

ABSTRACT
The Epstein-Barr virus (EBV) establishes a lifelong latent infection in humans. EBV infection of primary B cells causes cell activation and proliferation, a process driven by the viral latency III gene expression program, which includes EBV nuclear proteins (EBNAs), latent membrane proteins, and untranslated RNAs, including microRNAs. Some latently infected cells enter the long-lived memory B-cell compartment and express only EBNA1 transiently (Lat I) or no EBV protein at all (Lat 0). Targeting the molecular machinery that controls B-cell fate decisions, including the Bcl-2 family of apoptosis-regulating proteins, is crucial to the EBV cycle of infection. Here, we show that BIK (also known as NBK), which encodes a proapoptotic “sensitizer” protein, is repressed by the EBNA2-driven Lat III program but not the Lat I program. BIK repression occurred soon after infection of primary B cells by EBV but not by a recombinant EBV in which the EBNA2 gene had been knocked out. Ectopic BIK induced apoptosis in Lat III cells by a mechanism dependent on its BH3 domain and the activation of caspases. We show that EBNA2 represses BIK in EBV-negative B-cell lymphoma-derived cell lines and that this host-virus interaction can inhibit the proapoptotic effect of transforming growth factor β1 (TGF-β1), a key physiological mediator of B-cell homeostasis. Reduced levels of TGF-β1-associated regulatory SMAD proteins were bound to the BIK promoter in response to EBV Lat III or ectopic EBNA2. These data are evidence of an additional mechanism used by EBV to promote B-cell survival, namely, the transcriptional repression of the BH3-only sensitizer BIK.
IMPORTANCE Over 90% of adult humans are infected with the Epstein-Barr virus (EBV). EBV establishes a lifelong silent infection, with its DNA residing in small numbers of blood B cells that are a reservoir from which low-level virus reactivation and shedding in saliva intermittently occur. Importantly, EBV DNA is found in some B-cell-derived tumors in which viral genes play a key role in tumor cell emergence and progression. Here, we report for the first time that EBV can shut off a B-cell gene called BIK. When activated by a molecular signal called transforming growth factor β1 (TGF-β1), BIK plays an important role in killing unwanted B cells, including those infected by viruses. We describe the key EBV–B-cell molecular interactions that lead to BIK shutoff. These findings further our knowledge of how EBV prevents the death of its host cell during infection. They are also relevant to certain posttransplant lymphomas where unregulated cell growth is caused by EBV genes.
INTRODUCTION
Epstein-Barr virus (EBV) is a B lymphotropic human herpesvirus with oncogenic potential (for reviews, see references 1 and 2). Following primary infection, EBV establishes a lifelong latent infection in more than 90% of all adults, with intermittent virus shedding in very low levels in saliva. EBV persists in a quiescent state in circulating, resting, memory B cells. EBV is a potent transforming virus in vitro and efficiently infects resting B cells, leading to the outgrowth of permanently growing lymphoblastoid cell lines (LCLs), a process known as B-cell immortalization. The EBV nuclear antigen 2 (EBNA2) is a key viral latent protein that initiates and maintains the EBV latency III gene expression program (Lat III; also known as the latency growth program) seen in LCLs. This transcription pattern involves the expression of at least six viral nuclear proteins (including EBNA1, -2, -3A, -3B, -3C, and –LP), three integral latent membrane proteins (LMP1, -2A, and -2B), two small nonpolyadenylated RNAs known as EBER1 and EBER2, a set of poorly understood transcripts known as BARTs (for a review, see reference 3), and a large number of more recently discovered microRNAs (4) EBNA2 is a transcription factor that does not bind directly to DNA but is recruited to its sites of action through complex and cell context-dependent interactions with cellular proteins, including CBF1 (also known as RBP-Jκ, a nuclear adapter component of the cellular Notch signaling pathway) and others (for reviews, see references 5 and 6). Important positive transcriptional targets of EBNA2 are the EBV LMP1 (7) and cellular MYC (c-MYC) (8), both of which encode proteins that have major effects on cell phenotype (reviewed in references 9 and 10).
In vivo, the main targets of EBV are naive B cells and B cells that undergo affinity maturation in a germinal center (GC). GCs are structured microenvironments of secondary lymphoid tissues in which antigen-activated B cells undergo proliferation, class switch recombination (CSR), somatic hypermutation (SHM), antigen selection, and affinity maturation (for a review, see reference 11). The currently accepted explanation for EBV persistence in healthy immunocompetent hosts is referred to as the GC model. Following primary infection, the EBNA2-driven Lat III program induces host B cells to proliferate as infected blasts. Such cells are frequently detectable in tonsillar tissues from patients with the acute symptomatic primary EBV infection known as infectious mononucleosis (IM) (12–14). Although this cell pool is efficiently targeted by the cytotoxic T cell (CTL) response in immunocompetent hosts, due to the immunogenicity of viral proteins, some infected cells transit the GC and enter into the long-lived memory B-cell compartment by exploiting normal B-cell biological processes. EBNA2 expression is shutoff during GC transit, and cells with a more restricted viral protein pattern, which includes EBNA1, LMP1 and LMP2 (known as latency II, or Lat II; also known as the default program), are detectable. Latently infected memory B cells exiting the GC express either no viral proteins at all (latency 0, or Lat 0) or only EBNA1 transiently (latency I, or Lat I) during rare mitoses and are therefore considered the site of long-term persistence due to immune invisibility and virus quiescence (15). Signals that promote the induction of B-cell terminal differentiation can also initiate virus lytic reactivation in a small subset of these cells, leading to the release of infectious virus particles. The latter are then either shed or go on to infect new naive B cells, thus completing the cycle. EBV production in infected epithelial cells also occurs and may serve to amplify the level of infectious virus particles at the point of entry or exit. EBV-associated B-cell malignancies arise from infected cells at different stages of the B-cell differentiation pathway. Thus, EBV-associated endemic Burkitt's lymphoma (BL) cells are believed to be of GC origin and the majority express the Lat I transcription program (16); Hodgkin's lymphoma (HL) malignant cells are thought to be derived from atypical post-GC cells and in EBV-positive cases they express Lat II (17); EBV-positive posttransplant lymphomas (PTLs) in immunosuppressed patients arise from virus-transformed B cells expressing the Lat III program that have escaped effective T-cell surveillance (18).
The strategic inhibition of B-cell apoptosis is central to EBV biology and is likely to also play a role in the development of EBV-related diseases (for reviews, see references 19 to 21). In the GC environment, only those B cells that express the highest-affinity immunoglobulins are rescued from stringent proapoptotic pathways that signal through transforming growth factor β (TGF-β) (22, 23), FAS (24, 25), and B-cell receptors (26). Bcl-2 proteins are critical for setting the threshold of resistance to apoptosis and initiating the apoptotic cascade, and members are grouped primarily by reference to distinct Bcl-2 homology (BH) domains (for a review, see reference 27). The so-called BH3-only proteins are proapoptotic and bind via their short α-helical BH3 domain to prosurvival Bcl-2 family members, and this interaction is required for their ability to kill cells (28). BH3-only proteins are classified into two groups, namely, activators (BIM, BID, and PUMA) capable of directly activating BAX and BAK and sensitizers (BIK, BMF, BAD, and NOXA) that interact with antiapoptotic Bcl-2 family members, thereby sensitizing cells to proapoptotic triggers. BH3-only proteins are subject to stringent control but become transcriptionally upregulated and/or posttranslationally modified in response to proapoptotic signals, thereby gaining their full apoptotic potential (29). BIK (Bcl2 interacting killer; also known as NBK), the founding member of the BH3-only group, is a potent inducer of apoptosis that can trigger through both p53-dependent and -independent pathways (30–34). BIK selectively inhibits the prosurvival BCL-XL, BFL-1, and BCL-w (35) and has been shown to sensitize tumor cells to apoptosis mediated by various therapeutic agents (36–38) by a mechanism that is dependent on its BH3 domain (39).
Several published observations have suggested that BIK plays a key role in B-cell homeostasis. BIK is upregulated in B cells following antigen receptor stimulation (40, 41) and is critical to the apoptotic selection of mature B lymphocytes. More recently, the mechanism of action of TGF-β in GC-derived centroblasts and BL-derived cell lines has been shown to involve BIK upregulation (22). We report here for the first time that BIK is a negative transcriptional target of EBV and is repressed by the EBNA2-driven Lat III program, independently of c-MYC. BIK repression occurred soon after infection of primary B cells by wild-type EBV but not by a recombinant EBV in which the EBNA2 gene had been knocked out. Furthermore, BIK repression was mediated by EBNA2 in EBV-negative B-cell lines, and this was effected at the level of the SMAD/BIK promoter complex. BIK induced apoptosis in Lat III cell lines by a mechanism dependent on its BH3 domain and the activation of caspases. EBNA2 antagonized TGF-β1-mediated BIK upregulation and induction of the intrinsic apoptotic program. These observations are evidence of an additional mechanism used by EBV to inhibit apoptosis during B-cell infection, namely, the transcriptional repression of a BH3-only sensitizer, the cellular proapoptotic BIK.
MATERIALS AND METHODS
Cell lines, B-cell isolation, and infection with EBV.
DG75, BL41, and Ramos are EBV-negative BL-derived cell lines; MUTU-I and KEM-BL are EBV+ BLs and express the EBV Lat I transcriptional program; MUTU-III and AG876 are EBV+ BLs that express the Lat III program; Oku-BL is an EBV+ BL-derived cell line that expresses a Wp-restricted latency program (expressing EBNA1, EBNA3A, -3B, -3C, and -LP and BHRF1) (42). IB4, IARC 171, IARC 290B, X50-7, and OKU-LCL are EBV+ LCLs; BJAB is an EBV-negative B-lymphoma cell line; BL41-B95-8 and BL41-P3HR1 are BL41 cells infected with wild-type EBV or an EBV strain (P3HR1) carrying an EBNA2-spanning genomic deletion, respectively; Daudi is an EBV-positive (EBNA2-deleted) BL (43–49). All cell lines were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The conditional LCL ER/EB2-5, its derivative P493-6, and the stable transfectants DG75-tTA-EBNA2, DG75-tTA-LMP1, and BL41-K3 and associated EBNA2/LMP1 induction or EBV growth program/ER-EBNA2 chimeric protein activation procedures have been described elsewhere (50–54). DG75 SM296D6 is an ER-EBNA2-expressing subclone of DG75, and DG75 SM296D3 is its clonal derivative in which both copies of the CBF1 gene have been inactivated by somatic knockout (55). The lentivirus-transduced ER/EB2-5 cell pools have been described elsewhere (56).
The EBNA2-deleted recombinant EBV (EBV EBNA2-KO) used (49) is derived from the original B95-8 2089 wild-type control (EBV wt) (57). Production of both viruses in HEK 293 producer cells was induced by transfection with BZLF1, and BALF4 expression vectors and supernatants containing virus were harvested and purified by density gradient centrifugation (Optiprep; Axis Shield) (58). Virus titrations were carried out by quantitative PCR (qPCR) as described previously (59). Primary B cells were positively selected from apheresis cones (National Health Service Blood and Transplant [NHSBT], Birmingham, United Kingdom) by using CD19 Dynabeads (Invitrogen), followed by a detachment step and then assessment for purity as described elsewhere (60). Isolated resting B cells were incubated with virus preparations at a multiplicity of infection (MOI) of 100. Infection was assessed by immunofluorescence cell staining with JF186 monoclonal antibody to detect EBNA-LP expression 2 days postinfection, at which time the cells were 70 to 75% EBNA-LP positive.
RNA assays and Western blot assays.
RNA was extracted from cell lines or EBV-infected B cells by using an RNeasy kit (Qiagen) and then DNase treated with a DNA-free kit (Ambion) according to the manufacturer's instructions. Reverse transcription (RT) was done using Sensiscript reverse transcriptase (Qiagen), and BIK and GAPDH mRNAs were detected by RT-qPCR using TaqMan assay reagents (Hs00154189_m1 and Hs99999905_ml, respectively; Applied Biosystems). All RT-qPCR data were analyzed as described previously (61, 62), and relative transcript levels were determined after coamplification and normalization to GAPDH transcript levels. The RNase protection assay (RPA) and Western blotting procedures used have been described elsewhere (63). The following primary antibodies were used: anti-BIK (557040; BD Biosciences), anti-SMAD3 (ab28379; Abcam), anti-SMAD4 (ab3219; Abcam), anti-β-actin (A1978, clone AC-15; Sigma-Aldrich), anti-EBNA2 (PE2; Dako Cytomation), anti-LMP1 (CS1-4 ab78113; Abcam), anti-EBNA-LP (JF186; reference 64), anti-c-Myc and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (N-262 [sc-764] and FL-335 [sc-25778]; Santa Cruz Biotechnology, respectively). The quantities of protein loaded for Western blot assays were normalized by probing for β-actin or GAPDH.
RNA interference, plasmids, and transfections.
Small interfering RNA (siRNA) knockdown experiments were performed with the Nucleofector device II (Lonza) using the following siRNA reagents (from Applied Biosystems): anti-BIK siRNA si1989 and anti-BIK siRNA si1990 (4390824), Silencer negative control siRNA (AM4611), and anti-SMAD3 siRNA56 and anti-SMAD3 siRNA57 (4390827). The plasmids pSGEBNA2, pSGEBNA2WW323SR, pcDNA3-HA-BIK, and pcDNA3-HA-BIKΔBH3 have been described elsewhere (39, 65). Transfection of cell lines with plasmids was done by electroporation using a Gene Pulser II (Bio-Rad) and Ingenio electroporation solution transfection reagent (MIR 50118; Mirus). All transfection results presented were compiled from three independent experiments.
Apoptosis assay.
Cells were seeded at 5 × 105 cells/ml in 2% FBS-supplemented medium prior to treatment with TGF-β1 (GF111; Merck Millipore). Cell viability and the onset of apoptosis was monitored using an Annexin-phycoerythrin (PE) apoptosis detection kit (559763; BD Biosciences), which contains recombinant Annexin V-fluorochrome PE conjugate and the vital dye 7-amino-actinomycin (7-AAD), followed by flow cytometry (FACSCalibur; BD Biosciences) and CellQest software. Data for at least 10,000 cells were collected for each analysis, and two-dimensional plots of 7-AAD versus PE were generated. Other reagents used were N-benzyloxycarbonyl-Val-Ala-Asp (OMe)-fluoromethyl ketone (zVAD-fmk; 219007; Merck) and MG132 (C2211; Sigma-Aldrich).
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed using a ChIP kit (ab500; Abcam) according to the manufacturer's instructions. In brief, chromatin/DNA complexes were extracted from 3 × 106 cells. Chromosomal DNA was sheared using a sonifier (Branson 450) to an optimal DNA fragment size of 200 to 1,000 bp. Equal samples of sonicated chromatin were then individually immune precipitated with the ChIP-grade antibodies Ab28379 (anti-SMAD3), Ab3219 (anti-SMAD4), and isotype control IgG (Abcam). The relative levels of BIK promoter present in each immunoprecipitate were then determined following amplification by PCR of a 420-bp fragment located upstream of the BIK transcription start site, by using the primer sequences 5′-GGAGGCCCTAGAAGAAAAGACTAC-3′ and 5-GGAACAGAGGAGGTAAAGTGTGAT-3′ (22). The primers used to amplify a portion of the GAPDH promoter were 5′-AGCTCAGGCCTCAAGACCTT-3′ and 5′-AAGAAGATGCGGCTGACTGT-3′ (Human ChIP-seq grade GAPDH TSS primers; Diagenode). A 1/100 portion of the precipitated chromatin was used for PCR.
RESULTS
BIK is downregulated in cell lines expressing the EBV Lat III but not the EBV Lat I program.
We first investigated if BIK was regulated by EBV, and to this end, BIK protein levels were profiled in a range of well-studied B-cell lines. BIK was detected in BL-derived cell lines that were either EBV negative or EBV positive but expressed the Lat I program, in which EBNA1 is the only detectable viral protein (Fig. 1A). In contrast, BIK mRNA and protein levels were repressed in LCLs and EBV-positive Lat III BLs, both of which express the full spectrum of EBV latent gene products (Fig. 1A and B). Interestingly, BIK levels remained elevated in the BL cell lines Daudi and BL41-P3HR1, both of which contain EBV genomes that harbor deletions spanning the EBNA2 coding sequence, and also in OKU-BL, which exhibits a Wp-restricted latency gene expression pattern in which EBNA2 is not expressed (42).
FIG 1.
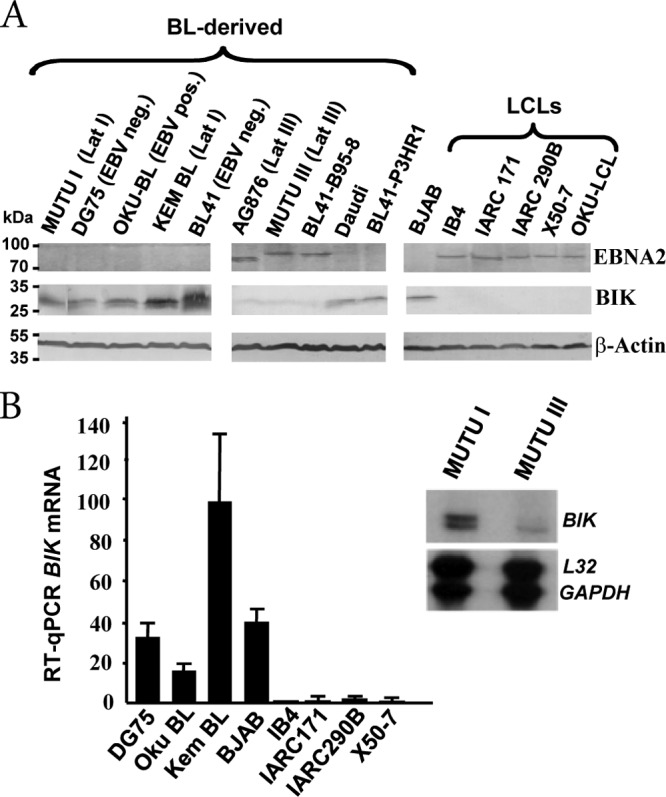
Expression of BIK in a panel of lymphoma-derived B-cell lines and LCLs. (A) Western blot analysis showing EBNA2, BIK, and β-actin levels, indicated to the right of each panel. The EBV and Lat program status for each BL-derived cell line is given in brackets. OKU-BL is EBV positive and exhibits a Wp-restricted latency gene expression pattern in which EBNA2 is not expressed. BL41-B95-8 is a subclone of BL41 that was infected with the EBV B95-8 strain and expresses EBNA2; Daudi and BL41-P3HR1 are EBV-positive EBNA2 deletion-containing cell lines. BJAB is a non-BL EBV-negative B-lymphoma cell line. AG876 expresses type II EBNA2, which has a lower molecular weight than type I EBNA2. (B) Comparative BIK mRNA levels in a panel of B-cell lines. The bar graph shows RT-qPCR data. Relative BIK transcript levels were determined after coamplification and normalization to GAPDH transcript levels. The image on the right is of an RNase protection assay (RPA) autoradiogram and shows comparative BIK, L32, and GAPDH transcript levels in the isogenic EBV-positive BLs MUTU I (Lat I) and MUTU III (Lat III).
BIK is repressed by the EBV Lat III program in a conditional LCL.
In LCLs, EBNA2 drives the EBV growth program, and we therefore investigated if BIK was also a negative target of EBV in this context. ER/EB2-5 is a conditional LCL in which the function of an estrogen receptor-EBNA2 fusion protein (and therefore the proliferative and growth transformation effects of EBV) is dependent on β-estradiol (50). It can be seen in Fig. 2A and B that inactivation of chimeric EBNA2 led to BIK induction in ER/EB2-5 and that readdition of β-estradiol restored BIK repression. It has been shown elsewhere that the effects of β-estradiol withdrawal can be reversed in this setting upon introduction of wild-type EBNA2 (66) or partially reversed with the intracellular domain of Notch1 (Notch1IC), a cellular functional homologue of EBNA2 (56). Here, trans-complementation of ER/EB2-5 following lentivirus transduction with EBNA2 or high levels of Notch1IC also maintained BIK transcriptional repression in the absence of β-estradiol (Fig. 2A). Elsewhere, BIK repression has been reported in response to estrogen signaling in a breast cancer-derived cell line (MCF7) (67). This possibility can be excluded in the present study, however, as BIK repression was observed in both the ER/EB2-5 trans-complementation and DG75-tTA-EBNA2 induction experiments (see Fig. 5, below), neither of which involved the use of β-estradiol. c-MYC is a key direct target of EBNA2 in LCLs (8), and enforced c-MYC expression at high levels is sufficient to drive B-cell proliferation in the absence of EBNA2 and LMP1 (68). P493-6 is an ER/EB2-5 derivative in which exogenous c-MYC is negatively regulated by tetracycline, thus permitting the c-MYC growth program to be uncoupled from that of EBV (54). Here, we observed that the steady-state levels of BIK mRNA and protein were significantly higher in P493-6 cells proliferating due to c-MYC (− β-estradiol/− TET) than in their EBV-driven counterparts (+ β-estradiol/+ TET, which behaved like the parental ER/EB2-5 cell line) (Fig. 2C). This was reminiscent of the BIK repression seen in EBV-driven LCLs, in contrast to BL type 1 cell lines, which are driven to proliferate by c-MYC (Fig. 1A). Overall, these results showed that BIK is a negative transcriptional target of the EBNA2-driven Lat III program in LCL and that a contribution of c-MYC to BIK repression can be excluded in this context.
FIG 2.
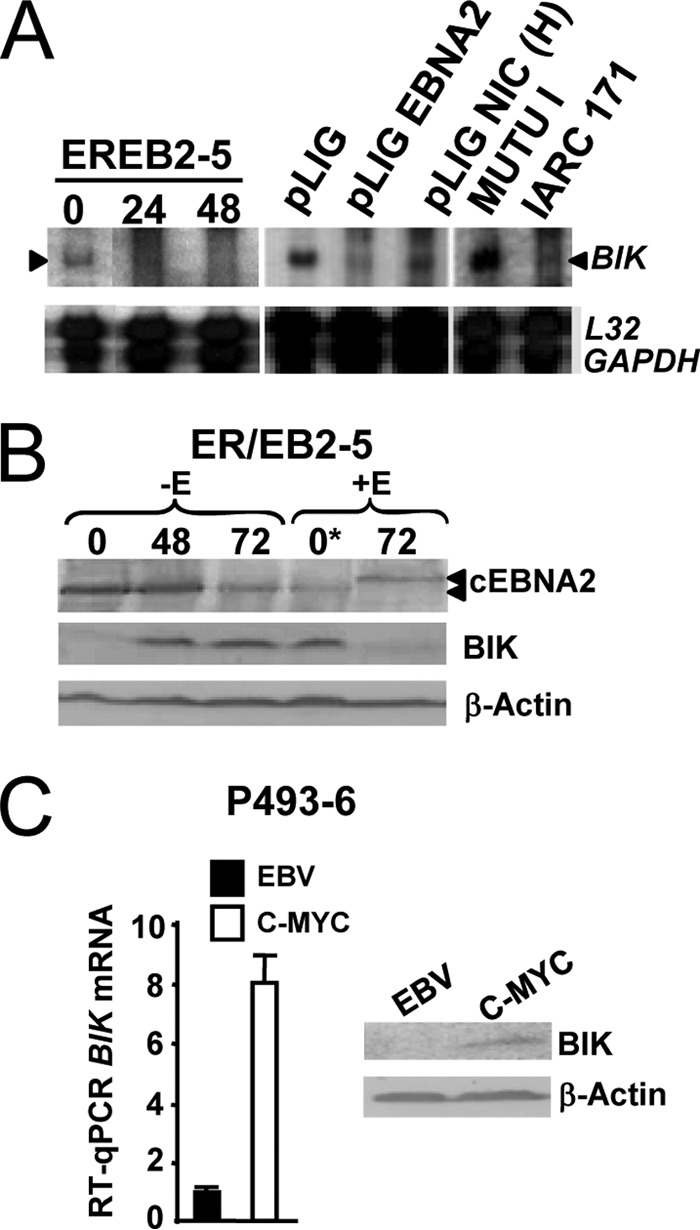
BIK is repressed by the EBNA2-driven Lat III program in a conditional LCL. (A) RPA autoradiogram of processed RNA samples from ER/EB2-5 cells that were first starved of β-estradiol (0) and then rescued by either reculturing in β-estradiol and sampled for RNA analysis at various time points (indicated in hours, above) or by transduction with lentiviral vectors expressing either EBNA2 (pLIG-EBNA2) or high levels of human Notch1IC [pLIG-NIC(H)]. RNA samples from cycling MUTU I and IARC171 cells were also processed as controls. (B) Western blot showing BIK protein levels in response to activation of chimeric EBNA2 (cEBNA2). +/−E means +/− β-estradiol. Sampling time points following removal or addition of β-estradiol are indicated in hours above each lane (0*, the starting time point at which β-estradiol was reintroduced following 72 h without E). The anticipated migration shift of cEBNA2 in response to β-estradiol is evident (arrows, lane +E, 72 h). (C) P493-6 cells (an ER/EB2-5 subclone) were divided and cultured separately to permit cycling on the EBV Lat III program (+β-estradiol/+TET) or c-MYC growth program (−β-estradiol/−TET) and sampled for RNA and protein. RT-qPCR (left) and Western blotting (right) showed steady-state BIK mRNA and protein levels in P493-6 cells driven to proliferate due to EBV Lat III (EBV) or ectopic c-MYC (c-MYC).
FIG 5.

R-SMADs are key regulators of BIK and are modulated by EBV Lat III in a conditional LCL and by ectopic EBNA2 in EBV-negative B cells. (A) Ramos and BJAB were transfected with anti-SMAD3 siRNAs (siRNA56 and siRNA57) and nonspecific control siRNA (siNC). Twenty-four hours later, cells were treated with either 10 ng/ml of TGF-β1 or vehicle for a further 4 h, harvested, and analyzed by RT-qPCR for BIK mRNA levels. The BIK transcript level in siNC-transfected/−TGF-β1 cells was set to 1, and other values are presented relative to that. The statistical comparisons shown were made with the BIK transcript level in the corresponding siNC-transfected TGF-β-treated control. Data are means ± standard deviations. *, P ≤ 0.05. (B) Western blotting for SMAD3, BIK, and β-actin using protein extracts from the same experiment as shown in panel A. (C) LCL ER/EB2-5 cells were cultured in the presence or absence of β-estradiol (E + and −) and harvested for total RNA and protein 48 and 72 h later (values indicated underneath). Shown are RT-qPCR results for BIK mRNA (graph on left) and Western blot analysis results for SMAD3 (image on right). (D) ChIP analysis showing the relative SMAD3 and SMAD4 levels bound to the endogenous BIK promoter. Samples of sonicated chromatin were prepared from ER/EB2-5 cells that had been cultured with or without β-estradiol (E) for both 48 and 72 h (+ or − E) (values underneath the graph). These were then incubated separately with anti-SMAD3, anti-SMAD4, or isotype control antibody (control IgG). Input DNA and DNA isolated from immune-precipitated material (target-enriched DNA or isotype control-enriched DNA) were amplified by qPCR with primers designed to amplify a 420-bp SBE-containing sequence from the BIK promoter (pBIK). An irrelevant target DNA sequence (from the GAPDH promoter; pGAPDH) was also amplified independently from the same samples. Levels of promoter-bound SMAD3 and SMAD4 are expressed as percentages of the total input. Statistical comparisons were made between β-estradiol-treated or untreated samples taken at the same time points. The data shown were compiled from three experiments. Means ± standard deviations are shown. *, P ≤ 0.05, **, P = 0.001 to 0.01. (E and F) ChIP analysis results, showing the relative SMAD3 and SMAD4 levels bound to the endogenous BIK promoter in Ramos (E) and BJAB (F) following transfection with effector plasmids (samples bracketed together underneath each graph) and treatment with TGF-β1. Forty-eight hours after transfection, cells were treated with or without 10 ng/ml TGF-β1 for a duration of 4 h. Cells were then harvested, and ChIP was performed as described for panel D, targeting the same regions of the BIK and GAPDH promoters. Levels of promoter-bound SMAD3 and SMAD4 are expressed as percentages of the total input. Statistical comparisons were made relative to the corresponding pSG-transfected/TGF-β1-treated samples. The data shown were compiled from three experiments. Values are means ± standard deviations. *, P ≤ 0.05; **, P = 0.001 to 0.01. (G) Western blotting results, showing endogenous SMAD3 levels in BJAB cells 48 h after transfection with effector plasmids (names given above each lane) and treatment with or without TGF-β1 at 10 ng/ml (+ and − underneath the blots).
BIK repression occurs following EBV infection of primary B cells in vitro by a mechanism requiring EBNA2.
In order to investigate BIK expression during an EBV infection in vitro, isogenic populations of freshly isolated primary B cells were separately infected with wild-type EBV (EBV wt) or a recombinant EBV in which the EBNA2 gene had been knocked out (EBV EBNA2-KO) (Fig. 3A). Western blot analysis using protein extracts sampled at various time points following infection confirmed EBNA2 expression only when wild-type EBV was used (Fig. 3B). EBNA2 was detectable as early as 6 h following infection and at all time points thereafter. A concomitant decrease in BIK protein levels was observed in response to infection with EBV wt but not EBV EBNA2-KO. Furthermore, BIK repression was clearly in evidence as early as 6 h after infection. Conversely, BIK levels were seen to increase starting at 24 h following infection with EBV EBNA2-KO and to increase further at 48 h and again at 72 h (Fig. 3B). Elsewhere, this EBV EBNA2-KO was shown to express EBNA1, -LP, -3A, and -3C and BHRF1 at 24 h following infection and also LMP1 (detectable at 3 days postinfection) (69). We concluded, therefore, that BIK repression occurs following EBV infection of primary B cells in vitro by a mechanism requiring EBNA2. Furthermore, the experiment also suggested that EBNA2 expression serves to prevent an increase in BIK levels that would otherwise occur following EBV infection.
FIG 3.
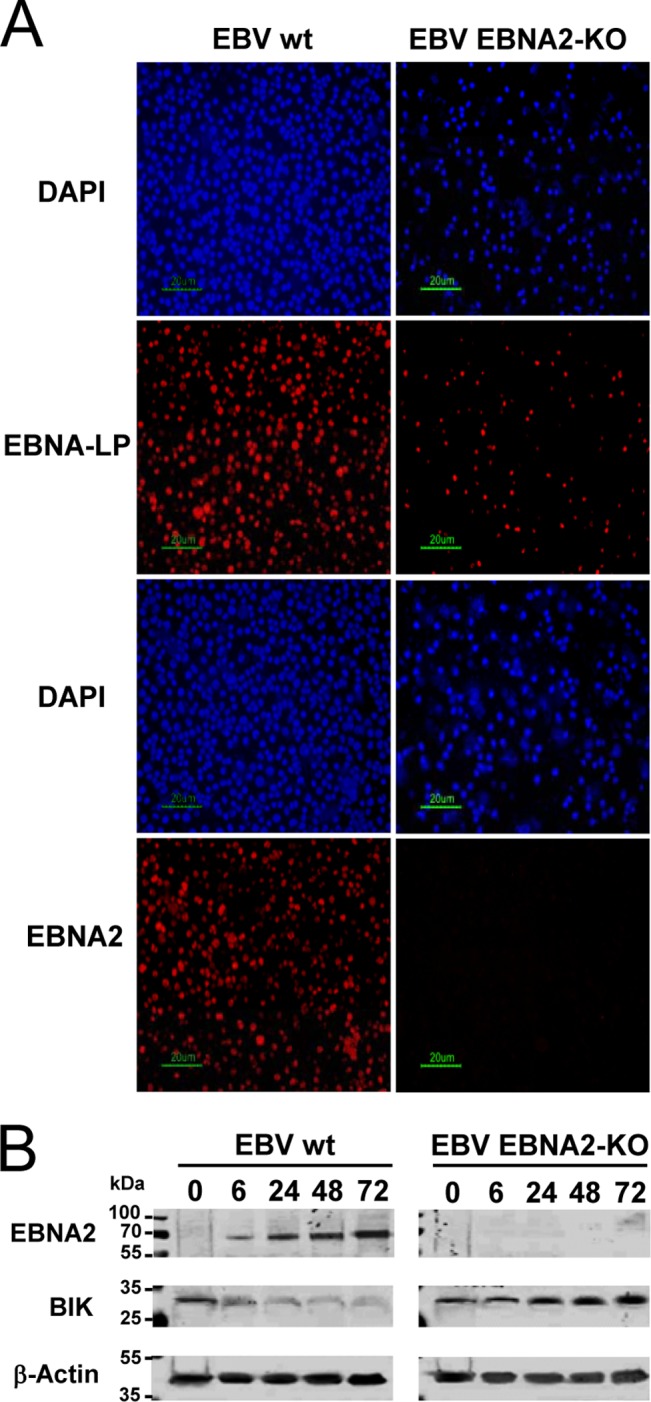
BIK is repressed by EBNA2 following EBV infection of primary B cells in vitro. (A) EBV latent antigen expression in primary B cells infected with either a wild-type EBV strain or a recombinant EBV strain in which the EBNA2 gene was knocked out (EBV EBNA2-KO). Immunofluorescence staining was performed for EBNA-LP or EBNA2 (red staining) at 48 h postinfection. 4′,6-Diamidino-2-phenylindole (DAPI) counterstaining (blue) shows all the nuclei in the field. (B) Western blots showing EBNA2, BIK, and β-actin levels following the infections of panel A. The numbers above each lane represent the time points (in hours) at which total cellular proteins were harvested after infection.
EBNA2 represses BIK in BL cell lines.
Sustained BIK expression in the Daudi, BL41-P3HR1, and OKU-BL cell lines pointed to a role for EBNA2 in BIK repression. This possibility was therefore investigated using BL-derived transfectants that express either chimeric estrogen receptor-EBNA2 (ER-EBNA2), whose function is dependent on β-estradiol (BL41-K3 and BL41-P3HR1-9A) (50, 51, 53) or that can be induced to express EBNA2 in response to the removal of tetracycline (DG75-tTA-EBNA2) (52). In all cases, activation or induction of EBNA2 led to the transcriptional repression of BIK (Fig. 4A and B). In contrast BIK was not repressed in response to the induction of LMP1 in a stable DG75 transfectant (DG75-tTA-LMP1) (52). A role for c-MYC in BIK repression is unlikely here, as both genes are coexpressed in EBV-negative and EBV Lat 1 cell lines. Furthermore, EBNA2 has been shown to negatively regulate c-MYC in BL41-K3 but not in BJAB-K3 cells, which do not carry the BL-associated t(8;14) chromosomal translocation (55, 70), yet we observed BIK repression in both cases (BJAB-K3 results not shown). We also observed a decrease in BIK mRNA levels following the addition of β-estradiol to an ER-EBNA2-expressing subclone of DG75 (SM296D3), in which both copies of the CBF1 gene had been inactivated by somatic knockout (Fig. 4C) (55). These results demonstrated that BIK is transcriptionally downregulated by EBNA2 in EBV-negative BL lines and following trans-complementation of the EBNA2 genomic deletion in the EBV-infected BL41-P3HR1, and that neither c-MYC nor CBF1 plays a significant role in this regard.
FIG 4.
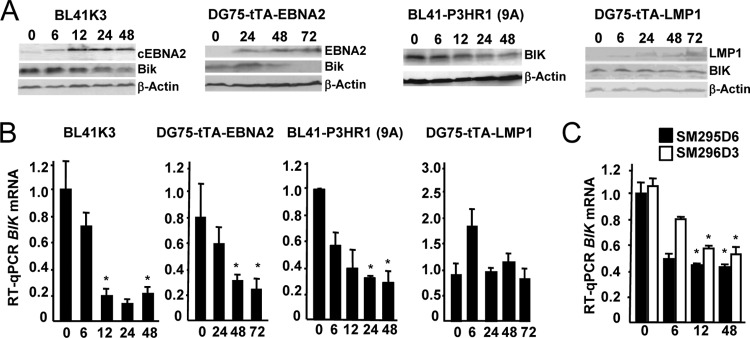
EBNA2 transcriptionally represses BIK in EBV-negative B-cell lines. (A) Western blot analyses of EBNA2 or chimeric EBNA2 (cEBNA2), LMP1, BIK, and β-actin by using protein extracts prepared from the cell lines named above the corresponding panel of blots. BL41K3 and BL41-P3HR1 (9A) are stable transfectants of BL41 and BL41-P3HR1, respectively, that express a chimeric estrogen receptor-EBNA2 whose function is dependent on β-estradiol (cEBNA2; shown for BL41K3 only). The numbers above these two panels are the times (in hours) following the addition of β-estradiol to the cultures. DG75-tTA-EBNA2 and DG75-tTA-LMP1 are stable transfectants of DG75 that can be induced to express EBNA2 and LMP1, respectively, by reculturing the cells in the absence of tetracycline (times in hours following removal of tetracycline are indicated above each lane). (B) The corresponding BIK mRNA levels from triplicate sets of RNAs from the experiments shown in panel A, determined by RT-qPCR. The times (expressed in hours) following cEBNA2 activation or EBNA2/LMP1 induction are given underneath each bar chart. BIK transcript levels were normalized to that of GAPDH. Data are means ± standard deviations. *, P < 0.05; statistical comparisons were made between each starred time point and the 0-h time point. (C) RT-qPCR showing BIK mRNA levels following the addition of β-estradiol (expressed in hours, underneath) to SM295D6 and SM296D3, both ER-EBNA2-expressing subclones of DG75. In SM296D3, both copies of the CBF1 gene have been inactivated by somatic knockout. BIK transcript levels were normalized to that of GAPDH and then plotted relative to the value obtained with SM295D6 (arbitrarily assigned a value of 1). Data are means ± standard deviations. *, P < 0.05; statistical comparisons were made between each starred time point and the corresponding 0-h time point for the same cell line.
Reduced levels of SMAD proteins are bound to the BIK promoter upon activation of the EBV Lat III program or expression of ectopic EBNA2.
TGF-β1 is a physiological mediator of GC B-cell homeostasis through cell type-specific induction of apoptosis (for a review, see reference 71). TGF-β1-driven BIK expression is associated with the recruitment of regulatory SMAD proteins (R-SMADs), the primary mediators of canonical TGF-β1 signaling, to a functional SMAD-binding element (SBE) present on the human BIK promoter (22). Here, we show that SMAD3 knockdown with siRNAs led to decreased basal levels of BIK mRNA and protein and an inhibition of BIK induction by TGF-β1 in both Ramos and BJAB cells (Fig. 5A and B), thus confirming an essential role for SMAD3 as a positive transcriptional regulator that sets the threshold level of BIK in this cell context. Furthermore, BIK repression by the EBV Lat III program in ER/EB2-5 cells occurred concomitantly with a decrease in total SMAD3 levels (Fig. 5C). Using ChIP assays, we observed reduced levels of SMAD3 and SMAD4 bound to the BIK promoter in cycling ER/EB2-5 cells following activation of ER-EBNA2 (Fig. 5D). No changes in SMAD3/4 binding to the GAPDH promoter were seen in the same experiment, demonstrating specificity. Furthermore, decreased levels of SMAD3 and SMAD4 were bound to the BIK promoter in the presence of TGF-β1 when either ectopic EBNA2 or EBNA2WW323SR was expressed in Ramos and BJAB cells (Fig. 5E and F). Again, no changes in SMAD3/4 binding to the GAPDH promoter were observed under the same conditions (Fig. 5E; data not shown for BJAB). Total SMAD3 levels were also decreased in the presence of EBNA2 or EBNA2WW323SR following treatment of BJAB with TGF-β1 (Fig. 5G).
Ectopic BIK induces apoptosis in EBV Lat III cell lines by a mechanism dependent on its BH3 domain and the activation of caspases.
BIK is proapoptotic in mature B lymphocytes (41), and we therefore asked if the reintroduction of this protein would have a negative impact on the survival of B cells proliferating due to EBV. In a control experiment, the 7-AAD/Annexin V staining profile of the IB4 LCL was first established by fluorescence-activated cell sorting (FACS) analysis in response to the apoptosis-inducing proteasome inhibitor MG132 (72). MG132 efficiently induced apoptosis in IB4 cells, and this effect was inhibited by the broad-spectrum caspase inhibitor zVAD-fmk (Fig. 6A). Elsewhere, MG132 has been shown to induce the accumulation of BIK, but not other Bcl-2 family proteins, in a range of cancer cell lines (73). IB4 cells were then transiently transfected with a plasmid expressing hemagglutinin (HA)-tagged BIK (HA-Bik) together with a green fluorescent protein (GFP) expression plasmid (pMaxGFP; Amaxa GmbH), and the survival profile of GFP-expressing cells was analyzed 6 h later. Exogenous BIK rapidly induced apoptotic death in transfected cells in a dose-dependent manner (Fig. 6B). Furthermore, this effect was significantly reduced upon deletion of the BIK BH3 domain and virtually absent when empty vector or the antiapoptotic BFL-1 was substituted as the effector (Fig. 6B; BFL-1 results not shown). It can be seen that zVAD-fmk efficiently inhibited BIK-induced apoptosis in IB4 (Fig. 6C), in agreement with previous observations that the activation of caspases are key downstream events during BIK-induced cell death (74–76). Cell survival data obtained following transfections of other EBV Lat III-expressing cell lines (including ER/EB2-5 and AG876) consistently demonstrated BH3-dependent death due to ectopic BIK (data not shown).
FIG 6.
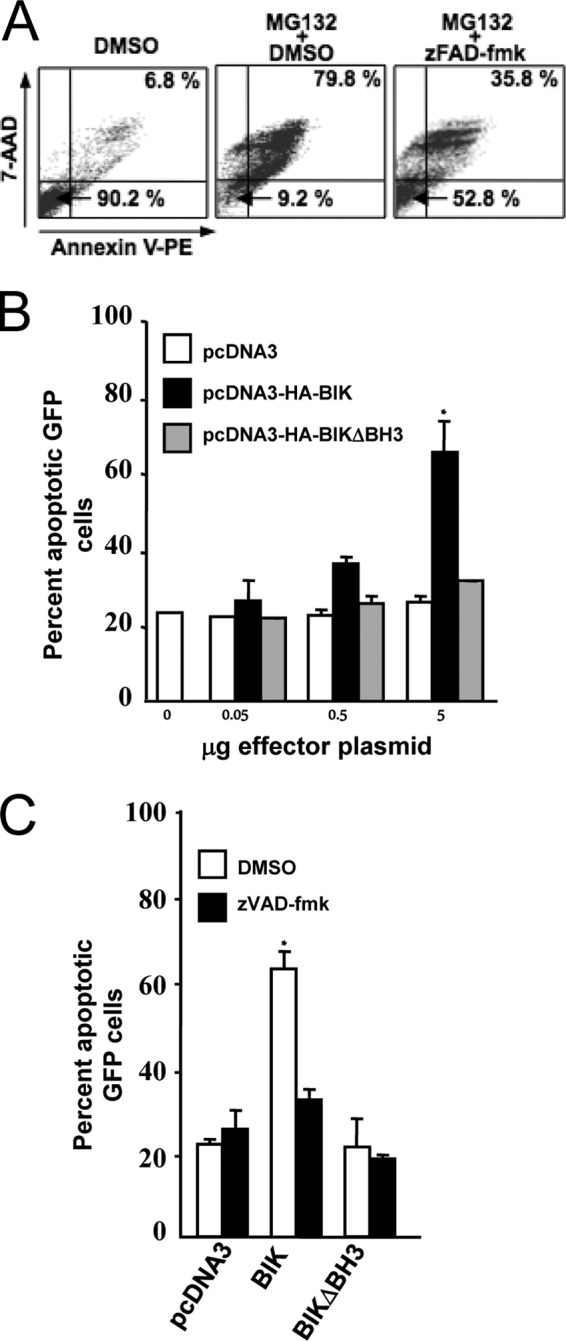
Ectopic BIK induces apoptosis in the LCL IB4 by a mechanism dependent on its BH3 domain and the activation of caspases. (A) Representative IB4 cell viability FACS profiles. IB4 cells were treated with dimethyl sulfoxide (DMSO; vehicle) or the apoptosis-inducing proteasome inhibitor MG132 (15 mM) alone or in combination with the pan-caspase inhibitor zVAD-fmk (50 mM) or vehicle (DMSO). Twelve hours later, cells were then double-stained with Annexin V/7-AAD, and survival profiles were monitored by FACS. Viable cells (Annexin V− and 7-AAD−) and late-stage apoptotic cells (Annexin V+ and 7-AAD+) are represented in the bottom left and top right quadrants, respectively. Data for 10,000 cells were collected in each case, and the percentages of the total population in these quadrants are shown. (B) Dose-dependent induction of apoptosis by ectopic BIK in IB4. IB4 cells were cotransfected with 2 μg of pMaxGFP together with pcDNA3, pCDNA3-HABIK, or pcDNA3-HABIKDBH3 (quantities of effector plasmids used are indicated underneath). In all cases, the total amount of DNA used was kept constant at 7 μg by adding an appropriate amount of pcDNA3. Six hours later, cells were washed twice with PBS, and the survival profiles of GFP-expressing populations were determined as for panel A following 7-AAD/Annexin V staining. Data are means ± standard deviations. *, P < 0.05. The results shown were compiled from three separate transfections. (C) BIK-induced apoptosis is inhibited by the pan-caspase inhibitor z-VAD-fmk. IB4 cells were transiently cotransfected as described for panel B and then immediately either treated or untreated with of 50 mM zVAD-fmk. Cell viability was analyzed 3 h later by 7-AAD/Annexin V staining as described for panel A. The percentage of GFP-expressing cells in late apoptosis was then plotted. Data are means ± standard deviations. *, P < 0.05. The results shown were generated from three separate transfections.
BIK repression by EBNA2 antagonizes TGF-β1-induced apoptosis in B-cell lines.
Some EBV− BL and EBV+ BL Lat I cell lines are highly sensitive to TGF-β1, whereas LCLs and EBV+ BL Lat III cells are protected from its antiapoptotic and antiproliferative activities (77–81). As BIK expression has been shown here to follow this pattern, i.e., repressed in LCLs and BL Lat III cell lines while it is upregulated in EBV-negative and BL Lat I cell lines (Fig. 1), we therefore investigated a possible functional role for BIK downregulation by EBNA2. We first confirmed that BIK knockdown with siRNAs could antagonize both TGF-β1-mediated BIK induction and apoptosis in the EBV-negative BL Ramos line, and we also verified this in a second EBV-negative non-BL line, BJAB (Fig. 7A and B). Furthermore, transient transfection of Ramos and BJAB with plasmids expressing ectopic EBNA2 or EBNA2WW323SR (a non-CBF-1-binding EBNA2 [65]) led to the inhibition of BIK upregulation by TGF-β1 (Fig. 7C) and rescued Ramos cells from the proapoptotic effect of TGF-β1 (Fig. 7D). The ability of the above EBNA2 mutant to repress BIK corroborated the result seen using the DG75 CBF1 somatic knockout cell line (Fig. 4C). In summary, these findings strongly suggested that BIK downregulation by EBV is a key host-virus interaction that is modulated at the level of the R-SMAD/BIK promoter complex and that these events contribute to resistance to the antiapoptotic effects of TGF-β1 seen in cells expressing EBNA2.
FIG 7.

Transient BIK knockdown and ectopic EBNA2 antagonize TGF-β1-induced apoptosis. (A) Ramos and BJAB cells were transfected with anti-BIK siRNAs (si1989 and si1990) and negative control siRNA (siNC) and then either treated with TGF-β1 (10 ng/ml) or vehicle. Relative BIK mRNA and BIK protein levels were determined 24 h later by RT-qPCR (graph on left) and Western blotting (image on right). Fold differences were calculated relative to the siNC-transfected control (assigned a value of 1). RT-qPCR data are means ± standard deviations. *, P ≤ 0.05; **, P = 0.001 to 0.01; statistical comparisons were made between each effector siRNA (+TGF-β1) and TGF-β1-treated siNC. (B) Survival profiles from cells transfected and treated as described for panel A were determined by double-staining with Annexin V/7-AAD followed by FACS. The bar chart shows the percentages of viable cells. The percentage of viable cells following transfection with siNC was set to 100%, and other values are presented relative to that. BIK knockdown with si1989 and si1990 (in the absence of TGF-β1) reduced the extent of cell death associated with the transfection procedure itself. Data are means ± standard deviations. **, P = 0.001 to 0.01. (C) Ramos and BJAB cells were transfected with 1 μg of pSG5, pEBNA2 (pE2), or pSGEBNA2WW323SR (pE2m). Forty-eight hours later, cells were treated with TGF-β1 (10 ng/ml) and relative BIK mRNA levels were determined 24 h later by RT-qPCR (bar charts on left). Data are means ± standard deviations. **, P = 0.001 to 0.01. The corresponding EBNA2, BIK, and β-actin protein levels were also determined by Western blotting (panels on right). The effector plasmids used for transfection and the presence/absence of TGF-β1 (+/−) are indicated above each lane. Protein extract from IB4 cells (not treated with TGF-β1) was loaded as a control for EBNA2 expression. (D) Survival profiles of Ramos cells that were transfected and treated as described for panel C were obtained by double-staining with Annexin V/7-AAD followed by FACS. The bar chart shows the percentages of viable cells. Data are means ± standard deviations. **, P = 0.001 to 0.01.
DISCUSSION
Here, we report for the first time a direct link between BIK, a BH3-only sensitizer protein, and EBV. The only studies to date associating BIK and EBV concerned the EBV protein BHRF1. This viral Bcl-2 homologue has been shown to bind BAK and also a subset of BH3-only activators, but not BH3-only sensitizers, including BIK (82, 83). BAK inactivation therefore, and not direct interaction with BIK, corroborates an earlier finding where BHRF1 was shown to inhibit apoptosis induced by ectopic BIK (84, 85).
EBV− and EBV+ Lat I BLs do not express high levels of BCL-2, BCL-XL, or MCL-1, all of which are known to counter BIK-induced apoptosis (82, 86, 87). Inactivating BIK mutations are a frequent feature of human peripheral B-cell lymphomas with GC/post-GC origins (88), but to our knowledge, data for BL have not been reported. Our analysis of cDNA sequences generated from two EBV-positive (Akata and MUTU III) and two EBV-negative (BL41 and DG75) BL cell lines did not reveal mutations in the BIK open reading frame, however (data not shown).
BL cell lines are derived from centroblasts differentiating within GCs and are highly sensitive to TGF-β-induced apoptosis (23, 79, 89). The demonstration of BIK repression by the EBV Lat III but not the Lat I gene expression program is consistent with observations made elsewhere on increased resistance to TGF-β in BLs (80, 90). Various mechanisms by which EBV confers resistance to TGF-β have been proposed (for a review, see reference 19), including a decrease in the level of TGF-β receptors (78, 79, 91). Elsewhere, however, it has been shown that the EBV Lat III program, but not c-MYC, preferentially protects P493-6 cells from the antiproliferative effect of TGF-β1 (92). Furthermore, the same study ruled out the abolition of TGF-β1 apoptotic signaling, cyclin D2, EBV lytic cycle activation, and secondary genetic events as potential contributory factors. BIK repression due to EBV Lat III (but not c-MYC) in P493-6 cells (Fig. 2C) therefore occurs in the presence of a functioning TGF-β1 signaling pathway. Some LCLs have been shown to produce TGF-β yet are resistant to its effects (93, 94). As an additional mechanism of antagonism to TGF-β, the EBV-BIK interaction may therefore further desensitize the virus-infected cell to the TGF-β autoregulatory feedback loop and provide a survival advantage during the expansion of the infected B-cell population.
EBNA2 has been shown to inhibit Nurr77-induced apoptosis by directly interacting with that protein (95, 96) and to also upregulate the antiapoptotic BFL-1 (97). EBNA2 expression is invariably accompanied by LMP1 during EBV infection and almost always so in EBV-associated disease settings. Modest sensitization to TGF-β following treatment with antisense oligodeoxynucleotides to LMP1 has been shown elsewhere for LCLs (98), although others have found no evidence to suggest that LMP1 plays a role in blocking TGF-β-mediated responses in B cells (79). LMP1 induction of Id1/repression of ATF3 has been shown to inhibit TGF-β-mediated cytostasis in epithelial cells (99). We did not detect BIK expression in nasopharyngeal carcinoma-derived C33A cells in the presence or absence of LMP1 (data not shown) (100). We also noted BIK transcriptional repression in a range of Hodgkin/Reed-Sternberg (H/RS)-derived cell lines, irrespective of EBV status (EBV− lines were L428, L1236, KMH2; EBV+ line was L591; KMH2-EBV was EBV− but infected with EBV in vitro, noting that neither EBV+ H/RS clone reflected the EBV gene expression pattern of primary H/RS cells [data not shown]).
Here, we have shown that infection of primary B cells in vitro leads to BIK repression by an EBNA2-dependent mechanism. The EBNA2-driven Lat III program promotes B-cell growth transformation and immortalization, and the EBV/BIK interactions described here may play an important role in that context and in disease settings where EBNA2 is expressed, such as EBV-associated posttransplant lymphoproliferative disease. Regulated BIK expression is critical for the selection of mature B lymphocytes (41), and this is likely due to its ability to inhibit BCL-XL, whose function is key to GC cell survival. Elsewhere, gene expression profiling of B cells during stages of GC transit (naive to centroblast [CB] to memory cells) showed that genes known to exert proapoptotic functions, including BIK and the FAS CD95 receptor, are upregulated in the CB (8.5- and 17-fold, respectively) relative to naive B cells and remain expressed at similar levels in the emerging memory B cells (101). The transition from CB to memory cells was characterized by a return to a phenotype similar to that of naive B cells except for an apoptotic program primed for both death and survival (101). Cells expressing the EBV Lat III program are present in and restricted to the naive B-cell subset of healthy tonsils, however (102). The loss of EBNA2 expression in vivo during GC transit implies that an EBNA2-independent mechanism(s) is required to maintain BIK repression in that setting, opening up the possibility that EBNA2-induced stable epigenetic changes or other EBV gene products play a role in that regard. This interpretation, however, implies that ER/EB2-5 cells, in which BIK is derepressed following EBV Lat III inactivation, do not completely recapitulate a true naive B cell as such, as has been noted elsewhere (103), and highlights the need for further studies using infected primary material.
In this study, both the presence of a TGF-β-activated SBE on the BIK promoter and a key role for SMAD3 in regulating both endogenous and TGF-β-1-induced BIK levels were confirmed. We showed that an EBV/BIK interaction exists, that it is mediated by EBNA2, and that it involves an overall reduction in the level of SMAD3 bound to this upstream regulatory element. In additional mechanistic studies, we did not consistently observe trans-repression by EBNA2 of a 1.9-kb BIK promoter fragment containing the SBE (bp −1710/+203) [104]) following extensive promoter-reporter cotransfection assays using EBV-negative BL cell lines, nor did we observe differences in the stability of BIK mRNA in the presence or absence of activated chimeric EBNA2 in ER/EB2-5 (data not shown). Others have reported BIK transcriptional silencing due to hypermethylation (38, 105); however, we did not detect BIK derepression in LCLs in response to known inhibitors of methylation (data not shown). These results indicate that BIK modulation by EBNA2 is likely to also involve a role for more distal or downstream/intronic transcriptional regulatory elements in addition to the SMAD/BIK promoter interactions described here.
blk (BIK-like killer; also known as mouse BIK) is considered the murine orthologue of human BIK, on the basis of its location in syntenic regions, gene organization, and nucleic acid sequence as well as amino acid sequence similarity. Mice with a heritable defect resulting in elevated levels of BIK RNA have been shown to have higher levels of apoptosis in splenic B cells, and normal B-cell development was restored by BCL-XL overexpression (106). In another study, B cells from BIK−/− knockout mice developed and reproduced normally, and deletion of this gene was shown to have little effect on the sensitivity of murine cells to apoptotic stimuli (40), including p53 overexpression (33). Murine and human BIK respond differently to stress stimuli, however (40, 75), and distinctions between the functions of these orthologues may be explained by substantial differences: (i) in structure, as mouse and human BIK proteins are only 43% identical, despite having similar gene structures (107), (ii) in expression, because unlike its human counterpart, mouse BIK is largely restricted to hematopoietic and endothelial cells, implying a difference in regulation of expression (40), and (iii) in response to TGF-β, as the regulation of these genes is crucially different in that the SMAD-binding regions in the human BIK promoter are not conserved in mouse or rat (22), indicating that BIK is unlikely to be involved in TGF-β-regulated B-cell homeostasis in mice.
A recent mathematical description of the current model for EBV persistence makes a case for the EBV cycle of infection being the basis for persistence rather than EBV quiescence in the memory B-cell compartment (15). Although the cellular responses that lead to BIK-mediated death remain incompletely characterized, one identified trigger is the shutoff of protein synthesis due to viral infection, a process induced by the EBV early lytic gene BGLF5 (82, 108, 109). Interestingly, the EBV antiapoptotic Bcl-2 homologues, BHRF1 and BALF1, are transiently expressed immediately following EBV infection and are essential for B-cell immortalization, but they become dispensable once latent infection is established (57). It may therefore be the case that negative transcriptional modulation of BIK by EBNA2 supersedes these early events and extends this survival advantage, thus favoring immortalization, persistence, and potentially lymphomagenesis.
ACKNOWLEDGMENTS
We are most grateful to B. Kempkes for the P493-6 and ER/EB2-5 cell lines and for total RNA from the DG75 clones SM295D6 and SM296D3. We thank A. Gordadze and P. Ling for the generous gift of lentivirus-transduced ER/EB2-5 cell pools. We are grateful to G. Chinnadurai for pcDNA3-HA-BIK and pcDNA3-HA-BIK-ΔBH3 and to D. Hayward for pSGEBNA2 and pSGEBNA2WW323SR.
This work was funded by research grants from the Health Research Board (HRB RP2005/212, Ireland) (D.W.) and Cancer Research Ireland (CRI02WAL; D.W. and B.N.D). R.H. was funded under the Program for Research in Third Level Institutions (PRTLI) Cycle 4. The PRTLI is cofunded through the European Regional Development Fund (ERDF), part of the European Union Structural Funds Program 2007–2013.
Footnotes
Published ahead of print 19 February 2014
REFERENCES
- 1.Pattle SB, Farrell PJ. 2006. The role of Epstein-Barr virus in cancer. Expert Opin. Biol. Ther. 6:1193–1205. 10.1517/14712598.6.11.1193 [DOI] [PubMed] [Google Scholar]
- 2.Kieff E, Rickinson AB. 2007. Epstein-Barr virus, p 2603–2654 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 5th ed. Lippincott-Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3.Young LS, Arrand JR, Murray PG. 2007. EBV gene expression and regulation, p 461–489 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 4.Lopes LF, Ruiz Miyazawa KW, de Almeida ER, Serafim KG, de Almeida Gualtieri K, Costa IC, Felipe I, Pavanelli WR, Watanabe MA. 2013. Epstein-Barr virus (EBV) microRNAs: involvement in cancer pathogenesis and immunopathology. Int. Rev. Immunol. 32:271–281. 10.3109/08830185.2012.748053 [DOI] [PubMed] [Google Scholar]
- 5.Zimber-Strobl U, Strobl LJ. 2001. EBNA2 and Notch signalling in Epstein-Barr virus mediated immortalization of B lymphocytes. Semin. Cancer Biol. 11:423–434 http://dx.doi.org/10.1006/scbi.2001.0409 [DOI] [PubMed] [Google Scholar]
- 6.Hayward SD. 2004. Viral interactions with the Notch pathway. Semin. Cancer Biol. 14:387–396 http://dx.doi.org/10.1016/j.semcancer.2004.04.018 [DOI] [PubMed] [Google Scholar]
- 7.Wang F, Tsang SF, Kurilla MG, Cohen JI, Kieff E. 1990. Epstein-Barr virus nuclear antigen 2 transactivates latent membrane protein LMP1. J. Virol. 64:3407–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, Kempkes B. 1999. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J. Virol. 73:4481–4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soni V, Cahir-McFarland E, Kieff E. 2007. LMP1 TRAFficking activates growth and survival pathways. Adv. Exp. Med. Biol. 597:173–187. 10.1007/978-0-387-70630-6_14 [DOI] [PubMed] [Google Scholar]
- 10.Fernandez D, Sanchez-Arevalo VJ, de Alboran IM. 2012. The role of the proto-oncogene c-myc in B lymphocyte differentiation. Crit. Rev. Immunol. 32:321–334. 10.1615/CritRevImmunol.v32.i4.30 [DOI] [PubMed] [Google Scholar]
- 11.Allen CD, Okada T, Cyster JG. 2007. Germinal-center organization and cellular dynamics. Immunity 27:190–202. 10.1016/j.immuni.2007.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. 1995. Epstein-Barr virus in B-cell non-Hodgkin's lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J. Pathol. 175:263–271 [DOI] [PubMed] [Google Scholar]
- 13.Kurth J, Spieker T, Wustrow J, Strickler GJ, Hansmann LM, Rajewsky K, Kuppers R. 2000. EBV-infected B cells in infectious mononucleosis: viral strategies for spreading in the B cell compartment and establishing latency. Immunity 13:485–495. 10.1016/S1074-7613(00)00048-0 [DOI] [PubMed] [Google Scholar]
- 14.Anagnostopoulos I, Hummel M, Kreschel C, Stein H. 1995. Morphology, immunophenotype, and distribution of latently and/or productively Epstein-Barr virus-infected cells in acute infectious mononucleosis: implications for the interindividual infection route of Epstein-Barr virus. Blood 85:744–750 [PubMed] [Google Scholar]
- 15.Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. 2013. The pathogenesis of Epstein-Barr virus persistent infection. Curr. Opin. Virol. 3:227–232. 10.1016/j.coviro.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowe M, Kelly GL, Bell AI, Rickinson AB. 2009. Burkitt's lymphoma: the Rosetta Stone deciphering Epstein-Barr virus biology. Semin. Cancer Biol. 19:377–388. 10.1016/j.semcancer.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuppers R. 2002. Molecular biology of Hodgkin's lymphoma. Adv. Cancer Res. 84:277–312. 10.1016/s0065-230x(02)84009-x [DOI] [PubMed] [Google Scholar]
- 18.Young L, Alfieri C, Hennessy K, Evans H, O'Hara C, Anderson KC, Ritz J, Shapiro RS, Rickinson A, Kieff E. 1989. Expression of Epstein-Barr virus transformation-associated genes in tissues of patients with EBV lymphoproliferative disease. N. Engl. J. Med. 321:1080–1085 [DOI] [PubMed] [Google Scholar]
- 19.Spender LC, Inman GJ. 2011. Inhibition of germinal centre apoptotic programs by Epstein-Barr virus. Adv. Hematol. 2011:829525. 10.1155/2011/829525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4:757–768. 10.1038/nrc1452 [DOI] [PubMed] [Google Scholar]
- 21.Kutok JL, Wang F. 2006. Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. 1:375–404. 10.1146/annurev.pathol.1.110304.100209 [DOI] [PubMed] [Google Scholar]
- 22.Spender LC, O'Brien DI, Simpson D, Dutt D, Gregory CD, Allday MJ, Clark LJ, Inman GJ. 2009. TGF-beta induces apoptosis in human B cells by transcriptional regulation of BIK and BCL-XL. Cell Death Differ. 16:593–602. 10.1038/cdd.2008.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inman GJ, Allday MJ. 2000. Apoptosis induced by TGF-beta 1 in Burkitt's lymphoma cells is caspase 8 dependent but is death receptor independent. J. Immunol. 165:2500–2510 [DOI] [PubMed] [Google Scholar]
- 24.Klein U, Dalla-Favera R. 2008. Germinal centres: role in B-cell physiology and malignancy. Nat. Rev. Immunol. 8:22–33. 10.1038/nri2217 [DOI] [PubMed] [Google Scholar]
- 25.Hao Z, Duncan GS, Seagal J, Su YW, Hong C, Haight J, Chen NJ, Elia A, Wakeham A, Li WY, Liepa J, Wood GA, Casola S, Rajewsky K, Mak TW. 2008. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity 29:615–627. 10.1016/j.immuni.2008.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romero-Camarero I, Jiang X, Natkunam Y, Lu X, Vicente-Duenas C, Gonzalez-Herrero I, Flores T, Garcia JL, McNamara G, Kunder C, Zhao S, Segura V, Fontan L, Martinez-Climent JA, Garcia-Criado FJ, Theis JD, Dogan A, Campos-Sanchez E, Green MR, Alizadeh AA, Cobaleda C, Sanchez-Garcia I, Lossos IS. 2013. Germinal centre protein HGAL promotes lymphoid hyperplasia and amyloidosis via BCR-mediated Syk activation. Nat. Commun. 4:1338. 10.1038/ncomms2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cory S, Huang DC, Adams JM. 2003. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22:8590–8607. 10.1038/sj.onc.1207102 [DOI] [PubMed] [Google Scholar]
- 28.Willis SN, Adams JM. 2005. Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 17:617–625 http://dx.doi.org/10.1016/j.ceb.2005.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cory S, Adams JM. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2:647–656. 10.1038/nrc883 [DOI] [PubMed] [Google Scholar]
- 30.Real PJ, Sanz C, Gutierrez O, Pipaon C, Zubiaga AM, Fernandez-Luna JL. 2006. Transcriptional activation of the proapoptotic bik gene by E2F proteins in cancer cells. FEBS Lett. 580:5905–5909 http://dx.doi.org/10.1016/j.febslet.2006.08.088 [DOI] [PubMed] [Google Scholar]
- 31.Hur J, Bell DW, Dean KL, Coser KR, Hilario PC, Okimoto RA, Tobey EM, Smith SL, Isselbacher KJ, Shioda T. 2006. Regulation of expression of BIK proapoptotic protein in human breast cancer cells: p53-dependent induction of BIK mRNA by fulvestrant and proteasomal degradation of BIK protein. Cancer Res. 66:10153–10161. 10.1158/0008-5472.CAN-05-3696 [DOI] [PubMed] [Google Scholar]
- 32.Bartke T, Siegmund D, Peters N, Reichwein M, Henkler F, Scheurich P, Wajant H. 2001. p53 upregulates cFLIP, inhibits transcription of NF-κB-regulated genes and induces caspase-8-independent cell death in DLD-1 cells. Oncogene 20:571–580. 10.1038/sj.onc.1204124 [DOI] [PubMed] [Google Scholar]
- 33.Mathai JP, Germain M, Marcellus RC, Shore GC. 2002. Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene 21:2534–2544. 10.1038/sj/onc/1205340 [DOI] [PubMed] [Google Scholar]
- 34.Chinnadurai G, Vijayalingam S, Rashmi R. 2008. BIK, the founding member of the BH3-only family proteins: mechanisms of cell death and role in cancer and pathogenic processes. Oncogene 27(Suppl. 1):S20–S29. 10.1038/onc.2009.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17:393–403. 10.1016/j.molcel.2004.12.030 [DOI] [PubMed] [Google Scholar]
- 36.Daniel PT, Pun KT, Ritschel S, Sturm I, Holler J, Dorken B, Brown R. 1999. Expression of the death gene Bik/Nbk promotes sensitivity to drug-induced apoptosis in corticosteroid-resistant T-cell lymphoma and prevents tumor growth in severe combined immunodeficient mice. Blood 94:1100–1107 [PubMed] [Google Scholar]
- 37.Zou Y, Peng H, Zhou B, Wen Y, Wang SC, Tsai EM, Hung MC. 2002. Systemic tumor suppression by the proapoptotic gene bik. Cancer Res. 62:8–12 [PubMed] [Google Scholar]
- 38.Sturm I, Stephan C, Gillissen B, Siebert R, Janz M, Radetzki S, Jung K, Loening S, Dorken B, Daniel PT. 2006. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 13:619–627. 10.1038/sj.cdd.4401782 [DOI] [PubMed] [Google Scholar]
- 39.Tong Y, Yang Q, Vater C, Venkatesh LK, Custeau D, Chittenden T, Chinnadurai G, Gourdeau H. 2001. The pro-apoptotic protein, Bik, exhibits potent antitumor activity that is dependent on its BH3 domain. Mol. Cancer Ther. 1:95–102 [PubMed] [Google Scholar]
- 40.Coultas L, Bouillet P, Stanley EG, Brodnicki TC, Adams JM, Strasser A. 2004. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programd death. Mol. Cell. Biol. 24:1570–1581. 10.1128/MCB.24.4.1570-1581.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang A, Clark EA. 2001. Involvement of Bik, a proapoptotic member of the Bcl-2 family, in surface IgM-mediated B cell apoptosis. J. Immunol. 166:6025–6033 [DOI] [PubMed] [Google Scholar]
- 42.Kelly G, Bell A, Rickinson A. 2002. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat. Med. 8:1098–1104. 10.1038/nm758 [DOI] [PubMed] [Google Scholar]
- 43.Andersson ML, Stam NJ, Klein G, Ploegh HL, Masucci MG. 1991. Aberrant expression of HLA class-I antigens in Burkitt lymphoma cells. Int. J. Cancer 47:544–550 [DOI] [PubMed] [Google Scholar]
- 44.Ben-Bassat H, Goldblum N, Mitrani S, Goldblum T, Yoffey JM, Cohen MM, Bentwich Z, Ramot B, Klein E, Klein G. 1977. Establishment in continuous culture of a new type of lymphocyte from a “Burkitt like” malignant lymphoma (line D.G.-75). Int. J. Cancer 19:27–33 [DOI] [PubMed] [Google Scholar]
- 45.Gregory CD, Rowe M, Rickinson AB. 1990. Different Epstein-Barr virus-B cell interactions in phenotypically distinct clones of a Burkitt's lymphoma cell line. J. Gen. Virol. 71:1481–1495 [DOI] [PubMed] [Google Scholar]
- 46.Klein G, Giovanella B, Westman A, Stehlin JS, Mumford D. 1975. An EBV-genome-negative cell line established from an American Burkitt lymphoma; receptor characteristics. EBV infectibility and permanent conversion into EBV-positive sublines by in vitro infection. Intervirology 5:319–334 [DOI] [PubMed] [Google Scholar]
- 47.Menezes J, Leibold W, Klein G, Clements G. 1975. Establishment and characterization of an Epstein-Barr virus (EBC)-negative lymphoblastoid B cell line (BJA-B) from an exceptional, EBV-genome-negative African Burkitt's lymphoma. Biomedicine 22:276–284 [PubMed] [Google Scholar]
- 48.Rowe M, Rooney CM, Edwards CF, Lenoir GM, Rickinson AB. 1986. Epstein-Barr virus status and tumour cell phenotype in sporadic Burkitt's lymphoma. Int. J. Cancer 37:367–373 [DOI] [PubMed] [Google Scholar]
- 49.Kelly GL, Milner AE, Tierney RJ, Croom-Carter DS, Altmann M, Hammerschmidt W, Bell AI, Rickinson AB. 2005. Epstein-Barr virus nuclear antigen 2 (EBNA2) gene deletion is consistently linked with EBNA3A, -3B, and -3C expression in Burkitt's lymphoma cells and with increased resistance to apoptosis. J. Virol. 79:10709–10717. 10.1128/JVI.79.16.10709-10717.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kempkes B, Spitkovsky D, Jansen-Durr P, Ellwart JW, Kremmer E, Delecluse HJ, Rottenberger C, Bornkamm GW, Hammerschmidt W. 1995. B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J. 14:88–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kempkes B, Zimber-Strobl U, Eissner G, Pawlita M, Falk M, Hammerschmidt W, Bornkamm GW. 1996. Epstein-Barr virus nuclear antigen 2 (EBNA2)-oestrogen receptor fusion proteins complement the EBNA2-deficient Epstein-Barr virus strain P3HR1 in transformation of primary B cells but suppress growth of human B cell lymphoma lines. J. Gen. Virol. 77:227–237 [DOI] [PubMed] [Google Scholar]
- 52.Floettmann JE, Ward K, Rickinson AB, Rowe M. 1996. Cytostatic effect of Epstein-Barr virus latent membrane protein-1 analyzed using tetracycline-regulated expression in B cell lines. Virology 223:29–40 [DOI] [PubMed] [Google Scholar]
- 53.Kempkes B, Pawlita M, Zimber-Strobl U, Eissner G, Laux G, Bornkamm GW. 1995. Epstein-Barr virus nuclear antigen 2-estrogen receptor fusion proteins transactivate viral and cellular genes and interact with RBP-J kappa in a conditional fashion. Virology 214:675–679 [DOI] [PubMed] [Google Scholar]
- 54.Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, Eick D, Kohlhuber F. 1999. Control of cell growth by c-Myc in the absence of cell division. Curr. Biol. 9:1255–1258 [DOI] [PubMed] [Google Scholar]
- 55.Maier S, Santak M, Mantik A, Grabusic K, Kremmer E, Hammerschmidt W, Kempkes B. 2005. A somatic knockout of CBF1 in a human B-cell line reveals that induction of CD21 and CCR7 by EBNA-2 is strictly CBF1 dependent and that downregulation of immunoglobulin M is partially CBF1 independent. J. Virol. 79:8784–8792. 10.1128/JVI.79.14.8784-8797.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gordadze AV, Peng R, Tan J, Liu G, Sutton R, Kempkes B, Bornkamm GW, Ling PD. 2001. Notch1IC partially replaces EBNA2 function in B cells immortalized by Epstein-Barr virus. J. Virol. 75:5899–5912. 10.1128/JVI.75.13.5899-5912.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Altmann M, Hammerschmidt W. 2005. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 3(12):e404. 10.1371/journal.pbio.0030404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shannon-Lowe C, Adland E, Bell AI, Delecluse HJ, Rickinson AB, Rowe M. 2009. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: viral genome expression, genome maintenance, and genome amplification. J. Virol. 83:7749–7760. 10.1128/JVI.00108-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shannon-Lowe C, Baldwin G, Feederle R, Bell A, Rickinson A, Delecluse HJ. 2005. Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J. Gen. Virol. 86:3009–3019. 10.1099/vir.0.81153-0 [DOI] [PubMed] [Google Scholar]
- 60.Heath E, Begue-Pastor N, Chaganti S, Croom-Carter D, Shannon-Lowe C, Kube D, Feederle R, Delecluse HJ, Rickinson AB, Bell AI. 2012. Epstein-Barr virus infection of naive B cells in vitro frequently selects clones with mutated immunoglobulin genotypes: implications for virus biology. PLoS Pathog. 8(5):e1002697. 10.1371/journal.ppat.1002697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loughran ST, Campion EM, D'Souza BN, Smith SM, Vrzalikova K, Wen K, Murray PG, Walls D. 2011. Bfl-1 is a crucial pro-survival nuclear factor-κB target gene in Hodgkin/Reed-Sternberg cells. Int. J. Cancer 129:2787–2796. 10.1002/ijc.25950 [DOI] [PubMed] [Google Scholar]
- 62.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔCT) method. Methods 25:402–408 http://dx.doi.org/10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 63.D'Souza BN, Edelstein LC, Pegman PM, Smith SM, Loughran ST, Clarke A, Mehl A, Rowe M, Gelinas C, Walls D. 2004. Nuclear factor kappa B-dependent activation of the antiapoptotic bfl-1 gene by the Epstein-Barr virus latent membrane protein 1 and activated CD40 receptor. J. Virol. 78:1800–1816. 10.1128/JVI.78.4.1800-1816.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Finke J, Rowe M, Kallin B, Ernberg I, Rosen A, Dillner J, Klein G. 1987. Monoclonal and polyclonal antibodies against Epstein-Barr virus nuclear antigen 5 (EBNA-5) detect multiple protein species in Burkitt's lymphoma and lymphoblastoid cell lines. J. Virol. 61:3870–3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ling PD, Rawlins DR, Hayward SD. 1993. The Epstein-Barr virus immortalizing protein EBNA-2 is targeted to DNA by a cellular enhancer-binding protein. Proc. Natl. Acad. Sci. U. S. A. 90:9237–9241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gordadze AV, Poston D, Ling PD. 2002. The EBNA2 polyproline region is dispensable for Epstein-Barr virus-mediated immortalization maintenance. J. Virol. 76:7349–7355. 10.1128/JVI.76.14.7349-7355.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hur J, Chesnes J, Coser KR, Lee RS, Geck P, Isselbacher KJ, Shioda T. 2004. The Bik BH3-only protein is induced in estrogen-starved and antiestrogen-exposed breast cancer cells and provokes apoptosis. Proc. Natl. Acad. Sci. U. S. A. 101:2351–2356. 10.1073/pnas.0307337101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Polack A, Hortnagel K, Pajic A, Christoph B, Baier B, Falk M, Mautner J, Geltinger C, Bornkamm GW, Kempkes B. 1996. c-myc activation renders proliferation of Epstein-Barr virus (EBV)-transformed cells independent of EBV nuclear antigen 2 and latent membrane protein 1. Proc. Natl. Acad. Sci. U. S. A. 93:10411–10416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tierney RJ, Kao KY, Nagra JK, Rickinson AB. 2011. Epstein-Barr virus BamHI W repeat number limits EBNA2/EBNA-LP coexpression in newly infected B cells and the efficiency of B-cell transformation: a rationale for the multiple W repeats in wild-type virus strains. J. Virol. 85:12362–12375. 10.1128/JVI.06059-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klein G, Lindahl T, Jondal M, Leibold W, Menezes J, Nilsson K, Sundstrom C. 1974. Continuous lymphoid cell lines with characteristics of B cells (bone-marrow-derived), lacking the Epstein-Barr virus genome and derived from three human lymphomas. Proc. Natl. Acad. Sci. U. S. A. 71:3283–3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schuster N, Krieglstein K. 2002. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 307:1–14. 10.1007/s00441-001-0479-6 [DOI] [PubMed] [Google Scholar]
- 72.Magae J, Illenye S, Tejima T, Chang YC, Mitsui Y, Tanaka K, Omura S, Heintz NH. 1997. Transcriptional squelching by ectopic expression of E2F-1 and p53 is alleviated by proteasome inhibitors MG-132 and lactacystin. Oncogene 15:759–769 [DOI] [PubMed] [Google Scholar]
- 73.Zhu H, Zhang L, Dong F, Guo W, Wu S, Teraishi F, Davis JJ, Chiao PJ, Fang B. 2005. Bik/NBK accumulation correlates with apoptosis-induction by bortezomib (PS-341, Velcade) and other proteasome inhibitors. Oncogene 24:4993–4999. 10.1038/sj.onc.1208683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Germain M, Mathai JP, Shore GC. 2002. BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J. Biol. Chem. 277:18053–18060. 10.1074/jbc.M201235200 [DOI] [PubMed] [Google Scholar]
- 75.Mathai JP, Germain M, Shore GC. 2005. BH3-only BIK regulates BAX,BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J. Biol. Chem. 280:23829–23836. 10.1074/jbc.M500800200 [DOI] [PubMed] [Google Scholar]
- 76.Zhao X, Sun Y, Yu H, Ye L, Zhang L, Lu J, Yuan Y, Qian G, Ge S. 2007. Apoptosis induced by BIK was decreased with RNA interference of caspase-12. Biochem. Biophys. Res. Commun. 359:896–901 http://dx.doi.org/10.1016/j.bbrc.2007.05.175 [DOI] [PubMed] [Google Scholar]
- 77.Kehrl JH, Taylor AS, Delsing GA, Roberts AB, Sporn MB, Fauci AS. 1989. Further studies of the role of transforming growth factor-beta in human B cell function. J. Immunol. 143:1868–1874 [PubMed] [Google Scholar]
- 78.Kumar A, Rogers T, Maizel A, Sharma S. 1991. Loss of transforming growth factor beta 1 receptors and its effects on the growth of EBV-transformed human B cells. J. Immunol. 147:998–1006 [PubMed] [Google Scholar]
- 79.Inman GJ, Allday MJ. 2000. Resistance to TGF-β1 correlates with a reduction of TGF-β type II receptor expression in Burkitt's lymphoma and Epstein-Barr virus-transformed B lymphoblastoid cell lines. J. Gen. Virol. 81:1567–1578 [DOI] [PubMed] [Google Scholar]
- 80.Altiok A, Ehlin-Henriksson B, Klein E. 1993. Correlation between the growth-inhibitory effect of TGF-beta 1 and phenotypic characteristics in a panel of B-cell lines. Int. J. Cancer 55:137–140 [DOI] [PubMed] [Google Scholar]
- 81.Schrantz N, Blanchard DA, Auffredou MT, Sharma S, Leca G, Vazquez A. 1999. Role of caspases and possible involvement of retinoblastoma protein during TGF-beta-mediated apoptosis of human B lymphocytes. Oncogene 18:3511–3519 [DOI] [PubMed] [Google Scholar]
- 82.Shimazu T, Degenhardt K, Nur-E-Kamal A, Zhang J, Yoshida T, Zhang Y, Mathew R, White E, Inouye M. 2007. NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes Dev. 21:929–941. 10.1101/gad.1522007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kvansakul M, Wei AH, Fletcher JI, Willis SN, Chen L, Roberts AW, Huang DC, Colman PM. 2010. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 6(12):e1001236. 10.1371/journal.ppat.1001236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, Ebb RG, Subramanian T, Chittenden T, Lutz RJ. 1995. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene 11:1921–1928 [PubMed] [Google Scholar]
- 85.Elangovan B, Chinnadurai G. 1997. Functional dissection of the pro-apoptotic protein Bik. Heterodimerization with anti-apoptosis proteins is insufficient for induction of cell death. J. Biol. Chem. 272:24494–24498 [DOI] [PubMed] [Google Scholar]
- 86.D'Souza B, Rowe M, Walls D. 2000. The bfl-1 gene is transcriptionally upregulated by the Epstein-Barr virus LMP1, and its expression promotes the survival of a Burkitt's lymphoma cell line. J. Virol. 74:6652–6658. 10.1128/JVI.74.14.6652-6658.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gillissen B, Essmann F, Hemmati PG, Richter A, Richter A, Oztop I, Chinnadurai G, Dorken B, Daniel PT. 2007. Mcl-1 determines the Bax dependency of Nbk/Bik-induced apoptosis. J. Cell Biol. 179:701–715. 10.1083/jcb.200703040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arena V, Martini M, Luongo M, Capelli A, Larocca LM. 2003. Mutations of the BIK gene in human peripheral B-cell lymphomas. Genes Chromosomes Cancer 38:91–96. 10.1002/gcc.10245 [DOI] [PubMed] [Google Scholar]
- 89.Schrantz N, Bourgeade MF, Mouhamad S, Leca G, Sharma S, Vazquez A. 2001. p38-mediated regulation of an Fas-associated death domain protein-independent pathway leading to caspase-8 activation during TGF-beta-induced apoptosis in human Burkitt lymphoma B cells BL41. Mol. Biol. Cell 12:3139–3151. 10.1091/mbc.12.10.3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Altiok A, Bejarano MT, Ruscetti F, Altiok E, Klein G, Klein E. 1991. Effect of transforming growth factor-beta 1 and -beta 2 on the proliferation of Burkitt lymphoma and lymphoblastoid cell lines. Growth Factors 4:117–128 [DOI] [PubMed] [Google Scholar]
- 91.Fukuda M, Kurosaki H, Sairenji T. 2006. Loss of functional transforming growth factor (TGF)-β type II receptor results in insensitivity to TGF-β1-mediated apoptosis and Epstein-Barr virus reactivation. J. Med. Virol. 78:1456–1464. 10.1002/jmv.20719 [DOI] [PubMed] [Google Scholar]
- 92.Horndasch M, Raschke EE, Bommer G, Schuhmacher M, Dumont E, Kuklik-Roos C, Eick D, Kempkes B. 2002. Epstein-Barr virus antagonizes the antiproliferative activity of transforming growth factor-beta but does not abolish its signaling. Int. J. Cancer 101:442–447. 10.1002/ijc.10626 [DOI] [PubMed] [Google Scholar]
- 93.Rochford R, Cannon MJ, Sabbe RE, Adusumilli K, Picchio G, Glynn JM, Noonan DJ, Mosier DE, Hobbs MV. 1997. Common and idiosyncratic patterns of cytokine gene expression by Epstein-Barr virus transformed human B cell lines. Viral Immunol. 10:183–195 [DOI] [PubMed] [Google Scholar]
- 94.Wroblewski JM, Copple A, Batson LP, Landers CD, Yannelli JR. 2002. Cell surface phenotyping and cytokine production of Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines (LCLs). J. Immunol. Methods 264:19–28 http://dx.doi.org/10.1016/S0022-1759(01)00565-8 [DOI] [PubMed] [Google Scholar]
- 95.Lee JM, Lee KH, Farrell CJ, Ling PD, Kempkes B, Park JH, Hayward SD. 2004. EBNA2 is required for protection of latently Epstein-Barr virus-infected B cells against specific apoptotic stimuli. J. Virol. 78:12694–12697. 10.1128/JVI.78.22.12694-12697.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee JM, Lee KH, Weidner M, Osborne BA, Hayward SD. 2002. Epstein-Barr virus EBNA2 blocks Nur77-mediated apoptosis. Proc. Natl. Acad. Sci. U. S. A. 99:11878–11883. 10.1073/pnas.182552499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pegman PM, Smith SM, D'Souza BN, Loughran ST, Maier S, Kempkes B, Cahill PA, Simmons MJ, Gelinas C, Walls D. 2006. Epstein-Barr virus nuclear antigen 2 trans-activates the cellular antiapoptotic bfl-1 gene by a CBF1/RBPJ kappa-dependent pathway. J. Virol. 80:8133–8144. 10.1128/JVI.00278-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kenney JL, Guinness ME, Reiss M, Lacy J. 2001. Antisense to the Epstein-Barr virus (EBV)-encoded latent membrane protein 1 (LMP-1) sensitizes EBV-immortalized B cells to transforming growth factor-beta and chemotherapeutic agents. Int. J. Cancer 91:89–98. [DOI] [PubMed] [Google Scholar]
- 99.Lo AK, Dawson CW, Lo KW, Yu Y, Young LS. 2010. Upregulation of Id1 by Epstein-Barr virus-encoded LMP1 confers resistance to TGF-beta-mediated growth inhibition. Mol. Cancer 9:155. 10.1186/1476-4598-9-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miller WE, Earp HS, Raab-Traub N. 1995. The Epstein-Barr virus latent membrane protein 1 induces expression of the epidermal growth factor receptor. J. Virol. 69:4390–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad, Miljkovic JV, Cattoretti G, Califano A, Dalla-Favera R. 2003. Gene expression dynamics during germinal center transit in B cells. Ann. N. Y. Acad. Sci. 987:166–172. 10.1111/j.1749-6632.2003.tb06045.x [DOI] [PubMed] [Google Scholar]
- 102.Joseph AM, Babcock GJ, Thorley-Lawson DA. 2000. Cells expressing the Epstein-Barr virus growth program are present in and restricted to the naive B-cell subset of healthy tonsils. J. Virol. 74:9964–9971. 10.1128/JVI.74.21.9964-9971.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spender LC, Cornish GH, Sullivan A, Farrell PJ. 2002. Expression of transcription factor AML-2 (RUNX3, CBFα-3) is induced by Epstein-Barr virus EBNA-2 and correlates with the B-cell activation phenotype. J. Virol. 76:4919–4927. 10.1128/JVI.76.10.4919-4927.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Verma S, Budarf ML, Emanuel BS, Chinnadurai G. 2000. Structural analysis of the human pro-apoptotic gene Bik: chromosomal localization, genomic organization and localization of promoter sequences. Gene 254:157–162 http://dx.doi.org/10.1016/S0378-1119(00)00276-6 [DOI] [PubMed] [Google Scholar]
- 105.Milutinovic S, Brown SE, Zhuang Q, Szyf M. 2004. DNA methyltransferase 1 knock down induces gene expression by a mechanism independent of DNA methylation and histone deacetylation. J. Biol. Chem. 279:27915–27927. 10.1074/jbc.M312823200 [DOI] [PubMed] [Google Scholar]
- 106.Amanna IJ, Dingwall JP, Hayes CE. 2003. Enforced bcl-xL gene expression restored splenic B lymphocyte development in BAFF-R mutant mice. J. Immunol. 170:4593–4600 [DOI] [PubMed] [Google Scholar]
- 107.Hegde R, Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. 1998. Blk, a BH3-containing mouse protein that interacts with Bcl-2 and Bcl-xL, is a potent death agonist. J. Biol. Chem. 273:7783–7786 [DOI] [PubMed] [Google Scholar]
- 108.Subramanian T, Vijayalingam S, Lomonosova E, Zhao LJ, Chinnadurai G. 2007. Evidence for involvement of BH3-only proapoptotic members in adenovirus-induced apoptosis. J. Virol. 81:10486–10495. 10.1128/JVI.00808-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rowe M, Glaunsinger B, van Leeuwen D, Zuo J, Sweetman D, Ganem D, Middeldorp J, Wiertz EJ, Ressing ME. 2007. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. U. S. A. 104:3366–3371. 10.1073/pnas.0611128104 [DOI] [PMC free article] [PubMed] [Google Scholar]