

ABSTRACT
Coxsackievirus A9 (CVA9) is a member of the human enterovirus B species in the Enterovirus genus of the family Picornaviridae. According to earlier studies, CVA9 binds to αVβ3 and αVβ6 integrins on the cell surface and utilizes β2-microglobulin, dynamin, and Arf6 for internalization. However, the structures utilized by the virus for internalization and uncoating are less well understood. We show here, based on electron microscopy, that CVA9 is found in multivesicular structures 2 h postinfection (p.i.). A neutral red labeling assay revealed that uncoating occurs mainly around 2 h p.i., while double-stranded RNA is found in the cytoplasm after 3 h p.i. The biogenesis of multivesicular bodies (MVBs) is crucial for promoting infection, as judged by the strong inhibitory effect of the wild-type form of Hrs and dominant negative form of VPS4 in CVA9 infection. CVA9 infection is dependent on phospholipase C at the start of infection, whereas Rac1 is especially important between 1 and 3 h p.i., when the virus is in endosomes. Several lines of evidence implicate that low pH does not play a role in CVA9 infection. The infection is not affected by Bafilomycin A1. In addition, CVA9 is not targeted to acidic late endosomes or lysosomes, and the MVBs accumulating CVA9 have a neutral pH. Thus, CVA9 is the second enterovirus demonstrated so far, after echovirus 1, that can trigger neutral MVBs, which are important for virus infection.
IMPORTANCE We demonstrate here that the enterovirus coxsackievirus A9 (CVA9) uses a nonclathrin and nonacidic pathway to infect cells. CVA9 does not accumulate in conventional late endosomes or lysosomes. We found that inhibitors of phospholipase C (PLC), Rac1, and the Na+/H+ exchanger decreased CVA9 infection. The PLC inhibitor acts on early entry, the Rac1 inhibitor acts between 1 and 3 h, when the virus is in endosomes, and the Na+/H+ exchange inhibitor acts during various steps during the virus life cycle. The infection depends on the formation of novel neutral multivesicular bodies (MVBs), which accumulate CVA9 during the first hours of entry. Thus, CVA9 is the second enterovirus demonstrated so far, after echovirus 1, that can trigger formation of neutral MVBs. The data show that these enteroviruses favor nonacidic conditions and complex MVBs to promote virus infection.
INTRODUCTION
Human enteroviruses (HEVs) belong to the Picornaviridae family and have an approximately 7.5-kb-long single-stranded RNA genome with positive polarity. The genome is enclosed in an icosahedral protein capsid (30 nm in diameter), consisting of 60 copies of each of the four different structural proteins (VP1 to -4). HEVs are important human pathogens and cause illnesses ranging from the common cold to paralysis and other infections of the central nervous system, carditis, and severe disease syndromes in newborns. HEVs are classified into four species (A to D) on the basis of sequence analysis (1). Of the coxsackie A viruses (CVAs), type 9 is the only one that belongs to the HEV-B species (2). CVA9 particle has an extension containing an RGD (arginine-glycine-aspartic acid) motif at the C terminus of the VP1 protein that is not found in other HEVs except for echovirus 9 strain Barty (3).
CVA9 has been studied quite extensively by using molecular biology methods and in animal models. It has been shown that the virus attaches to αV integrins on the cell surface in several cell lines (4–6), including the lung carcinoma A549 cells used in this study. Other receptor candidates, such as glucose-regulated protein 78 A and a subunit of major histocompatibility complex class I (MHC-I) antigen (7), as well as β2-microglobulin (8), have also been proposed to be involved in CVA9 entry. After primary attachment, CVA9 does not appear to be bound to αV integrins during internalization (4), in contrast to another enterovirus, echovirus 1 (EV1), which is internalized in complex with its receptor, α2β1 integrin (9).
Most animal viruses take advantage of the host cell's endocytic mechanisms for internalization, and viruses have evolved to use one or more receptors and entry mechanisms (10). After arrival in the endosome lumen, viruses can be, for example, exposed to changes in pH and ions and proteolytic events that may initiate the uncoating process (11, 12). These events trigger changes in the virus particle that lead to the delivery of the genome into the cell for further replication.
From our previous studies, we know that β2-microglobulin, dynamin, and Arf6 play roles in the entry process of CVA9 (13). Recent structural studies also revealed details about the uncoating process of CVA9 (14). However, the cellular structures used by CVA9 for internalization and uncoating are still poorly known. Recently, we showed that another enterovirus, EV1, does not enter the host cell in acidic endosomes (15, 16). Instead, EV1 enters multivesicular bodies (MVBs) that morphologically resemble acidic endosomes, even though they are biochemically different structures. This was a striking finding, as the MVBs are supposed to exhibit low pH, and acidity has been suggested to contribute to their biogenesis (17, 18). As both EV1 and CVA9 are acid-stable enteroviruses, we wanted to see if these viruses exhibit similarities in their internalization processes and, more importantly, if CVA9 could also induce the biogenesis of neutral MVBs. We show here that after uptake from the plasma membrane, CVA9 accumulates in nonacidic MVBs and that those structures are needed in CVA9 infection.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
The human lung carcinoma A549 cell line was obtained from the American Type Culture Collection (ATCC). The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal bovine serum (FBS) supplemented with penicillin and streptomycin. CVA9 (Griggs strain) (2, 19) was propagated in A549 cells and purified in sucrose gradients as described previously (20). Culture medium for virus infections was supplemented with 1% FBS. For all infection studies, a multiplicity of infection (MOI) of 10 was used. Typically, this MOI leads to 50 to 70% infection in A549 cells.
Polyclonal rabbit antiserum against CVA9 was produced as described earlier (21), and mouse monoclonal antibody (MAb) K6 against the virus (22) was obtained from Lucia Fiore (Instituto Superiore di Sanita, Rome, Italy). Alexa Fluor 488 (AF-488)- and AF-555-labeled anti-mouse and anti-rabbit secondary antibodies and the ProLong Gold antifade reagent were obtained from Invitrogen. In addition, the following antibodies were used: J2, recognizing double-stranded RNA (dsRNA; MAb; catalog number 10010500; English & Scientific Consulting Kft.); early endosome antigen 1 (EEA1; MAb; catalog number 610457; BD Transduction Laboratories); Lamp1, to identify late endosomes and lysosomes (MAb; sc-20011; Santa Cruz Biotechnology); Rab7, to identify a member of the Rab family of small guanosine triphosphatases (GTPases) Rab7 (polyclonal antibody produced in rabbit; catalog number R4779; Sigma-Aldrich); phalloidin-tetramethylrhodamine B isothiocynate for labeling filamentous actin (catalog number P1951; Sigma); anti-green fluorescent protein (GFP; IgG fraction produced in rabbit) and LysoTracker Green for labeling acidic structures (product numbers A11122 and L7526, respectively; Invitrogen). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate–low-density lipoprotein (DiI-LDL) was obtained from Seppo Ylä-Herttuala (University of Eastern Finland, Kuopio, Finland).
Immunofluorescence and confocal microscopy.
A549 cells were grown on coverslips to subconfluency. CVA9 was first bound to the cells for 45 min on ice in DMEM containing 1% FBS, then the cells were washed, and after incubation at 37°C (in 1% DMEM) they were fixed with 4% paraformaldehyde (PFA) for 20 min, permeabilized with 0.2% Triton X-100 for 5 min, and stained with antibodies (dilutions made in 3% bovine serum albumin [BSA]-phosphate-buffered saline [PBS]). In the preinternalization labeling assays, CVA9 capsids were labeled with primary and secondary antibodies prior to the incubation. The cells were mounted using ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole and examined with an Olympus microscope IX81 with a FluoView-1000 confocal setup or a Zeiss Cell Observer wide-field microscope.
Neutral red-CVA9 assay.
A Neutral red-CVA9 (NR-CVA9) assay was conducted in A549 cells that were infected in the presence of 10 μg/ml of NR (catalog number 101369; Merck). The virus was released after overnight infection by freeze-thawing the cells three times and harvesting by centrifugation. NR-CVA9 was used without further purification. A549 cells were incubated with NR-CVA9 for 45 min on ice, washed, and incubated at 37°C in the darkness. At the indicated time points, the cells were exposed to light for 10 min at room temperature and subsequently transferred back to 37°C. The control cells were not exposed to the light reaction. At 6 h postinfection (p.i.), the cells were labeled for immunofluorescence (see “Immunofluorescence and confocal microscopy,” above). The proportion of infected cells that reacted with the anti-CVA9 antibody was then calculated. In a subsequent NR-CVA9 assay, the Rac1 inhibitor NSC23766 (see “Chemical inhibitors,” below, and Table 1) or dimethyl sulfoxide (DMSO) was added to A459 cells prior to incubation. At the indicated time points, the cells were exposed to light for 10 min at room temperature, after which the medium was changed back to 1% DMEM and the cells were transferred back to 37°C.
TABLE 1.
Pharmacological inhibitors used in this study
| Inhibitor | Effect | Concn used |
|---|---|---|
| U-73122 | Phospholipase C inhibition | 10 μM |
| Wortmannin | Phosphoinositide-3-kinase inhibition | 100 nM |
| NSC23766 | Rac1 inhibition | 100 μM |
| IPA-3 | Pak1 inhibition | 5 μM |
| EIPA | Na+/H+ exchange inhibition | 100 μM |
| Nocodazole | Microtubule depolymerization | 33 μM |
| Bafilomycin A1 | Vacuolar ATPase inhibition | 50 nM |
| LY290042 | Phosphoinositide-3-kinase inhibition | 50 μM |
TCID50 assay.
For the 50% tissue culture infective dose (TCID50) assay, first the A549 cells were grown on 24-well plates to subconfluency. CVA9 was bound to cells for 45 min on ice in DMEM containing 1% FBS. Then, the cells were washed and incubated at 37°C for the indicated times, after which they were scraped and pelleted. The cell-virus pellets were stored at −20°C and later dissolved in PBS (containing 0.5 mM MgCl2). For the TCID50 assay, A549 cells were seeded in a 96-well plate and after overnight incubation the earlier-prepared cell-virus extracts were added to the first well and then serially diluted using 1:4 dilutions. The plates were incubated at 37°C for 2 days. To fix and label the extract mixtures in the plates, a 0.2% crystal violet stain (containing 5% formalin and 10% ethanol) was added to the wells. After incubation at room temperature, the wells were washed with H2O and air dried. The TCID50/ml values were calculated by using the Reed-Muench formula.
Chemical inhibitors.
A549 cells were incubated at 37°C in DMEM supplemented with 1% FBS and 10 μM U-73122 (product number 662035; Calbiochem), 100 nM Wortmannin (product number 681675; Calbiochem), 100 μM NSC 23766 (product number 2161; Tocris Bioscience), 5 μM IPA 3 (product number 3622; Tocris Bioscience), 100 μM 5-(N-ethyl-N-isopropyl) amiloride (EIPA; product number A3085; Sigma), 33 μM Nocodazole (product number M1404; Sigma), 50 nM Bafilomycin A1 (product number 196000; Calbiochem), or 50 μM LY294002 (product number 440202; Calbiochem). The inhibitors were added to cells 30 min before the infection assay or at the indicated time points postinfection, depending on the assay. After 6 h, the proportion of the cells containing virus capsid protein were calculated. The cell viability after the drug treatments was tested in a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cell growth assay (product number CT02; Millipore) according to the manufacturer's instructions.
RT-PCR.
The control and CVA9-infected A549 cells were collected from the plates 6 h p.i., pelleted, and stored at −70°C until analyzed. Methods for the RNA quantification have been described earlier (23). The cell pellets were subjected to automatic nucleic acid extraction in a MagNA Pure 96 instrument (product item 06541089001; Roche Applied Science). An aliquot of each extract was reverse transcribed with 1.2 μM antisense primer (5′-GAAACACGGACACCCAAAGTA) or sense primer (5′-CGGCCCCTGAATGCGGCTAA) or without a primer, using 20 U RevertAid H Minus Moloney murine leukemia virus reverse transcriptase (RT) and 4 U RiboLock RNase inhibitor (Fermentas). From the 20-μl RT reaction mixture, a 5-μl sample was used in a PCR with 600 nM each primer and Maxima SYBR master mix (Fermentas). Amplifications were run on a Rotor-Gene 6000 (Corbett Research) with the following cycling conditions: 95°C for 10 min; 40 cycles of 95°C for 15 s, 64 to 55°C (touch-down 1°C/cycle for the first 10 s) for 30 s, 72°C for 40 s; and final melt at 72 to 95°C, 1°C/5 s. A dilution series of a plasmid containing CVA9 cDNA (2) was used to generate a standard curve. The copy numbers of the genomic and complementary strands were obtained by PCR after the RT reaction with the antisense primer and with the sense primer, respectively. The copy number observed after PCR of the RT reaction mixture without a primer was deducted from the assay values to correct for the primer-independent signal.
EM.
For visualization of the internalized CVA9 particles (see Fig. 3B and D, below), prior to the incubation at 37°C A549 cells were labeled with anti-CVA9 rabbit polyclonal antibody against the viral capsid proteins and, subsequently, with protein A (PA)-gold (diameter, 10 nm; Cell Microscopy Center, Utrecht, Netherlands). The samples were processed for electron microscopy (EM) as described previously (24). Briefly, the cells were fixed in 4% PFA containing 0.1% glutaraldehyde in 50 mM Tris buffer, pH 7.6, at room temperature for 1 h or at 4°C overnight. After 1% osmiumtetroxide (in H2O) treatment, the cells were dehydrated, stained with 2% uranylacetate, and embedded in LX-112 Epon (product number 21210; Ladd Research).
FIG 3.

CVA9 accumulates in MVBs. (A) After CVA9 binding onto A549 cells, the virus was labeled with primary anti-CVA9 capsid antibody followed by fluorescent secondary label (Pre-Int). The 6-h infection was carried out, and the infection rates (cells producing CVA9 capsid protein at 6 h p.i.) were quantified. The experiment was performed three times, and the means are shown (± standard errors [SE]). (B) For EM, the virus and antibody binding was as performed as described for panel A, except that the fluorescent secondary label was replaced with protein A (PA)-gold particles (diameter, 10 nm). After 5 min of incubation at 37°C, the samples were prepared for EM (epon embedding). The arrowhead indicates the presence of CVA9 label in sections. Bar, 100 nm. (C) A549 cells were infected with CVA9, and after 5 and 15 min at 37°C, the samples were fixed and labeled with anti-CVA9 capsid antibody (green) and tetramethyl rhodamine isothiocyanate phalloidin (red). Bars, 5 μm. (D) Sample preparations were prepared as described for panel B. The asterisks indicate the presence of CVA9 label in the sections after 30 min and 2 h of incubation. Bars, 200 nm. (E) The ratio of CVA9-positive structures with (black bars) or without (gray bars) ILVs was quantified from more than 300 cells. (F and G) Prior to the 30 min or 2 h of incubation at 37°C, A549 cells were labeled with anti-CVA9 antibody (green). After the incubations, samples were fixed and labeled with EEA1 or Lamp1 antibodies (red). Bars, 10 μm.
In addition to this monolayer preparation, EM samples were also prepared by cell pelleting. A549 cells, grown on plates, were washed with 0.1 M phosphate buffer and fixed with 2.5% glutaraldehyde in the buffer at room temperature for 10 min. The samples were centrifuged (6,000 rpm, 30 min) and washed three times with 0.1 M phosphate buffer and two times with H2O. The pellets were then embedded into 2.5% agarose in H2O at 37°C. The pelleted samples in agarose were fixed again and stained with 1% osmium tetroxide in H2O at room temperature for 30 min. After three washes, the samples were stained with 1% uranyl acetate in H2O at room temperature for 30 min, washed again, and dehydrated with acetone (two times with 30%, 50%, 70%, and 100% for 10 min). Before proceeding to the 100% LX-112 Epon embedding step, the samples were preembedded with a mixture of Epon in 100% acetone (equal volumes) at room temperature for 45 min. The monolayer and pellet samples were cut, and the section images were observed by using a Jeol 1400 microscope.
For cryo-immuno-EM (see Fig. 7A to C, below), cells were treated as described by Slot and Geuze (25). Briefly, nontransfected and transfected A549 cells were infected with CVA9. The cells were then fixed in 4% PFA in 0.1 M phosphate buffer at room temperature, after which the cells were scraped from the substrate and further fixed for 1 h. After pelleting, the cells were immersed in 12% gelatin in PBS, cut into small pieces, placed in 2.3 M sucrose in PBS at 4°C, and frozen in liquid nitrogen. Thin cryosections were cut with a Leica Ultracut UCT microtome. For double immunolabeling, the sections were first incubated in 5% BSA, 0.1% gelatin in PBS. Antibodies and gold conjugates were diluted in 0.1% BSA in PBS. All washings were performed in 0.1% BSA-C in PBS. After blocking as described above, sections were exposed to the primary antibodies (rabbit anti-CVA9 and rabbit anti-GFP) for 60 min each (with a blocking step in between). After washing, mixtures with PA-gold complex (diameters, 5 and 10 nm, respectively) were incubated. The controls were prepared by carrying out the labeling procedure without primary antibody. The sections were embedded in methylcellulose and examined with a Jeol 1400 microscope.
FIG 7.

WT-Hrs and DN-Vps4 transfections restrict CVA9-MVB formation. (A) A549 cells were infected with CVA9 for 2 h after which the cells were prepared for cryo-immuno-EM. Thin frozen sections were then labeled for CVA9 capsid protein (PA-gold; diameter, 5 nm; asterisks) and GFP (PA-gold; diameter, 10 nm). Bars, 100 nm. (B and C) A549 cells were transfected with the DN mutant of Vps4 (VPS4-E235Q–GFP; DN-Vps4 (B) or WT-Hrs (C). After 48 h for expression, the cells were infected with CVA9 for 2 h and then prepared for cryo-immuno-EM. Thin frozen sections were then labeled for CVA9 capsid protein (PA-gold; diameter, 5 nm; asterisks) and GFP (PA-gold; diameter, 10 nm). Bars, 100 nm.
Transfections.
FuGENE HD transfection reagent (product number E2311; Promega) was used for transfections of subconfluent A549 cells according to the manufacturer's instructions. The cells were used for experiments after an expression time of 48 h. The Hrs-WT–GFP fusion was obtained from Sylvie Urbé (Cellular and Molecular Physiology, University of Liverpool, Liverpool, United Kingdom), the VPS4-E235Q–GFP plasmid was obtained from Harald Stenmark (Department of Biochemistry, Institute for Cancer Research, The Norwegian Radium Hospital, Oslo, Norway), the Rab5-Q79L–yellow fluorescent protein (YFP) and Rab5-S34N–enhanced green fluorescent protein (EGFP) fusions were obtained from Lucas Pelkmans (Institute of Molecular Life Sciences, University of Zurich, Zurich, Switzerland), and the Rab5-WT–GFP fusion was obtained from Miguel Seabra (Faculty of Medicine, National Heart and Lung Institute, Imperial College, London, United Kingdom).
DiI-LDL assay.
A549 cells were grown on coverslips to subconfluency. CVA9 was first bound to the cells for 45 min on ice in DMEM containing 1% FBS. After removing the excess virus, the cells were incubated in 1% DMEM complemented with 30 μg/ml of DiI-LDL for 2 h. After fixation and permeabilization, the cells were stained with CVA9 anti-capsid, anti-Lamp1, and fluorescent secondary antibodies.
LysoTracker green assay.
One hour after CVA9 inoculation, LysoTracker Green was added to the A549 cells and the mixture was incubated for 1 h at 37°C. CVA9 was labeled prior to internalization, which was initiated by moving A549 cells to a preheated (37°C) microscope sample stage. Localization of LysoTracker Green and CVA9 capsid proteins was followed during the infection in live cells by using the Zeiss LSM510 confocal setup with CO2-independent medium (Life Technologies) containing 1% FBS.
Intraendosomal pH measurements.
Intraendosomal pH measurements were conducted as previously described (15). Briefly, CVA9 was first bound to A549 cells on ice, followed by incubation with rabbit polyclonal anti-CVA9 antibody. Then, equal amounts of goat anti-rabbit–fluorescein and goat anti-rabbit AF-555 conjugates were bound to the cells on ice, and the cells were moved to 37°C for 4 h. The cells were kept at 37°C in the Zeiss LSM510 confocal setup under CO2-independent medium containing 1% FBS. Confocal sections were taken 1, 2, and 3 h p.i. To construct a pH titration curve, cell membranes were permeabilized with 20 μM nigericin in pH standard buffer solutions [150 mM KCl, 5 mM glucose, and 15 mM Tris (pH 7.5 and 7.0) or 15 mM 2-(N-morpholino)ethanesulfonic acid (pH 6.5)]. Altogether, 30 cells in each case from two independent experiments were scanned, and the ratio of fluorescein/AF-555 fluorescence was determined for each section with the program BioImageXD (26).
Data analysis and processing of the microscopy data.
An open source software package, BioImageXD (26), was used for the confocal image quantifications. Levels for the laser power, detector amplification, and optical sections were optimized for each channel before starting the imaging. In the dsRNA assay, altogether 20 cells from two independent experiments, 10 cells from each, and from all different time points were randomly selected and optically sectioned using a confocal microscope. For measuring of the number, size, and intensities of dsRNA objects, the following BioImageXD protocol was used. First, volumes were filtered with a difference-of-Gaussians filter to suppress noise and enhance regions of interest. Next, a manually selected threshold level was applied to produce binary image data. Finally, individual objects were found for analysis by computing a Euclidean distance transform on binary data, inverting the result, and applying the watershed transform from local minimal positions. Depending on the experiment, objects smaller than 0, 6, or 10 voxels were removed.
To quantify the level of colocalization, altogether 20 cells from two independent experiments, 10 cells from each, were randomly selected and optically sectioned by using a confocal microscope. Colocalization was evaluated from the center slice images or from the projection of the cell by examination of the merged images, and further analysis was performed with the BioImageXD program. The images were preprocessed with a difference-of-Gaussians filter to remove noise and enhance regions of interest before the colocalization analysis. Thresholds for the analysis were adjusted manually to eliminate fluorescence originating from the background and from diffuse staining. Signal overlap was expressed separately for both of the channels, as a proportion of the intensity colocalizing with the other channel. Statistical significance of observed colocalization was calculated by using Costes algorithm (27), embedded within the software, and only colocalizations with zero coincidence probabilities were taken into account (i.e., P = 1.00).
For measuring the intensities of the vesicles in the pH experiment, a multistep segmentation protocol implemented within BioImageXD software was used. Images were first smoothed with the Gaussian filter and binary by using an adaptively calculated threshold value (mean + 5) from 10 lateral pixel size radius regions. Objects were defined using the Euclidean distance transform and watershed transform as explained above. Finally, all objects of sizes smaller than 5 voxels were removed from the analysis to remove noise and small debris.
Statistical testing.
A t test was used for pairwise statistical comparison between samples. For proportions or ratios, a t test was applied after an arcsine square root transformation of the original variable to convert the binomial distribution of the data to follow a normal distribution. For testing the means between samples from EM, a binomial t test was applied.
RESULTS
Uncoating and replication of CVA9.
In order to determine the time course of CVA9 uncoating in A549 cells, we used a Neutral red-labeled virus (NR-CVA9) (28). Light exposure causes irreversible cross-linking of the labeled viral genome, thus inhibiting the viral uncoating process. If the viruses have not undergone uncoating before the light reaction, no new virus will be synthesized, whereas uncoated viruses will initiate a replication cycle that includes expression of VP1. If the light exposure was performed 0 or 15 min p.i., the cells did not produce VP1 at 6 h p.i. When the light exposure was performed at 30 min or 1 h p.i., less than 20% of the cells were infected at 6 h p.i., suggesting that the uncoating process had started but was not yet at its peak (Fig. 1A). When the light treatment was carried out at 2 and 4 h p.i., around 60 to 75% of the cells expressed VP1 at 6 h after infection. The results suggest that the majority of CVA9 capsid uncoating takes place around 2 h after virus attachment.
FIG 1.

Uncoating and replication of CVA9 in A549 cells. (A) The infection rate (the proportion of cells producing viral capsid protein 6 h p.i.) after incubation with Neutral Red (NR)-labeled CVA9. The cells were exposed to light treatment at different time points, and the infection was continued to 6 h p.i. The control cells (Ctrl) were infected and incubated in the darkness. The results are averages of two independent experiments (± standard errors [SE]) for 600 cells counted. (B) The number of dsRNA structures in cells. A549 cells were infected with CVA9; samples were fixed at different time points and labeled with anti-dsRNA and anti-CVA9 capsid antibodies. The results are averages of two independent experiments (± SE). At each time point, 20 cells were imaged. (C) Representative confocal images of CVA9-infected A549 cells at 3, 4, and 5 h p.i., labeled with anti-CVA9 capsid (green) and anti-dsRNA (red) antibodies. Bars, 10 μm. (D) The TCID50/ml values at different time points p.i. A549 cells were infected with CVA9, collected at different time points, and serially diluted on top of A549 monolayers. After 2 days of incubation, TCID50/ml values were calculated.
To investigate the timing of active replication, we fixed the infected cells at different time points and labeled them with both a CVA9 (anti-capsid) antibody and a J2 antibody. The J2 antibody recognizes dsRNA that is the primary element in the infection cycle. The mock-infected cells were negative for J2 labeling (Fig. 1B). The first visible dsRNA-containing structures appeared in cells at 3 h p.i. (Fig. 1B and C), and they appeared in scattered locations near but not colocalizing with the structures positive for CVA9 capsid protein (Fig. 1C). The number of dsRNA structures (Fig. 1B and C) in addition to the volume and intensity increased as the infection progressed (data not shown).
Furthermore, we performed a TCID50 assay to evaluate the timing of infective particle formation. Infected A549 cells were collected at different time points, fragmented, serially diluted, and cultured on top of a new monolayer of A549 cells. After 2 days of incubation, the cells were fixed and labeled with crystal violet dye, and TCID50/ml values were calculated. The results showed that as the entry and uncoating processes begin, the CVA9 particles start to lose their infectivity after 1 h (Fig. 1A and D). After the 3-h time point, at the same time when dsRNA structures appear in the cytoplasm (Fig. 1B and C), the TCID50/ml values correspondingly increased (Fig. 1D). Taken together, we conclude that the uncoating of the majority of CVA9 particles takes place around 2 h p.i., and active replication (visible dsRNA structures) of the virus and new infective particle formation occur after 3 h p.i.
Effects of chemical inhibitors of endocytosis on CVA9 infection.
Earlier studies suggested that components belonging to the clathrin pathway are not involved in CVA9 entry (13). However, there is still very little understanding of CVA9 internalization, and we wanted to investigate in further detail some players that have effects on EV1 entry, e.g., phospholipase C (PLC), Pak1, Rac1, and the Na+/H+ exchanger (29). We decided to use chemical inhibitors that have been widely used to downregulate the action of these proteins. The inhibitors used in this study and their mode of action are shown in Table 1. First, we determined which inhibitors affected CVA9 infection. We treated the A549 cells with the chemical inhibitors for 30 min prior to CVA9 binding on ice. After removing the unbound virus, the cells were incubated at 37°C with the inhibitors. At 6 h p.i., the cells were fixed and labeled with the primary anti-CVA9 antibody and fluorescent secondary antibody. The proportion of A549 cells full of virus capsid proteins was calculated in different situations (Fig. 2A). U-73122, NSC23766, and EIPA caused statistically highly significant (P = 0.0, 0.0004, and 0.0, respectively) decreased in infection, whereas Wortmannin and IPA-3 had no significant effects. To evaluate the cell viability after the drug treatments, a colorimetric MTT assay (Millipore) was performed. The drug treatments did not cause significant cell death (data not shown), indicating that the observed changes in virus proliferation were due to inhibitory effects of the drugs.
FIG 2.
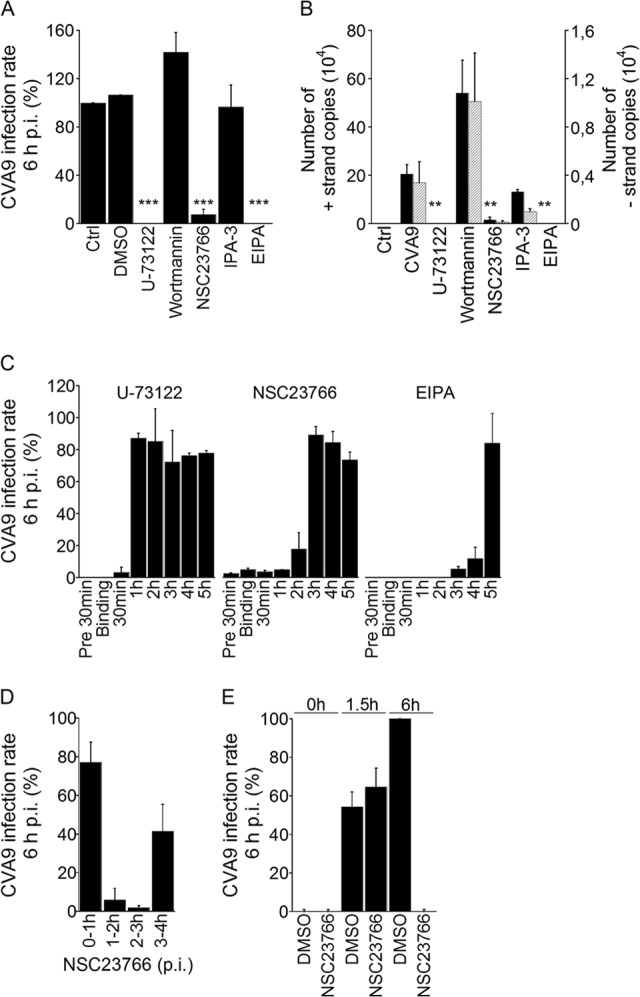
Effects of endocytosis inhibitors on CVA9 infection. (A and B) A549 cells were preincubated with inhibitors (Table 1) that were also present throughout the experiment. After virus binding, the cells were incubated for 6 h. (A) The infection percentages (cells producing high amounts of CVA9 capsid protein at 6 h p.i.) of control (Ctrl and DMSO) and inhibitor-treated cells were quantified. The experiment was performed three times, and the means are shown (± standard errors [SE]). Statistical significance was calculated with a paired-sample t test (after arcsine square root transformation). ***, P < 0.001. (B) The presence of genomic (+) and complementary (−) RNA strands was analyzed with RT-PCR. The results are averages of three parallel samples (± SE). Statistical significance was calculated with a paired-sample t test. **, P < 0.01. (C) The inhibitors were introduced to the cells at different time points postinfection, and the incubation with the inhibitors was carried out until 6 h p.i., after which the infection percentages were quantified. The experiment was performed two times, and the means are shown (± SE). (D) The Rac1 inhibitor (NSC23766) was introduced to the cells at different time points. After the 1-h treatment, the medium was replaced with fresh 1% DMEM and the incubation was carried out until 6 h p.i., after which the infection percentages were quantified. The experiment was performed two times, and the means are shown (± SE). (E) The NR-CVA9 was bound to A549 cells in the darkness. The Rac1 inhibitor (NSC23766) or DMSO (a control) were introduced to the cells, and cells were incubated in the darkness for 0, 1.5, or 6 h. At these time points, the 10-min light reactions were carried out, the drug was replaced with fresh medium, and cells were transferred back into the incubator for the remaining incubation time (6 h). The infection percentages were quantified at each time point. The experiment was performed two times, and the means are shown (± SE).
Next, we carried out an RT-PCR assay to find out whether the drugs affected new viral RNA production. In correspondence with the immunofluorescent (IF) assay results described above, U-73122, NSC23766, and EIPA caused statistically significant decreases (P = 0.006, 0.01, and 0.006, respectively) in genomic strand production (Fig. 2B). The same inhibitors seemed to decrease the production of the complementary strand also, although the reductions were not statistically significant. As in the IF assay (Fig. 2A), Wortmannin and IPA-3 had no statistically significant effects on virus RNA production (Fig. 2B). To verify the Wortmannin result, we also used the drug LY290042, another inhibitor of phosphatidylinositol 3-kinase (PI3-kinase), which gave similar results (data not shown). The results from both the IF and RT-PCR assays thus suggested that PLC, Rac1, and Na+/H+ exchanger inhibitors decrease CVA9 infection in A549 cells.
In order to clarify more carefully the temporal regulation of PLC, Rac1, and Na+/H+ exchanger inhibitors in CVA9 infection, we performed a more detailed time course inhibitory assay. In this assay, the drugs were introduced to the cells at different time points postinfection, and incubation with the inhibitors was carried out until 6 h p.i. After fixation, the VP1 protein was labeled with fluorescent antibody and the proportions of A549 cells containing newly synthesized virus proteins were calculated under different conditions. As shown in Fig. 2C, the inhibitory effect of U-73122 was limited to early stages of infection (before 1 h p.i.). In the case of NSC23766, the inhibitory effect seemed to be limited to time points before 3 h p.i. (Fig. 2C). EIPA, on the other hand, disrupted the infection more or less throughout the experiment (Fig. 2C).
To better elucidate the time point when the Rac1 inhibitor affects CVA9 infection, the 1-h interval treatments with NSC23766 were carried out over 6 h of infection. The virus bound onto the cells without the inhibitor. If the Rac1 inhibitor treatment were performed at 0 to 1 h p.i. or later, NSC23766 was replaced with fresh 1% DMEM; at the 6-h time point, only a 20% decrease in the infection rate was observed, relative to the DMSO control (set as 100%) (Fig. 2D). This result also confirmed the known fact that the inhibitory effect of NSC23766 is reversible. If the NSC23766 treatment were carried out between the 1- to 2-h or 2- to 3-h time points, CVA9 infection rates were dramatically decreased (Fig. 2D). The drug treatment at 3 to 4 h p.i. (Fig. 2D) confirmed the previous results (Fig. 2C; NSC23766 chart), indicating that after the 3-h time point NSC23766 lost its inhibitory effect. These results suggested to us that Rac1 is somehow involved in the CVA9 infection process at time points between 1 and 3 h.
As the inhibitory effect of NSC23766 seemed to coincide with the time points most important for the CVA9 uncoating process, we carried out a variation of the NR assay with this drug. In this experiment, NR-CVA9 bound to A549 cells on ice in the dark. After removing the unbound virus, the Rac1 inhibitor (NSC23766) or DMSO (control) was introduced to the cells, and cells were incubated in the dark for 0, 1.5, or 6 h. At these time points, the 10-min light reactions were carried out, the drug was replaced with fresh 1% DMEM without the inhibitor, and cells were put back into the incubator for the remaining time (total incubation time, 6 h). After fixation, the cells were labeled with the primary anti-CVA9 antibody and fluorescent secondary antibody, and the proportions of A549 cells containing newly synthesized virus proteins were calculated. As shown in Fig. 2E, if the light reaction was executed at the 0-h time point, no newly synthetized VP1 proteins were observed after 6 h of incubation in either DMSO- or NSC23766-treated cells, as expected (control experiments). In addition, if the NSC23766 was kept on the cells throughout the assay, the virus infection was totally inhibited (Fig. 2E, NSC23766 at 6 h), as already shown in Fig. 2A and B for the other assays. Furthermore, DMSO-treated cells were infected after 6 h, as expected (Fig. 2D). Interestingly, if the light reaction was executed at the 1.5-h time point, both DMSO- and NSC23766-treated cells were infected with similarly lower efficiencies at 6 h p.i. The magnitude of infection was in good agreement with the uncoating results obtained and illustrated in Fig. 1A. The results therefore suggest that the Rac1 inhibitor NSC23766 does not affect the uncoating of CVA9, and this leaves us with an interesting suggestion, that Rac1 may exert its effect through other means than uncoating when the virus resides in endosomes.
CVA9 accumulates in MVBs.
In order to investigate CVA9 infection at the ultrastructural level, we used EM methods. CVA9 was first bound onto the surface of A549 cells on ice, and the virus particles were labeled with an anti-capsid antibody and PA-gold particles (diameter, 10 nm). Cell-bound CVA9, attached to antibody and PA-gold, was then internalized at 37°C, and the cells were fixed at various time points. We confirmed by fluorescence microscopy methods that this preinternalization labeling of the virus maintained to a large extent its infectivity (Fig. 3A). At 5 min, CVA9 particles were found mostly on the plasma membrane, usually near membrane extensions (Fig. 3B). After 15 min, the virus was still found mainly attached to these ruffles in several areas (data not shown). When the CVA9 particles and cellular actin were labeled with fluorescent dyes, these ruffles were typically actin positive (Fig. 3C).
We were interested in the nature of the structures where CVA9 accumulated during the entry process. The EM assay indicated that at 30 min, around 28% of CVA9 particles were internalized and could be found in vesicles close to the plasma membrane (Fig. 3D) and that about half of the vesicular structures had visible intraluminal vesicles (ILVs) (Fig. 3E). At the 30-min time point, the colocalization of CVA9 capsid protein, labeled with IF methods before virus internalization, with early endosome (EEA1) antibody, was negligible (Fig. 3F). EM images taken at 2 h p.i. revealed that internalized CVA9 was then mostly found in MVBs (Fig. 3D and E). The MVBs resembled typical multivesicular structures, with several small ILVs and occasional larger vesicles. Strikingly, at the 2-h time point, the preinternalization labeled CVA9 did not colocalize with the lysosomal marker Lamp1 (Fig. 3G). Quantification of the proportions of the CVA9-containing endosomes with or without ILVs showed that the number of multivesicular structures containing CVA9 increased significantly (P < 0.001) between 30 min and 2 h (Fig. 3E). Uninfected cells, treated with primary and secondary antibodies in the EM assay, showed negligible PA-gold background. These results demonstrated that during the internalization process, CVA9 accumulates in MVBs, which interestingly seems to be distinct from EEA1- and Lamp1-positive structures.
CVA9 is not targeted to EEA1- or Lamp1/Rab7-positive structures, and the infection is microtubule independent.
Although the CVA9-MVBs in the EM images closely resembled the acidic early and late endosomes, they did not seem to colocalize with the canonical endosomes in the IF images (Fig. 3D, F, and G). Therefore, the role of early and late endosomes in CVA9 infection was studied further, and the presence of the virus particles in these structures was quantified. The A549 cells were infected with CVA9, and between 15 to 120 min of incubation, cells were fixed and labeled with CVA9 anticapsid antibody and early endosome (EEA1) or late endosome/lysosome (Lamp1 and Rab7) antibodies. In addition, multiple transfections were prepared to investigate the role of Rab5 in CVA9 infection, and the colocalization between CVA9 and DiI-LDL during the internalization process was determined.
During 15 to 60 min of incubation, a low amount of the CVA9 particles (labeled after fixation) colocalized with EEA1-positive structures (Fig. 4A). Only a few vesicles showed colocalization in the IF images, and it seemed that even the low amount of colocalization was somewhat exaggerated, due to close apposition of the separate vesicles and limited resolution of the confocal microscopy images, leading to some false colocalization. In addition to EEA1 labeling, we were interested in the role of Rab5 in CVA9 infection. The A549 cells were transfected with wild-type (WT)-Rab5–GFP, constitutively active Rab5 (Rab5-Q79L–GFP), or dominant negative (DN)-Rab5 (Rab5-S34N–GFP); after 48 h of expression the cells were infected with CVA9 for 6 h, and virus capsid protein production in transfected A549 cells was determined. The constitutively active mutant had little effect on CVA9 infection, whereas the DN Rab5 (Rab5-S34N–GFP) construct inhibited CVA9 infection in a statistically significant manner (P = 0.009) (Fig. 4B).
FIG 4.

CVA9 infection is not associated with the lysosomal pathway. (A) A549 cells were infected with CVA9, and after 15, 30, or 60 min of incubation, the samples were fixed and labeled with anti-CVA9 capsid antibody (green) and EEA1 antibody (red). Quantification of colocalization of CVA9 with EEA1 was done from projection stacks of single cells by using the BioImageXD software (see Materials and Methods). Altogether, 30 cells from three independent experiments were analyzed. The results are shown as mean values (± standard errors [SE]). Bars, 10 μm. (B) A549 cells were transfected with a wild-type Rab5 (Rab5-WT–GFP; WT in the chart), constantly active Rab5 construct (Rab5-Q79L–GFP; Q79L in the chart), or a dominant negative Rab5 construct (Rab5-S34N–GFP; S34N in the chart). After 48 h for expression, the cells were infected with CVA9 and the infection percentage (cells producing CVA9 capsid protein at 6 h p.i.) of transfected cells was quantified. The results are mean values of two independent experiments (± SE), and statistical significance was calculated with a paired-sample t test (after arcsine square root transformation). **, P < 0.01. (C and D) A549 cells were treated and analyzed as described for panel A. After 30, 60, and 120 min of incubation, the samples were fixed and labeled with anti-CVA9 capsid antibody (green) and Lamp1 or Rab7 antibodies (red). Altogether, 30 cells from three independent experiments were analyzed. The results are shown as mean values (± SE). Bars, 10 μm. (E) CVA9 was internalized into A549 cells with DiI-LDL (30 μg/ml) for 2 h. The samples were fixed and labeled with anti-CVA9 capsid (green) and Lamp1 antibodies (blue); DiI-LDL is seen in red. The colocalized voxels between CVA9 and DiI-LDL or Lamp1 and DiI-LDL were done from projection stacks and using the BioImageXD software.
Colocalization of virus particles with Lamp1-positive structures was quite low, reaching a maximum 25% at the 2-h time point (Fig. 4C), suggesting that CVA9 mainly associates with structures other than Lamp1-positive endosomes. Furthermore, the colocalization of CVA9 with Rab7 was even lower (<10%) during the 30- to 120-min incubation periods, indicating that CVA9 does not traffic through late endosomes (Fig. 4D). According to our previous observations, and as stated above, close apposition of the endosomal vesicles between virus signal with Lamp1 may cause a false-positive signal due to limited resolution under confocal microscopy (16). As seen in Fig. 4C and D, Lamp1 vesicles were more crowded in the perinuclear region than Rab7-positive structures, which may be the reason why CVA9 colocalization with Lamp1 seems to be higher than with Rab7. In order to verify the negligible use of the clathrin pathway, we examined simultaneous entry of CVA9 with DiI-LDL, which is the canonical marker of the clathrin pathway, which ends eventually in lysosomes. Co-uptake of DiI-LDL and CVA9 resulted in clearly distinct endosomes in the cytoplasm after 2 h (Fig. 4E). Several DiI-LDL vesicles costained with Lamp1, whereas CVA9 stayed separate, further demonstrating that CVA9 acts separate from the clathrin-dependent pathway.
The microtubule-depolymerizing agent Nocodazole (Noc) blocks transport of the cargo from early to late endosomes, resulting in the accumulation of cargo in earlier structures in the acidic pathway (30). We thus decided to use this drug to inhibit CVA9 entry to late endosomes in order to evaluate whether the virus transfer to late endosomes is important for CVA9 infection. We treated A549 cells similarly as in the chemical inhibitor assay above (see Materials and Methods). The results showed that Noc treatment did not inhibit infection during the 6 h of incubation (Fig. 5A). We confirmed the breakdown of the microtubule network after Noc treatment based on diffuse cytoplasmic labeling of tubulin monomers when we used IF labeling (data not shown). The cell viability after the drug treatments was verified by the colorimetric MTT assay (Millipore) (data not shown). Next, we tested whether Noc treatment caused an accumulation of CVA9 label in early endosomes, which would be expected if late endosomes were used in the viral infectious pathway. A549 cells were treated with Noc for 30 min prior to CVA9 binding, and after 1 h of incubation the cells were fixed and labeled with CVA9 anticapsid antibody and EEA1 antibody. As Fig. 5B shows, such accumulation did not occur. In addition, we studied whether Noc treatment affected CVA9 colocalization with Lamp1. The results showed that Noc treatment did not statistically significantly decrease CVA9 colocalization with Lamp1 (P = 0.62) (Fig. 5B). In conclusion, since Noc treatment did not inhibit CVA9 infection or affect colocalization with EEA1, the results further confirmed that CVA9 internalization is not dependent on the canonical entry pathway (from early to late endosome and lysosome).
FIG 5.
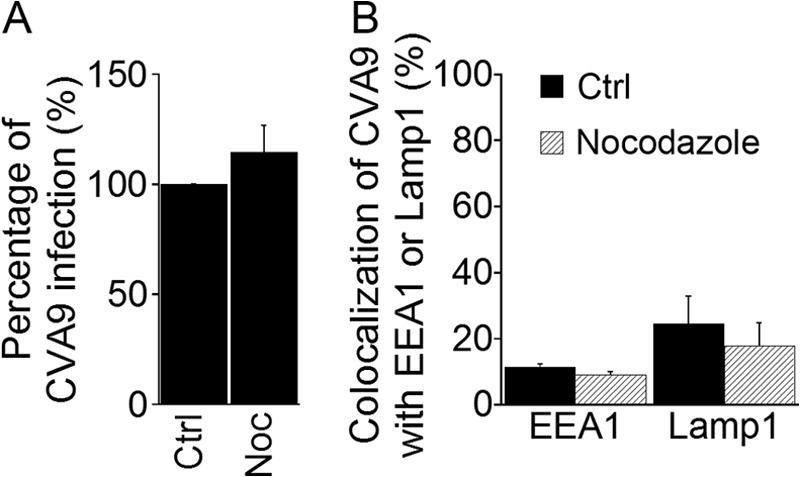
Microtubules are not needed in CVA9 infection. (A) A549 cells were preincubated with nocodazole (Noc) (see Table 1). After CVA9 binding, cells were incubated for 6 h. The inhibitor was present throughout the experiment. The proportions of infected cells (cells producing CVA9 capsid protein 6 h p.i.) among control (Ctrl) and inhibitor-treated cells were quantified. The experiment was repeated three times, and the mean values are shown (± standard errors [SE]). Statistical significance was calculated with a paired-sample t test (after arcsine square root transformation). (B) A549 cells were preincubated with Noc, and the cells were infected with CVA9. After a 1-h (EEA1) or 2-h (Lamp1) incubation, the samples were fixed and labeled with anti-CVA9 and EEA1 or Lamp1 antibodies, respectively. Quantification of colocalization of CVA9 with EEA1 and Lamp1 was done from confocal projection stacks of single cells by using BioImageXD software. Thirty cells from each of the three independent experiments were analyzed. The results are shown as mean values (± SE).
MVBs are important for CVA9 infection.
In mammalian cells, MVBs are form via progressive involution of the endosomal limiting membrane in a process catalyzed by the endosomal sorting complex required for transport (ESCRT) (31, 32). One of the first components that acts in the complex set of events leading to protein sorting to MVBs is ESCRT 0, the hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) (33). Hrs binds directly to the limiting membrane of the endosome and recruits other ESCRT components to the site. Since earlier results suggested that overexpression of WT-Hrs causes a block of epidermal growth factor receptor (EGFr) transport, possibly by titration of interacting factors required downstream of Hrs (e.g., ESCRTs) or due to problems in the release of Hrs from endosomal membranes (34), we were interested in whether Hrs overexpression had an effect on CVA9 infection. We transfected A549 cells with Hrs-WT–GFP or a control GFP plasmid, and after 48 h of expression the cells were infected with CVA9 for 6 h and virus capsid protein was labeled. In control cells, transfected with the control GFP plasmid, the cytoplasm contained large amounts of virus capsid proteins after 6 h p.i. (Fig. 6A). In contrast, the overexpression of WT-Hrs–GFP had a strong and statistically significant (P = 0.0007) inhibitory effect on VP1 production (Fig. 6B). In WT-Hrs-transfected cells, CVA9 label was found in vesicle-like structures, whereas in normally infected cells (Fig. 6A) the whole cytoplasm was full of newly synthesized capsid protein and we could not identify vesicle-like structures. For visualization purposes when using the confocal microscope, the imaging thresholds were set differently to visualize the low amount of input virus after transfection (Fig. 6B and C) and to visualize the much higher amount of newly synthetized capsid protein in infected cells (Fig. 6A).
FIG 6.
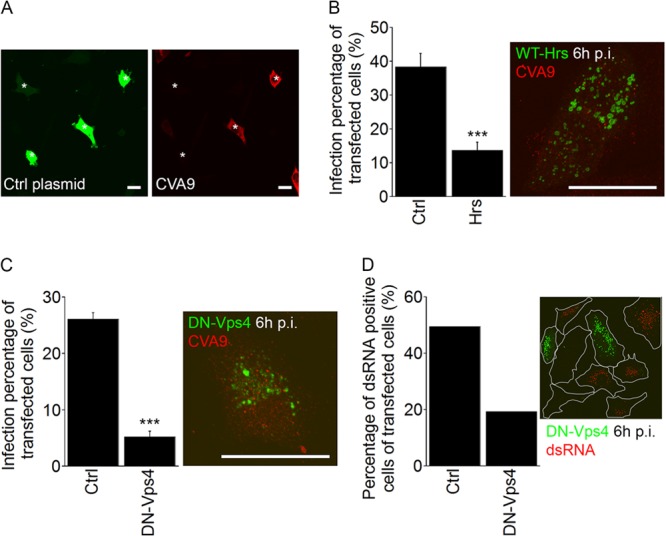
The ESCRT complex is needed for successful CVA9 infection. (A) Fluorescent images of GFP-transfected cells (Ctrl plasmid, green) and CVA9-infected cells (red) at 6 h p.i. The asterisks represent transfected cells. Bars, 30 μm. (B) A549 cells were transfected with a wild-type plasmid of Hrs (Hrs-WT–GFP; Hrs in the chart) or the control GFP construct (Ctrl). After 48 h for expression, cells were infected with CVA9, and the infection percentages (cells producing CVA9 capsid protein 6 h p.i.) of transfected cells were quantified 6 h p.i. The results are mean values of two independent experiments (± standard errors [SE]), and statistical significance was calculated with a paired-sample t test (after arcsine square root transformation). ***, P < 0.001. In the maximum intensity projection of confocal sections (time point, 6 h p.i.), the Hrs-WT–GFP was visualized in green and CVA9 was visualized in red. Bar, 30 μm. (C) The dominant negative mutant of Vps4 (VPS4-E235Q–GFP; DN-Vps4 in the chart) and the control GFP construct (Ctrl) were transfected in cells. Sample preparation and analysis were performed as described for Fig. 6B. The results are mean values of two independent experiments (± SE), and statistical significance was calculated with a paired-sample t test (after arcsine square root transformation). ***, P < 0.001. Maximum intensity projection of confocal sections of a cell expressing VPS4-E235Q–GFP (DN-VPS4, green) at 6 h p.i. CVA9 is shown in red. Bar, 30 μm. (D) The transfection was performed as for panel C. After CVA9 infection (6 h), the samples were labeled with anti-dsRNA antibody, and the percentages of dsRNA-positive cells within the transfected cells were calculated. In both cases, more than 50 cells were quantified.
We previously showed that the last component of ESCRTs, the ATPase Vps4 (35), inhibits EV1 infection and EV1-MVB function (15). Therefore, we also wanted to examine the role of Vps4 in CVA9 infection. A549 cells were transfected with DN-Vps4 (VPS4-E235Q–GFP) or a control GFP plasmid. As with the Hrs transfection experiment mentioned above, after 48 h of expression, the cells were processed for CVA9 infection and capsid protein labeling by IF methods. The overexpression of VPS4-E235Q–GFP had a strong and statistically significant (P = 2.3E−05) inhibitory effect on VP1 production (Fig. 6C). As with the WT-Hrs transfection described above, CVA9 label in DN-Vps4-transfected cells was found in vesicle-like structures. Besides the VP1 labeling, we also labeled the dsRNA in cells transfected with VPS4-E235Q–GFP or control plasmid. The confocal images and analyses based on those images indicated that in VPS4-E235Q-GFP-expressing cells, the amount of dsRNA structures was clearly decreased (Fig. 6D).
In addition to the infectivity assays descripted above, we used cryo-immuno-EM to verify whether virus label was present in MVBs during normal infection versus after overexpression of WT-Hrs or transfection with DN-Vps4 (VPS4-E235Q–GFP). Nontransfected and transfected A549 cells were infected with CVA9, and cryo-immuno-EM-samples were prepared. From thin sections, CVA9 capsid protein and GFP were labeled with PA-gold (diameter, 5 and 10 nm, respectively). In nontransfected cells, MVBs positive for CVA9 label were frequently found after 2 h (Fig. 7A). In contrast, in cells transfected with DN-Vps4 (VPS4-E235Q–GFP; verified by GFP labeling), CVA9 was observed in small vesicles with no clear accumulation of ILVs (Fig. 7B). Likewise, after overexpression of WT-Hrs, the transfected cells showed high amounts of smaller vesicles in the cytoplasm, and in those cells CVA9 was not observed in MVBs (Fig. 7C). Together with the infectivity assay results (Fig. 6A to D), these results showed that at least two players (Hrs and Vps4) are involved in the MVB formation process during CVA9 infection and thus directly indicated that ESCRTs are needed for formation of virus-driven MVBs.
Endosomes accumulating CVA9 are neutral.
In order to study in more detail the relations of CVA9 to acidic structures and whether acidity is at all needed for CVA9 infection, we used several approaches that exploited chemical inhibitors and direct intraendosomal measurements of acidity. A vacuolar proton ATPase (v-ATPase) is known to establish an acidic pH in the lumen of endocytic organelles, and the pH gradually proceeds to lower values from early endosomes to late endosomes and lysosomes (36). Bafilomycin A1 (Baf) is a specific v-ATPase inhibitor, and it has been shown to inhibit vesicle budding from early endosomes (37) and the transport from late endosomes to lysosomes (38). We thus decided to use this drug to inhibit acidification of endosomes during CVA9 entry. We treated A549 cells similarly as for the first chemical inhibitor assay (see Materials and methods) and observed that, although the Baf treatment slightly decreased the infection rate of CVA9, the virus maintained its level of infectivity to a large extent (Fig. 8A). In addition, we performed an RT-PCR assay for the Baf-treated cells, and no decrease in the genomic or complementary strand production was observed (Fig. 8B). To verify that Baf treatment elevated the pH in endocytic compartments, an intraendosomal measurement assay was carried out for the control protein EGFr, which is known to accumulate in late endosomes and lysosomes (16). The cell viability after the drug treatments was verified by the colorimetric MTT assay (Millipore).
FIG 8.
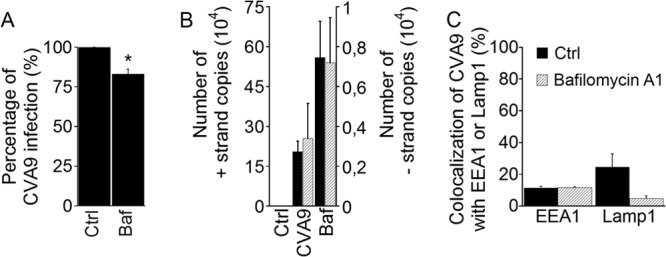
Bafilomycin A1 does not inhibit CVA9 infection. (A) A549 cells were preincubated with Baf (see Table 1). After CVA9 binding, the cells were incubated for 6 h in the presence of inhibitor. The proportions of infected cells (cells producing CVA9 capsid protein at 6 h p.i.) of control (Ctrl) and inhibitor-treated (Baf) cells were quantified. The experiment was repeated three times, and the mean values are shown (± standard errors [SE]). Statistical significance was calculated with a paired-sample t test (after arcsine square root transformation). *, P < 0.05. (B) A549 cells were preincubated with Baf, and the cells were infected with CVA9. After 6 h, samples were prepared for RT-PCR. The presence of genomic (+) and complementary (−) RNA strands was analyzed with RT-PCR. The RT-PCR results are averages of three parallel samples (± SE). Statistical significance was calculated with a paired-sample t test. (C) A549 cells were pre-incubated with Baf, after which the A549 cells were infected with CVA9 and after incubation for 1 h (for EEA1 labeling) or 2 h (for Lamp1 labeling), cells were fixed and labeled with anti-CVA9 and EEA1 or Lamp1 antibodies. Quantification of colocalization of CVA9 with EEA1 and Lamp1 was done from confocal projection stacks of single cells by using BioImageXD software. Thirty cells from each of the three independent experiments were analyzed. The results are shown as mean values (± SE).
In HeLa cells, Baf arrests the transport of markers in early endosomes, in the canonical early endosomes-to-late endosomes-to-lysosomes route (39). Thus, we next tested whether Baf treatment caused an accumulation of CVA9 label in early endosomes. A549 cells were treated with Baf for 30 min prior to CVA9 binding, after which the cells were infected with the virus; after 60 min of incubation, the cells were fixed and labeled with CVA9 anticapsid antibody and EEA1 antibody. As shown in Fig. 8C (EEA1 bars), such accumulation did not occur. In addition, we studied whether Baf treatment affected CVA9 colocalization with Lamp1. The results showed that the colocalization between CVA9 capsid protein and Lamp1 labels decreased, although not in a statistically significant manner (P = 0.06).
As the Baf result described above suggested that the CVA9 entry pathway is not dependent on vesicle acidification, we wanted to determine directly whether the endosomes used by CVA9 are at all acidic. We started by live imaging of CVA9 with LysoTracker green, which is used as a live probe that accumulates in acidic structures. After CVA9 binding, capsid protein labeling, and internalization for 1 h at 37°C, LysoTracker green was added to the cells for 1 h at 37°C. Our earlier results (Fig. 3A) already demonstrated that this kind of preinternalization labeling of the virus does not inhibit the CVA9 infection rate significantly. LysoTracker green labeling showed a bright signal in a high number of endosomes, probably representing both late endosomes and lysosomes (Fig. 9A). Despite the high number of acidic endosomes, there was no apparent colocalization with CVA9-positive endosomes (Fig. 9B). These results thus further confirmed that CVA9 does not accumulate in acidic structures during a 3.5-h live imaging period.
FIG 9.
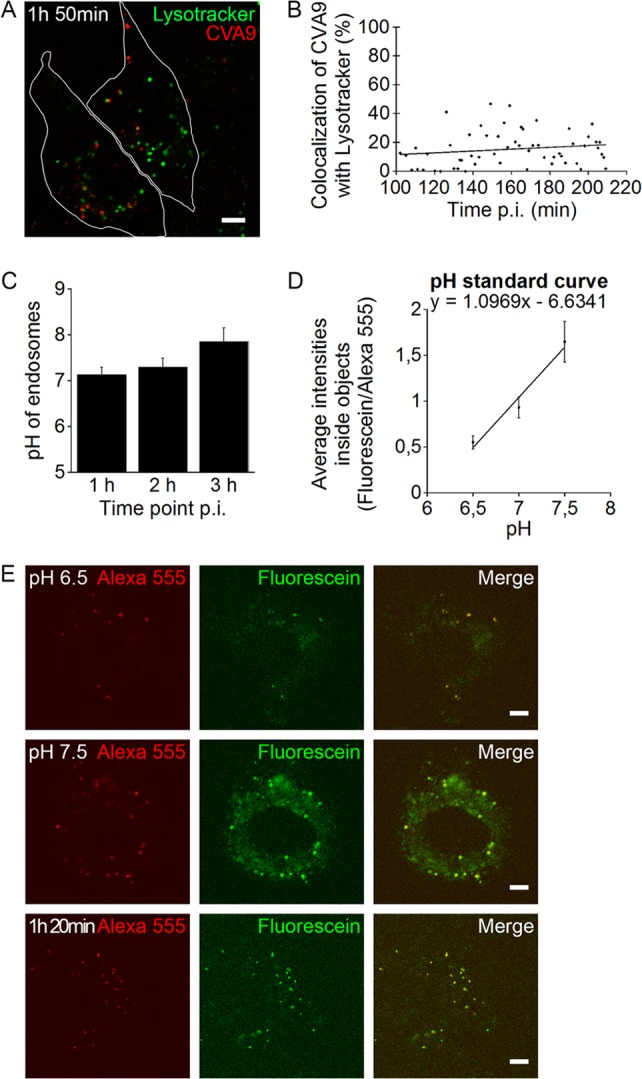
CVA9 structures are nonacidic. (A and B) After CVA9 binding, the virus capsid was labeled (red) and the internalization was initiated by moving A549 cells to a preheated (37°C) microscope sample stage. After 1 h, LysoTracker green was added to the cells for 1 h. Localization of LysoTracker green- and CVA9-positive structures was followed during CVA9 infection in live cells. Bar, 5 μm. (A) A representative confocal projection stack image taken at 1 h 50 min p.i. Bar, 10 μm. (B) Quantifications of colocalization of CVA9 with LysoTracker green were done from projection stacks of single cells by using the BioImageXD software. Altogether, 60 individual cells were analyzed. (C, D, and E) A549 cells were sequentially incubated on ice with primary and secondary antibodies to label plasma membrane-bound CVA9. The secondary antibody was conjugated with pH-sensitive FITC (fluorescein; green) and pH-resistant AF-555 (red). The internalization was started by moving the cells to 37°C, and the cells were imaged live with a confocal microscope. (C) The ratios between FITC and AF-555 signals with or without CVA9 were measured from each time point and compared with the pH standard curve. Error bars represent standard errors of three independent experiments (30 cells measured from each). (D) The pH standard curve was acquired by measuring ratios of virus-bound dyes in the presence of different pH buffer solutions supplemented with nigericin. Error bars represent standard errors of three independent experiments (30 cells measured from each). (E) Images represent single confocal sections of cells, showing the two dyes in endosomes at pH 6.5 and 7.5 and after 1 h 20 min p.i. Bars, 5 μm.
In order to gain more accurate information of the pHs of the endosomes along the infectious CVA9 pathway, we performed an intraendosomal pH measurement assay (15, 16). In the assay, a pH-stable AF-555 conjugate and a pH-sensitive fluorescein isothiocyanate (FITC) conjugate were introduced to forming CVA9-positive endosomes. The fluorescence intensity ratio between FITC and AF-555 was measured from internalized CVA9 vesicles at different time points and compared against a pH standard curve (Fig. 9D) that was prepared in the same live assay. The assay showed in three consecutive assays that the pH of CVA9 vesicles stayed remarkably neutral at time points 1, 2, and 3 h p.i. (Fig. 9C). As shown in Fig. 9E, the fluorescence signal for the fluorescein conjugate was remarkably lower at pH 6.5 than pH 7.5. The results thus demonstrated altogether that CVA9 does not accumulate in acidic endosomes.
DISCUSSION
In spite of extensive molecular studies of CVA9, the structures involved in the infectious entry and uncoating processes have remained unclear. We recently reported that CVA9 does not use clathrin- or caveolin1-associated pathways to infect A549 cells (13). Here we have shown that CVA9 enters endosomes that gradually develop into nonacidic multivesicular endosomes. These results are remarkably similar to those we observed with another enterovirus, EV1 (15, 16), and thus suggest that these two acid-stable enteroviruses may also bear other similarities in theur mechanisms of internalization, uncoating, and genome release from endosomes.
According to the results presented here, uncoating of CVA9 in A549 cells is initiated at 30 min p.i. The major part of uncoating takes place around 2 h, when the virus is found in MVBs, which was confirmed by EM. The first signs of active replication (dsRNA) were observed at 3 h p.i., and the formation of new, infective CVA9 particles occurred after the 3-h time point. The uncoating and replication time schedule is very similar to what we found for EV1. In SAOS-α2 cells, the majority of EV1 uncoating took place after 1 h (9, 40), and the increase of positive- and negative-strand viral RNA was detectable around 2 to 3 h (15, 23, 41). Inspections of the data accumulated from other enteroviruses suggest some further similarities, although there are clearly cell-line-dependent differences as well. In HeLa cells, poliovirus (PV) RNA release is rapid and efficient and begins already after 10 min p.i. (28). On the other hand, in polarized human brain microvascular endothelial cells, PV remains on the cell surface longer, as it starts to enter the cytoplasm at 2.5 h p.i., and the uncoating takes place after 4 h (42).
The effects of inhibitory drugs on infection showed clear similarities with EV1 but also some interesting differences. Similar to EV1, CVA9 infection was affected by inhibitors of PLC, Rac1, and an Na+/H+ exchanger but, in contrast, was not affected by the inhibitors of PI3K and Pak1. As with EV1, the effect of PLC inhibition was restricted to early time points of infection (before 1 h). We showed previously that PLC inhibition causes an arrest of EV1 and fluid-phase uptake on the plasma membrane (29). Most probably, this is the case also with CVA9, considering that PLC inhibition was without effect during uncoating of the virus. EIPA has been used as one major indicator of macropinocytosis in numerous virus studies. It exerts its effect already during early entry, leaving many viruses and ligands undergoing macropinocytosis on the plasma membrane or, as it was shown for CVA9 and EV1, in peripheral early-type vesicles (13, 29). However, as seen from our detailed time course studies, EIPA seems to have several inhibitory effects all the way through virus infection and not only during virus entry, which makes it difficult pinpoint the point of action. It has been shown that EIPA inhibits the intracellular replication of human rhinovirus 2 and coxsackievirus B3 (43, 44), and in the case of human rhinovirus 2, the viral release process is also inhibited in EIPA-treated cells. EIPA was recently shown to cause local changes in the Na+ and H+ gradients in vesicles and prevent Rac1 and Cdc42 activation (45).
The effects on Rac1 signaling by EIPA may actually explain some of the unexpected results that we observed with the Rac1 inhibitor at a later step for both the EIPA and Rac1 inhibitors. Our previous results with EV1 suggested that Rac1 is involved during early entry, leading to downstream Pak1 activation and use of CtBP/BARS for macropinocytic entry on the plasma membrane (29, 46). Now, with CVA9, it is clear that Rac1 inhibition was most effective in the postentry, endosomal step. Furthermore, CVA9 infection was not dependent on Pak1, suggesting that early downstream activation of Pak1 via Rac1 is not needed. What could be the role of Rac1 in the endosomal structures? Unexpectedly, the Rac1 inhibitor did not disturb the viral uncoating process, which was verified in an NR-based uncoating assay. Rac1 is a small GTPase, which plays a fundamental role in a variety of cellular processes: actin cytoskeletal reorganization, cell transformation, the introduction of DNA synthesis, and cell migration, to name a few (47). Actin nucleation and polymerization have been shown to control endosome biogenesis, even beyond the first entry step, indicating that actin may have direct effects on cytoplasmic endosomes, presumably driving membrane remodeling (48). Here, our results suggest that Rac1 activation is needed after the viral uncoating process. The exact role of Rac1 in the endosomes during CVA9 infection is a subject for further studies.
We were especially interested in the endosomes that accumulated CVA9 during the first 2 h of infection, because the majority of CVA9 uncoating takes place during that time period and because the production of dsRNA has not yet started in the cytoplasm. Several lines of evidence demonstrated that MVBs are important structures in CVA9 infection. First of all, the EM analysis showed the majority of the CVA9-positive structures were multivesicular at 2 h p.i. Furthermore, between 30 min and 2 h, the proportion of CVA9 vesicles with ILVs increased. The most direct evidence for the importance of the MVBs to CVA9 infection comes from the studies with the ESCRT machinery. Both the overexpression of the first component of this machinery, Hrs, as well as expression of the DN mutant of the last component (Vps4) had a strong effect on CVA9 infectivity, indicating that infection does not occur without MVBs. This is similar to what we found for EV1 previously (15). In addition, cryo-immuno-EM showed that in cells transfected with either WT-Hrs or DN-Vps4 (VPS4-E235Q–GFP), CVA9 was found in endosomes without ILVs. The mechanistic understanding of the possible role of ILVs in EV1 and CVA9 infections remains a subject for further studies.
Since the most studied pathway leading to MVBs is the acidic canonical endosomal pathway (early to late endosomes and lysosomes), the dogma has been that the MVBs are mostly acidic. However, we showed previously and extensively for EV1 that acidity did not play a role for EV1 infection and that the MVBs used by EV1 were close to neutral in acidity (15, 16, 29). We demonstrated here that CVA9 does not enter the conventional acidic endosomes and, further, that it does not require an acidic environment for initiation of infection: (i) Baf treatment did not decrease viral RNA or protein production, (ii) CVA9 did not colocalize with EEA1, Lamp1, internalized DiI-LDL, or Rab7, and (iii) the extents of colocalization with these structures were not changed after the Baf or Noc treatment. The findings are similar to what we obtained for EV1 infection (16, 29, 41) but, interestingly, these results bear similarities also with PV infection. Coyne et al. (42) showed that vesicles containing PV, VP1, or the PV receptor did not colocalize with EEA1, Rab7, or the late endosome marker LAMP-II. In addition, the productive RNA release and infectivity of PV were not dependent on the acidic environment of late endosomes (28, 49–51). Moreover, endocytosis of yet another enterovirus, coxsackievirus B3, and its infectivity are suggested to be independent of clathrin, caveolin, and endosomal acidification in HeLa cells (52). Thus, it is becoming more evident that some enteroviruses have similarities in their entry and that acidity may not play a role in their infectious cycle.
In conclusion, based on the findings in this report, we generated a model for CVA9 internalization and intracellular CVA9 structures that are important in the infection process (Fig. 10). CVA9 infection starts by virus uptake associated with plasma membrane ruffles. Inhibition of PLC activity is most effective during the earlier time points of virus uptake. Internalized CVA9 is found in endosomes that gradually form ESCRT-driven intraluminal vesicles, leading to neutral MVBs. The majority of uncoating occurs around 2 h p.i., when the bulk of CVA9 is in MVBs. Rac1 inhibition affects CVA9 infection in the endosomal step, after which replication starts in the cytoplasm and leads to accumulation of dsRNA structures and later newly synthesized viral capsid proteins and infective virus particles.
FIG 10.

Model of the CVA9 internalization pathway. CVA9 infection starts with virus uptake associated with plasma membrane ruffles. Inhibition of PLC activity prevents early entry of CVA9. CVA9 is internalized to endosomes, which gradually form ESCRT-driven intraluminal vesicles, leading to neutral MVBs. The majority of uncoating occurs around 2 h p.i., when CVA9 is in MVBs. Rac1 inhibition affects CVA9 infection in the endosomal step, after which the replication starts in the cytoplasm and leads to accumulation of dsRNA structures and later the newly synthetized viral capsid proteins and infective virus particles.
ACKNOWLEDGMENTS
We thank Lucia Fiore (Instituto Superiore di Sanita, Rome, Italy), Sylvie Urbé (Cellular and Molecular Physiology, University of Liverpool, Liverpool, United Kingdom), Harald Stenmark (The Norwegian Radium Hospital, Oslo, Norway), Lucas Pelkmans (Institute of Molecular Life Sciences, University of Zurich, Zurich, Switzerland), and Miguel Seabra (Faculty of Medicine, National Heart and Lung Institute, Imperial College, London, United Kingdom) for providing us with K6 CVA9 antibody, Hrs-WT–GFP, VPS4-E235Q–GFP, Rab5-Q79L–YFP, and Rab5-S34N–EGFP and Rab5-WT–GFP, respectively. We thank the Biocenter Oulu EM Core Facility for cutting and labeling thin frozen sections and Epon samples.
The work was supported by grants from the Academy of Finland (grants 119354 and 257125), Sigrid Juselius Foundation, National Graduate School on Nanoscience, and Ellen and Artturi Nyyssönen Foundation.
Footnotes
Published ahead of print 26 February 2014
REFERENCES
- 1.Hyypiä T, Hovi T, Knowles NJ, Stanway G. 1997. Classification of enteroviruses based on molecular and biological properties. J. Gen. Virol. 78:1–11 [DOI] [PubMed] [Google Scholar]
- 2.Chang KH, Auvinen P, Hyypiä T, Stanway G. 1989. The nucleotide sequence of coxsackievirus A9; implications for receptor binding and enterovirus classification. J. Gen. Virol. 70:3269–3280 [DOI] [PubMed] [Google Scholar]
- 3.Nelsen-Salz B, Eggers HJ, Zimmermann H. 1999. Integrin αvβ3 (vitronectin receptor) is a candidate receptor for the virulent echovirus 9 strain Barty. J. Gen. Virol. 80:2311–2313 [DOI] [PubMed] [Google Scholar]
- 4.Heikkilä O, Susi P, Stanway G, Hyypiä T. 2009. Integrin αVβ6 is a high-affinity receptor for coxsackievirus A9. J. Gen. Virol. 90:197–204. 10.1099/vir.0.004838-0 [DOI] [PubMed] [Google Scholar]
- 5.Roivainen M, Piirainen L, Hovi T, Virtanen I, Riikonen T, Heino J, Hyypiä T. 1994. Entry of coxsackievirus A9 into host cells: specific interactions with alpha v beta 3 integrin, the vitronectin receptor. Virology 203:357–365. 10.1006/viro.1994.1494 [DOI] [PubMed] [Google Scholar]
- 6.Williams CH, Kajander T, Hyypiä T, Jackson T, Sheppard D, Stanway G. 2004. Integrin alpha v beta 6 is an RGD-dependent receptor for coxsackievirus A9. J. Virol. 78:6967–6973. 10.1128/JVI.78.13.6967-6973.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Triantafilou K, Fradelizi D, Wilson K, Triantafilou M. 2002. GRP78, a coreceptor for coxsackievirus A9, interacts with major histocompatibility complex class I molecules which mediate virus internalization. J. Virol. 76:633–643. 10.1128/JVI.76.2.633-643.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Triantafilou M, Triantafilou K, Wilson KM, Takada Y, Fernandez N, Stanway G. 1999. Involvement of β2-microglobulin and integrin αvβ3 molecules in the coxsackievirus A9 infectious cycle. J. Gen. Virol. 80:2591–2600 [DOI] [PubMed] [Google Scholar]
- 9.Marjomäki V, Pietiäinen V, Matilainen H, Upla P, Ivaska J, Nissinen L, Reunanen H, Huttunen P, Hyypiä T, Heino J. 2002. Internalization of echovirus 1 in caveolae. J. Virol. 76:1856–1865. 10.1128/JVI.76.4.1856-1865.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu. Rev. Biochem. 79:803–833. 10.1146/annurev-biochem-060208-104626 [DOI] [PubMed] [Google Scholar]
- 11.Fuchs R, Blaas D. 2010. Uncoating of human rhinoviruses. Rev. Med. Virol. 20:281–297. 10.1002/rmv.654 [DOI] [PubMed] [Google Scholar]
- 12.Scott CC, Gruenberg J. 2011. Ion flux and the function of endosomes and lysosomes. pH is just the start: the flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. Bioessays 33:103–110. 10.1002/bies.201000108 [DOI] [PubMed] [Google Scholar]
- 13.Heikkilä O, Susi P, Tevaluoto T, Härmä H, Marjomäki V, Hyypiä T, Kiljunen S. 2010. Internalization of coxsackievirus A9 is mediated by β2-microglobulin, dynamin, and Arf6 but not by caveolin-1 or clathrin. J. Virol. 84:3666–3681. 10.1128/JVI.01340-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shakeel S, Seitsonen JJ, Kajander T, Laurinmäki P, Hyypiä T, Susi P, Butcher SJ. 2013. Structural and functional analysis of coxsackievirus A9 integrin αvβ6 binding and uncoating. J. Virol. 87:3943–3951. 10.1128/JVI.02989-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karjalainen M, Rintanen N, Lehkonen M, Kallio K, Mäki A, Hellström K, Siljamäki V, Upla P, Marjomäki V. 2011. Echovirus 1 infection depends on biogenesis of novel multivesicular bodies. Cell. Microbiol. 13:1975–1995. 10.1111/j.1462-5822.2011.01685.x [DOI] [PubMed] [Google Scholar]
- 16.Rintanen N, Karjalainen M, Alanko J, Paavolainen L, Mäki A, Nissinen L, Lehkonen M, Kallio K, Cheng RH, Upla P, Ivaska J, Marjomäki V. 2012. Calpains promote α2β1 integrin turnover in nonrecycling integrin pathway. Mol. Biol. Cell 23:448–463. 10.1091/mbc.E11-06-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanson PI, Cashikar A. 2012. Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 28:337–362. 10.1146/annurev-cellbio-092910-154152 [DOI] [PubMed] [Google Scholar]
- 18.Matsuo H, Chevallier J, Mayran N, Le Blanc I, Ferguson C, Faure J, Blanc NS, Matile S, Dubochet J, Sadoul R, Parton RG, Vilbois F, Gruenberg J. 2004. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 303:531–534. 10.1126/science.1092425 [DOI] [PubMed] [Google Scholar]
- 19.Hendry E, Hatanaka H, Fry E, Smyth M, Tate J, Stanway G, Santti J, Maaronen M, Hyypiä T, Stuart D. 1999. The crystal structure of coxsackievirus A9: new insights into the uncoating mechanisms of enteroviruses. Structure. 7:1527–1538 [DOI] [PubMed] [Google Scholar]
- 20.Abraham G, Colonno RJ. 1984. Many rhinovirus serotypes share the same cellular receptor. J. Virol. 51:340–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pulli T, Roivainen M, Hovi T, Hyypiä T. 1998. Induction of neutralizing antibodies by synthetic peptides representing the C terminus of coxsackievirus A9 capsid protein VP1. J. Gen. Virol. 79:2249–2253 [DOI] [PubMed] [Google Scholar]
- 22.Buttinelli G, Donati V, Ruggeri FM, Joki-Korpela P, Hyypiä T, Fiore L. 2003. Antigenic sites of coxsackie A9 virus inducing neutralizing monoclonal antibodies protective in mice. Virology 312:74–83 http://dx.doi.org/10.1016/S0042-6822(03)00182-X [DOI] [PubMed] [Google Scholar]
- 23.Upla P, Marjomäki V, Nissinen L, Nylund C, Waris M, Hyypiä T, Heino J. 2008. Calpain 1 and 2 are required for RNA replication of echovirus 1. J. Virol. 82:1581–1590. 10.1128/JVI.01375-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Upla P, Marjomäki V, Kankaanpää P, Ivaska J, Hyypiä T, Van Der Goot FG, Heino J. 2004. Clustering induces a lateral redistribution of alpha 2 beta 1 integrin from membrane rafts to caveolae and subsequent protein kinase C-dependent internalization. Mol. Biol. Cell 15:625–636. 10.1091/mbc.E03-08-0588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slot JW, Geuze HJ. 2007. Cryosectioning and immunolabeling. Nat. Protoc. 2:2480–2491. 10.1038/nprot.2007.365 [DOI] [PubMed] [Google Scholar]
- 26.Kankaanpää P, Paavolainen L, Tiitta S, Karjalainen M, Päivärinne J, Nieminen J, Marjomäki V, Heino J, White DJ. 2012. BioImageXD: an open, general-purpose and high-throughput image-processing platform. Nat. Methods 9:683–689. 10.1038/nmeth.2047 [DOI] [PubMed] [Google Scholar]
- 27.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. 2004. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 86:3993–4003. 10.1529/biophysj.103.038422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM. 2007. Imaging poliovirus entry in live cells. PLoS Biol. 5(7):e183. 10.1371/journal.pbio.0050183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karjalainen M, Kakkonen E, Upla P, Paloranta H, Kankaanpää P, Liberali P, Renkema GH, Hyypiä T, Heino J, Marjomäki V. 2008. A Raft-derived, Pak1-regulated entry participates in α2β1 integrin-dependent sorting to caveosomes. Mol. Biol. Cell 19:2857–2869. 10.1091/mbc.E07-10-1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gruenberg J, Griffiths G, Howell KE. 1989. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J. Cell Biol. 108:1301–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gruenberg J, Stenmark H. 2004. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 5:317–323. 10.1038/nrm1360 [DOI] [PubMed] [Google Scholar]
- 32.Hirsch JG, Fedorko ME, Cohn ZA. 1968. Vesicle fusion and formation at the surface of pinocytic vacuoles in macrophages. J. Cell Biol. 38:629–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raiborg C, Malerod L, Pedersen NM, Stenmark H. 2008. Differential functions of Hrs and ESCRT proteins in endocytic membrane trafficking. Exp. Cell Res. 314:801–813. 10.1016/j.yexcr.2007.10.014 [DOI] [PubMed] [Google Scholar]
- 34.Urbe S, Sachse M, Row PE, Preisinger C, Barr FA, Strous G, Klumperman J, Clague MJ. 2003. The UIM domain of Hrs couples receptor sorting to vesicle formation. J. Cell Sci. 116:4169–4179. 10.1242/jcs.00723 [DOI] [PubMed] [Google Scholar]
- 35.Katzmann DJ, Odorizzi G, Emr SD. 2002. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3:893–905. 10.1038/nrm973 [DOI] [PubMed] [Google Scholar]
- 36.Futai M, Oka T, Sun-Wada G, Moriyama Y, Kanazawa H, Wada Y. 2000. Luminal acidification of diverse organelles by v-ATPase in animal cells. J. Exp. Biol. 203:107–116 [DOI] [PubMed] [Google Scholar]
- 37.Clague MJ, Urbe S, Aniento F, Gruenberg J. 1994. Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J. Biol. Chem. 269:21–24 [PubMed] [Google Scholar]
- 38.van Deurs B, Holm PK, Kayser L, Sandvig K. 1995. Delivery to lysosomes in the human carcinoma cell line HEp-2 involves an actin filament-facilitated fusion between mature endosomes and preexisting lysosomes. Eur. J. Cell Biol. 66:309–323 [PubMed] [Google Scholar]
- 39.Bayer N, Schober D, Prchla E, Murphy RF, Blaas D, Fuchs R. 1998. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: implications for viral uncoating and infection. J. Virol. 72:9645–9655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siljamäki E, Rintanen N, Kirsi M, Upla P, Wang W, Karjalainen M, Ikonen E, Marjomäki V. 2013. Cholesterol dependence of collagen and echovirus 1 trafficking along the novel α2β1 integrin internalization pathway. PLoS One 8(2):e55465. 10.1371/journal.pone.0055465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pietiäinen V, Marjomäki V, Upla P, Pelkmans L, Helenius A, Hyypiä T. 2004. Echovirus 1 endocytosis into caveosomes requires lipid rafts, dynamin II, and signaling events. Mol. Biol. Cell 15:4911–4925. 10.1091/mbc.E04-01-0070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coyne CB, Kim KS, Bergelson JM. 2007. Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J. 26:4016–4028. 10.1038/sj.emboj.7601831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gazina EV, Harrison DN, Jefferies M, Tan H, Williams D, Anderson DA, Petrou S. 2005. Ion transport blockers inhibit human rhinovirus 2 release. Antiviral Res. 67:98–106. 10.1016/j.antiviral.2005.05.003 [DOI] [PubMed] [Google Scholar]
- 44.Harrison DN, Gazina EV, Purcell DF, Anderson DA, Petrou S. 2008. Amiloride derivatives inhibit coxsackievirus B3 RNA replication. J. Virol. 82:1465–1473. 10.1128/JVI.01374-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S. 2010. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 188:547–563. 10.1083/jcb.200908086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liberali P, Kakkonen E, Turacchio G, Valente C, Spaar A, Perinetti G, Bockmann RA, Corda D, Colanzi A, Marjomäki V, Luini A. 2008. The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J. 27:970–981. 10.1038/emboj.2008.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bosco EE, Mulloy JC, Zheng Y. 2009. Rac1 GTPase: a “Rac” of all trades. Cell. Mol. Life Sci. 66:370–374. 10.1007/s00018-008-8552-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morel E, Parton RG, Gruenberg J. 2009. Annexin A2-dependent polymerization of actin mediates endosome biogenesis. Dev. Cell 16:445–457. 10.1016/j.devcel.2009.01.007 [DOI] [PubMed] [Google Scholar]
- 49.Doedens J, Maynell LA, Klymkowsky MW, Kirkegaard K. 1994. Secretory pathway function, but not cytoskeletal integrity, is required in poliovirus infection. Arch. Virol. Suppl. 9:159–172 [DOI] [PubMed] [Google Scholar]
- 50.Gromeier M, Wetz K. 1990. Kinetics of poliovirus uncoating in HeLa cells in a nonacidic environment. J. Virol. 64:3590–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez L, Carrasco L. 1993. Entry of poliovirus into cells does not require a low-pH step. J. Virol. 67:4543–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel KP, Coyne CB, Bergelson JM. 2009. Dynamin- and lipid raft-dependent entry of decay-accelerating factor (DAF)-binding and non-DAF-binding coxsackieviruses into nonpolarized cells. J. Virol. 83:11064–11077. 10.1128/JVI.01016-09 [DOI] [PMC free article] [PubMed] [Google Scholar]