

ABSTRACT
Foot-and-mouth disease virus (FMDV) causes a highly contagious, debilitating disease in cloven-hoofed animals with devastating economic consequences. To survive in the host, FMDV has evolved to antagonize the host type I interferon (IFN) response. Previous studies have reported that the leader proteinase (Lpro) and 3Cpro of FMDV are involved in the inhibition of type I IFN production. However, whether the proteins of FMDV can inhibit type I IFN signaling is less well understood. In this study, we first found that 3Cpro of FMDV functioned to interfere with the JAK-STAT signaling pathway. Expression of 3Cpro significantly reduced the transcript levels of IFN-stimulated genes (ISGs) and IFN-stimulated response element (ISRE) promoter activity. The protein level, tyrosine phosphorylation of STAT1 and STAT2, and their heterodimerization were not affected. However, the nuclear translocation of STAT1/STAT2 was blocked by the 3Cpro protein. Further mechanistic studies demonstrated that 3Cpro induced proteasome- and caspase-independent protein degradation of karyopherin α1 (KPNA1), the nuclear localization signal receptor for tyrosine-phosphorylated STAT1, but not karyopherin α2, α3, or α4. Finally, we showed that the protease activity of 3Cpro contributed to the degradation of KPNA1 and thus blocked STAT1/STAT2 nuclear translocation. Taken together, results of our experiments describe for the first time a novel mechanism by which FMDV evolves to inhibit IFN signaling and counteract host innate antiviral responses.
IMPORTANCE We show that 3Cpro of FMDV antagonizes the JAK-STAT signaling pathway by blocking STAT1/STAT2 nuclear translocation. Furthermore, 3Cpro induces KPNA1 degradation, which is independent of proteasome and caspase pathways. The protease activity of 3Cpro contributes to the degradation of KPNA1 and governs the ability of 3Cpro to inhibit the JAK-STAT signaling pathway. This study uncovers a novel mechanism evolved by FMDV to antagonize host innate immune responses.
INTRODUCTION
Foot-and-mouth disease (FMD) is a highly contagious disease with high morbidity in cloven-hoofed animals, including important livestock species such as cattle and swine. FMD is caused by FMD virus (FMDV), which belongs to the Aphthovirus genus of the Picornaviridae family (1, 2). The genome of FMDV is about 8.5 kb in length, with only one open reading frame (ORF) to encode a polyprotein. Through the process of cleavage, which is conducted by three virus-encoded proteinases, leader (Lpro), 2A, and 3Cpro, the viral polyprotein is processed into precursors and individual structural and nonstructural proteins later on (2, 3).
The type I interferon (IFN) family is recognized as an essential component of the innate immune response and the first line of defense against virus infection. Type I IFNs, including IFN-α and IFN-β, are known to be important for triggering a robust host response against viral infection (4). Initially, virus invasion is detected by the sensors of the immune system. Pathogen-associated molecular patterns (PAMPs) are sensed by host pattern recognition receptors (PRRs) (5, 6). This process activates host protein signaling cascades, followed by the activation of transcription factors, including interferon regulatory factor 3 (IRF3), NF-κB, and ATF-2/c-JUN (7). The cooperation of these factors leads to the expression of type I IFNs (8). After secretion, type I IFNs bind to a common heterodimeric receptor composed of IFN-α/β receptor 1 (IFNAR1) and IFN-α/β receptor 2 (IFNAR2) on adjacent cell surfaces to activate the Janus kinase (JAK) family and the signal transducers and activators of transcription (STATs) family. Upon JAK1 and tyrosine kinase 2 (JAK1/Tyk2)-mediated tyrosine phosphorylation, STAT1 and STAT2 heterodimerize and translocate to the nucleus, where they bind to IFN regulatory factor 9 (IRF9) to form the transcription complex IFN-stimulated gene factor 3 (ISGF3). ISGF3, in turn, sequence-specifically binds to an IFN-stimulated response element (ISRE) that is present in numerous type I IFN-stimulated genes (ISGs), such as double-stranded RNA-dependent protein kinase R (PKR), 2′,5′-oligoadenylate synthetase (OAS), myxovirus resistance 1 (Mx1), IFN-stimulated gene 15 (ISG15), and ISG56, many of which exhibit antiviral activity (9–13).
Picornaviruses have developed mechanisms to counteract the host innate immune systems, and such viral antagonists have been identified, including 2A protease and 3C protease of enterovirus 71 (EV71) (14–17), 3C protease of coxsackievirus B3 (CVB3) (18), and 3C protease of hepatitis A virus (HAV) (19, 20). For FMDV, previous studies have shown that it has developed the ability to counteract the host innate immune response. The viral proteases Lpro and 3Cpro, whose primary functions are to process viral polyprotein, have been found to antagonize the host type I IFN response. Lpro reduces the level of immediate-early induction of IFN-β mRNA and ISGs such as PKR, OAS, and Mx1 mRNAs in swine cells (21). To be more specific, in FMDV-infected cells, Lpro brings about the degradation of not only NF-κB's subunit p65/RelA (22) but also IRF3/IRF7, both of which play important roles in the production of IFN (23). 3Cpro has recently been shown to cleave the nuclear transcription factor kappa B (NF-κB) essential modulator (NEMO) and impair the ability of NEMO to activate downstream IFN production and to participate in the MDA-5/RIG-I (retinoic acid-inducible protein I) pathway (24). However, whether FMDV inhibits the type I IFN signaling pathway remains unclear.
In this study, we provide evidence that FMDV 3Cpro inhibits the JAK-STAT signaling pathway by blocking the nuclear translocation of STAT1/STAT2. 3Cpro induced degradation of karyopherin α1 (KPNA1), and the protease activity of 3Cpro contributed to the degradation and thus blocked STAT1/STAT2 nuclear translocation. These data uncover a novel mechanism evolved by FMDV to antagonize host innate immune responses.
MATERIALS AND METHODS
Cells and chemicals.
HeLa cells (NIH AIDS Research and Reference Reagent Program, Germantown, MD, USA), BHK-21 cells (ATCC CCL-10), and porcine kidney (PK-15) cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA), 2 mM l-glutamine, 100 U of penicillin/ml, and 100 μg of streptomycin/ml in a humidified incubator with 5% CO2 at 37°C. IFN-β (Calbiochem) was used for IFN stimulation. The proteasome inhibitor MG132 and the caspase inhibitor zVAD-FMK (carboxybenzyl-Val-Ala-Asp-fluoromethylketone) were obtained from Beyotime (China).
Plasmids.
The eukaryotic expression vector pXJ41 is a derivative of pXJ40 in which the polylinker region was modified (25, 26). The plasmid pISRE-Luc contains ISRE binding sequence upstream of the luciferase (Luc) reporter gene (Stratagene, La Jolla, CA, USA). The pRL-TK plasmid (Promega, Madison, WI, USA) contains the Renilla luciferase reporter under the control of the herpes simplex virus thymidine kinase (HSV-tk) promoter, serving as an internal control. The FMDV 3ABC, 3A, 3B, and 3C genes were amplified from the cDNA of FMDV type O, strain Tibet/CHA/99 (GenBank accession number AJ539138), with primers containing restriction sites for HindIII or KpnI to facilitate directional cloning, fused with a N-terminal FLAG tag (for 3ABC, 3A, and 3B) or a C-terminal hemagglutinin (HA) tag (for 3ABC and 3C). The PCR fragments were cloned into the pXJ41 mammalian expression vector to obtain plasmids pXJ41-3ABC, pXJ41-3A, pXJ41-3B, and pXJ41-3C. Specific mutations (H46Y, D84N, C163G, and H205R) were introduced to pXJ41-3C or an FMDV infectious cDNA clone of FMDV type O, strain Tibet/CHA/99, using a QuikChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA), with modifications as described elsewhere (26). Primers used in cloning and specific mutations are listed in Table 1. Based on the KPNA1, -2, -3, and -4 gene sequences (GenBank accession numbers BC090864, NM_002266, NM_002267 and NM_002268, respectively), four pairs of PCR primers were designed (Table 1). cDNAs encoding KPNA1, -2, -3, and -4 were amplified by reverse transcription-PCR (RT-PCR) using total RNA extracted from HeLa cells and cloned into pCMV-Tag 1 containing an N-terminal FLAG tag (Stratagene, La Jolla, CA, USA). pXJ41-NS1 expressing swine influenza virus (SIV) nonstructural protein 1 (NS1) and pXJ41-GST expressing glutathione S-transferase (GST) were used as positive and negative controls, respectively, as described elsewhere (27). All of the expression plasmids were sequenced to confirm the correct tandem in-frame insertion of individual genes.
TABLE 1.
Primers used in the study
| Primer name | Sequence (5′–3′)a | Restriction site (promoter or tag) | Purpose |
|---|---|---|---|
| 3ABC-Fwd | GCGAAGCTTCCACCATGGATTACAAGGATGACGACGATAAGATCTCAATTCCTTCCC | HindIII (FLAG) | FMDV 3ABC amplification |
| 3ABC-Rev | GCGGGTACCTATTAAGCGTAATCAGGAACGTCGTAAGGGTACTCGTGGTGTGGTTCG | KpnI (HA) | |
| 3A-Rev | GCGGGTACCTATTATTCAGCTTGTGGTTGTTC | KpnI | FMDV 3A amplification |
| 3B-Fwd | GCGAAGCTTCCACCATGGATTACAAGGATGACGACGATAAGGGACCCTACACCGGT | HindIII (FLAG) | FMDV 3B amplification |
| 3B-Rev | GCGGGTACCTATTACTCAGTGACAATCAAG | KpnI | |
| 3C-Fwd | GCGAAGCTTCCACCATGGCTAGTGGTGCTCCCCCGACT | HindIII | FMDV 3C amplification |
| 3C-H46Y-Fwd | TACCTTGTTCCTCGTTATCTTTTCGCAGAGAAG | Site 46 of 3C mutagenesis | |
| 3C-H46Y-Rev | CTTCTCTGCGAAAAGATAACGAGGAACAAGGTA | ||
| 3C-D84N-Fwd | CAGGACATGCTCTCAAACGCCGCGCTCATGGTG | Site 84 of 3C mutagenesis | |
| 3C-D84N-Rev | CACCATGAGCGCGGCGTTTGAGAGCATGTCCTG | ||
| 3C-C163G-Fwd | ACCAAGGCGGGTTACGGTGGAGGAGCCGTTCTT | Site 163 of 3C mutagenesis | |
| 3C-C163G-Rev | AAGAACGGCTCCTCCACCGTAACCCGCCTTGGT | ||
| 3C-H205R-Fwd | CTTAAAATGAAGGCACGCATCGATCCCGAACCA | Site 205 of 3C mutagenesis | |
| 3C-H205R-Fwd | TGGTTCGGGATCGATGCGTGCCTTCATTTTAAG | ||
| KPNA1-Fwd | GGCGGATCCACCATGGATTACAAGGATGACGACGATAAGATGACCACCCCAGGAAA | BamHI (FLAG) | KPNA1 amplification |
| KPNA1-Rev | GCGCTCGAGTCAAAGCTGGAAACCT | XhoI | |
| KPNA2-Fwd | GGCGGATCCACCATGGATTACAAGGATGACGACGATAAGATGTCCACCAACGAGA | BamHI (FLAG) | KPNA2 amplification |
| KPNA2-Rev | GCGCTCGAGCTAAAAGTTAAAGGT | XhoI | |
| KPNA3-Fwd | GGCGCGGCCGCCACCATGGATTACAAGGATGACGACGATAAGATGGCCGAGAACCCC | NotI (FLAG) | KPNA3 amplification |
| KPNA3-Rev | GCGCTCGAGTTAAAAATTAAATTC | XhoI | |
| KPNA4-Fwd | GGCGCGGCCGCCACCATGGATTACAAGGATGACGACGATAAGATGGCGGACAACGAG | NotI (FLAG) | KPNA4 amplification |
| KPNA4-Rev | GCGCTCGAGCTAAAACTGGAACCCTTCT | XhoI | |
| GAPDH-Fwd | ATGGCCTTCCGTGTCC | GAPDH amplification | |
| GAPDH-Rev | GTGGGTGTCGCTGTTGA | ||
| ISG15-Fwd | GACAAATGCGACGAACCTC | ISG15 amplification | |
| ISG15-Rev | CCGCTCACTTGCTGCTT | ||
| ISG56-Fwd | CCTCCTTGGGTTCGTCTACA | ISG56 amplification | |
| ISG56-Rev | GGCTGATATCTGGGTGCCTA | ||
| Mx1-Fwd | CCATCGGAATCTTGACG | Mx1 amplification | |
| Mx1-Rev | GCTTCGGACAGGCTCAG | ||
| OAS-Fwd | CTCAAGAGCCTCATCCGC | OAS amplification | |
| OAS-Rev | GACCGTCCGAAATCCCT | ||
| PKR-Fwd | TAACGAGAAGGCGGAGCG | PKR amplification | |
| PKR-Rev | CCATTTGGATGAAAAGGCACT | ||
| FMDV-A-Fwd | TATGGCGCGCCTAATACGACTCACTATAGGTTGAAAGGGGGCGTTAGGG | AscI (T7) | FMDV fragment A amplification |
| FMDV-A-Rev | TGAGCGGCCGCCTCAGGTGTTTTG | NotI | |
| FMDV-Am-Fwd | ACTGGCGAGTGCTAGC*AACAGCACTGTTGCT | NheI | NheI site mutagenesis |
| FMDV-Am-Rev | AGCAACAGTGCTGTTG*CTAGCACTCGCCAGT | NheI | |
| FMDV-B-Fwd | GAGGCGGCCGCTCACTGCATTCAT | NotI | FMDV fragment B amplification |
| FMDV-B-Rev | TGTCCCGGGAGAGTTTTCTCTCTGA | XmaI | |
| FMDV-C-Fwd | TCTCCCGGGACACAAGGCGAGTGAT | XmaI | FMDV fragment C amplification |
| FMDV-C-Rev | GCGGCGGCGTTAATTAATTTTTTTTTTTTTTT | PacI |
Restriction endonuclease sites are underlined. The Flag or HA tag or T7 promoter is in boldface. The mutated sites are in boldface and underlined. The asterisk refers to the mutated nucleotide of FMDV type O, strain Tibet/CHA/99, sequence.
Real-time PCR.
To determine the effect of FMDV 3Cpro on the transcript levels of ISGs, HeLa cells were seeded in 24-well plates and incubated until cells reached approximately 70 to 80% confluence. Cells were transfected with 0.5 μg of the empty vector pXJ41, pXJ41-GST, pXJ41-NS1, pXJ41-3ABC, pXJ41-3A, pXJ41-3B, or pXJ41-3C using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). At 24 h posttransfection, cells were treated with IFN-β (Calbiochem) at a concentration of 1,000 U/ml for 5 h. The total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. RNA was further purified by using an RNeasy Minikit, including a DNase digestion step (Qiagen, Chatsworth, CA, USA). One microgram of purified RNA was reverse transcribed to cDNA using an oligo(dT) primer and SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA), and 1 μl cDNA was subsequently used for SYBR green PCR assay (Applied Biosystems) as previously described (24, 28). Real-time PCR primers used in this study are listed in Table 1. The abundances of individual mRNA transcripts in each sample were assayed three times and normalized to the level of human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA (as an internal control). Relative transcript levels were quantified by the 2−ΔΔCT (where CT is threshold cycle) method and are shown as fold change relative to the level for the mock-treated control untransfected cells.
Luciferase reporter gene assay.
HeLa cells were seeded in 12-well plates and grown to 70 to 80% confluence. Cells were transfected with 0.5 μg of the empty vector pXJ41, pXJ41-GST, pXJ41-NS1, pXJ41-3ABC, pXJ41-3A, pXJ41-3B, pXJ41-3C, or individual mutants of pXJ41-3C and 0.5 μg of pISRE-Luc along with 0.05 μg of pRL-TK using Lipofectamine 2000. At 24 h posttransfection, cells were incubated with 1,000 U/ml of IFN-β for 16 h. Luciferase activities were measured using a dual-luciferase reporter assay kit (Promega, Madison, WI, USA) according to the manufacturer's protocol. The values were normalized with respect to Renilla luciferase activities. Then the results were expressed as relative luciferase activities, which are shown as fold change relative to the mock-treated control untransfected cells. All assays were repeated at least three times, with each experiment performed in triplicate.
Western blotting assay.
HeLa or PK-15 cells were lysed in cell lysis buffer supplemented with phenylmethylsulfonyl fluoride (PMSF; Beyotime, China). The samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. Briefly, the cell lysates were resolved in a 10% polyacrylamide gel. Separated proteins were then transferred onto a nitrocellulose membrane and probed with antibody (Ab) against STAT1 (Cell Signaling, Danvers, MA, USA), STAT2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-STAT1 (Tyr701, herein named STAT1-Y701; Cell Signaling), phospho-STAT2 (Tyr690, herein named STAT2-Y690; Cell Signaling), HA (Sigma-Aldrich, St. Louis, MO, USA), FLAG (Sigma-Aldrich), heat shock protein 90 (HSP90; Santa Cruz Biotechnology), poly(ADP-ribose) polymerase (PARP; Santa Cruz Biotechnology), KPNA1 (Santa Cruz Biotechnology), mouse anti-3C serum [prepared by immunizing mice with purified FMDV 3Cpro expressed by pET-32a(+) vector in Escherichia coli BL21 [laboratory stock], or β-actin antibody (Santa Cruz Biotechnology). Specific reaction products were detected with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG and goat anti-mouse IgG (Boster, Wuhan, China). The membranes were developed using SuperSignal West Pico Chemiluminescent Substrate according to the manufacturer's suggestions (Pierce, Rockford, IL, USA). Digital signal acquisition and analysis were conducted by the Quantity One program, version 4.6 (Bio-Rad).
IP.
For immunoprecipitation (IP), HeLa cells were transfected with empty vector pXJ41 or pXJ41-3C. At 24 h posttransfection, cells were treated with IFN-β at 1,000 U/ml for 1 h. The cells were lysed with cell lysis buffer supplemented with PMSF (Beyotime, China). The lysates were clarified by centrifugation at 12,000 × g for 10 min at 4°C. Antibody against STAT1 or STAT2 was added to the supernatant. IP with protein A/G-agarose (Beyotime, China) was performed according to the manufacturer's instructions. The IP samples with antibody against STAT1 were subjected to Western blotting with STAT2-Y690 antibody. The IP samples with antibody against STAT2 were subjected to Western blotting with STAT1-Y701 antibody.
Indirect immunofluorescence.
HeLa cells were seeded directly onto coverslips, cultured overnight, and transfected with plasmid pXJ41, pXJ41-3C, or individual mutants of pXJ41-3C. At 18 h posttransfection, IFN-β was added to the cells at a final concentration of 1,000 U/ml. One hour after IFN-β treatment, cells were washed twice in ice-cold phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS at 4°C for 1 h. Cells were then washed again three times with ice-cold PBS and permeabilized with 0.5% Triton X-100 for 15 min. The coverslips were then incubated with anti-STAT1 or STAT2 polyclonal Ab (1:1,000) and anti-HA tag monoclonal antibody (MAb; 1:800) in PBS for 1 h. After three PBS washes, coverslips were incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG(H+L) and Alexa Fluor 594-conjugated goat anti-rabbit IgG(H+L) antibodies at room temperature for 1 h. Then the coverslips were washed three times and treated with 4′,6′-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) to view the nuclei, followed by five washes. The coverslips were then mounted with mounting buffer (60% glycerol and 0.1% sodium azide in PBS) and observed under an Olympus BX51 inverted fluorescence microscope. The number of cells showing STAT1 or STAT2 nuclear localization was determined by counting 100 cells each in random microscopic fields. Each experiment was conducted in triplicate and repeated three times.
Subcellular fractionation.
The nuclear fraction was extracted from HeLa cells using a nuclear/cytosol fractionation kit (BioVision, Mountain View, CA, USA). Cells were transfected with empty vector pXJ41, pXJ41-3C, or individual mutants of pXJ41-3C. At 18 h posttransfection, cells were treated with IFN-β at 1,000 U/ml for 1 h. Cell collection, lysis, and subcellular fractionation were performed according to the manufacturer's instructions, with minor modifications. Briefly, HeLa cells were collected in PBS and centrifuged at 4°C for 5 min at 5,000 × g in a microcentrifuge (Eppendorf 5415R). Cell pellets were resuspended in cytosolic extraction buffer A (CEB-A) and incubated for 10 min on ice prior to addition of CEB-B. The lysates were centrifuged at 4°C for 5 min at 12,000 rpm in a microcentrifuge, and the supernatants were kept as the cytoplasmic fractions. The nuclear pellet was resuspended in nuclear extraction buffer (NEB) and vortexed for 30 s. This step was repeated every 10 min five times. The nuclear pellet was centrifuged at 4°C for 10 min at 12,000 rpm, and the supernatant was kept as a nuclear fraction. The cytoplasmic and nuclear fractions were subjected to Western blotting. Antibodies against β-actin, HSP90, and PARP were used to assess the successful fractionation.
Construction of a full-length cDNA clone and rescue of FMDV.
The infectious cDNA clone of FMDV type O, strain Tibet/CHA/99, was constructed and rescued as described before (29). Briefly, the complete viral genome of FMDV type O, strain Tibet/CHA/99, was divided into three fragments flanked by unique restriction sites. Viral RNA was extracted from the supernatant of BHK-21 cells infected with FMDV type O, strain Tibet/CHA/99, using a QIAamp Viral RNA Kit (Qiagen). cDNA synthesis was performed with SuperScript III Reverse Transcriptase (Invitrogen). Then three fragments, A, B, and C, were individually PCR amplified with three pairs of PCR primers (FMDV-A-Fwd/FMDV-A-Rev, FMDV-B-Fwd/FMDV-B-Rev, and FMDV-C-Fwd/FMDV-C-Rev) (Table 1) using Pfu Ultra High-Fidelity DNA Polymerase AD (Stratagene) according to the manufacturer's protocol. The first fragment A containing the T7 promoter was digested with AscI and NotI and cloned into pBluescript II SK+ vector to produce pSK-A. Then, the genetic marker of NheI was introduced to pSK-A with the primer pair FMDV-Am-Fwd/FMDV-Am-Rev to generate pSK-Am (Table 1), using a QuikChange XL site-directed mutagenesis kit as described elsewhere (26). The fragments B and C were assembled in one plasmid, vector pMD18-T (TaKaRa Biotechnology Co. Ltd., Dalian, China), to produce pT-BC. Then fragments B and C were digested from pT-BC and cloned into pSK-Am using NotI and PacI sites to obtain pSK-AmBC covering the full-length cDNA of FMDV type O, strain Tibet/CHA/99.
The plasmid pSK-AmBC was linearized with PacI. Purified, linearized DNAs served as templates for in vitro transcription using an mMESSAGE mMACHINE T7 Ultra Kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. The RNA pellet was resuspended in water and used for transfection of BHK-21 cells. BHK-21 cells grown to 80% confluence in six-well plates were transfected with 5 μg of RNA and 10 μl of DMRIE-C (1,2-dimyristyloxypropyl-3-dimethyl-hydroxy ethyl ammonium bromide and cholesterol; Invitrogen). After 4 h of exposure to DMRIE-C and RNA, the monolayers were washed, and fresh medium was added. Supernatants from cells at 48 h posttransfection were serially passaged on PK-15 cells (five passages, each for 2 days).
RESULTS
FMDV 3Cpro interferes with ISG mRNA synthesis.
Type I IFN signaling brings about strong expression of a variety of cellular genes, including ISG15, ISG56, Mx1, OAS, and PKR (9–13). HeLa cells were transfected with pXJ41, pXJ41-GST, pXJ41-NS1, pXJ41-3ABC, pXJ41-3A, pXJ41-3B, or pXJ41-3C and treated with IFN-β at 24 h posttransfection. pXJ41-NS1 was used as a positive control, which is known to inhibit downstream activation of the IFN-β promoter by associating with RIG-I (retinoic acid-inducible protein I) (30) and disrupting IFN-β signaling through reducing the IFN-inducible phosphorylation of STATs (31), whereas pXJ41 and pXJ41-GST served as negative controls (27, 32). The mRNA levels of ISGs were analyzed by real-time RT-PCR 5 h after IFN-β treatment.
As seen in Fig. 1A, the mRNA levels of ISG15, ISG56, Mx1, OAS, and PKR in IFN-treated cells transfected with pXJ41 increased 24.7-, 339.9-, 534.2-, 43.2-, and 27.1-fold, respectively, in comparison with the levels in mock-treated cells. In contrast, in HeLa cells transfected with pXJ41-3ABC/pXJ41-3C after IFN stimulation, the mRNA levels of ISG15, ISG56, Mx1, OAS, and PKR were 5.4-/18.8-, 11.4-/26.4-, 9.1-/16.6-, 7.2-/21.2-, and 4.5-/7.2-folds lower than those in the pXJ41-transfected cells, respectively. Expression of 3Cpro showed a 2- to 4-fold greater inhibitory effect than 3ABC. However, such an inhibitory effect was not observed in cells expressing 3A and 3B. In 3ABC-transfected cells, 3A, 3AB, and 3ABC were detected by Western blotting using FLAG antibody (Fig. 1B), while 3C and 3ABC were detected with HA antibody (Fig. 1C). From the view of band intensities, 3ABC is mainly cleaved into 3A and 3C (Fig. 1B and C). We cannot detect 3B because 3B was not fused with tags in the 3ABC construct. As 3ABC of FMDV is eventually cleaved into 3A, 3B, and 3Cpro (2), it is 3Cpro which contributes to 3ABC inhibiting the transcript levels of ISGs. As we expected, the expression of SIV NS1 significantly inhibited ISG mRNA synthesis while the expression of GST showed no inhibitory effect.
FIG 1.
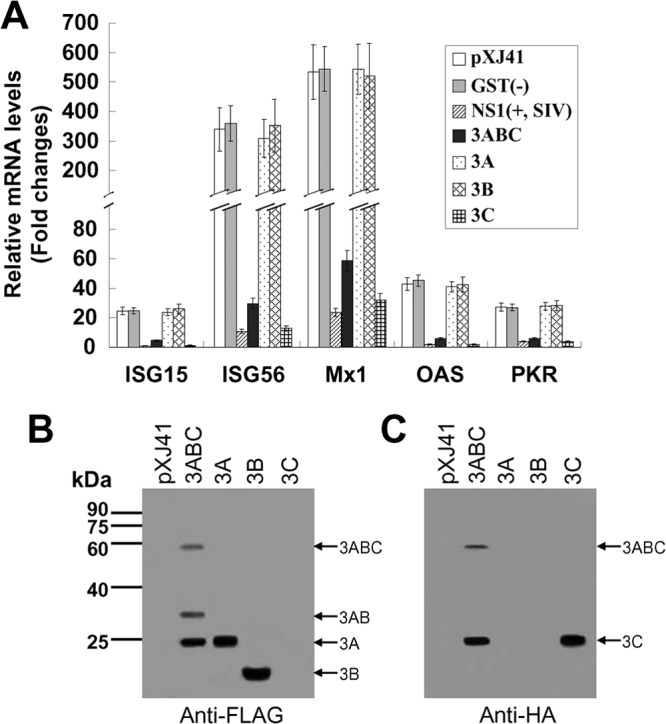
(A) FMDV 3Cpro reduces the transcript levels of ISGs. HeLa cells were transfected with pXJ41, pXJ41-GST, pXJ41-NS1, pXJ41-3ABC, pXJ41-3A, pXJ41-3B, or pXJ41-3C and treated with 1,000 U/ml of IFN-β at 24 h posttransfection. Five hours later, the transcript levels of ISGs were analyzed by real-time RT-PCR. pXJ41-GST and pXJ41-NS1 were used as negative and positive controls, respectively. Relative transcript levels are shown as relative fold changes in comparison with the level for the mock-treated control of untransfected cells. The data represent the means of three independent experiments, with each experiment performed in triplicate. Data represent the means ± the standard deviations (error bars) of three experiments. Expression of 3ABC, 3A, 3B, or 3C protein in HeLa cells transfected with pXJ41-3ABC, pXJ41-3A, pXJ41-3B, or pXJ41-3C was detected by Western blotting using FLAG antibody (B) or HA antibody (C).
FMDV 3Cpro inhibits ISRE promoter activity.
Once IFN is released from cells, it binds to an interferon receptor, and signaling occurs through the JAK-STAT pathway, resulting in the activation of antiviral genes containing ISREs in their promoters (11). To further investigate the effects of FMDV 3Cpro on IFN-β-induced ISRE-mediated activation, cells were cotransfected with the indicated plasmids (Fig. 2A), along with ISRE-luciferase and Renilla luciferase reporters. pXJ41-NS1 was used as a positive control, and pXJ41 and pXJ41-GST were used as negative controls. Upon IFN-β treatment, 3A- and 3B-expressing cells induced luciferase activity at levels similar to those of the negative controls. However, the ISRE promoter activity in FMDV 3ABC-expressing cells was 5.1-fold lower than that in pXJ41-transfected cells, while in 3Cpro-expressing cells it was approximately 8.6-fold lower than that in pXJ41-transfected cells, in a manner similar to pXJ41-NS1 (Fig. 2A). In addition, the expression of 3Cpro and 3ABC inhibited ISRE promoter activities in a dose-dependent manner, while 3Cpro showed a much stronger inhibitory effect than 3ABC (Fig. 2B). The results provide evidence that FMDV 3Cpro has the responsibility for 3ABC suppressing the activation of the ISRE promoter.
FIG 2.

Suppression of ISRE promoter activity by FMDV 3Cpro. (A) HeLa cells were seeded in 12-well plates and cotransfected with 0.5 μg of empty vector pXJ41, pXJ41-GST, pXJ41-NS1, pXJ41-3ABC, pXJ41-3A, pXJ41-3B, or pXJ41-3C and 0.5 μg of pISRE-Luc along with 0.05 μg of pRL-TK as an internal control. pXJ41-GST and pXJ41-NS1 were used as negative and positive controls, respectively. At 24 h posttransfection, cells were incubated with 1,000 U/ml of IFN-β for 16 h. Cells were lysed, and reporter expression was examined using a dual-luciferase reporter assay kit (Promega). The values were normalized with respect to Renilla luciferase activities. Then the results were expressed as relative luciferase activities, which are shown as relative fold change in comparison with the mock-treated control of untransfected cells. Expression of 3ABC, 3A, 3B, or 3C protein in HeLa cells transfected with pXJ41-3ABC, pXJ41-3A, pXJ41-3B, or pXJ41-3C was detected by Western blotting using FLAG or HA antibody. All assays were repeated at least three times, with each experiment performed in triplicates. (B) 3Cpro inhibited ISRE promoter activities in a dose-dependent manner. HeLa cells were cotransfected with 0 μg, 0.125 μg, 0.25 μg, or 0.5 μg of empty vector pXJ41, pXJ41-3ABC, pXJ41-3A, pXJ41-3B, or pXJ41-3C and 0.5 μg of pISRE-Luc along with 0.05 μg of pRL-TK. A luciferase reporter gene assay was conducted as described above. Expression of 3ABC, 3A, 3B, or 3C protein in HeLa cells transfected with pXJ41-3ABC, pXJ41-3A, pXJ41-3B, or pXJ41-3C was detected by Western blotting using FLAG or HA antibody. The data represent the means of three independent experiments, with each experiment performed in triplicate. Error bars indicate the standard deviations of three experiments.
As FMDV 3Cpro contributed to the 3ABC-mediated inhibition of mRNA synthesis for ISGs and ISRE promoter activity and because 3A and 3B showed no inhibition, they were not investigated any further. We then focused on the mechanism of FMDV 3Cpro interfering with the JAK-STAT pathway.
The catalytic triad residues H46, D84, and C163 of 3Cpro are essential for FMDV 3Cpro to suppress ISRE promoter activities.
Previous reports have shown that the catalytic triad of H46, D84, and C163 of FMDV 3Cpro played essential roles in its enzyme activity (33, 34). Mutations at any of these residues (Y for H46 [H46Y], D84N, or C163G) eliminated the capacity of 3Cpro to process the P1, P2, or P3 substrate in trans, but the 3Cpro-H205R mutant had no influence (34). To determine whether the protease activity of 3Cpro was involved in the type I IFN signaling pathway, the catalytic triad mutants were made, and a luciferase assay was performed. As shown in Fig. 3, compared to the pXJ41 vector control, the wild-type (WT) 3Cpro and 3Cpro-H205R mutant strongly suppressed ISRE promoter activities. In contrast, the H46Y, D84N, and C163G mutants had no suppressive effect, demonstrating that the catalytic triad of H46, D84, and C163 of 3Cpro is essential for FMDV 3Cpro to antagonize the type I IFN signaling pathway.
FIG 3.
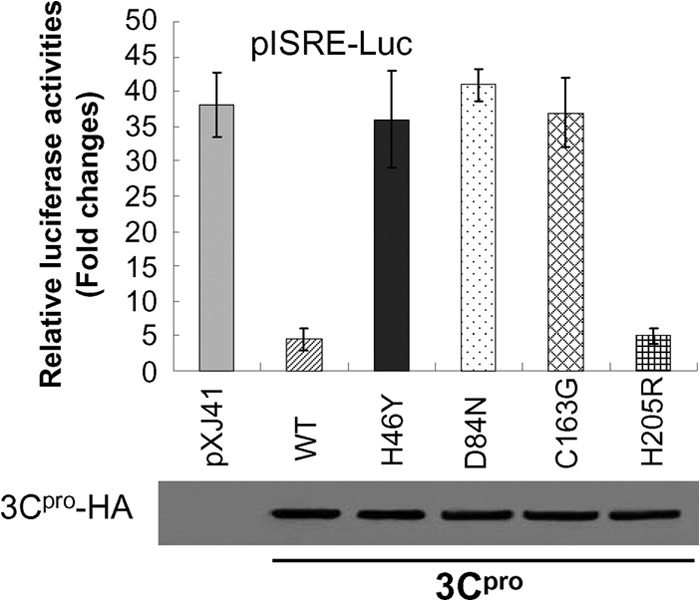
The catalytic triad H46, D84, and C163 of 3Cpro played essential roles in FMDV 3Cpro suppressing ISRE promoter activity. HeLa cells were transfected with 0.5 μg of empty vector pXJ41, pXJ41-3C, or individual mutants of pXJ41-3C and 0.5 μg of pISRE-Luc along with 0.05 μg of pRL-TK. At 24 h posttransfection, cells were incubated with 1,000 U/ml of IFN-β for 16 h. A luciferase reporter gene assay was conducted as described above. Expression of 3Cpro was also shown. The data represent the means of three independent experiments, with each experiment performed in triplicate. Error bars indicate the standard deviations of three experiments.
FMDV 3Cpro does not alter the protein level, phosphorylation, or heterodimerization of STAT1 and STAT2.
To examine if STATs were expressed normally in the presence of 3Cpro, HeLa cells were transfected with pXJ41-3C and stimulated with IFN-β, followed by Western blotting. While the STAT2 protein level increased considerably in pXJ41-transfected cells upon IFN stimulation (Fig. 4A, STAT2, and B), consistent with the previous report (35), the STAT2 protein in 3Cpro-expressing cells did not show a significant difference compared to nonexpressing cells. In addition, no reduction was observed in the level of STAT1 protein by FMDV 3Cpro expression (Fig. 4A, STAT1, and B). These results indicate that FMDV 3Cpro does not induce STAT1 and STAT2 degradation.
FIG 4.
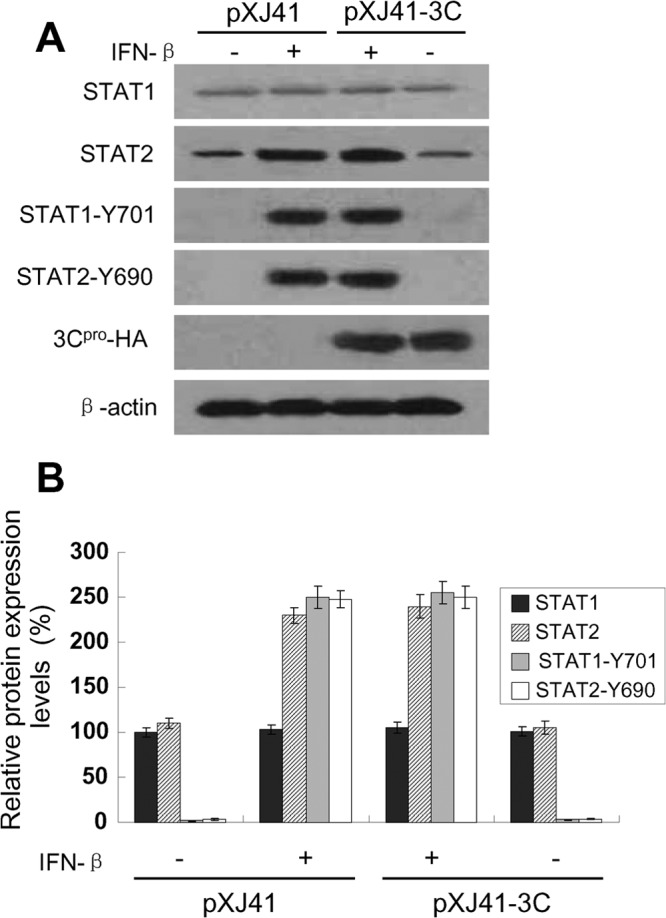
Protein level and phosphorylation status of STAT1 and STAT2 in FMDV 3Cpro-expressing cells after IFN-β stimulation. (A) HeLa cells were transfected with empty vector pXJ41 or pXJ41-3C. At 24 h posttransfection, cells were either left untreated (−) or treated (+) with 1,000 U/ml of IFN-β for 1 h and harvested for Western blotting with antibody against STAT1, STAT2, phospho-STAT1 (STAT1-Y701), phospho-STAT2 (STAT2-Y690) or HA, as indicated. The same blot was incubated with β-actin antibody as a protein loading control. The data presented here are results from one experiment of three Western blotting experiments. (B) Densitometry analysis of the digital image from three independent experiments. The band intensities are shown as the relative protein expression levels, normalized with β-actin. Error bars indicate the standard deviations of three experiments.
After type I IFNs bind to their receptors (IFNARs), STATs are phosphorylated and dimerized to enter into the nucleus; thus phosphorylation-dependent activation of STATs is critical to mediate IFN-inducible antiviral responses (36). To explore whether FMDV 3Cpro interfered with the phosphorylation of STAT1 and STAT2, lysates from 3Cpro-expressing cells were subjected to Western blotting using phospho-STAT1 (STAT1-Y701) and phospho-STAT2 (STAT2-Y690) antibodies, respectively. The levels of phosphorylated STAT1 and phosphorylated STAT2 were greatly increased after IFN-β treatment, and no difference was found between 3Cpro-expressing cells and nonexpressing cells (Fig. 4A, STAT1-Y701 and STAT2-Y690, and B). In mock-treated cells, the phosphorylation of STAT1 and STAT2 was below the detection level. The results show that FMDV 3Cpro does not alter the phosphorylation status of STAT1 and STAT2 after IFN-β stimulation.
Once activation is triggered by IFN, STAT1 and STAT2 form a heterodimer and associate with IRF9 to assemble the ISGF3 complex (11, 35). In our experiments, FMDV 3Cpro did not alter the phosphorylation of STAT1 and STAT2, and thus whether 3Cpro affected the heterodimerization of STAT1 and STAT2 was examined. IP with STAT1 antibody followed by immunoblotting with STAT2-Y690 antibody showed the presence of phosphorylated STAT2 in the samples. Similarly, IP with STAT2 antibody and blotting with STAT1-Y701 antibody showed the presence of phosphorylated STAT1 in the samples. No obvious difference was observed between the IFN-treated cells transfected with pXJ41 and pXJ41-3C (Fig. 5A and B). The data suggest that the heterodimerization of STAT1 and STAT2 in 3Cpro-expressing cells is not remarkably affected.
FIG 5.

Detection of STAT1/STAT2 heterodimer formation in HeLa cells after IFN-β treatment. (A) Cells were transfected with empty vector pXJ41 or pXJ41-3C. At 24 h posttransfection, cells were either left untreated (−) or treated (+) with 1,000 U/ml of IFN-β for 1 h and lysed for IP and Western blotting (WB), as indicated on the figure. Expression of 3Cpro is also shown. β-Actin was used as a loading control. The data presented here were results from one experiment of three IP and Western blotting experiments. (B) Densitometry analysis of the digital image from three independent experiments. The band intensities are shown as relative protein expression levels. Error bars indicate the standard deviations of three experiments.
Inhibition of STAT1/STAT2 nuclear translocation by FMDV 3Cpro.
Once the ISGF3 complex is formed, it translocates to the nucleus to initiate gene transcription by binding to ISREs. In our study, ISRE promoter activity was reduced significantly compared to the negative control in the presence of 3Cpro, and 3Cpro did not alter the protein levels, phosphorylation, or heterodimerization of STAT1 and STAT2. Thus, the nuclear translocation of STAT1/STAT2 was examined. HeLa cells were transfected with pXJ41, pXJ41-3C, or individual mutants and treated with IFN-β at 18 h posttransfection; then an immunofluorescence assay (IFA) was performed. In pXJ41-transfected cells or cells expressing WT 3Cpro without stimulation, STAT1 was diffused in the cytoplasm and nucleus (Fig. 6A, first and third rows ). After IFN-β stimulation, STAT1 was predominantly found in the nucleus in pXJ41-transfected cells (Fig. 6A, second row), indicating efficient translocation to the nucleus. However, the majority of the STAT1 remained in the cytoplasm of cells expressing WT 3Cpro and the 3Cpro-H205R mutant even after IFN-β treatment (Fig. 6A, WT +IFN and H205R +IFN). Interestingly, the 3Cpro mutants of H46Y, D84N, and C163G no longer inhibited STAT1 nuclear translocation (Fig. 6A, H46Y, D84N, and C163G +IFN). The mainly STAT1 nuclear localization of cells was also determined by counting 100 cells each in random microscopic fields. In pXJ41-transfected cells or cells expressing WT 3Cpro without IFN stimulation, STAT1 was not primarily localized in the nucleus. After IFN-β stimulation, STAT1 translocated to the nucleus in 90.0% of pXJ41-transfected cells. In 3Cpro H46Y, D84N, and C163G mutant-expressing cells, STAT1 translocated to the nucleus in 89.0%, 91.0%, and 90.0% of cells, respectively. In contrast, in WT 3Cpro-expressing and 3Cpro-H205R mutant-expressing cells, STAT1 translocated to the nucleus only in 10.0% and 9.0% of cells, respectively (Fig. 6B). Cell staining with STAT2 antibody showed results similar to those with STAT1 antibody (data not shown).
FIG 6.

(A) FMDV 3Cpro inhibits nuclear translocation of STAT1, as observed by fluorescence microscopy. HeLa cells were transfected with pXJ41, pXJ41-3C, or individual mutants of pXJ41-3C. At 18 h posttransfection, cells were either left untreated (−) or treated (+) with 1,000 U/ml of IFN-β for 1 h and fixed, and an IFA was conducted as indicated in Materials and Methods. Yellow arrows indicate WT- or mutated 3Cpro-expressing cells, and white arrows indicate untransfected cells. (B) The percentage of STAT1 nuclear localization of HeLa cells transfected with pXJ41, pXJ41-3C, or individual mutants. The number of STAT1 nuclear localization cells was determined by counting 100 cells each in random microscopic fields. Each experiment was conducted in triplicate and repeated three times. Error bars indicate the standard deviations of three experiments.
This observation was further confirmed by nuclear and cytoplasmic fractionation of the cells. Antibodies against STAT1-Y701 and STAT2-Y690 were used to detect the presence of the phosphorylated proteins in those two fractions. In pXJ41-transfected cells, more STAT1-Y701 and STAT2-Y690 were found in the nuclear fraction than in the cytoplasmic fraction after IFN-β treatment. Similarly, in H46Y, D84N, and C163G mutated 3Cpro-expressing cells after IFN-β stimulation most STAT1-Y701 and STAT2-Y690 proteins were detected in the nuclear fraction. In contrast, in cells expressing the WT 3Cpro and 3Cpro-H205R mutant, STAT1-Y701 and STAT2-Y690 were predominantly found in the cytoplasmic fraction rather than in the nuclear fraction, indicating that WT 3Cpro and 3Cpro-H205R prevent STAT1/STAT2 nuclear translocation (Fig. 7A, STAT1-Y701 and STAT2-Y690). HSP90 and PARP, the cytoplasmic and nuclear protein markers, respectively, remained in their respective fractions, eliminating the possible cross-contamination of both fractions (Fig. 7A, HSP90 and PARP). FMDV 3Cpro was detected in both the cytoplasm and nucleus (Fig. 7A, 3Cpro-HA), which was consistent with the results observed by fluorescence microscopy (Fig. 6A). β-Actin is a cytoskeletal protein and mainly distributed in the cytoplasm (Fig. 7A). Densitometry analysis of the digital images of the Western blotting results showed that 76.0% of STAT1-Y701 and 79.0% of STAT2-Y690 were detected in the nuclear fraction of pXJ41-transfected cells, while only 25.0% and 26.0% of STAT1-Y701 and 29.0% and 28.0% of STAT2-Y690 were detected in the nuclear fraction of cells expressing the WT 3Cpro and 3Cpro-H205R mutant, respectively. Similar to cells transfected with pXJ41, in H46Y-, D84N-, and C163G-mutated 3Cpro-expressing cells, 77.0%, 76.5%, and 77.2% of STAT1-Y701 and 78.5%, 78.8%, and 79.5% of STAT2-Y690, respectively, were detected in the nuclear fraction (Fig. 7B). The fractionation results are in line with the observation from IFAs demonstrating that FMDV 3Cpro blocks STAT1/STAT2 nuclear translocation and that the catalytic triad H46, D84, and C163 of 3Cpro are critical for 3Cpro-mediated inhibition of STAT1/STAT2 nuclear translocation. These results are consistent with our earlier observation that the catalytically inactive 3Cpro mutants (H46Y, D84N, and C163G) lost their capacity to suppress ISRE promoter activities (Fig. 3).
FIG 7.
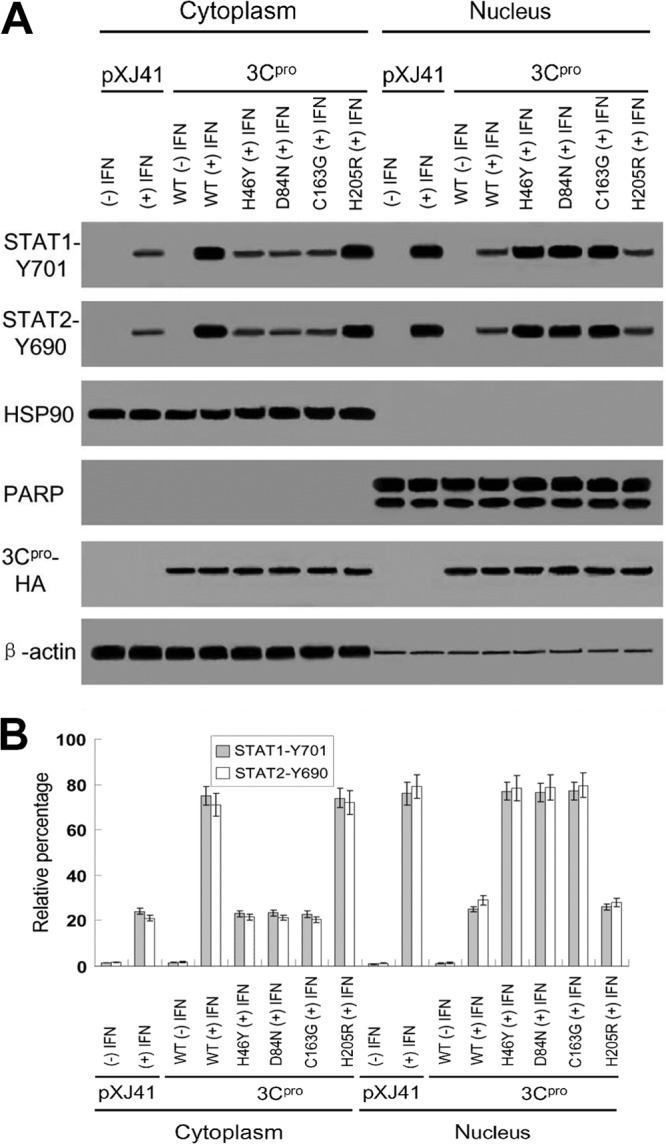
(A) Phosphorylated STAT1 and STAT2 in nuclear and cytoplasmic fractions. Subcellular fractionation of HeLa cells, 1 h after IFN-β treatment, was performed for nuclear and cytoplasmic fractions, followed by Western blotting using STAT1-Y701 antibody, STAT2-Y690 antibody, anti-HSP90 antibody as a cytoplasmic marker, and anti-PARP antibody as a nuclear protein marker, as indicated. HA antibody was used to detect FMDV 3Cpro expression (3Cpro-HA), and β-actin antibody was used as a loading control. (B) Densitometry analysis of the digital image of phosphorylated STAT1 and STAT2 in nuclear and cytoplasmic fractions from three independent experiments. The band intensities of each fraction are shown as the relative percentages of the total density of corresponding cytoplasmic and nuclear fractions. Normalization for cytoplasmic and nuclear fractions was done with HSP90 and PARP, respectively. Error bars indicate the standard deviations of three experiments. −, without IFN-β treatment; +, with IFN-β treatment.
KPNA1 degradation and 3Cpro protease activity.
Phosphorylated STAT1 accumulates in the nucleus via interaction with a specific nuclear localization signal receptor, karyopherin α1 (KPNA1) (37, 38). Our finding that the protease activity of FMDV 3Cpro was critical for the blocking STAT1/STAT2 nuclear translocation (Fig. 6 and 7) raised a possibility that 3Cpro may target a nuclear localization signal receptor for degradation. We examined whether KPNA1 was degraded in cells expressing 3Cpro. HeLa cells were cotransfected with pXJ41-3C and plasmids expressing KPNA1, KPNA2, KPNA3, or KPNA4. At 24 h posttransfection, cells were lysed, and Western blotting was done individually with antibody against FLAG, HA, or β-actin. The results show that FMDV 3Cpro specifically induces KPNA1 degradation but that KPNA2, KPNA3, and KPNA4 are not affected (Fig. 8A and B).
FIG 8.

FMDV 3Cpro induces KPNA1 degradation. (A) FMDV 3Cpro had no effects on KPNA2, KPNA3, or KPNA4. HeLa cells were cotransfected with pXJ41-3C and FLAG-tagged KPNA1, KPNA2, KPNA3, or KPNA4 plasmids. pXJ41 empty vector was used as a control. At 24 h posttransfection, cells were lysed with the cell lysis buffer supplemented with PMSF. Western blotting was done individually with antibody against FLAG, HA, or β-actin. The data presented here are results from one experiment of three Western blotting experiments. (B) Densitometry analysis of the digital image of FLAG-tagged KPNA1, KPNA2, KPNA3, or KPNA4 from three independent experiments. The band intensities of blotting with FLAG antibody are shown as the relative protein expression levels, normalized with β-actin. Error bars indicate the standard deviations of three experiments. (C) KPNA1 degradation in 3Cpro-expressing cells was independent of the proteasome and caspase pathways. HeLa cells were transfected with pXJ41 empty vector or pXJ41-3C. At 18 h posttransfection, MG132 or zVAD-FMK was added. Six hours later, the cells were harvested, and the endogenous KPNA1 was analyzed by Western blotting using antibody against KPNA1, HA, or β-actin. The data presented here are the results from one experiment of three Western blotting experiments. (D) Densitometry analysis of the digital image of KPNA1 from three independent experiments. The band intensities of blotting with KPNA1 antibody are shown as the relative protein expression levels, normalized with β-actin. Data represent the means ± the standard deviations (error bars) of three experiments.
To further investigate whether the KPNA1 reduction was due to degradation in a proteasome- or caspase-dependent manner, HeLa cells were transfected with pXJ41-3C, and the proteasome inhibitor MG132 or the caspase inhibitor zVAD-FMK was added to cells at 18 h posttransfection. Cells were harvested 6 h later, and the endogenous KPNA1 was examined. As shown in Fig. 8C and D, neither MG132 nor zVAD-FMK inhibited 3Cpro-mediated KPNA1 degradation, indicating that the KPNA1 degradation observed in 3Cpro-expressing cells is independent of the proteasome and caspase pathways.
Since KPNA1 degradation was not proteasome or caspase dependent, we next examined whether the protease activity of 3Cpro induced KPNA1 degradation directly. HeLa cells were transfected with pXJ41, pXJ41-3C, or individual mutants of pXJ41-3C, and the endogenous KPNA1 was examined by Western blotting. While WT 3Cpro and 3Cpro-H205R induced KPNA1 degradation, the catalytically inactive 3Cpro mutants H46Y, D84N, and C163G were unable to cause KPNA1 degradation (Fig. 9A and B). These results may explain why the WT 3Cpro and 3Cpro-H205R mutant prevent STAT1/STAT2 nuclear translocation, while the 3Cpro H46Y, D84N, and C163G mutants exert no inhibitory effects on the nuclear translocation of STAT1/STAT2 (Fig. 6 and 7).
FIG 9.

The protease activity of FMDV 3Cpro is responsible for KPNA1 degradation. (A) HeLa cells were transfected with empty vector pXJ41, pXJ41-3C, or individual mutants of pXJ41-3C. After 24 h, the endogenous KPNA1 was tested by Western blotting using antibody against KPNA1, HA, or β-actin. The data presented here are results from one experiment of three Western blotting experiments. (B) Densitometry analysis of the digital image of KPNA1 from three independent experiments. The band intensities of blotting with KPNA1 antibody are shown as the relative protein expression levels, normalized with β-actin. Data represent the means ± the standard deviations (error bars) of three experiments.
KPNA1 degradation in FMDV-infected cells.
KPNA1 degradation was further examined in the context of FMDV infection. To this end, PK-15 cells were infected at a multiplicity of infection (MOI) of 1.0 with FMDV type O, strain Tibet/CHA/99, and harvested at 12, 18, and 24 h postinfection (hpi) for Western blotting. Compared to mock-infected cells, endogenous KPNA1 levels in FMDV-infected cells were reduced at 12 hpi and were considerably decreased at 18 and 24 hpi (Fig. 10A and B). These results were consistent with the observation in cells expressing 3Cpro in gene transfection experiments (Fig. 8 and 9).
FIG 10.
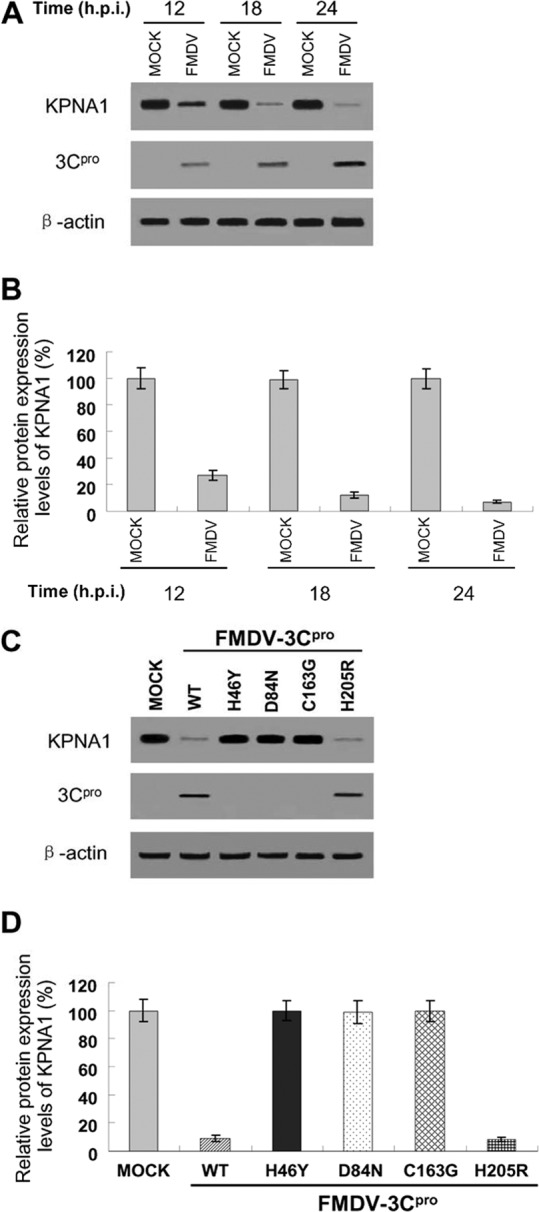
Reduction of endogenous KPNA1 levels by infection with FMDV or rescued FMDV using an infectious cDNA clone. (A) PK-15 cells were infected at an MOI of 1.0 with FMDV type O, strain Tibet/CHA/99, and harvested at the indicated times for Western blotting using antibody against KPNA1, mouse anti-3C serum, or β-actin. The data presented here are results from one experiment of three Western blotting experiments. (B) Densitometry analysis of the digital image of KPNA1 in PK-15 cells infected with FMDV from three independent experiments. The band intensities of blotting with KPNA1 antibody are shown as the relative protein expression levels, normalized with β-actin. Data are the means ± the standard deviations (error bars) of three experiments. (C) Supernatants from BHK-21 cells transfected with RNA of individual FMDV infectious cDNA clones were used to infect PK-15 cells at an MOI of 1.0. An equal volume was used to infect PK-15 cells for infectious cDNA clones harboring the 3C mutation H46Y, D84N, or C163G, which were completely unable to produce virus. PK-15 cells were harvested at 24 hpi for Western blotting using antibody against KPNA1, mouse anti-3C serum, or β-actin. The data presented here are results from one experiment of three Western blotting experiments. (D) Densitometry analysis of the digital image of KPNA1 in PK-15 cells infected with rescued FMDV using an infectious cDNA clone from three independent experiments. The band intensities of blotting with KPNA1 antibody are shown as the relative protein expression levels, normalized with β-actin. Data are the means ± the standard deviations (error bars) of three experiments.
We also constructed an infectious cDNA clone using FMDV type O, strain Tibet/CHA/99. The H46Y, D84N, and C163G mutations were introduced into the FMDV infectious cDNA clone, but no viruses were rescued, demonstrating that the catalytic triad residues H46, D84, and C163 of 3Cpro are critical for FMDV growth. The mutant virus FMDV-3Cpro-H205R was rescued and induced KPNA1 degradation at a level similar to that of FMDV-WT-3Cpro (Fig. 10C and D), which further confirmed the results from pXJ41-3C and individual mutants of pXJ41-3C in gene transfection experiments (Fig. 9A and B).
DISCUSSION
The IFN system is an extremely powerful antiviral response that is capable of controlling most, if not all, virus infections in the absence of adaptive immunity (39). In order to evade the IFN response, many viruses have evolved to counteract the host innate immunity. As the pathogen of FMD, one of the major causes of great economic loss of cloven-hoofed animals, FMDV has adopted strategies to evade the host innate immune response (21–24). In the present study, we first provide evidence that FMDV 3Cpro interferes with the IFN signaling pathway by degrading KPNA1 and thus blocking STAT1/STAT2 nuclear translocation. Further, we show that the protease activity of 3Cpro governs the ability of 3Cpro to inhibit the JAK-STAT pathway.
Previous studies have demonstrated that FMDV 3Cpro inhibits the production pathway of type I IFN (24). To determine whether FMDV 3Cpro could antagonize the signaling pathway, we explored the JAK-STAT pathway by expressing 3Cpro in cells. It appears that 3Cpro contributes to 3ABC-mediated inhibition of the transcript levels of ISGs and ISRE promoter activity (Fig. 1 and 2). Then we examined the inhibitory mechanism of FMDV 3Cpro interfering with the JAK-STAT pathway.
Viruses may inhibit the IFN-activated JAK-STAT signaling pathway by degrading STATs (40–42), blocking STAT phosphorylation (43–45), inhibiting STAT1/STAT2 nuclear translocation (35, 38, 46–54), or inhibiting a process after nuclear translocation of ISGF3 (28, 55). We demonstrated that FMDV 3Cpro does not affect the amounts of STAT1 and STAT2 in IFN-β-treated cells, nor does it alter the IFN-induced phosphorylation of those proteins (Fig. 4). Furthermore, 3Cpro does not change the heterodimerization of STAT1 and STAT2 (Fig. 5). Interestingly, 3Cpro blocks STAT1/STAT2 nuclear translocation, and the protease activity of 3Cpro is associated with the inhibition of STAT1/STAT2 nuclear translocation (Fig. 3, 6, and 7).
Multiple levels of regulation occur during ISGF3 translocation to the nucleus. After being phosphorylated, STAT1 exposes a nuclear localization signal (NLS) for recruitment of importin-5, also called KPNA1 (37). Each of the karyopherins (designated KPNA1 to KPNA4) recognizes NLS sites, which promotes nuclear translocation of ISGF3 (10, 56). Some viruses are known to dampen the trafficking of STAT1 by interference with one or multiple importins. For example, NSP2 of Venezuelan equine encephalitis virus and VP24 of Ebola virus specifically interact with the nuclear importin karyopherin α1 (38, 49, 50). The hepatitis B virus (HBV) polymerase competitively binds to protein kinase C-δ (PKC-δ) and importin-α5, thereby inhibiting STAT1 nuclear translocation (46). The severe acute respiratory syndrome (SARS) coronavirus ORF6 protein alone blocks STAT1 nuclear translocation by tethering karyopherin alpha 2 (KPNA2) (47), and a recent study showed that porcine respiratory and reproductive virus Nsp1β blocks ISGF3 nuclear translocation by inducing KPNA1 degradation in a ubiquitin-proteasome manner (54). Our data showed that the protease activity is critical for FMDV 3Cpro-mediated prevention of STAT1/STAT2 nuclear translocation. While 3Cpro expression does not alter the protein levels, phosphorylation, or heterodimerization of STAT1 and STAT2, it is possible that FMDV 3Cpro may target an NLS receptor(s) for degradation, and 3Cpro indeed induces KPNA1 degradation. However, no degradation was seen for KPNA2, KPNA3, or KPNA4 (Fig. 8A and B). Moreover, the 3Cpro-mediated KPNA1 degradation is proteasome and caspase independent (Fig. 8C and D).
KPNA1 degradation was specifically attributable to the protease activity of FMDV 3Cpro as the catalytically inactive 3Cpro mutants (H46Y, D84N, and C163G) were unable to induce KPNA1 degradation (Fig. 9). As a result, the H46Y, D84N, and C163G mutants did not prevent STAT1/STAT2 nuclear translocation and thus suppress the ISRE promoter activities (Fig. 3, 6, and 7). The degradation of endogenous KPNA1 was shown in PK-15 cells after FMDV infection (Fig. 10A and B), which validates our observation on the KPNA1 degradation during infection. The H46Y, D84N, C163G, and H205R mutations of 3Cpro were also introduced into the FMDV infectious cDNA clone. The mutant virus FMDV-3Cpro-H205R grew to similar titers and induced KPNA1 degradation at levels similar to FMDV-WT-3Cpro (Fig. 10C and D). However, the transfections performed with infectious clones harboring the 3C mutation H46Y, D84N, or C163G were completely unable to produce virus, and as a result, we cannot do animal experiments to study the mechanism further.
In summary, we have first elucidated the mechanism of FMDV 3Cpro mediated-inhibition of the JAK-STAT pathway. The KPNA1 degradation by 3Cpro presents the first example of a viral immune evasion mechanism that involves direct degradation of KPNA1 by a viral protease, not by a proteasome- and caspase-dependent pathway. Our results provide a new insight into understanding how viral proteins of FMDV function as inhibitors of type I IFN signaling.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation (31100119, 31170146, and 31370189), the Special Fund for Agro-scientific Research in the Public Interest (201303046), the Research Award Fund for Outstanding Young Scientist of Shandong Province (BS2012NY012), a grant from the China Postdoctoral Science Foundation (2012M521341), and the Shandong Province Postdoctoral Special Fund for Innovative Projects (201203036).
We thank Daniel L. Rock from the Department of Pathobiology, University of Illinois at Urbana-Champaign, Urbana, IL, for critically reviewing the manuscript.
Footnotes
Published ahead of print 19 February 2014
REFERENCES
- 1.Fry EE, Stuart DI, Rowlands DJ. 2005. The structure of foot-and-mouth disease virus. Curr. Top. Microbiol. Immunol. 288:71–101. 10.1007/3-540-27109-0_4 [DOI] [PubMed] [Google Scholar]
- 2.Grubman MJ, Baxt B. 2004. Foot-and-mouth disease. Clin. Microbiol. Rev. 17:465–493. 10.1128/CMR.17.2.465-493.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belsham GJ. 2005. Translation and replication of FMDV RNA. Curr. Top. Microbiol. Immunol. 288:43–70. 10.1007/3-540-27109-0_3 [DOI] [PubMed] [Google Scholar]
- 4.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559–568. 10.1038/nri2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137. 10.1038/ni1303 [DOI] [PubMed] [Google Scholar]
- 6.Takeuchi O, Akira S. 2007. Recognition of viruses by innate immunity. Immunol. Rev. 220:214–224. 10.1111/j.1600-065X.2007.00562.x [DOI] [PubMed] [Google Scholar]
- 7.Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y. 2012. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J. Virol. 86:3839–3850. 10.1128/JVI.06466-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yie J, Senger K, Thanos D. 1999. Mechanism by which the IFN-beta enhanceosome activates transcription. Proc. Natl. Acad. Sci. U. S. A. 96:13108–13113. 10.1073/pnas.96.23.13108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aaronson DS, Horvath CM. 2002. A road map for those who don't know JAK-STAT. Science 296:1653–1655. 10.1126/science.1071545 [DOI] [PubMed] [Google Scholar]
- 10.Banninger G, Reich NC. 2004. STAT2 nuclear trafficking. J. Biol. Chem. 279:39199–39206. 10.1074/jbc.M400815200 [DOI] [PubMed] [Google Scholar]
- 11.Darnell JE, Jr, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421. 10.1126/science.8197455 [DOI] [PubMed] [Google Scholar]
- 12.Shuai K, Liu B. 2003. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 3:900–911. 10.1038/nri1226 [DOI] [PubMed] [Google Scholar]
- 13.Tang X, Gao JS, Guan YJ, McLane KE, Yuan ZL, Ramratnam B, Chin YE. 2007. Acetylation-dependent signal transduction for type I interferon receptor. Cell 131:93–105. 10.1016/j.cell.2007.07.034 [DOI] [PubMed] [Google Scholar]
- 14.Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B, Wang J. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 84:8051–8061. 10.1128/JVI.02491-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 85:8811–8818. 10.1128/JVI.00447-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu J, Yi L, Zhao J, Yu J, Chen Y, Lin MC, Kung HF, He ML. 2012. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J. Virol. 86:3767–3776. 10.1128/JVI.06687-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z. 2013. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 9:e1003231. 10.1371/journal.ppat.1003231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB. 2011. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 7:e1001311. 10.1371/journal.ppat.1001311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qu L, Feng Z, Yamane D, Liang Y, Lanford RE, Li K, Lemon SM. 2011. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 7:e1002169. 10.1371/journal.ppat.1002169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. 2007. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. U. S. A. 104:7253–7258. 10.1073/pnas.0611506104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Los Santos T, de Avila Botton S, Weiblen R, Grubman MJ. 2006. The leader proteinase of foot-and-mouth disease virus inhibits the induction of beta interferon mRNA and blocks the host innate immune response. J. Virol. 80:1906–1914. 10.1128/JVI.80.4.1906-1914.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Los Santos T, Diaz-San Segundo F, Grubman MJ. 2007. Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J. Virol. 81:12803–12815. 10.1128/JVI.01467-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D, Fang L, Luo R, Ye R, Fang Y, Xie L, Chen H, Xiao S. 2010. Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem. Biophys. Res. Commun. 399:72–78. 10.1016/j.bbrc.2010.07.044 [DOI] [PubMed] [Google Scholar]
- 24.Wang D, Fang L, Li K, Zhong H, Fan J, Ouyang C, Zhang H, Duan E, Luo R, Zhang Z, Liu X, Chen H, Xiao S. 2012. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 86:9311–9322. 10.1128/JVI.00722-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao JH, Davidson I, Matthes H, Garnier JM, Chambon P. 1991. Cloning, expression, and transcriptional properties of the human enhancer factor TEF-1. Cell 65:551–568. 10.1016/0092-8674(91)90088-G [DOI] [PubMed] [Google Scholar]
- 26.Du Y, Zuckermann FA, Yoo D. 2010. Myristoylation of the small envelope protein of porcine reproductive and respiratory syndrome virus is non-essential for virus infectivity but promotes its growth. Virus Res. 147:294–299. 10.1016/j.virusres.2009.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song C, Krell P, Yoo D. 2010. Nonstructural protein 1α subunit-based inhibition of NF-κB activation and suppression of interferon-β production by porcine reproductive and respiratory syndrome virus. Virology 407:268–280. 10.1016/j.virol.2010.08.025 [DOI] [PubMed] [Google Scholar]
- 28.Paulus C, Krauss S, Nevels M. 2006. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc. Natl. Acad. Sci. U. S. A. 103:3840–3845. 10.1073/pnas.0600007103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu G, Liu Z, Xie Q, Chen Y, Bao H, Chang H, Liu X. 2004. Generation of an infectious cDNA clone of an FMDV strain isolated from swine. Virus Res. 104:157–164. 10.1016/j.virusres.2004.04.002 [DOI] [PubMed] [Google Scholar]
- 30.Mibayashi M, Martínez-Sobrido L, Loo YM, Cárdenas WB, Gale M, Jr, García-Sastre A. 2007. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 81:514–524. 10.1128/JVI.01265-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jia D, Rahbar R, Chan RW, Lee SM, Chan MC, Wang BX, Baker DP, Sun B, Peiris JS, Nicholls JM, Fish EN. 2010. Influenza virus non-structural protein 1 (NS1) disrupts interferon signaling. PLoS One 5:e13927. 10.1371/journal.pone.0013927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han M, Du Y, Song C, Yoo D. 2013. Degradation of CREB-binding protein and modulation of type I interferon induction by the zinc finger motif of the porcine reproductive and respiratory syndrome virus Nsp1α subunit. Virus Res. 172:54–65. 10.1016/j.virusres.2012.12.012 [DOI] [PubMed] [Google Scholar]
- 33.Birtley JR, Knox SR, Jaulent AM, Brick P, Leatherbarrow RJ, Curry S. 2005. Crystal structure of foot-and-mouth disease virus 3C protease. New insights into catalytic mechanism and cleavage specificity. J. Biol. Chem. 280:11520–11527. 10.1074/jbc.M413254200 [DOI] [PubMed] [Google Scholar]
- 34.Grubman MJ, Zellner M, Bablanian G, Mason PW, Piccone ME. 1995. Identification of the active-site residues of the 3C protease of foot-and-mouth disease virus. Virology 213:581–589. 10.1006/viro.1995.0030 [DOI] [PubMed] [Google Scholar]
- 35.Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. 2010. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J. Virol. 84:11045–11055. 10.1128/JVI.00655-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, Al Ghonaium A, Tufenkeji H, Frayha H, Al-Gazlan S, Al-Rayes H, Schreiber RD, Gresser I, Casanova JL. 2003. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33:388–391. 10.1038/ng1097 [DOI] [PubMed] [Google Scholar]
- 37.McBride KM, Banninger G, McDonald C, Reich NC. 2002. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-alpha. EMBO J. 21:1754–1763. 10.1093/emboj/21.7.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF. 2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 80:5156–5167. 10.1128/JVI.02349-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47. 10.1099/vir.0.83391-0 [DOI] [PubMed] [Google Scholar]
- 40.Kumar A, Bühler S, Selisko B, Davidson A, Mulder K, Canard B, Miller S, Bartenschlager R. 2013. Nuclear localization of dengue virus nonstructural protein 5 does not strictly correlate with efficient viral RNA replication and inhibition of type I interferon signaling. J. Virol. 87:4545–4557. 10.1128/JVI.03083-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parisien JP, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. 2002. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J. Virol. 76:4190–4198. 10.1128/JVI.76.9.4190-4198.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ulane CM, Horvath CM. 2002. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 304:160–166. 10.1006/viro.2002.1773 [DOI] [PubMed] [Google Scholar]
- 43.Devaux P, von Messling V, Songsungthong W, Springfeld C, Cattaneo R. 2007. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology 360:72–83. 10.1016/j.virol.2006.09.049 [DOI] [PubMed] [Google Scholar]
- 44.Dong C, Zafrullah M, Mixson-Hayden T, Dai X, Liang J, Meng J, Kamili S. 2012. Suppression of interferon-α signaling by hepatitis E virus. Hepatology 55:1324–1332. 10.1002/hep.25530 [DOI] [PubMed] [Google Scholar]
- 45.Laurent-Rolle M, Boer EF, Lubick KJ, Wolfinbarger JB, Carmody AB, Rockx B, Liu W, Ashour J, Shupert WL, Holbrook MR, Barrett AD, Mason PW, Bloom ME, García-Sastre A, Khromykh AA, Best SM. 2010. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 84:3503–3515. 10.1128/JVI.01161-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J, Wu M, Zhang X, Zhang W, Zhang Z, Chen L, He J, Zheng Y, Chen C, Wang F, Hu Y, Zhou X, Wang C, Xu Y, Lu M, Yuan Z. 2013. Hepatitis B virus polymerase impairs interferon-α-induced STAT activation through inhibition of importin-α5 and protein kinase C-δ. Hepatology 57:470–482. 10.1002/hep.26064 [DOI] [PubMed] [Google Scholar]
- 47.Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. 2007. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81:9812–9824. 10.1128/JVI.01012-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kopecky-Bromberg SA, Martínez-Sobrido L, Frieman M, Baric RA, Palese P. 2007. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 81:548–557. 10.1128/JVI.01782-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Montgomery SA, Johnston RE. 2007. Nuclear import and export of Venezuelan equine encephalitis virus nonstructural protein 2. J. Virol. 81:10268–10279. 10.1128/JVI.00371-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. 2007. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J. Virol. 81:13469–13477. 10.1128/JVI.01097-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodriguez JJ, Horvath CM. 2004. Host evasion by emerging paramyxoviruses: Hendra virus and Nipah virus V proteins inhibit interferon signaling. Viral. Immunol. 17:210–219. 10.1089/0882824041310568 [DOI] [PubMed] [Google Scholar]
- 52.Shaw ML, García-Sastre A, Palese P, Basler CF. 2004. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J. Virol. 78:5633–5641. 10.1128/JVI.78.11.5633-5641.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takayama I, Sato H, Watanabe A, Omi-Furutani M, Sugai A, Kanki K, Yoneda M, Kai C. 2012. The nucleocapsid protein of measles virus blocks host interferon response. Virology 424:45–55. 10.1016/j.virol.2011.12.011 [DOI] [PubMed] [Google Scholar]
- 54.Wang R, Nan Y, Yu Y, Zhang YJ. 2013. Porcine reproductive and respiratory syndrome virus Nsp1β inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-α1 degradation. J. Virol. 87:5219–5228. 10.1128/JVI.02643-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vidy A, El Bougrini J, Chelbi-Alix MK, Blondel D. 2007. The nucleocytoplasmic rabies virus P protein counteracts interferon signaling by inhibiting both nuclear accumulation and DNA binding of STAT1. J. Virol. 81:4255–4263. 10.1128/JVI.01930-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chook YM, Blobel G. 2001. Karyopherins and nuclear import. Curr. Opin. Struct. Biol. 11:703–715. 10.1016/S0959-440X(01)00264-0 [DOI] [PubMed] [Google Scholar]