovirus (CMV), pseudorabies virus (PRV), Sindbis virus, and VSV. Furthermore, exposure of normal human cell types to exogenous interferon evoked no attenuation of parvoviral infection. In transformed human melanoma, sarcoma, and glioma, IFN had different degrees of effectiveness at blocking VSV infection; however, in no case did IFN protect these cells against infection by parvovirus. Parvoviral progeny production was also unaffected by IFN pretreatment in human glioma and melanoma lines. Overall, our findings argue against the relevance of innate immunity to the phenomenon of parvoviral oncoselectivity in most human cells. Furthermore, although the apparent lack of parvoviral interaction with the innate immune system in human cells may have exceptions, our results demonstrate that in human tumors with residual IFN pathway function, the ability of parvoviruses to replicate irrespective of the presence of interferon may constitute an advantage in their use as therapeutic agents compared to the use of IFN-sensitive viruses.
MATERIALS AND METHODS
Cell culture.
Normal human glial cell cultures, isolated through explant cultures and tested for immunoreactivity to glial fibrillary acidic protein (GFAP; specific for astrocytes rather than hematopoietic microglia), have been described previously (27). Normal human fibroblasts were purchased from Cambrex (Walkersville, MD). SW-R and SW-S are single-cell-derived subclones of human synovial sarcoma SW-982 which are VSV resistant and VSV susceptible, respectively, and were produced and characterized earlier (28). U87 and A172 are human glioma cell lines originally obtained from ATCC (Manassas, VA) (8). The preceding cells were all cultured in Dulbecco modified Eagle medium with 10% fetal bovine serum (FBS), penicillin-streptomycin, nonessential amino acids, and 25 mM HEPES. Normal human melanocytes were obtained from the Specimen Research Core of the Yale Specialized Program of Research Excellence (SPORE) and were derived from newborn foreskin tissue as described previously (30) and maintained as a primary culture in Opti-MEM (Life Technologies, Grand Island, NY) supplemented with 5% FBS, penicillin-streptomycin, basic fibroblast growth factor (10 ng/ml; ConnStem, Cheshire, CT), heparin (1 ng/ml; Sigma, St. Louis, MO), dibutyryl cyclic AMP (100 μM; Sigma), and 1-methyl-3-(2-methylpropyl)-7H-purine-2,6-dione (100 μM; Sigma). YUSIV, YUSIK, and YURIF are primary human melanoma cultures obtained from the Yale SPORE in Skin Cancer and cultured in Opti-MEM with 5% FBS and penicillin-streptomycin. Frozen normal human PBMCs (hPBMCs) were obtained from Stem Cell Technologies (Vancouver, Canada), recovered according to the manufacturer's protocol, and cultured in RPMI (Gibco) with 10% heat-inactivated FBS and 1% penicillin-streptomycin. All cells were incubated in a humidified atmosphere containing 5% CO2 at 37°C.
Viruses.
Parvoviruses MVMp, LuIII, and H-1, as previously employed (8), were grown on HeLa cells, and plaque titers were determined on 324K transformed human kidney fibroblasts (a gift from Peter Tattersall, Yale University [31]), using neutral red stain to visualize plaques at 6 days postinfection (dpi). VSV-G/GFP, a vesicular stomatitis virus expressing a G glycoprotein-green fluorescent protein (GFP) fusion reporter protein (32), was grown on BHK cells, and titers were determined using fluorescence microscopy to visualize plaques at 24 h postinfection (hpi). Sindbis virus expressing GFP (32) was also grown on BHK cells, and titers were determined by visualizing GFP at 24 hpi. Human cytomegalovirus (hCMV) expressing GFP (32) was grown on BJ human primary foreskin fibroblasts (a gift from Peter Tattersall originally obtained from ATCC [33]), titers were determined, and GFP-positive plaques were visualized at 6 dpi. Pseudorabies virus (PRV) expressing red fluorescent protein (RFP) (32) was grown on porcine PK15 cells, titers were determined, and RFP-positive plaques were visualized at 24 hpi.
scAAV-tdTomato.
A self-complementary (sc) adeno-associated virus type 2 (AAV-2) plasmid featuring a mutation in one inverted terminal repeat (ITR) to allow packaging of a palindromic self-complementary genome (34) was a gift from Reed Clark (Nationwide Children's Hospital, Columbus, OH). tdTomato, a red fluorophore, was cloned from pCMV-tdTomato (Clontech) into the scAAV-2 plasmid in front of the CMV promoter and between the ITRs. This plasmid was used by the Viral Vector Core at Nationwide Children's Hospital (Columbus, OH) to generate recombinant AAV vector and determine the titer of the genome.
Host cell gene expression analysis by qPCR.
Triplicate wells were seeded at 2.0E5 to 2.5E5 cells/well (fibroblasts and glia), 4E5 cells/well (melanocytes), or 3E6 cells/well (hPBMCs) and then infected with each virus at the multiplicity of infection (MOI) specified in the text. RNA was extracted at the indicated time points postinfection using an RNeasy+ minikit (Qiagen, Hilden, Germany) and reverse transcribed with random hexamer priming using a High-Capacity cDNA reverse transcription kit (Life Technologies). TaqMan gene expression assays (Life Technologies) for human IFN-β, MX1, oligoadenylate synthetase (OAS), IFIT-1, protein kinase R (PKR), and β-actin were acquired and employed as recommended by the manufacturer (Life Technologies). Quantitative PCRs (qPCRs) were run on an iCycler-IQ instrument (Bio-Rad, Hercules, CA), and results were analyzed with iCycler software. Each individual sample was assessed in triplicate PCRs with 20-μl reaction mixtures employing the iTaq Universal Probes Supermix (Bio-Rad); results were internally normalized to β-actin expression levels in that sample, also measured in triplicate.
Analysis of infectivity by immunofluorescence.
Cultured cells in triplicate wells were infected at the multiplicity indicated in the text. At 24 hpi, cells infected with parvovirus LuIII, MVMp, or H-1 were fixed in 2% paraformaldehyde (20 min), rinsed with phosphate-buffered saline (PBS), permeabilized by washing 5 times for 15 min each time in PBS with 0.1% l-lysine, 1% bovine serum albumin, 0.4% Triton-X, blocked in washing buffer plus 2% normal horse serum (NHS), and exposed to primary antibody in blocking solution. Primary antibody was rabbit polyclonal antibody directed against viral NS proteins (35). Cells were rinsed in PBS 3 times for 15 min each time, exposed to donkey antirabbit antibody conjugated to Alexa Fluor 488 (Invitrogen, Carlsbad, CA) in PBS with 0.4% Triton-X, and washed in PBS. The infectivity of VSV and AAV-T (an AAV-2-encapsidated vector that we engineered and that encodes the fluorescent marker tdTomato under the control of the CMV promoter) was determined at specified time points by assessing the percentage of cells expressing fluorescent GFP or tdTomato, respectively. Approximately 500 cells were analyzed in each well. Phase-contrast and fluorescent images were captured with an Olympus Optical IX71 fluorescence microscope (Tokyo, Japan) with a SPOT RT camera (Diagnostic Instruments, Sterling Heights, MI).
Analysis of replication.
A total of 100,000 cells (A172, U87, YUSIK, or YURIF) were seeded per well in triplicate wells and infected the next day with LuIII or VSV at an MOI of 1.0 PFU/cell. At 24 hpi, supernatants of VSV-infected cells were harvested for analysis of the titer on BHK cells. At 72 hpi, supernatants of LuIII-infected cells were harvested, and cells were scraped into 150 μl of 50 mM Tris, 0.5 mM EDTA, pH 8.7. Cell harvests were subjected to three rounds of freeze-thawing (in a dry ice-ethanol bath and room temperature), followed by centrifugation to remove debris. Ten percent of these cellular viral extracts were combined with 10% of the supernatant from the same well to generate from each well a representative sample of parvoviral progeny, whose plaque titers were determined on 324K cells as described above.
RESULTS
Parvovirus infection does not induce an IFN-β response in normal human glia, fibroblasts, or melanocytes.
Cellular detection of a diverse number of viruses results in upregulation of the expression of type I IFNs, specifically, IFN-β (an exception is plasmacytoid dendritic cells [pDCs], which typically upregulate IFN-α) (6, 36). Secreted IFN-β acts in an autocrine or paracrine manner to activate type I IFN receptors on the cell surface, which in turn upregulate the expression of interferon-stimulated genes (ISGs); many of these genes have direct antiviral effects. We tested the ability of oncolytic parvoviruses MVMp and LuIII to upregulate the levels of mRNA encoding IFN-β and MX1, which is among the most strongly upregulated ISGs (37), in three different normal primary human cells. Primary human glia, fibroblasts, and melanocytes were infected (MOI, 5 PFU/cell) with parvoviruses MVMp and LuIII. Control cells were infected with VSV as a positive-control virus known to produce robust IFN responses (38). RNA harvested at 36 hpi from triplicate wells was subjected to reverse transcription and qPCR. As substantial cell death is found 48 h after inoculation with parvoviruses (12, 14), to avoid complications in interpretation we focused on infection periods of shorter duration. The mRNA levels measured for uninfected and infected cells relative to the β-actin levels for both cell types are shown in Fig. 1. VSV induced robust IFN and ISG responses in glia (a >1,900-fold increase in IFN-β, a >600-fold increase in MX1), fibroblasts (a >900-fold increase in IFN-β, a >40-fold increase in MX1), and melanocytes (a >180-fold increase in IFN-β, a >6-fold increase in MX1). As an additional positive control, we verified that MVMp (5 PFU/cell) was capable of producing an IFN response in hPBMCs, as has been reported (23). We found a modest statistically significant 4.5-fold elevation of IFN-β levels at 24 hpi with MVMp (P < 0.05). In contrast, in all instances, infection by LuIII or MVMp failed to alter IFN-β levels or MX1 concentrations from those observed in uninfected glia, fibroblasts, and melanocytes (all comparisons were nonsignificant by analysis of variance [ANOVA], and the level of IFN-β mRNA was below the detection limit for both uninfected and parvovirus-infected glia). Thus, in three varieties of normal primary human cells, we found no evidence for parvovirus LuIII or MVMp stimulating a canonical IFN response.
FIG 1.

No IFN response to parvoviral infection in three normal human cell types. Normal human glia (A), human fibroblasts (B), or human melanocytes (C) were left uninfected (Uninf.) or were infected with the indicated viruses at 5 PFU/cell. mRNA levels for IFN-β and for the interferon-stimulated gene MX1 were determined for samples taken at 36 hpi, and the ratio of the IFN-β and MX-1 mRNA levels to the level of β-actin expression was determined. n.d., none detected; n.s., not significant by ANOVA. Error bars, SEMs.
Levels of IFN-β and four ISGs are unaltered in human glia after LuIII parvovirus infection.
In the experiment described above, IFN-β mRNA levels were below the qPCR detection limit in both uninfected and LuIII-infected human glia, leaving open the possibility that the levels had been altered to some degree by infection. We repeated this part of the experiment on a larger scale, allowing us to measure the level of IFN-β in uninfected cells and thereby calculate the fold change induced by infection. We were also able to measure the levels of additional ISGs, including OAS, IFIT-1, and PKR, to assess more carefully the possibility of downstream effects on ISG expression that any enhancement of IFN-β secretion, even minimal, may stimulate. Human glia from triplicate cultures, uninfected or infected with LuIII or VSV at an MOI of 5 PFU/cell, were harvested at 24 hpi, and the levels were normalized to those of β-actin; the fold induction for each gene relative to that for uninfected cells appears in Fig. 2. For all five genes tested, the mRNA levels in LuIII-infected cells did not differ significantly from the levels in uninfected cells (by ANOVA); thus, all fold change ratios were very close to 1.0 (10°). These data are evidence for the absence of any IFN response to LuIII infection in glia. In contrast, VSV infection induced dramatic changes in the IFN-β level (>10,000-fold increase), robust increases in MX1, OAS, and IFIT-1 expression levels (over 100-fold increases), and a modest statistically significant 5-fold increase in PKR expression at 18 hpi.
FIG 2.
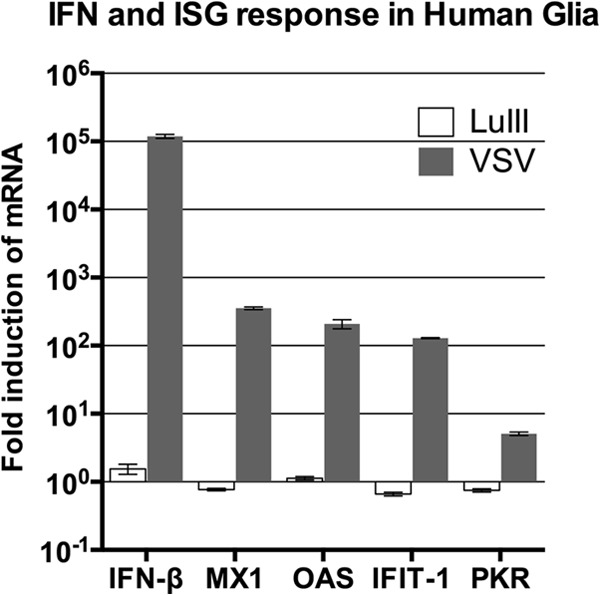
Detailed analysis of ISG response to infection of human glia. Cells were left uninfected or were infected with LuIII or VSV at 5 PFU/cell. At 18 hpi (VSV) and 24 hpi (LuIII), cells were harvested for analysis of mRNA levels of IFN-β and of the interferon-stimulated genes MX1, OAS, IFIT1, and PKR. All values were normalized to those for β-actin and are expressed as the fold induction over that for uninfected cells. Error bars, SEMs.
Infection of human glia with LuIII at a high MOI, in contrast to infection with other viruses, does not stimulate IFN-β expression.
There are a variety of intracellular virus-sensing proteins for different classes of virus (39). To test if human glial cells retain an intact viral detection capability across a panel of viruses with different structures, we infected triplicate wells with 3 viruses, in addition to parvovirus LuIII (a nonenveloped single-stranded DNA virus) and positive-control rhabdovirus VSV (an enveloped negative-stranded RNA virus): herpesviruses hCMV and PRV (both enveloped, double-stranded DNA viruses) and the alphavirus Sindbis virus (a nonenveloped positive-stranded RNA virus). The IFN-β mRNA levels at 24 hpi are shown in Fig. 3. hCMV and PRV infection both resulted in greater than 100-fold increases in IFN-β levels; Sindbis virus infection boosted IFN-β levels over 1,000-fold, and VSV infection increased IFN-β levels over 100,000-fold. Still, IFN-β mRNA levels were not significantly different (ANOVA) between uninfected cells and cells infected with LuIII, even at the higher MOI used in this experiment (10 PFU/cell, equal to approximately 7,000 genomes/cell). Thus, despite the ability to respond to a variety of viruses by upregulating IFN-β, normal human glia showed no increase in IFN-β levels even after infection with parvovirus LuIII at a high MOI.
FIG 3.
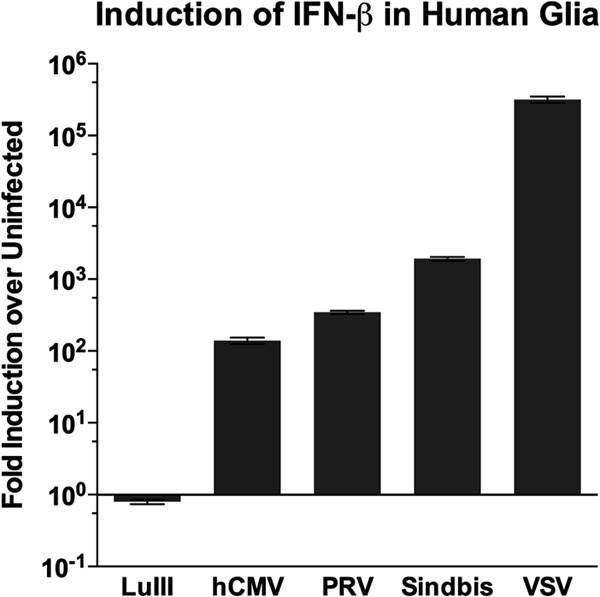
High-MOI LuIII parvovirus infection (10 PFU/cell) evokes no IFN response in human glial cells responsive to 4 unrelated viruses. Normal human glial cells were uninfected or were infected at 10 PFU/cell with LuIII, hCMV, PRV, Sindbis virus, or VSV. At 24 hpi, RNA was harvested and the IFN-β mRNA level was determined and normalized to the β-actin mRNA level. For each virus, the fold induction of IFN-β relative to that for uninfected cells is shown. Error bars, SEMs.
Exogenous IFN preexposure does not protect human glia, fibroblasts, or melanocytes from parvoviral infection.
The preceding experiments demonstrated the inefficacy of parvoviruses at stimulating an IFN response in a variety of normal untransformed human cells. We next sought to assess the degree to which IFN pretreatment of cells with intact IFN signaling pathways can block parvoviral infection. This is physiologically relevant because in vivo, cells may be exposed to exogenous, paracrine IFN secreted by other cells (such as leukocytes) even if they themselves do not secrete IFN in response to infection.
To determine if IFN preexposure protected cells from parvoviral infection, we initially compared the infectivity of three parvoviruses (LuIII, MVMp, and H-1) in IFN-unexposed versus IFN-exposed cells by measuring the percentage of cells infected at 24 hpi. Normal human glia and fibroblasts in triplicate wells were pretreated (or not) with universal IFN-αA/D (100 U/ml for 4 h), then infected with H-1, LuIII, or MVMp (5 PFU/cell), and then fixed at 24 hpi and immunostained for early parvoviral protein NS1; the IFN-sensitive virus VSV (encoding the GFP reporter) was used as a control for the efficacy of IFN exposure of these cells at stimulating an effective antiviral response. Results and representative micrographs for fibroblasts are shown in Fig. 4. Whereas IFN pretreatment abolished VSV infectivity at 24 hpi in both cell types, the infectivity of LuIII, MVM, and H-1 was unaffected by IFN pretreatment; no significant difference in the percentage of cells infected was observed in any case (ANOVA). Similarly, in normal human melanocytes, the infectivity of H-1 was unaffected by IFN pretreatment (data not shown). These results are consistent with the view that in these normal human cells, stimulation of the type I IFN receptor and the resulting antiviral ISG response, despite being highly protective against VSV infectivity, have no inhibitory effect on the early steps of parvoviral infection up to and including early gene expression.
FIG 4.

Parvoviral infection of normal cells with and without IFN preexposure. Normal human glia (A) and fibroblasts (B and C) were left untreated or were preexposed to exogenous IFN. Cells were infected with LuIII, MVMp, H-1, or VSV at 5 PFU/cell and fixed at 24 hpi. VSV was visualized by use of a GFP reporter, and parvovirus-infected cells were visualized by NS1 immunostaining. Error bars, SEMs. * and n.s., significant and nonsignificant, respectively, by ANOVA.
LuIII preinfection of normal human cells does not affect subsequent infection with VSV.
The results presented above suggest either that parvoviruses fail to be recognized by PRRs in these cells or that they are recognized but somehow interdict signaling that would normally lead to subsequent IFN and ISG expression. If the parvoviruses blocked IFN responses generally, we might find that preinfection of cells with a parvovirus could enhance infection by highly IFN-sensitive viruses, such as VSV. Therefore, we tested if parvovirus preinfection facilitated subsequent infection with VSV, in which restriction in normal human cells is IFN mediated. However, when we preinfected normal human fibroblasts and glia with LuIII at 10 PFU/cell for 24 h and then infected them with VSV, we could detect no significant effect of LuIII preinfection on VSV infectivity at 10 hpi (Fig. 5). These results suggest that parvoviruses do not block IFN responses but, rather, escape detection.
FIG 5.
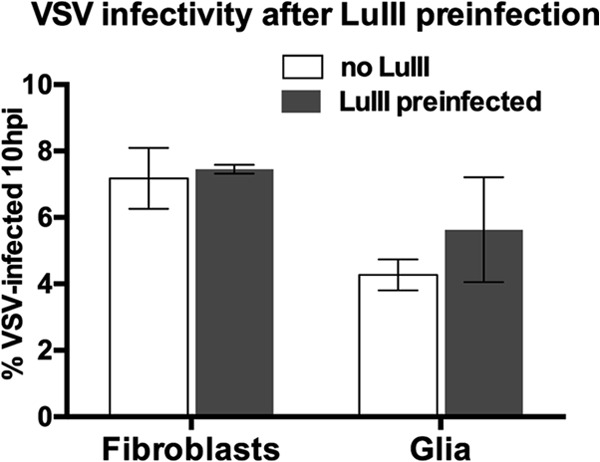
Preinfection with LuIII for 24 h does not diminish VSV infectivity. Human fibroblasts or glia were LuIII preinfected (10 PFU/cell) or not for 24 h and then inoculated with VSV. The percentage of GFP-positive (VSV-infected) cells was determined at 10 hpi. Error bars, SEMs.
A human tumor line with endogenously active IFN signaling is resistant to VSV but not to parvoviruses.
We next extended our analysis of parvoviral sensitivity to the effects of exogenous IFN in oncogenically transformed cells. Initially, we employed two cell lines subcloned from human synovial sarcoma SW982 that we previously isolated and characterized: SW-R and SW-S (28). The VSV-resistant SW-R cells have endogenously active IFN signaling and upregulated ISG expression, whereas cells of the VSV-susceptible SW-S line have a constitutively low level of expression of ISGs which can be upregulated by exogenous IFN. SW-R cells were found to be fully resistant to VSV as well as to Sindbis virus and hCMV (28).
In triplicate cultures of untreated SW-R cells, untreated SW-S cells, and SW-S cells pretreated with universal IFN-αA/D (100 U/ml for 4 h), we measured infectivity at 24 hpi for VSV, LuIII, and a parvovirus-based vector, AAV-T. AAV-T is an AAV-2-encapsidated vector that we engineered and that encodes the fluorescent marker tdTomato under the control of the CMV promoter (see Materials and Methods). Adeno-associated viruses, parvoviruses of the Dependovirus genus, have a genomic strategy and life cycle very similar to those of members of the related Parvovirus genus (MVM, LuIII, H-1); thus, we would anticipate the interaction of these viruses with IFN pathways to have similarities. Untreated SW-S cells, susceptible to VSV infection, were rendered highly VSV resistant by pretreatment with IFN, consistent with an intact ISG response. In contrast, IFN pretreatment of SW-S cells had no significant effect on the infectivity of LuIII or on that of AAV-T. Importantly, SW-R cells with constitutive ISG upregulation, despite being highly VSV resistant, had the same susceptibility to LuIII or AAV-T as their VSV-susceptible SW-S cell counterparts (Fig. 6A and B). These data present complementary lines of evidence that the presence of an otherwise effective IFN-stimulated antiviral state does not protect these transformed cells from parvoviral infection.
FIG 6.
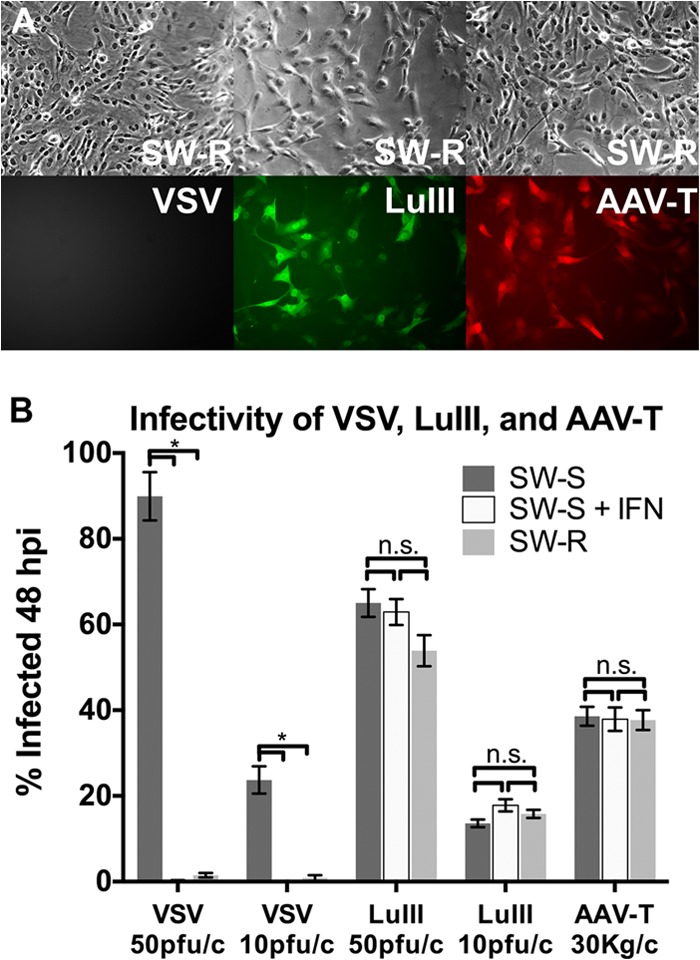
Infectivity of parvovirus LuIII and parvoviral vector AAV-T are unimpaired in human sarcoma cells that are protected from VSV by either intrinsic IFN (SW-R) or extrinsic IFN (SW-S + IFN). (A) Micrographs show SW-R cells, cells of an IFN-secreting cell line that is highly VSV resistant but susceptible to parvoviruses LuIII and AAV-T. (B) Measured infectivity of VSV, LuIII, and AAV-T in VSV-sensitive sarcoma SW-S cells either with IFN (+IFN) or without IFN pretreatment and in VSV-resistant sarcoma SW-R cells. n.s., not significant; *, significant by ANOVA. Error bars, SEMs; pfu/c, number of PFU per cell; Kg/c, number of kilogenomes per cell.
Oncogenically transformed cells protected from VSV by IFN are not protected from parvovirus infection by IFN.
The oncoselectivity of VSV is largely a function of the impaired IFN signaling of many oncogenically transformed cells (25, 40). However, the antiviral response machinery retains various degrees of functionality among different tumor cells, resulting in different degrees of VSV susceptibility. To determine if this variability in IFN functionality among tumors also influences susceptibility to parvoviral infection, we selected a human melanoma line (YUSIV) that can be protected from VSV by IFN (IFN responsive) and a human glioma line (U87) that is minimally protected from VSV by IFN (IFN resistant). IFN-pretreated cells (exposed to IFN-αA/D at 200 U/ml for 6 h) or untreated cells were infected with VSV, LuIII, or AAV-T, and the percentage of infected cells was determined at 24 hpi; results are shown in Fig. 7. IFN pretreatment completely abrogated VSV infection in the YUSIV melanoma, showing that this tumor retains a degree of IFN pathway functionality. YUSIV was not, however, protected from LuIII or AAV-T infection by IFN pretreatment (Fig. 7A and B). Human glioma U87, in contrast, was not protected from VSV infection by IFN pretreatment, as expected. In U87, as in YUSIV, the infectivity of LuIII and AAV-T was unaffected by IFN pretreatment (Fig. 7C). These results are consistent with the idea that impairment of the antiviral response in transformed cells is not necessary for optimal parvoviral infectivity.
FIG 7.
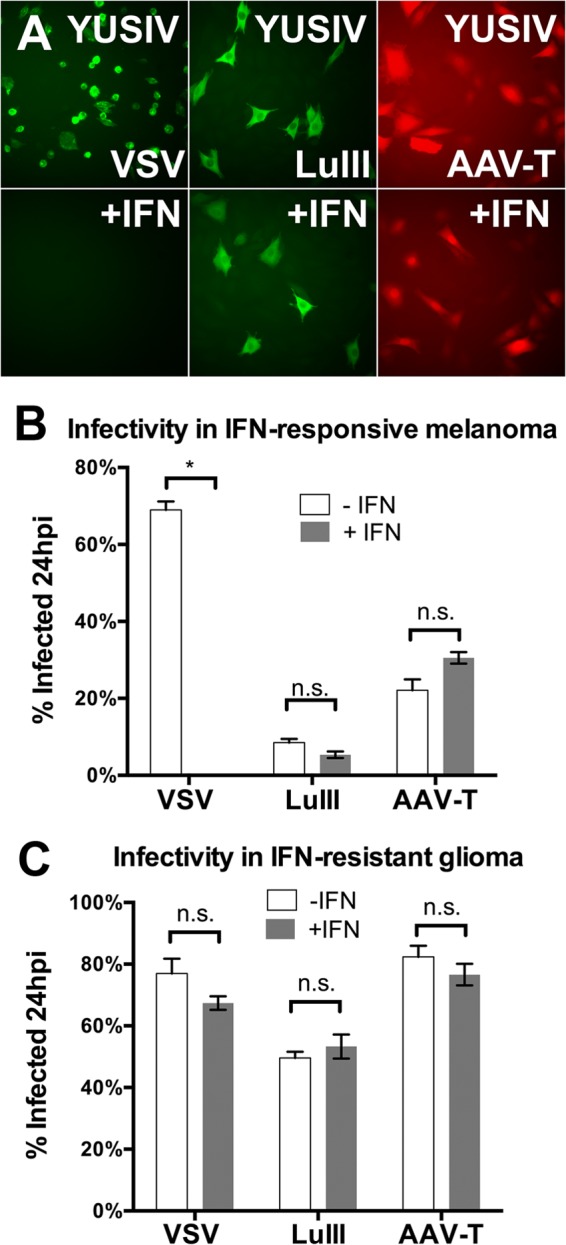
The infectivity of parvoviruses LuIII and AAV-T is unaffected by IFN in human tumors regardless of IFN status. IFN pretreatment protected human melanoma YUSIV cells (A and B) (5 PFU/cell) but not human glioma U87 cells (C) from VSV. The infectivity of LuIII (20 PFU/cell) and AAV (20,000 genomes/cell) was unaffected by IFN pretreatment in both tumor types (B and C). Error bars, SEMs. * and n.s., significant and nonsignificant, respectively, by ANOVA.
Parvoviruses MVMp, LuIII, and H-1 demonstrate oncoselectivity independently of the effects of IFN.
In addition to VSV, there are a number of other oncolytic viruses that are IFN susceptible, whether innately (e.g., Newcastle disease virus [17, 41]) or by design (e.g., attenuated measles virus [17]), allowing the targeting of cancers with IFN signaling deficiencies. This raises the question of whether IFN signaling impairment plays a role in the oncoselectivity of parvoviruses as well.
To address this question in human cells, we used normal human glia and human glioma U87; normal glial cells are IFN responsive, and U87 gliomas are IFN nonresponsive, at least with respect to VSV, as assessed by the effect of exogenous IFN on VSV infectivity (Fig. 4 and 7). We tested the three most commonly studied oncolytic parvoviruses, H-1, MVM, and LuIII, for their infectivity at 24 hpi in these two cell types with and without IFN pretreatment (100 U for 4 h). Results are shown in Fig. 8A and are replotted in Fig. 8B. Fig. 8A shows that IFN pretreatment had no significant effect on parvoviral infectivity for parvovirus MVMp, H-1, or LuIII in either cell type. The replot of the data in Fig. 8B makes clear that a significant infectivity preference for U87 over normal glia, i.e., oncoselectivity, was demonstrated by all parvoviruses. The magnitude of this oncoselectivity would be enhanced by IFN pretreatment if IFN pathways had inhibited parvovirus infection in normal cells but not in transformed cells; instead, the magnitude of oncoselectivity was not significantly affected when cells were pretreated with IFN. Thus, these data are consistent with the possibility that any differential IFN responsiveness between normal and transformed cells is not relevant to oncoselective parvoviral infectivity.
FIG 8.
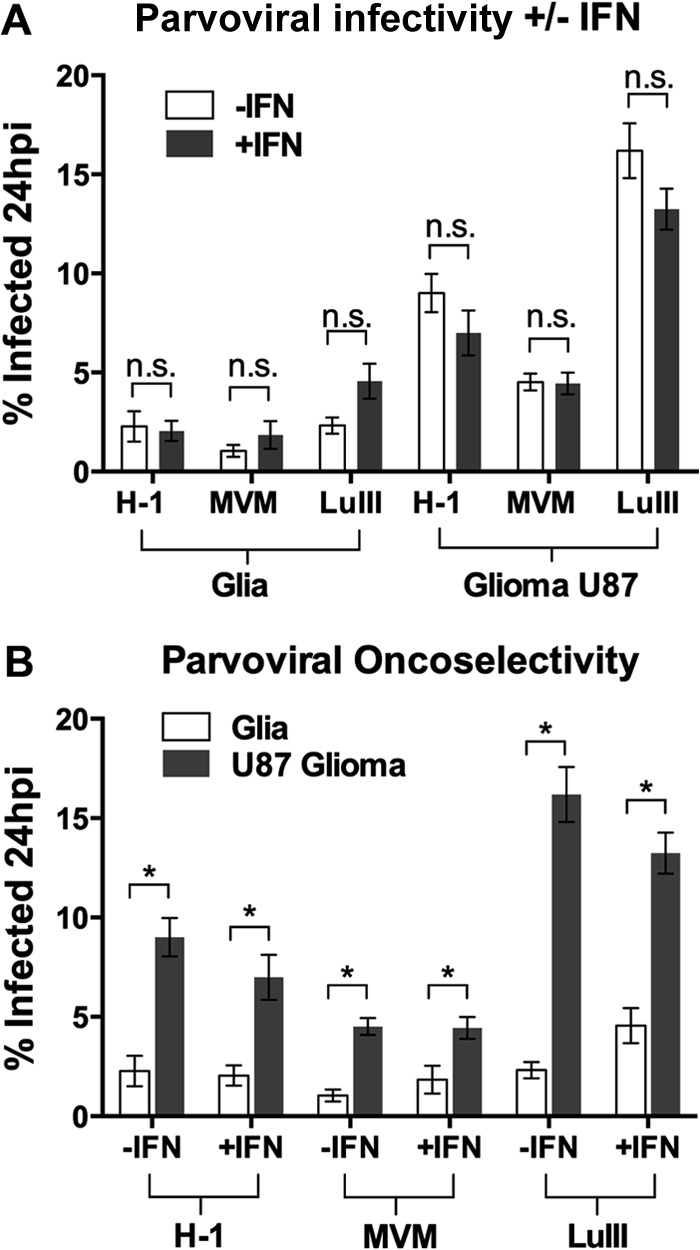
Parvoviral selectivity for transformed glioma over normal human glia is unaffected by IFN pretreatment. Human glia or glioma U87 cells were left untreated (−IFN) or were pretreated with interferon (+IFN; 100 U/ml for 4 h) and then infected with parvovirus H-1, MVM, or LuIII at 0.1 PFU/cell. The percentage of infected cells was determined at 24 hpi. The data are plotted in two formats: to illustrate that in no circumstance did IFN pretreatment affect infectivity (A) and to illustrate that the oncoselectivity of all three parvoviruses was unaltered by IFN pretreatment (B). Error bars, SEMs. * and n.s., significant and nonsignificant, respectively, by ANOVA.
IFN preexposure in tumors with intact IFN signaling does not diminish multistep parvoviral growth.
Our infectivity data show that the initial steps of parvoviral infection up to and including early viral gene expression are not affected by preinfection upregulation of IFN-stimulated pathways in a variety of normal and transformed human cells. We next tested if subsequent events in the parvoviral life cycle (genome replication, packaging, and export, culminating in viral progeny) might be impaired in human glioma and melanoma by preexposure to IFN.
To assess the efficiency of the parvoviral life cycle overall, we determined the titers of LuIII progeny (harvested from cells and the supernatant and combined in proportion) at 72 hpi. In parallel infections with VSV, the titers of progeny harvested at 24 hpi (from supernatants) were determined as an indicator of the integrity of the IFN antiviral response in these cells. Human melanoma-derived cell lines YUSIK and YURIF, as well as human glioma-derived cell lines U87 and A172, were all tested in triplicate; results are shown in Fig. 9. The production of VSV progeny titers at 24 hpi was reduced by IFN pretreatment in all four tumors, showing evidence of some degree of antiviral effector capacity in these cells; notably, the reduction in titer varied significantly among cells: >10,000-fold in YURIF cells, 5,000-fold in YUSIK cells, 1,400-fold in A172 cells, and 30-fold in U87 cells. The relatively small effect of IFN against VSV replication in U87 cells is consistent with previously reported findings (42), as well as with the finding that the IFN response in U87 cells is ineffective against the initiation of VSV infection (Fig. 7C). In all cell types, regardless of these variations in their capacity for an antiviral response, LuIII replication efficiency was unaffected by IFN pretreatment; at 72 hpi, progeny titers were not significantly different from those for untreated cells in any case (Fig. 9A). Together, these data are consistent with the idea that neither the early nor the later steps of the parvoviral life cycle are inhibited by IFN-mediated upregulation of antiviral effectors in these human tumor cell lines, all of which are competent for IFN-mediated inhibition of VSV replication to some degree. This supports the notion that, as an oncolytic virus, LuIII may have an advantage over VSV and other IFN-sensitive viruses in tumors that retain residual IFN functionality.
FIG 9.
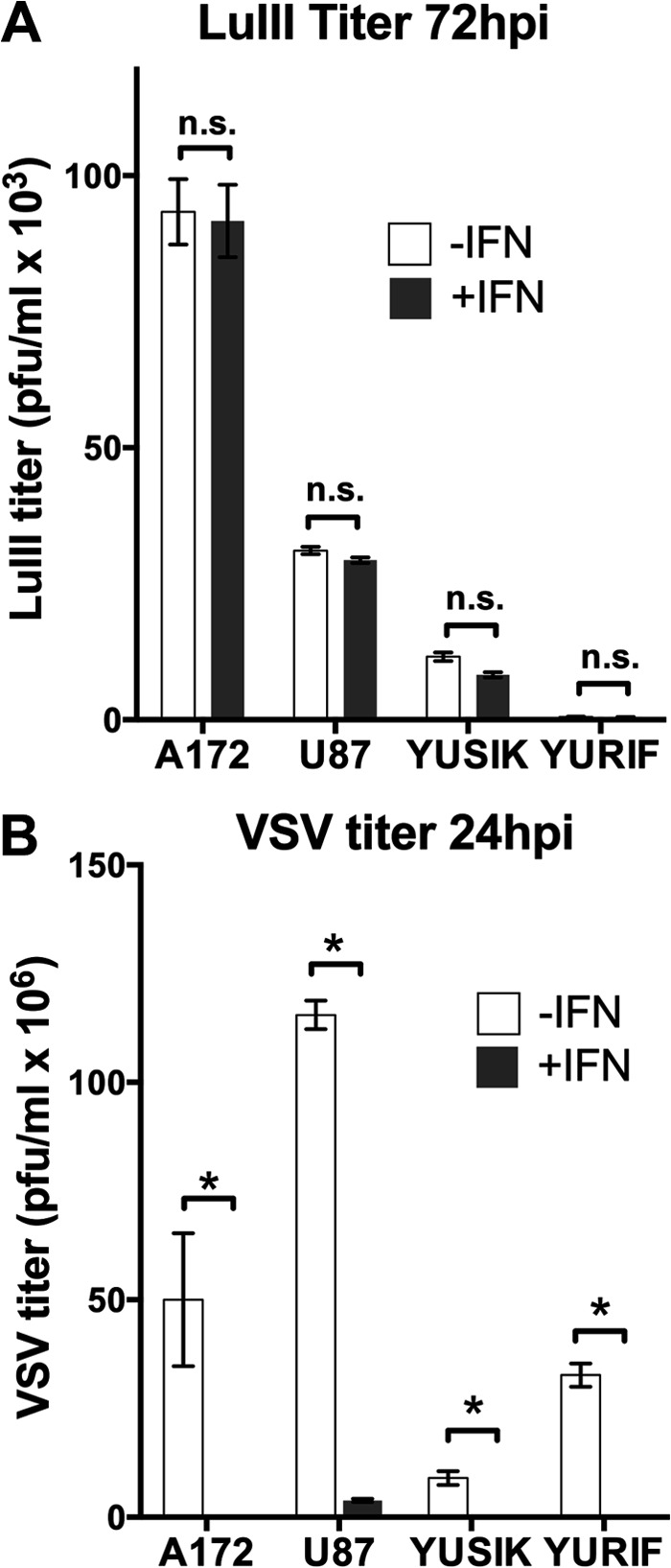
Parvovirus LuIII replication is not blocked by IFN pretreatment in a variety of human tumor-derived cell lines. Human gliomas (A172 and U87) and human melanomas (YUSIK and YURIF) were infected at 1 PFU/cell with LuIII (A) or VSV (B) either without IFN (−IFN) or with IFN (+IFN) pretreatment (100 U/ml for 4 h). At 24 hpi, supernatant from VSV-infected cells was harvested for determination of the titer of the VSV progeny (B). At 72 hpi, cells and supernatant from LuIII-infected cells were harvested for determination of the titer of LuIII progeny (A). Error bars, SEMs. * and n.s., significant and nonsignificant, respectively, by ANOVA.
DISCUSSION
In normal human glia, fibroblasts, and melanocytes, infection with parvoviruses LuIII and MVMp produced no measurable induction of IFN or ISGs; in contrast, other viruses that we tested generated robust IFN/ISG responses in these cells. Furthermore, exogenous IFN exerted no inhibitory action against infection by LuIII, MVM, or H1; this was the case in normal human cells as well as in human cancer cells, including glioma, sarcoma, and melanoma. This parvoviral resistance to IFN was particularly remarkable in light of the capacity of all the same cells to mount an IFN-mediated response that dramatically blocked VSV infection. Not only was the initiation of parvoviral infection unaffected by IFN exposure, but also parvoviral progeny production was unaffected in four cell types in which IFN was able to block VSV replication. Thus, after testing 10 different cell types, including normal and cancer cells with four different parvoviruses, we found no stimulation of an IFN response and no detectable antiparvoviral effect of IFN. This suggests that, in general, a wide variety of human cells neither respond to nor block parvovirus infection via IFN-mediated innate immunity.
Attachment of many parvoviruses to cells, resulting in endocytosis, is mediated by sialoglycoproteins abundant on most mammalian cells (1, 43), whereas host cell specificity is mediated by subsequent intracellular factors (33, 44). Mammalian cells exposed to high concentrations of parvoviruses internalize a significant proportion of the inoculum (between 25 and 50%) (33), representing an opportunity for recognition by the innate immune system. However, we found no evidence of such recognition in normal human nonimmune cells. The particle-to-infectivity ratio for parvoviruses is very high, approximately 700 genome-containing virions per PFU, on average (45), so that the multiplicities of purified virus that we employed for infection, 5 PFU/cell (Fig. 1 and 2) and 10 PFU/cell (Fig. 3), represent approximately 3,500 and 7,000 virions per cell, respectively. This high number of virions would seem to represent an amount sufficient to allow detection by the innate immune system. The time points that we chose for analysis of gene expression were based on the IFN responses to parvovirus infection observed from 24 to 36 h in hPBMCs (23) and MEFs (21).
An absence of an IFN response to parvoviruses was reported in some murine cells, including A9 transformed murine fibroblasts (21) and murine plasmacytoid dendritic cells (46). Our study may be the first to investigate the IFN response to autonomous parvoviruses in a variety of normal human cells. A subset of normal cells can mount an IFN response to parvoviruses, specifically, murine embryonic fibroblasts (21, 22) and hPBMCs (23). hPBMCs produced an increase in IFN-β transcript levels at 24 h postinfection with MVMp (23), data which we corroborated.
We did not find evidence for any ability of parvoviruses to block the IFN response that is triggered rapidly by VSV infection; 24 h of preinfection of fibroblasts or glia with LuIII did not affect subsequent VSV infectivity. These data argue in favor of parvoviruses evading detection by PRRs that stimulate IFN, rather than blocking the signaling downstream of PRRs that trigger IFN expression. There is some evidence that this evasion may be capsid mediated; DNA genome-containing virions can be translocated to the nucleus intact (1); thus, it may be that genome uncoating is an event typically restricted to the nucleus, as opposed to the endosomes or cytoplasm where DNA-sensing molecules of the innate immune system reside (47). In hPBMCs, which did respond to parvovirus infection, a partial inhibition of the IFN response could be generated by a Toll-like receptor 9 (TLR-9) antagonist oligonucleotide (iODN), implicating endosomal DNA sensor TLR-9 as a likely relevant PRR in this cell's response (23). iODN inhibition of the IFN response did not, however, enhance parvovirus infection of these cells; thus, there was no evidence of innate immunity directly counteracting parvoviral infection, consistent with our findings. Since the only cell populations within hPBMCs known to possess functional TLR-9 pathways are B cells and plasmacytoid dendritic cells (pDCs) (48) and since B cells secrete tumor necrosis factor alpha and interleukin-6 rather than IFN upon TLR-9 stimulation (49), pDCs may be the predominant producers of the IFN response in hPBMCs (23). However, bone marrow-derived murine pDCs did not respond to MVMp infection (46). Interestingly, purified naked MVMp genomic single-stranded DNA did stimulate IFN-α secretion in murine pDCs, suggesting MVMp capsid-mediated physical protection of the DNA genome from PRRs or capsid-mediated trafficking of viral PAMPs away from PRRs (46). Among different cell types, parvoviral DNA exposure outside the capsid may vary in degree and in location (50). In addition to endosomal TLR-9, which is preferentially expressed in pDCs (51), candidate PRRs for potentially detecting parvoviral DNA include cytoplasmic DAI (ZBP1), DDX41, IFI16, and IFI204, which are found in a broader range of cell types (52).
Although the IFN response may have played a role in the oncoselectivity of parvoviruses when MEFs were compared to A9 transformed murine fibroblasts (21), we found no evidence for any IFN-mediated mechanism underlying the oncoselectivity of parvoviruses in the human cells tested here. Exogenous IFN did not affect the infectivity of parvoviruses in any cell tested. In addition, exogenous IFN had no effect on the rate of parvoviral progeny production in four cell types that could all be protected from VSV, supporting the argument that IFN-stimulated ISGs affect neither the early nor the late stages of the parvoviral life cycle. The magnitude of selectivity of parvovirus LuIII for glioma U87 cells over that for normal human glial cells was unaltered by pretreatment with IFN. A number of alternative mechanisms of parvoviral oncoselectivity that are unrelated to impairments in IFN signaling have been demonstrated to operate, including the oncoselectivity of the viral initiating promoter P4 and the oncoselective toxicity of the major viral nonstructural protein NS1 (14). The oncoselectivity of parvoviruses in nonleukocytic human cells and their transformed counterparts therefore does not appear to be related to the activity of IFN. Compared to other oncolytic viruses, the IFN indifference of parvoviruses may be a significant advantage in tumors with a residual or even a completely intact IFN response. Exogenous IFN had no effect on LuIII infectivity or replication rates in a number of melanomas and gliomas.
Furthermore, we found that the SW982 synovial sarcoma subclone line SW-R, which we reported earlier to be almost completely resistant to a number of viruses, including VSV, cytomegalovirus, and Sindbis virus, due to constitutive IFN activation (28), is nonetheless highly susceptible to infection by LuIII and AAV vectors. In fact, the infectivity of these parvoviruses in SW-R cells was no less than that in cells of the SW-S line, another SW982 subclone that is VSV susceptible and does not show constitutive IFN activation.
Interestingly, and consistent with our findings, a mechanism by which the innate immune system can counteract parvovirus infection may be one that is IFN independent. The most direct evidence for this is a report which shows that some element present in MVMp-infected murine NIH 3T3 fibroblasts (likely viral mRNA transcripts with tertiary structure) can rapidly activate the double-stranded RNA sensor PKR preexisting in the cytoplasm. PKR then phosphorylates the α subunit of eukaryotic initiation factor 2, an essential translational regulator, broadly inhibiting the initiation of translation and thereby abrogating parvoviral infection, all without the need for IFN-mediated upregulation of ISGs (53, 54). Whether this mechanism operates in any human cells remains to be determined; however, we saw no ability of exogenous IFN, which can upregulate PKR expression, to impair parvoviral infection. Still, this is an interesting area for further investigation in normal and transformed human cells.
Dependovirus AAV and recombinant AAV vectors have received more investigative attention than autonomous parvoviruses with regard to the innate immune response (55). Although the biology for these different genera cannot be presumed to be identical, some similarities might nevertheless be expected. TLR-9 mediated an IFN response to wild-type AAV in murine and human pDCs (56), consistent with results for autonomous parvoviruses in hPBMCs (23) but in contrast to the absence of a response to MVMp seen by Mattei et al. in murine pDCs (46). We found that exogenous IFN was unable to block infection of human cells by an AAV vector. We did not, however, investigate replication-competent wild-type AAV; interestingly, wild-type AAV-2 may stimulate IFN-β production at 24 hpi in human lung fibroblasts (HLFs), and TLR-9 appeared to be involved in detection (57). This contrast in findings, despite very similar experimental conditions (MOI, 2,500 genomes per cell, measurement of IFN at 24 hpi [57]), suggests that dependovirus genomes may be more exposed to TLR-9 detection in human fibroblasts than autonomous parvovirus genomes. Exogenous IFN was also capable of reducing the infectivity (2-fold) of wild-type AAV-2 (57), unlike what we and others (22) have observed for the AAV vector and for autonomous parvoviruses. The reasons why wild-type AAV may be more susceptible to the IFN-induced antiviral state than AAV vectors or autonomous parvoviruses remain unclear, and continued studies of dependoviruses in vitro and in vivo (58) will continue to complement studies of autonomous parvoviruses.
Autonomous parvoviruses failed to elicit IFN responses in a variety of normal nonimmune human cells otherwise capable of an IFN response. For normal human fibroblasts, melanocytes, and glia, as well as for human sarcoma, melanoma, and glioma, IFN exposure did not diminish susceptibility to parvoviral infection but did confer resistance to oncolytic VSV. The entire parvoviral life cycle was unaffected by IFN preexposure in multiple human tumors that we tested. In contrast to many other oncolytic viruses, impairment of innate immunity in tumors does not appear to play a role in parvoviral oncoselectivity. Rather than blocking signaling downstream of PRRs, there appears to be no interaction of parvoviruses with PRRs in nonimmune human cells. This, as well as the inability of exogenous IFN to diminish infection in tumors, may constitute a therapeutic advantage for oncolytic parvoviruses over other oncolytic viruses, particularly in tumors that retain a significant degree of innate antiviral activity. As oncolytic parvoviruses may have potential clinical utility against a variety of human tumors, the relationship of these viruses to the innate immune system in different cell types merits further exploration. Ultimately, it will be beneficial to understand the innate immune response to autonomous parvoviruses in the complete context of an animal host.
ACKNOWLEDGMENTS
We thank R. Halaban and the Yale SPORE in Skin Cancer for the generous contribution of melanomas and melanocytes, P. Tattersall for kindly sharing lab reagents and resources, M. Robek, G. Wollmann, and J. N. Davis for manuscript suggestions, and Y. Yang, J. N. Davis, and V. Rogulin for technical assistance.
Yale SPORE in Skin Cancer is supported by NCI grant 1 P50 CA121974. Grant support for this study was provided by NIH grants NS48454, CA175577, and CA124737 (to A.N.V.D.P.) and 1K08CA169005 (to J.C.P.).
Footnotes
Published ahead of print 19 February 2014
REFERENCES
- 1.Cotmore SF, Tattersall P. 2007. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 70:183–232. 10.1016/S0065-3527(07)70005-2 [DOI] [PubMed] [Google Scholar]
- 2.Farr GA, Zhang L-G, Tattersall P. 2005. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc. Natl. Acad. Sci. U. S. A. 102:17148–17153. 10.1073/pnas.0508477102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suikkanen S, Aaltonen T, Nevalainen M, Välilehto O, Lindholm L, Vuento M, Vihinen-Ranta M. 2003. Exploitation of microtubule cytoskeleton and dynein during parvoviral traffic toward the nucleus. J. Virol. 77:10270–10279. 10.1128/JVI.77.19.10270-10279.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ros C, Kempf C. 2004. The ubiquitin-proteasome machinery is essential for nuclear translocation of incoming minute virus of mice. Virology 324:350–360. 10.1016/j.virol.2004.04.016 [DOI] [PubMed] [Google Scholar]
- 5.Deleu L, Pujol A, Faisst S, Rommelaere J. 1999. Activation of promoter P4 of the autonomous parvovirus minute virus of mice at early S phase is required for productive infection. J. Virol. 73:3877–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47. 10.1099/vir.0.83391-0 [DOI] [PubMed] [Google Scholar]
- 7.Rommelaere J, Geletneky K, Angelova AL, Daeffler L, Dinsart C, Kiprianova I, Schlehofer JR, Raykov Z. 2010. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 21:185–195. 10.1016/j.cytogfr.2010.02.011 [DOI] [PubMed] [Google Scholar]
- 8.Paglino JC, Ozduman K, van den Pol AN. 2012. LuIII parvovirus selectively and efficiently targets, replicates in, and kills human glioma cells. J. Virol. 86:7280–7291. 10.1128/JVI.00227-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geletneky K, Kiprianova I, Ayache A, Koch R, Herrero y Calle M, Deleu L, Sommer C, Thomas N, Rommelaere J, Schlehofer JR. 2010. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro. Oncol. 12:804–814. 10.1093/neuonc/noq023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grekova SP, Aprahamian M, Daeffler L, Leuchs B, Angelova A, Giese T, Galabov A, Heller A, Giese NA, Rommelaere J, Raykov Z. 2011. Interferon γ improves the vaccination potential of oncolytic parvovirus H-1PV for the treatment of peritoneal carcinomatosis in pancreatic cancer. Cancer Biol. Ther. 12:888–895. 10.4161/cbt.12.10.17678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dempe S, Lavie M, Struyf S, Bhat R, Verbeke H, Paschek S, Berghmans N, Geibig R, Rommelaere J, Van Damme J, Dinsart C. 2012. Antitumoral activity of parvovirus-mediated IL-2 and MCP-3/CCL7 delivery into human pancreatic cancer: implication of leucocyte recruitment. Cancer Immunol. Immunother. 61:2113–2123. 10.1007/s00262-012-1279-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Angelova AL, Aprahamian M, Balboni G, Delecluse H-J, Feederle R, Kiprianova I, Grekova SP, Galabov AS, Witzens-Harig M, Ho AD, Rommelaere J, Raykov Z. 2009. Oncolytic rat parvovirus H-1PV, a candidate for the treatment of human lymphoma: in vitro and in vivo studies. Mol. Ther. 17:1164–1172. 10.1038/mt.2009.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dupont F. 2003. Risk assessment of the use of autonomous parvovirus-based vectors. Curr. Gene Ther. 3:567–582. 10.2174/1566523034578104 [DOI] [PubMed] [Google Scholar]
- 14.Nüesch JPF, Lacroix J, Marchini A, Rommelaere J. 2012. Molecular pathways: rodent parvoviruses—mechanisms of oncolysis and prospects for clinical cancer treatment. Clin. Cancer Res. 18:3516–3523. 10.1158/1078-0432.CCR-11-2325 [DOI] [PubMed] [Google Scholar]
- 15.Fuks F, Deleu L, Dinsart C, Rommelaere J, Faisst S. 1996. ras oncogene-dependent activation of the P4 promoter of minute virus of mice through a proximal P4 element interacting with the Ets family of transcription factors. J. Virol. 70:1331–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perros M, Deleu L, Vanacker JM, Kherrouche Z, Spruyt N, Faisst S, Rommelaere J. 1995. Upstream CREs participate in the basal activity of minute virus of mice promoter P4 and in its stimulation in ras-transformed cells. J. Virol. 69:5506–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wollmann G, Ozduman K, van den Pol AN. 2012. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J. 18:69–81. 10.1097/PPO.0b013e31824671c9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stojdl DF, Lichty BD, ten Oever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275. 10.1016/S1535-6108(03)00241-1 [DOI] [PubMed] [Google Scholar]
- 19.Chen H-M, Tanaka N, Mitani Y, Oda E, Nozawa H, Chen J-Z, Yanai H, Negishi H, Choi MK, Iwasaki T, Yamamoto H, Taniguchi T, Takaoka A. 2009. Critical role for constitutive type I interferon signaling in the prevention of cellular transformation. Cancer Sci. 100:449–456. 10.1111/j.1349-7006.2008.01051.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katsoulidis E, Kaur S, Platanias LC. 2010. Deregulation of interferon signaling in malignant cells. Pharmaceuticals 3:406–418. 10.3390/ph3020406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grekova S, Zawatzky R, Hörlein R, Cziepluch C, Mincberg M, Davis C, Rommelaere J, Daeffler L. 2010. Activation of an antiviral response in normal but not transformed mouse cells: a new determinant of minute virus of mice oncotropism. J. Virol. 84:516–531. 10.1128/JVI.01618-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mattei LM, Cotmore SF, Tattersall P, Iwasaki A. 2013. Parvovirus evades interferon-dependent viral control in primary mouse embryonic fibroblasts. Virology 442:20–27. 10.1016/j.virol.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raykov Z, Grekova SP, Hörlein R, Leuchs B, Giese T, Giese NA, Rommelaere J, Zawatzky R, Daeffler L. 2013. TLR-9 contributes to the antiviral innate immune sensing of rodent parvoviruses MVMp and H-1PV by normal human immune cells. PLoS One 8:e55086. 10.1371/journal.pone.0055086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barber GN. 2004. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 17:516–527. 10.1089/vim.2004.17.516 [DOI] [PubMed] [Google Scholar]
- 25.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 6:821–825. 10.1038/77558 [DOI] [PubMed] [Google Scholar]
- 26.Hastie E, Grdzelishvili VZ. 2012. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J. Gen. Virol. 93(Pt 12):2529–2545. 10.1099/vir.0.046672-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. 2010. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J. Virol. 84:1563–1573. 10.1128/JVI.02040-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paglino JC, van den Pol AN. 2011. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: rare resistance is overcome by blocking interferon pathways. J. Virol. 85:9346–9358. 10.1128/JVI.00723-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wollmann G, Davis JN, Bosenberg MW, van den Pol AN. 2013. Vesicular stomatitis virus variants selectively infect and kill human melanomas but not normal melanocytes. J. Virol. 87:6644–6659. 10.1128/JVI.03311-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tworkoski K, Singhal G, Szpakowski S, Zito CI, Bacchiocchi A, Muthusamy V, Bosenberg M, Krauthammer M, Halaban R, Stern DF. 2011. Phosphoproteomic screen identifies potential therapeutic targets in melanoma. Mol. Cancer Res. 9:801–812. 10.1158/1541-7786.MCR-10-0512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tattersall P, Bratton J. 1983. Reciprocal productive and restrictive virus-cell interactions of immunosuppressive and prototype strains of minute virus of mice. J. Virol. 46:944–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wollmann G, Tattersall P, van den Pol AN. 2005. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J. Virol. 79:6005–6022. 10.1128/JVI.79.10.6005-6022.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paglino J, Tattersall P. 2011. The parvoviral capsid controls an intracellular phase of infection essential for efficient killing of stepwise-transformed human fibroblasts. Virology 416:32–41. 10.1016/j.virol.2011.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray SJ, Foti SB, Schwartz JW, Bachaboina L, Taylor-Blake B, Coleman J, Ehlers MD, Zylka MJ, McCown TJ, Samulski RJ. 2011. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 22:1143–1153. 10.1089/hum.2010.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ball-Goodrich LJ, Tattersall P. 1992. Two amino acid substitutions within the capsid are coordinately required for acquisition of fibrotropism by the lymphotropic strain of minute virus of mice. J. Virol. 66:3415–3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haller O, Kochs G, Weber F. 2007. Interferon, Mx, and viral countermeasures. Cytokine Growth Factor Rev. 18:425–433. 10.1016/j.cytogfr.2007.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U. S. A. 95:15623–15628. 10.1073/pnas.95.26.15623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rieder M, Conzelmann K-K. 2009. Rhabdovirus evasion of the interferon system. J. Interferon Cytokine Res. 29:499–509. 10.1089/jir.2009.0068 [DOI] [PubMed] [Google Scholar]
- 39.Newton K, Dixit VM. 2012. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 4:a006049. 10.1101/cshperspect.a006049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barber GN. 2005. VSV-tumor selective replication and protein translation. Oncogene 24:7710–7719. 10.1038/sj.onc.1209042 [DOI] [PubMed] [Google Scholar]
- 41.Biswas M, Kumar SRP, Allen A, Yong W, Nimmanapalli R, Samal SK, Elankumaran S. 2012. Cell-type-specific innate immune response to oncolytic Newcastle disease virus. Viral Immunol. 25:268–276. 10.1089/vim.2012.0020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wollmann G, Robek MD, van den Pol AN. 2007. Variable deficiencies in the interferon response enhance susceptibility to vesicular stomatitis virus oncolytic actions in glioblastoma cells but not in normal human glial cells. J. Virol. 81:1479–1491. 10.1128/JVI.01861-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nam H-J, Gurda-Whitaker B, Gan WY, Ilaria S, McKenna R, Mehta P, Alvarez RA, Agbandje-McKenna M. 2006. Identification of the sialic acid structures recognized by minute virus of mice and the role of binding affinity in virulence adaptation. J. Biol. Chem. 281:25670–25677. 10.1074/jbc.M604421200 [DOI] [PubMed] [Google Scholar]
- 44.Spalholz BA, Tattersall P. 1983. Interaction of minute virus of mice with differentiated cells: strain-dependent target cell specificity is mediated by intracellular factors. J. Virol. 46:937–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lang SI, Boelz S, Stroh-Dege AY, Rommelaere J, Dinsart C, Cornelis JJ. 2005. The infectivity and lytic activity of minute virus of mice wild-type and derived vector particles are strikingly different. J. Virol. 79:289–298. 10.1128/JVI.79.1.289-298.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattei LM, Cotmore SF, Li L, Tattersall P, Iwasaki A. 2013. Toll-like receptor 9 in plasmacytoid dendritic cells fails to detect parvoviruses. J. Virol. 87:3605–3608. 10.1128/JVI.03155-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hornung V, Latz E. 2010. Intracellular DNA recognition. Nat. Rev. Immunol. 10:123–130. 10.1038/nri2690 [DOI] [PubMed] [Google Scholar]
- 48.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdörfer B, Giese T, Endres S, Hartmann G. 2002. Quantitative expression of Toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 168:4531–4537 [DOI] [PubMed] [Google Scholar]
- 49.Hanten JA, Vasilakos JP, Riter CL, Neys L, Lipson KE, Alkan SS, Birmachu W. 2008. Comparison of human B cell activation by TLR7 and TLR9 agonists. BMC Immunol. 9:39. 10.1186/1471-2172-9-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mani B, Baltzer C, Valle N, Almendral JM, Kempf C, Ros C. 2006. Low pH-dependent endosomal processing of the incoming parvovirus minute virus of mice virion leads to externalization of the VP1 N-terminal sequence (N-VP1), N-VP2 cleavage, and uncoating of the full-length genome. J. Virol. 80:1015–1024. 10.1128/JVI.80.2.1015-1024.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paludan SR, Bowie AG. 2013. Immune sensing of DNA. Immunity 38:870–880. 10.1016/j.immuni.2013.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burdette DL, Vance RE. 2013. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 14:19–26. 10.1038/ni.2491 [DOI] [PubMed] [Google Scholar]
- 53.Ventoso I, Berlanga JJ, Almendral JM. 2010. Translation control by protein kinase R restricts minute virus of mice infection: role in parvovirus oncolysis. J. Virol. 84:5043–5051. 10.1128/JVI.02188-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goubau D, Deddouche S, Reis e Sousa C. 2013. Cytosolic sensing of viruses. Immunity 38:855–869. 10.1016/j.immuni.2013.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rogers GL, Martino AT, Aslanidi GV, Jayandharan GR, Srivastava A, Herzog RW. 2011. Innate immune responses to AAV vectors. Front. Microbiol. 2:194. 10.3389/fmicb.2011.00194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu J, Huang X, Yang Y. 2009. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Invest. 119:2388–2398. 10.1172/JCI37607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laredj LN, Beard P. 2011. Adeno-associated virus activates an innate immune response in normal human cells but not in osteosarcoma cells. J. Virol. 85:13133–13143. 10.1128/JVI.05407-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jayandharan GR, Aslanidi G, Martino AT, Jahn SC, Perrin GQ, Herzog RW, Srivastava A. 2011. Activation of the NF-kappaB pathway by adeno-associated virus (AAV) vectors and its implications in immune response and gene therapy. Proc. Natl. Acad. Sci. U. S. A. 108:3743–3748. 10.1073/pnas.1012753108 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]