

ABSTRACT
Cleavage of the hemagglutinin (HA) by host proteases is essential for the infectivity of influenza viruses. Here, we analyzed the role of the serine protease TMPRSS2, which activates HA in the human respiratory tract, in pathogenesis in a mouse model. Replication of the human H7N9 isolate A/Anhui/1/13 and of human H1N1 and H3N2 viruses was compared in TMPRSS2 knockout (TMPRSS2−/−) and wild-type (WT) mice. Knockout of TMPRSS2 expression inhibited H7N9 influenza virus replication in explants of murine tracheas, bronchi, and lungs. H1N1 virus replication was also strongly suppressed in airway explants of TMPRSS2−/− mice, while H3N2 virus replication was only marginally affected. H7N9 and H1N1 viruses were apathogenic in TMPRSS2−/− mice, whereas WT mice developed severe disease with mortality rates of 100% and 20%, respectively. In contrast, all H3N2 infected TMPRSS2−/− and WT mice succumbed to lethal infection. Cleavage analysis showed that H7 and H1 are efficiently activated by TMPRSS2, whereas H3 is less susceptible to the protease. Our data demonstrate that TMPRSS2 is a host factor that is essential for pneumotropism and pathogenicity of H7N9 and H1N1 influenza virus in mice. In contrast, replication of H3N2 virus appears to depend on another, not yet identified protease, supporting the concept that human influenza viruses differ in protease specificity.
IMPORTANCE Cleavage of the hemagglutinin (HA) by host proteases is essential for the infectivity of influenza virus, but little is known about its relevance for pathogenesis in mammals. Here, we show that knockout mice that do not express the HA-activating protease TMPRSS2 are resistant to pulmonary disease with lethal outcome when infected with influenza A viruses of subtypes H7N9 and H1N1, whereas they are not protected from lethal H3N2 virus infection. These findings demonstrate that human influenza viruses differ in protease specificity, and that expression of the appropriate protease in respiratory tissues is essential for pneumotropism and pathogenicity. Our observations also demonstrate that HA-activating proteases and in particular TMPRSS2 are promising targets for influenza therapy.
INTRODUCTION
Worldwide circulation of influenza A viruses in a broad range of avian and mammalian hosts and their recurrent transmission among different species provides the basis for the emergence of novel influenza viruses that may cause outbreaks of severe respiratory disease or even provoke an influenza pandemic. The outbreak of an avian H7N9 influenza virus in China in 2013 that caused 137 confirmed human infections and 45 deaths (WHO; 52) once again demonstrated that the emergence of novel influenza viruses poses unpredictable challenges for public health (1–3). Phylogenetic analyses revealed that H7N9 has emerged through multiple reassortments, thereby obtaining its eight gene segments from at least three different avian influenza viruses (2, 3). Human H7N9 infections appeared to result from direct contact with infected poultry, but the viruses were shown to already possess molecular markers associated with adaptation to humans, including mutations in the surface glycoprotein hemagglutinin (HA) which increase the affinity to α2,6-linked sialic acid (human-type receptors) (4–9), as well as adaptive mutations in the polymerase subunit PB2 such as 627K and 701N, which are associated with enhanced polymerase activity and replication in mammalian cells (1, 2, 10, 11). Recent studies demonstrated that human H7N9 viruses can attach to epithelial cells of both the human upper and lower respiratory tracts and replicate efficiently in airway epithelial cells of humans and swine (12–14). Furthermore, these viruses can be easily transmitted by direct contact and less efficiently by respiratory droplets in the ferret model (4, 5, 14). As of January 2014, an increase of human H7N9 cases has been reported in China (ECDC; 53). Although there is no evidence for sustained human-to-human transmission thus far, the continuous circulation of the virus in avian populations carries the risk that a stable lineage that represents a continuous threat to public health and might possess pandemic potential will be established.
Influenza virus infection is initiated by HA through attachment to cell surface receptors and fusion of the viral lipid envelope and endosomal membranes upon endocytosis to release the virus genome into the cytosol. Posttranslational cleavage of HA by host proteases is a prerequisite for fusion activity and, thus, for virus infectivity. Most influenza viruses, including low-pathogenic avian influenza viruses (LPAIV) and human viruses, possess a single arginine (R) at the cleavage site and are activated by trypsin in vitro. Appropriate trypsin-like proteases are present in a restricted number of tissues, such as the respiratory or the intestinal tract, limiting the spread of infection (15, 16). We identified the human type II transmembrane serine proteases TMPRSS2 (transmembrane protease serine S1, member 2; also designated epitheliasin) and HAT (human airway trypsin-like protease; also designated TMPRSS11D) as HA-cleaving proteases present in the human airway epithelium (17). In contrast, highly pathogenic avian influenza viruses (HPAIV) of subtypes H5 and H7 contain a multibasic HA cleavage site (consensus sequence R-X-R/K-R) that is activated by ubiquitous expressed proteases furin and proprotein convertase 5/6 (PC5/6), supporting systemic infection with an often fatal outcome (18, 19). Novel human H7N9 viruses possess a monobasic HA cleavage site consistent with their low pathogenicity in poultry (2).
In this study, we investigated spread in the respiratory tract as well as the pathogenic potential of a human H7N9 virus (A/Anhui/1/13) in comparison to H1N1 and H3N2 viruses in TMPRSS2 knockout (TMPRSS2−/−) and wild-type (WT) mice. We show that knockout of TMPRSS2 protects the mice from lung infection with lethal outcome by H7N9 and H1N1 viruses. In contrast, knockout of TMPRSS2 did not affect pneumotropism and lethality of H3N2 virus in mice, indicating that another protease is responsible for activation of H3 HA. These data identify TMPRSS2 as a host cell factor essential for pathogenicity of H7N9 and H1N1 but not H3N2 virus in mice and may contribute to the development of specific protease inhibitors as a novel therapeutic approach against influenza.
(C.T. performed this work in partial fulfillment of the requirements for a Ph.D. degree from the Philipps-University Marburg, Germany.)
MATERIALS AND METHODS
Ethics statement.
All animal experiments were performed according to the guidelines of the German animal protection law. All animal protocols were approved by the relevant German authorities, Behörde für Stadtentwicklung und Umwelt Hamburg (34/13) and the Regierungspräsidium Gießen (A 59/2012). Mice were euthanized upon 25% weight loss according to the guidelines of the German animal protection law.
Mice.
TMPRSS2-deficient mice (TMPRSS2−/−, C57BL/6 background) were described previously (20). Homozygous TMPRSS2 knockout mice (TMPRSS2−/−) and wild-type (WT) littermates were bred and housed in the animal facility of the Philipps-University Marburg. Animal experiments were performed at the animal facilities of the Heinrich-Pette-Institute, Leibniz Institute for Experimental Virology in Hamburg.
Ex vivo organ cultures.
For preparation of ex vivo organ cultures, 10- to 18-week-old mice were euthanized by CO2 asphyxiation. The respiratory tract was rapidly excised, and the larynges, tracheas, bronchi, and lungs were dissected according to Fig. 1A. Surrounding tissues were removed, and explants of trachea, bronchi, and lung were separately maintained in 12-well plates (1 explant per well). Incubation and infection of the explants were performed in Dulbecco's modified Eagle medium (DMEM; Gibco) supplemented with 10% fetal calf serum (FCS; Gibco), 5% glutamine, 5% penicillin/streptomycin, and 1% nonessential amino acids (NEAA; Gibco) (referred to here as explant medium) at 37°C and 5% CO2.
FIG 1.
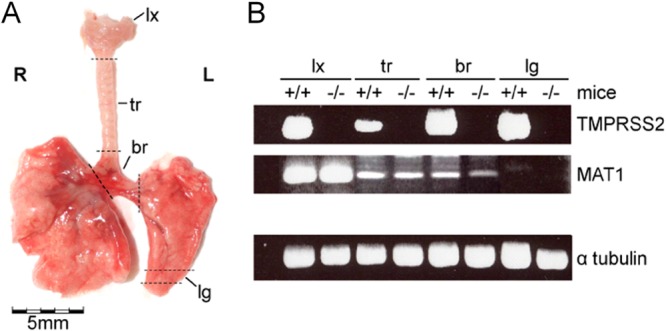
Expression of TMPRSS2 and MAT1 in ex vivo airway explants of wild-type and TMPRSS2 knockout mice. (A) Ventral view of the murine respiratory tract (R, right; L, left). The dashed lines delineate the larynx (lx), trachea (tr), bronchi (br), and lung (lg) explants. (B) RT-PCR analysis of MAT1- and TMPRSS2-specific mRNA in respiratory tissues of TMPRSS2 knockout (−/−) and wild-type (+/+) mice. Analysis of α-tubulin-specific mRNA was used as a control.
Cells and viruses.
Madin-Darby canine kidney (MDCK) cells were maintained in DMEM supplemented with 10% FCS (Gibco), glutamine, and antibiotics (growth medium). MDCK-MAT1 and MDCK-TMPRSS2 cells that express MAT1 or murine TMPRSS2 under doxycycline-dependent transcriptional activation were maintained in growth medium supplemented with 0.3 mg/ml Geneticin (Gibco) and 2 μg/ml puromycin (InvivoGen), and protease expression was induced by addition of 0.4 μg/ml doxycycline (InvivoGen). Infection experiments were performed using infection medium (DMEM supplemented with 0.1% bovine serum albumin [BSA], glutamine, and antibiotics). All cell growth and incubations were performed at 37°C and 5% CO2.
The influenza viruses used in this study were A/Anhui/1/13 (H7N9) (kindly provided by John McCauley, Division of Virology, MRC National Institute for Medical Research, London, United Kingdom), A/PuertoRico/8/34 (H1N1), A/Hamburg/NY1580/09 (H1N1pdm) and A/Aichi/2/68 (H3N2). H7N9, H3N2, and H1N1pdm were propagated in MDCK cells in infection medium containing 1 μg/ml tosyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma). H1N1 was propagated in 11-day-old embryonated chicken eggs. All experiments with H7N9 influenza virus were performed at biosafety level 3 (BSL3) facilities.
Antibodies.
A polyclonal antibody against HA of A/Anhui/1/13 (H7N9) was purchased from Sino Biological Inc. Rabbit serum against H1 was provided by Mikhail Matrosovich (Institute of Virology, Philipps-University Marburg), polyclonal rabbit sera against H3N2, H9N2, and H7N1 were derived from rabbits immunized with A/Aichi/2/68 (H3N2), A/Quail/Shantou/2061/00 (H9N2), and A/Chicken/Rostock/34 (H7N1; fowl plague virus [FPV]), respectively. Species-specific horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Dako.
RNA isolation and RT-PCR analysis.
Total RNA of murine respiratory tissues was extracted using the RNeasy minikit (Qiagen). Reverse transcription-PCR (RT-PCR) was carried out with 1 μg of total RNA by using the OneStep RT-PCR kit (Qiagen). H2O was used as a control. For detection of mRNA specific for TMPRSS2 or MAT1, specific sets of primers designed to amplify nucleotides (nt) 1 to 336 of MAT1-mRNA and nt 525 to 1473 of full-length murine TMPRSS2-mRNA, respectively, were used. RT-PCR of α-tubulin mRNA was used as an internal control. All primer sequences are available upon request.
Cloning of murine TMPRSS2 and MAT1 and generation of stable cell lines.
Full-length cDNAs of murine TMPRSS2 (GenBank accession number NM_015775.2) and MAT1 (GenBank accession number NM_145561.2) were cloned from total RNA of the lung and trachea, respectively, of C57BL/6 mice as described above using protease-specific primers. The cDNAs encoding murine TMPRSS2 and MAT1 were subcloned into the expression vector pTRE2pur (Clontech). All primer sequences are available upon request. Generation of MDCK-TMPRSS2 and MDCK-MAT1 was performed as previously described (21).
Multicycle virus replication and HA activation in MDCK-TMPRSS2 and MDCK-MAT1 cells.
To analyze virus activation and spread in MDCK-TMPRSS2 and MDCK-MAT1 cells, cells were seeded in 24-well plates and grown with or without 0.4 μg/ml doxycycline for 24 h. The cells were infected at a low multiplicity of infection (MOI) of 0.01 to 0.0001 and incubated for 24 h. Subsequently cells were fixed and immunostained against nucleoprotein (NP) as described previously (17). Briefly, cells were immunostained with a polyclonal rabbit serum against H9N2 virus, HRP-conjugated secondary antibodies, and subsequent incubation with the peroxidase substrate TrueBlue (KPL).
To analyze HA cleavage in protease-expressing cells, MDCK-TMPRSS2 and MDCK-MAT1 cells were grown in 6-well plates in the absence or presence of doxycycline for 24 h. Cells were infected at an MOI of 0.05 to 0.001 for 1 h, washed with phosphate-buffered saline (PBS) to remove the inoculum, and incubated for 40 h. Proteins in cell lysates or cell supernatants were analyzed by SDS-PAGE under reducing conditions and Western blotting using HA-specific antibodies and peroxidase-conjugated secondary antibodies as described previously (22). Proteins were visualized using the ChemiDoc XRS+ system with Image Lab software (Bio-Rad).
Infection and virus replication kinetics in ex vivo organ cultures.
For multicycle virus replication kinetics in ex vivo models of trachea, bronchi, and lung of TMPRSS2−/− and WT mice, explants were inoculated with 4 × 103 PFU of H7N9 or H1N1 or 105 PFU of H3N2 for 1 h. The inoculum was removed by careful washing, and infected explants were incubated in explant medium for 72 h. At 16, 24, 48, and 72 h postinfection, virus titers were determined as PFU/ml by plaque assay as described elsewhere (22).
Animal experiments.
All virus infection experiments in mice were performed at the animal facilities of the Heinrich-Pette-Institute, Leibniz Institute of Experimental Virology in Hamburg. All experiments followed the standard operating procedures of the approved biosafety protocols at biosafety level 2 (BSL2) and BSL3 (H7N9 influenza virus infections) animal facilities. WT (n = 16) and TMPRSS2−/− mice (n = 16) were intraperitoneally anesthetized with ketamine-xylazine (100 and 10 mg/kg, respectively) and intranasally inoculated with a volume of 50 μl of 105 PFU of H7N9, H1N1pdm, or H3N2 virus diluted in PBS. Mice receiving PBS only were used as control groups (n = 5). Animals were monitored for disease, survival, and weight loss for 14 days. On days 3 and 6 postinfection (p.i.), three animals were sacrificed, and tracheas and lungs were removed for subsequent analysis of virus titers and immunohistopathology as described previously (23).
Immunohistochemistry.
Tracheas and lungs of WT and TMPRSS2−/− mice (n = 3) were processed on day 3 p.i. for immunohistochemical staining (IHC) as described previously (23). Briefly, deparaffinized tissues were stained for viral antigens using an anti-FPV serum and a ZytoChem Plus HRP-3,3'-diaminobenzidine (DAB) broad-spectrum kit (Zytomed) according to the manufacturer′s protocols.
RESULTS
Expression of TMPRSS2 and MAT1 in airway tissues of WT and TMPRSS2−/− mice.
To study virus replication in respiratory tissues of TMPRSS2−/− and WT mice, we established ex vivo models of larynx, trachea, bronchus, and lung tissues (Fig. 1A). RT-PCR analysis using specific primer sets confirmed the presence of full-length TMPRSS2-mRNA in all organ explants of WT mice and its absence in TMPRSS2−/− mice (Fig. 1B). In addition, the presence of mRNA specific for the HAT-homologous protease murine airway trypsin-like protease 1 (MAT1) was analyzed in the different airway sections (24). MAT1 mRNA was present predominantly in the larynx and at lower levels in the trachea and bronchi but was not detected in the lung. These data indicate that WT mice express TMPRSS2 and MAT1 in the larynx, trachea, and bronchi but only TMPRSS2 in the lung. Accordingly, TMPRSS2−/− mice express MAT1 in the larynx, trachea, and bronchi but lack expression of the two proteases in the lungs.
H7N9 and H1N1 but not H3N2 influenza virus replication is inhibited in airway explants of TMPRSS2−/− mice.
To identify the role of TMPRSS2 in replication of A/Anhui/1/13 (H7N9) in mice, virus growth kinetics were examined in ex vivo models of the trachea, bronchi, and lung of WT and TMPRSS2−/− mice. Explants were infected and maintained under cell culture conditions for 72 h, and virus titers in the supernatants were determined at the indicated time points by plaque assay. As shown in Fig. 2, H7N9 virus replicated efficiently in tracheal, bronchial, and lung explants of WT mice. Interestingly, propagation of H7N9 was completely inhibited in all airway explants of TMPRSS2−/− mice. These findings demonstrate that TMPRSS2 is essential for replication of H7N9 influenza virus in murine respiratory cells.
FIG 2.

Replication of H7N9 and H1N1 but not H3N2 influenza virus is inhibited in airway explants of TMPRSS2 knockout mice. Growth kinetics of H7N9, H1N1, and H3N2 viruses in ex vivo organ cultures of TMPRSS2−/− and WT mice. Explants of murine trachea, bronchi, or lung were infected with 4 × 103 PFU H7N9 or H1N1 or 105 PFU of H3N2. At the indicated time points p.i., virus titers were determined by plaque assay. Error bars denote standard deviations.
Furthermore, replication of the human strains A/PR8/1/34 (H1N1) and A/Aichi/2/68 (H3N2) was analyzed in airway explants of TMPRSS2−/− and WT mice. Multicycle replication of H1N1 was observed in all explants from WT mice. In contrast, virus propagation was strongly suppressed (∼1,000- to 10,000-fold) in organ cultures of tracheas and bronchi from TMPRSS2−/− mice and was completely inhibited in lung explants (Fig. 2). Interestingly, H3N2 virus was able to replicate in tracheal and bronchial explants of both TMPRSS2−/− and WT mice. Propagation of H3N2 virus was reduced about 30- to 100-fold in the trachea but was only marginally affected in bronchial cultures of TMPRSS2−/− mice. We were unable to detect replication of H3N2 in lung explants, but the virus replicated efficiently in the lungs of infected mice (see Fig. 4 and 5). Taken together, these data demonstrate that TMPRSS2 supports efficient replication of H1N1 influenza virus in the murine trachea, bronchi, and lungs, whereas it is dispensable for H3N2 influenza virus replication in these tissues.
FIG 4.

TMPRSS2 is essential for pathogenicity of H7N9 and H1N1pdm, but not H3N2 virus in mice. Pathogenicity of H7N9, H1N1pdm, and H3N2 influenza virus in WT and TMPRSS2−/− mice. TMPRSS2−/− mice and wild-type littermates were infected with 105 PFU of the indicated virus. Survival (A) and weight loss (B) were monitored for 14 days. Mice receiving PBS were used as control. Error bars denote standard deviations.
FIG 5.
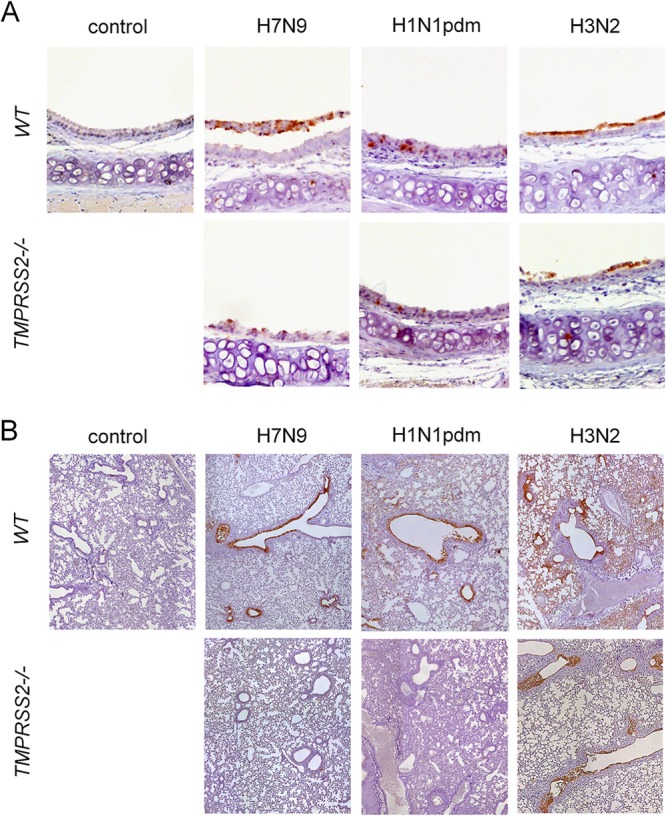
Knockout of TMPRSS2 prevents spread of H7N9 and H1N1pdm into the lung. Immunohistochemical analysis of tracheas and lungs of TMPRSS2−/− and WT mice infected with 105 PFU of H7N9, H1N1pdm, or H3N2 influenza virus was carried out. Control groups received PBS. Tracheas (A) and lungs (B) of mice (n = 3) were processed on day 3 p.i. for immunohistochemical stainings as described before (23). Subsequently, sections were counterstained with hematoxylin. Antigen-positive cells are red-brown.
Proteolytic activation of H7, H1, and H3 hemagglutinins by TMPRSS2 and MAT1.
To assess whether murine TMPRSS2 and MAT1 are able to activate H7, H1, and H3, the cDNAs encoding the proteases were cloned from murine lungs and tracheas, respectively, and used to generate stable MDCK-TMPRSS2 and MDCK-MAT1 cells with doxycycline-inducible protease expression. Cells were infected with H7N9 virus at a low MOI, and either cells were immunostained against the viral nucleoprotein at 24 h p.i., or cell lysates were subjected to SDS-PAGE and Western blot analysis at 40 h p.i. to examine cleavage of the HA0 precursor into the disulfide-linked subunits HA1 and HA2. As shown in Fig. 3A, multicycle replication of H7N9 was observed in both MAT1- and TMPRSS2-expressing cells, although foci of infection in MDCK-MAT1 cells were smaller than those in MDCK-TMPRSS2 cells. Western blot analysis confirmed cleavage of HA0 by coexpression of TMPRSS2 and slightly reduced levels of HA cleavage by coexpression of MAT1 (Fig. 3B). No HA1 was detected in cell lysates of infected cells, and at present it remains unclear whether HA1 is degraded or not recognized by the antibody.
FIG 3.
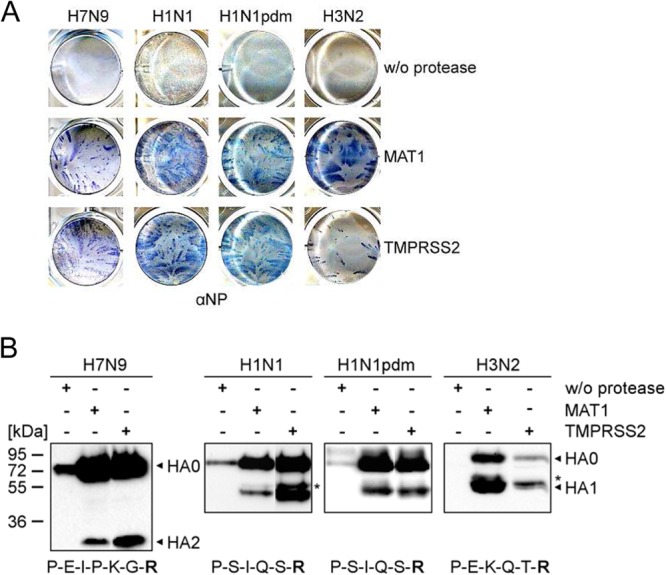
Proteolytic activation of H7N9, H1N1, and H3N2 influenza viruses by TMPRSS2 and MAT1 in vitro. (A) Multicycle virus replication in MDCK-TMPRSS2 and MDCK-MAT1 cells. MDCK cells that express either protease under doxycycline-induced transcriptional activation were infected with the indicated virus at a low MOI of 0.01 to 0.0001 and incubated for 24 h. Cells were fixed and immunostained against the viral nucleoprotein (NP). Cells without (w/o) induction of protease expression were used as controls. (B) Protease-expressing MDCK-TMPRSS2 and MDCK-MAT1 cells were infected with the indicated virus and incubated for 40 h. Virus-containing supernatants or cell lysates (H7N9-infected cells) were subjected to SDS-PAGE under reducing conditions and Western blotting using virus-specific antibodies. Cells without induction of protease expression were used as controls. Viral NP is indicated by an asterisk. The amino acid sequence at the HA cleavage site of each virus is indicated in the one-letter code.
In addition, proteolytic activation of influenza viruses of subtypes H1N1, H1N1pdm, and H3N2 was analyzed in MDCK-TMPRSS2 and MDCK-MAT1 cells. Spread of both H1N1 and H1N1pdm virus was similar in MAT1- and TMPRSS2-expressing cell lines, and released virus contained cleaved HA (Fig. 3). HA of H1N1 was more susceptible to cleavage by TMPRSS2, whereas HA of H1N1pdm was efficiently cleaved by both proteases. Interestingly, only small foci of H3N2 were visible in MDCK-TMPRSS2 cells, while large virus foci were observed in MDCK-MAT1 cells. H3 HA was efficiently cleaved by MAT1, supporting multicycle virus replication and large amounts of progeny virus in the cell supernatant, whereas only small amounts of virus were present in supernatants of TMPRSS2-expressing cells. MDCK cells lacking expression of either protease were used as controls. As expected, none of the viruses was able to undergo multicycle replication in these cells, and only HA0 was detected by immunoblotting (Fig. 3). These data show that H7N9 and H1N1 viruses are activated by TMPRSS2 and MAT1, while H3N2 virus is more efficiently activated by MAT1. Interestingly, the differences in susceptibility to TMPRSS2 correlate with the obtained virus growth kinetics in airway explants. The data suggest that MAT1 may support multicycle replication of H3N2 virus in tracheas and bronchi of TMPRSS2−/− mice and probably also WT mice. In contrast, although MAT1 is able to activate H7 and H1, it seems to be not or only barely sufficient to support activation of H7N9 and H1N1 viruses in murine respiratory cells.
H7N9 and H1N1pdm viruses are apathogenic, while H3N2 virus causes lethal infection in TMPRSS2−/− mice.
The data obtained here suggest that HA activation by TMPRSS2 might play an important role in the pathogenesis of H7N9 and H1N1 viruses in mice. We therefore infected TMPRSS2−/− and WT mice intranasally with a high virus dose (105 PFU), and survival and weight loss were monitored for 14 days. All WT mice infected with H7N9 displayed significant weight loss and succumbed to infection within 8 days (Fig. 4). Remarkably, TMPRSS2−/− mice neither lost weight nor developed symptoms of disease, and all animals survived. Immunohistochemical analysis of viral antigen expression on day 3 p.i. revealed prominent numbers of infected cells in the tracheas of WT mice but low numbers of infected cells in TMPRSS2−/− mice (Fig. 5A). Widespread staining of virus-positive cells was observed in lungs of WT mice, whereas no infected cells were visible in lungs of TMPRSS2−/− mice (Fig. 5B). Infection of WT mice with H1N1pdm also led to severe weight loss, and 20% of the animals died (Fig. 4). In contrast, all TMPRSS2−/− mice survived without displaying weight loss or signs of disease. TMPRSS2−/− mice presented reduced numbers of infected cells in the tracheas, and no virus-positive cells were detected in the lungs compared to large numbers of infected cells in the lungs of WT mice (Fig. 5).
In contrast, infection of mice with 105 PFU H3N2 virus resulted in 100% lethality in both WT and TMPRSS2−/− animals within 6 days after infection (Fig. 4). Immunohistochemical analysis revealed large numbers of virus-positive cells in the tracheas of WT and TMPRSS2−/− mice without marked differences (Fig. 5A), whereas slightly reduced viral antigen expression was observed in the lungs of TMPRSS2−/− mice compared to WT mice (Fig. 5B).
These observations demonstrate that knockout of the TMPRSS2 gene prevents spread of H7N9 and H1N1pdm viruses into the lung and development of disease in mice. In contrast, pneumotropism and pathogenicity of H3N2 virus do not depend on TMPRSS2, suggesting that, in this case, another protease is involved in virus activation.
DISCUSSION
The present study demonstrates that knockout of the type II transmembrane protease TMPRSS2 inhibits virus replication and pneumotropism and consequently pathogenicity of human H7N9 and H1N1 influenza viruses in mice. A/Anhui/1/13 (H7N9) virus isolated from a fatal human case has been shown to be highly pathogenic in mice (4, 6, 25, 26). Intriguingly, H7N9 was apathogenic in TMPRSS2−/− mice, whereas WT mice developed severe infection with 100% mortality. Similarly, H1N1pdm caused apathogenic infection in TMPRSS2−/− mice in contrast to severe infection with 20% mortality in WT mice. Growth kinetics in ex vivo airway models and immunohistochemical staining of viral antigen in infected mice demonstrated that knockout of TMPRSS2 prevents multicycle replication and virus spread into the lung. In contrast, replication and pathogenicity of H3N2 were only marginally affected by knockout of TMPRSS2, and both WT and TMPRSS2−/− mice succumbed to disease upon H3N2 infection.
Multicycle virus replication in protease-expressing MDCK cells revealed that H7N9 and H1N1 are activated by both murine TMPRSS2 and the HAT-homologous protease MAT1 in vitro, while H3N2 virus is efficiently activated only by MAT1. Consistent with previous studies (24, 27), we showed by RT-PCR analysis of MAT1-mRNA that MAT1 is expressed in the tracheas and bronchi but not in the lungs of mice. Therefore, MAT1 may support proteolytic activation of H3N2 virus in murine trachea and bronchi, but another protease is responsible for activation of this virus in the lung. Low levels of H1N1 replication in tracheal and bronchial explants of TMPRSS2−/− mice may also be due to MAT1 activity. However, the observations indicate that MAT1 is not crucial for proteolytic activation and spread of H7N9 and H1N1 influenza viruses in mice.
Explants of murine tracheas, bronchi, and lungs proved to be suitable models in our hands to investigate virus replication and tissue tropism in the respiratory tract. Multicycle replication of H7N9 and H1N1 in airway explants correlated with virus spread in vivo. The lack of H3N2 propagation in lung explants, however, was probably a matter of insufficient initial infection and maintenance of lung explants, since large numbers of virus-positive cells were detected in the lungs of infected mice.
Replication and virulence of H3N2 virus were independent of TMPRSS2 in mice, indicating that another protease can support activation of H3 HA. At present, it remains unknown which protease activates H3N2 in the lungs of mice. A number of proteases, including trypsin, plasmin, and kallikreins as well as tryptase Clara, have been demonstrated to cleave HA with monobasic cleavage site in vitro (28–31). A recent study reported that kallikrein-related peptidase 5 (KLK5) cleaves the HAs of different human H1 and H3 viruses, including strains A/PR8/1/34 (H1N1) and A/Aichi/2/68 (H3N2) in vitro (31). The TMPRSS2-related protease TMPRSS4 has been described to activate HA of the 1918 H1N1 pandemic virus and A/PR8/1/34 (H1N1) upon coexpression (32, 33). Furthermore, tryptase Clara purified from rat lung was able to cleave H3 of A/Aichi/2/68 (H3N2) in vitro; however, the genetic identity of the protease is still unknown (30). The observations here suggest that activation of H3N2 in WT and TMPRSS2−/− mice depends on a protease that does not support proteolytic activation of H1N1 and H7N9, and it remains to be determined whether one of the above-mentioned proteases is the right candidate.
Influenza virus HA is thought to be cleaved either at a single arginine-glycine bond or at a multibasic motif by trypsin-like proteases and furin, respectively. However, a number of studies, including the present one, demonstrate that the situation is more complex. Influenza strains can possess uncommon HA cleavage site motifs that facilitate cleavage by other or additional proteases. The influenza strain A/WSN/33 (H1N1), which is neurotropic in mice, contains the unusual H1 HA cleavage site I-Q-Y-R instead of I-Q-S-R (Fig. 3B) with a tyrosine (Y) in position P2 that was shown to facilitate efficient cleavage by plasmin, supporting proteolytic activation and virus spread to other tissues, including the brain (29, 34, 35). Many H9N2 influenza isolates from Asia and the Middle East possess di- or tribasic motifs R-S-S-R and R-S-R-R, respectively. In contrast to HPAIV of subtype H5 and H7, the cleavage sites of H9N2 viruses evolved by substitution and not by insertion of basic amino acids and are not cleaved by furin (36, 37). Recently, the type II transmembrane protease matriptase was shown to cleave H9 at R-S-S-R and R-S-R-R motifs (38). Matriptase is expressed in a wide range of tissues, including the kidney, and therefore may contribute to nephrotropism of H9N2 viruses in chickens. Importantly, the findings described here and by others (39, 40) reveal that HA with a monobasic cleavage site differs in protease specificity. A recent study assessed cleavage of the 16 HA subtypes by human TMPRSS2 and HAT in vitro and observed that cleavage efficiency can vary considerably among the subtypes (40). Furthermore, previous studies demonstrated that proteases secreted by some strains of Staphylococcus aureus are capable of cleaving influenza virus HA with a monobasic cleavage site (39). Interestingly, the susceptibility of different HAs to cleavage by bacterial proteases also varied among subtypes and virus strains. It will be of particular interest to unravel the protease specificity of HA with a monobasic cleavage site.
At present, further investigations are needed to understand the molecular basis of the differences in the protease specificity of H7N9 and H1N1 viruses on one hand and of H3N2 virus on the other hand. During the final preparation of our manuscript, a study by Hatesuer and coworkers that also investigated virus replication and pathogenicity of H1N1 and H3N2 but not H7N9 influenza viruses in mice lacking expression of TMPRSS2 was published (41). Consistent with our data, they found that knockout of TMPRSS2 inhibited replication and pathogenicity of H1N1 influenza virus, while H3N2 virus was still able to replicate and cause disease in TMPRSS2-deficient mice.
The physiological role of TMPRSS2 remains to be investigated. TMPRSS2 is associated with prostate carcinogenesis. The protease has been shown to be overexpressed in prostate cancer tissue (42, 43). In addition, fusion of the androgen-regulated TMPRSS2 promoter to different E-twenty-six (ETS) transcription factor genes has been established as a prognostic marker of prostate carcinogenesis (44). In the airways, TMPRSS2 might be involved in regulation of the airway surface liquid (ASL) volume by proteolytic cleavage of epithelial sodium channels (ENaCs) (45). Importantly, TMPRSS2−/− mice lack a discernible phenotype, suggesting functional redundancy in the host (20). Furthermore, there is evidence that TMPRSS2 might play a pathogenetic role in other respiratory infections, since it has been shown to activate also the fusion proteins of influenza B virus, human metapneumovirus, respiratory parainfluenza viruses, and different coronaviruses in vitro (46–49).
Already in the 1980s, Zhirnov and coworkers demonstrated that inhibition of HA cleavage by the natural protease inhibitor aprotinin suppresses influenza virus replication in vitro and in vivo (reviewed in reference 50). The identification of TMPRSS2 as an HA-activating protease provided a drug target for the development of specific protease inhibitors. Recent studies showed that inhibition of TMPRSS2 activity in human Calu-3 airway epithelial cells by peptide-conjugated phosphorodiamidate morpholino oligomers (PPMO) or peptide mimetic inhibitors of TMPRSS2 strongly suppresses human influenza virus replication (22, 47, 51). Interestingly, the combination of TMPRSS2 inhibitors and the neuraminidase (NA) inhibitor oseltamivir carboxylate was synergistic and efficiently blocked replication of H1N1pdm influenza virus in Calu-3 cells (47). Human H7N9 viruses have been shown to be sensitive to current NA inhibitors, and oseltamivir treatment protected mice from lethal H7N9 virus infection (6, 26). Therefore, the combination of TMPRSS2 inhibitors and NA inhibitors may provide a potential and novel approach for influenza chemotherapy.
In summary, the present study identified TMPRSS2 as a host cell factor essential for H7N9 and H1N1 influenza virus pneumotropism and pathogenicity in mice. Our findings highlight the crucial role of HA cleavage for influenza virus spread in the respiratory tract and provide support for the concept of targeting HA-activating proteases and in particular TMPRSS2 as therapeutic approach for influenza treatment.
ACKNOWLEDGMENTS
We thank John McCauley for providing the A/Anhui/1/13 (H7N9) virus. We thank Gundula Pilnitz-Stolze for assistance with immunohistochemical stainings. We thank the personnel from the animal facilities of the Institute of Virology at the Philipps-University in Marburg and the Heinrich-Pette-Institute, Leibniz Institute for Experimental Virology in Hamburg.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) SFB 593, the Emmy-Noether Programme (to G.G.) as well as grants from the Von-Behring Röntgen Stiftung (to E.B.-F.), the Universities of Giessen and Marburg Lung Center (UGMLC) (to E.B.-F.), the Fonds National de la Recherche, Luxembourg (to G.E.), and NCI grant P01CA097186 (to P.S.N.). The Heinrich-Pette-Institute is supported by the Free and Hanseatic City of Hamburg and the Federal Ministry of Health.
Footnotes
Published ahead of print 12 February 2014
REFERENCES
- 1.Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, Li Y, Yao X, Zhang Y, Wu H, Zheng S, Diao H, Xia S, Zhang Y, Chan KH, Tsoi HW, Teng JL, Song W, Wang P, Lau SY, Zheng M, Chan JF, To KK, Chen H, Li L, Yuen KY. 2013. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet 381:1916–1925. 10.1016/S0140-6736(13)60903-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, Xu X, Lu H, Zhu W, Gao Z, Xiang N, Shen Y, He Z, Gu Y, Zhang Z, Yang Y, Zhao X, Zhou L, Li X, Zou S, Zhang Y, Li X, Yang L, Guo J, Dong J, Li Q, Dong L, Zhu Y, Bai T, Wang S, Hao P, Yang W, Zhang Y, Han J, Yu H, Li D, Gao GF, Wu G, Wang Y, Yuan Z, Shu Y. 2013. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 368:1888–1897. 10.1056/NEJMoa1304459 [DOI] [PubMed] [Google Scholar]
- 3.Liu D, Shi W, Shi Y, Wang D, Xiao H, Li W, Bi Y, Wu Y, Li X, Yan J, Liu W, Zhao G, Yang W, Wang Y, Ma J, Shu Y, Lei F, Gao GF. 2013. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: phylogenetic, structural, and coalescent analyses. Lancet 381:1926–1932. 10.1016/S0140-6736(13)60938-1 [DOI] [PubMed] [Google Scholar]
- 4.Belser JA, Gustin KM, Pearce MB, Maines TR, Zeng H, Pappas C, Sun X, Carney PJ, Villanueva JM, Stevens J, Katz JM, Tumpey TM. 2013. Pathogenesis and transmission of avian influenza A (H7N9) virus in ferrets and mice. Nature 501:556–559. 10.1038/nature12391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richard M, Schrauwen EJ, de Graaf M, Bestebroer TM, Spronken MI, van Boheemen S, de Meulder D, Lexmond P, Linster M, Herfst S, Smith DJ, van den Brand JM, Burke DF, Kuiken T, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2013. Limited airborne transmission of H7N9 influenza A virus between ferrets. Nature 501:560–563. 10.1038/nature12476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe T, Kiso M, Fukuyama S, Nakajima N, Imai M, Yamada S, Murakami S, Yamayoshi S, Iwatsuki-Horimoto K, Sakoda Y, Takashita E, McBride R, Noda T, Hatta M, Imai H, Zhao D, Kishida N, Shirakura M, de Vries RP, Shichinohe S, Okamatsu M, Tamura T, Tomita Y, Fujimoto N, Goto K, Katsura H, Kawakami E, Ishikawa I, Watanabe S, Ito M, Sakai-Tagawa Y, Sugita Y, Uraki R, Yamaji R, Eisfeld AJ, Zhong G, Fan S, Ping J, Maher EA, Hanson A, Uchida Y, Saito T, Ozawa M, Neumann G, Kida H, Odagiri T, Paulson JC, Hasegawa H, Tashiro M, Kawaoka Y. 2013. Characterization of H7N9 influenza A viruses isolated from humans. Nature 501:551–555. 10.1038/nature12392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiong X, Martin SR, Haire LF, Wharton SA, Daniels RS, Bennett MS, McCauley JW, Collins PJ, Walker PA, Skehel JJ, Gamblin SJ. 2013. Receptor binding by an H7N9 influenza virus from humans. Nature 499:496–499. 10.1038/nature12372 [DOI] [PubMed] [Google Scholar]
- 8.Zhang Q, Shi J, Deng G, Guo J, Zeng X, He X, Kong H, Gu C, Li X, Liu J, Wang G, Chen Y, Liu L, Liang L, Li Y, Fan J, Wang J, Li W, Guan L, Li Q, Yang H, Chen P, Jiang L, Guan Y, Xin X, Jiang Y, Tian G, Wang X, Qiao C, Li C, Bu Z, Chen H. 2013. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science 341:410–414. 10.1126/science.1240532 [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Wang D, Gao R, Zhao B, Song J, Qi X, Zhang Y, Shi Y, Yang L, Zhu W, Bai T, Qin K, Lan Y, Zou S, Guo J, Dong J, Dong L, Zhang Y, Wei H, Li X, Lu J, Liu L, Zhao X, Li X, Huang W, Wen L, Bo H, Xin L, Chen Y, Xu C, Pei Y, Yang Y, Zhang X, Wang S, Feng Z, Han J, Yang W, Gao GF, Wu G, Li D, Wang Y, Shu Y. 2013. Biological features of novel avian influenza A (H7N9) virus. Nature 499:500–503. 10.1038/nature12379 [DOI] [PubMed] [Google Scholar]
- 10.Subbarao EK, London W, Murphy BR. 1993. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol. 67:1761–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabriel G, Dauber B, Wolff T, Planz O, Klenk HD, Stech J. 2005. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. U. S. A. 102:18590–18595. 10.1073/pnas.0507415102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knepper J, Schierhorn KL, Becher A, Budt M, Tönnies M, Bauer TT, Schneider P, Neudecker J, Rückert JC, Gruber AD, Suttorp N, Schweiger B, Hippenstiel S, Hocke AC, Wolff T. 2013. The novel human influenza A(H7N9) virus is naturally adapted to efficient growth in human lung tissue. mBio 4:e00601–13. 10.1128/mBio.00601-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Riel D, Leijten LM, de Graaf M, Siegers JY, Short KR, Spronken MI, Schrauwen EJ, Fouchier RA, Osterhaus AD, Kuiken T. 2013. Novel avian-origin influenza A (H7N9) virus attaches to epithelium in both upper and lower respiratory tract of humans. Am. J. Pathol. 183:1137–1143. 10.1016/j.ajpath.2013.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu H, Wang D, Kelvin DJ, Li L, Zheng Z, Yoon SW, Wong SS, Farooqui A, Wang J, Banner D, Chen R, Zheng R, Zhou J, Zhang Y, Hong W, Dong W, Cai Q, Roehrl MH, Huang SS, Kelvin AA, Yao T, Zhou B, Chen X, Leung GM, Poon LL, Webster RG, Webby RJ, Peiris JS, Guan Y, Shu Y. 2013. Infectivity, transmission, and pathology of human-isolated H7N9 influenza virus in ferrets and pigs. Science 341:183–186. 10.1126/science.1239844 [DOI] [PubMed] [Google Scholar]
- 15.Garten W, Klenk HD. 2008. Cleavage activation of the influenza virus hemagglutinin and its role in pathogenesis. Monogr. Virol. 27:156–167. 10.1159/000151618 [DOI] [Google Scholar]
- 16.Steinhauer DA. 1999. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258:1–20. 10.1006/viro.1999.9716 [DOI] [PubMed] [Google Scholar]
- 17.Böttcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 80:9896–9898. 10.1128/JVI.01118-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stieneke-Gröber A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, Klenk HD, Garten W. 1992. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 11:2407–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horimoto T, Nakayama K, Smeekens SP, Kawaoka Y. 1994. Proprotein-processing endoproteases PC6 and furin both activate hemagglutinin of virulent avian influenza viruses. J. Virol. 68:6074–6078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim TS, Heinlein C, Hackman RC, Nelson PS. 2006. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol. Cell. Biol. 26:965–975. 10.1128/MCB.26.3.965-975.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Böttcher E, Freuer C, Steinmetzer T, Klenk HD, Garten W. 2009. MDCK cells that express proteases TMPRSS2 and HAT provide a cell system to propagate influenza viruses in the absence of trypsin and to study cleavage of HA and its inhibition. Vaccine 27:6324–6329. 10.1016/j.vaccine.2009.03.029 [DOI] [PubMed] [Google Scholar]
- 22.Böttcher-Friebertshäuser E, Stein DA, Klenk HD, Garten W. 2011. Inhibition of influenza virus infection in human airway cell cultures by an antisense peptide-conjugated morpholino oligomer targeting the hemagglutinin- activating protease TMPRSS2. J. Virol. 85:1554–1562. 10.1128/JVI.01294-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otte A, Sauter M, Alleva L, Baumgarte S, Klingel K, Gabriel G. 2011. Differential host determinants contribute to the pathogenesis of 2009 pandemic H1N1 and human H5N1 influenza A viruses in experimental mouse models. Am. J. Pathol. 179:230–239. 10.1016/j.ajpath.2011.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen IA, Fassnacht M, Hahner S, Hammer F, Schammann M, Meyer SR, Bicknell AB, Allolio B. 2004. The adrenal secretory serine protease AsP is a short secretory isoform of the transmembrane airway trypsin-like protease. Endocrinology 145:1898–1905. 10.1210/en.2003-0930 [DOI] [PubMed] [Google Scholar]
- 25.Mok CK, Lee HH, Chan MC, Sia SF, Lestra M, Nicholls JM, Zhu H, Guan Y, Peiris JM. 2013. Pathogenicity of the novel A/H7N9 influenza virus in mice. mBio 4:e00362–13. 10.1128/mBio.00601-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baranovich T, Burnham AJ, Marathe BM, Armstrong J, Guan Y, Shu Y, Peiris JM, Webby RJ, Webster RG, Govorkova EA. The neuraminidase inhibitor oseltamivir is effective against A/Anhui/1/2013 (H7N9) influenza virus in a mouse model of acute respiratory distress syndrome. J. Infect. Dis., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sales KU, Hobson JP, Wagenaar-Miller R, Szabo R, Rasmussen AL, Bey A, Shah MF, Molinolo AA, Bugge TH. 2011. Expression and genetic loss of function analysis of the HAT/DESC cluster proteases TMPRSS11A and HAT. PLoS One 6:e23261. 10.1371/journal.pone.0023261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klenk HD, Rott R, Orlich M, Blödorn J. 1975. Activation of influenza A viruses by trypsin treatment. Virology 68:426–439. 10.1016/0042-6822(75)90284-6 [DOI] [PubMed] [Google Scholar]
- 29.Lazarowitz SG, Choppin PW. 1975. Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology 68:440–454. 10.1016/0042-6822(75)90285-8 [DOI] [PubMed] [Google Scholar]
- 30.Kido H, Okumura Y, Yamada H, Le TQ, Yano M. 2007. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr. Pharm. Des. 13:405–414. 10.2174/138161207780162971 [DOI] [PubMed] [Google Scholar]
- 31.Hamilton BS, Whittaker GR. 2013. Cleavage activation of human-adapted influenza virus subtypes by kallikrein-related peptidases 5 and 12. J. Biol. Chem. 288:17399–17407. 10.1074/jbc.M112.440362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, Takeda M, Bugge TH, Kim S, Park Y, Marzi A, Pöhlmann S. 2009. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 83:3200–3211. 10.1128/JVI.02205-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertram S, Glowacka I, Blazejewska P, Soilleux E, Allen P, Danisch S, Steffen I, Choi SY, Park Y, Schneider H, Schughart K, Pöhlmann S. 2010. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J. Virol. 84:10016–10025. 10.1128/JVI.00239-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goto H, Kawaoka Y. 1998. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 95:10224–10228. 10.1073/pnas.95.17.10224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun X, Tse LV, Ferguson AD, Whittaker GR. 2010. Modifications to the hemagglutinin cleavage site control the virulence of a neurotropic H1N1 influenza virus. J. Virol. 84:8683–8690. 10.1128/JVI.00797-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gohrbandt S, Veits J, Breithaupt A, Hundt J, Teifke JP, Stech O, Mettenleiter TC, Stech J. 2011. H9 avian influenza reassortant with engineered polybasic cleavage site displays a highly pathogenic phenotype in chicken. J. Gen. Virol. 92:1843–1853. 10.1099/vir.0.031591-0 [DOI] [PubMed] [Google Scholar]
- 37.Soda K, Asakura S, Okamatsu M, Sakoda Y, Kida H. 2011. H9N2 influenza virus acquires intravenous pathogenicity on the introduction of a pair of di-basic amino acid residues at the cleavage site of the hemagglutinin and consecutive passages in chickens. Virol. J. 8:64. 10.1186/1743-422X-8-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baron J, Tarnow C, Mayoli-Nüssle D, Schilling E, Meyer D, Hammami M, Schwalm F, Steinmetzer T, Guan Y, Garten W, Klenk HD, Böttcher-Friebertshäuser E. 2013. Matriptase, HAT, and TMPRSS2 activate the hemagglutinin of H9N2 influenza A viruses. J. Virol. 87:1811–1820. 10.1128/JVI.02320-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tashiro M, Ciborowski P, Reinacher M, Pulverer G, Klenk HD, Rott R. 1987. Synergistic role of staphylococcal proteases in the induction of influenza virus pathogenicity. Virology 157:421–430. 10.1016/0042-6822(87)90284-4 [DOI] [PubMed] [Google Scholar]
- 40.Galloway SE, Reed ML, Russell CJ, Steinhauer DA. 2013. Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: implications for host range and adaptation. PLoS Pathog. 9:e1003151. 10.1371/journal.ppat.1003151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pöhlman S, Schughart K. 2013. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 12:e1003774. 10.1371/journal.ppat.1003774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, Hood L, Nelson PS. 1999. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res. 59:4180–4184 [PubMed] [Google Scholar]
- 43.Lucas JM, True L, Hawley S, Matsumura M, Morrissey C, Vessella R, Nelson PS. 2008. The androgen-regulated type II serine protease TMPRSS2 is differentially expressed and mislocalized in prostate adenocarcinoma. J. Pathol. 215:118–125. 10.1002/path.2330 [DOI] [PubMed] [Google Scholar]
- 44.Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA, Schalken JA. 2009. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur. Urol. 56:275–286. 10.1016/j.eururo.2009.04.036 [DOI] [PubMed] [Google Scholar]
- 45.Donaldson SH, Hirsh A, Li DC, Holloway G, Chao J, Boucher RC, Gabriel SE. 2002. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 277:8338–8345. 10.1074/jbc.M105044200 [DOI] [PubMed] [Google Scholar]
- 46.Shirogane Y, Takeda M, Iwasaki M, Ishiguro N, Takeuchi H, Nakatsu Y, Tahara M, Kikuta H, Yanagi Y. 2008. Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J. Virol. 82:8942–8946. 10.1128/JVI.00676-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Böttcher-Friebertshäuser E, Lu Y, Meyer D, Sielaff F, Steinmetzer T, Klenk HD, Garten W. 2012. Hemagglutinin activating host cell proteases provide promising drug targets for the treatment of influenza A and B virus infections. Vaccine 30:7374–7380. 10.1016/j.vaccine.2012.10.001 [DOI] [PubMed] [Google Scholar]
- 48.Abe M, Tahara M, Sakai K, Yamaguchi H, Kanou K, Shirato K, Kawase M, Noda M, Kimura H, Matsuyama S, Fukuhara H, Mizuta K, Maenaka K, Ami Y, Esumi M, Kato A, Takeda M. 2013. TMPRSS2 is an activating protease for respiratory parainfluenza viruses. J. Virol. 87:11930–11935. 10.1128/JVI.01490-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S. 2013. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res. 100:605–614. 10.1016/j.antiviral.2013.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhirnov OP, Klenk HD, Wright PF. 2011. Aprotinin and similar protease inhibitors as drugs against influenza. Antiviral Res. 92:27–36. 10.1016/j.antiviral.2011.07.014 [DOI] [PubMed] [Google Scholar]
- 51.Meyer D, Sielaff F, Hammami M, Böttcher-Friebertshäuser E, Garten W, Steinmetzer T. 2013. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem. J. 452:331–343. 10.1042/BJ20130101 [DOI] [PubMed] [Google Scholar]
- 52.WHO. 25 October 2013. Number of confirmed human cases of avian influenza A(H7N9) reported to WHO. Report 10. WHO, Geneva, Switzerland: http://www.who.int/influenza/human_animal_interface/influenza_h7n9/10u_ReportWebH7N9Number.pdf [Google Scholar]
- 53.European Centre for Disease Prevention and Control. 27 January 2014. Updated rapid risk assessment: human infection with a novel avian influenza A(H7N9) virus, China, 3rd update. European Centre for Disease Prevention and Control, Stockholm, Sweden: http://www.ecdc.europa.eu/en/publications/Publications/influenza-AH7N9-China-rapid-risk-assessment-27-January-2014.pdf [Google Scholar]