

ABSTRACT
Acute HIV-1 infection is characterized by a type I interferon response, resulting in the induction of host restriction factors. HIV-1 has evolved to counteract these factors, and one such adaptation, the ability of Vpu to counteract BST2/tetherin, is associated with the evolution of simian immunodeficiency virus (SIVcpz) into pandemic group M human immunodeficiency virus type 1 (HIV-1). During transmission between individuals, very few viruses or even a single virus, the “transmitted/founder” (T/F) virus, gives rise to the new infection, but in the new host the selective pressure of the immune response yields the diverse “quasispecies” of chronic infection. Here we examine the functional characteristics of Vpu proteins encoded by T/F viruses compared to acute and chronic viruses from longitudinally sampled subjects. The studied T/F Vpu proteins showed a trend toward optimized CD4 downregulation compared to chronic Vpu proteins but did not differ substantially in their ability to downregulate BST2 or enhance virion release, although individual clones from each group were impaired in these activities. Analysis of the functionally impaired clones identified a C-terminal residue, W76, as important specifically for Vpu enhancement of virion release. Primary Vpu clones encoding a W76G polymorphism, or site-directed mutants encoding a W76G substitution, were impaired in their ability to enhance virion release, but they were not defective for BST2 surface downregulation. Conversely, the virion release function of impaired primary clones was restored by creating a G76W substitution. The identification of W76 as important for virion release enhancement that is independent of BST2 surface downregulation supports the potential to mechanistically separate these functions of Vpu.
IMPORTANCE To establish infection in a host, HIV-1 must evade the host's immune response, including the production of antiviral factors. HIV-1 encodes proteins that antagonize these defenses, including Vpu. Vpu counteracts the host protein BST2, which blocks the release of progeny viruses from the host cell. To determine the importance of Vpu activity to HIV-1 transmission, this study assessed the functionality of Vpu from viruses isolated soon after transmission (“transmitted/founder” viruses) compared to isolates from chronic infection. Although the anti-BST2 activity of Vpu proteins from the tested transmitted/founder viruses did not differ from the activity of the chronic Vpu proteins, the transmitted/founder Vpu proteins trended toward having superior activity against another host protein, CD4. Further, this study identified an amino acid near the C terminus of Vpu that is specifically important for Vpu's ability to enhance the release of progeny virus from the host cell, supporting the notion of a new mechanism for this function of Vpu.
INTRODUCTION
Early infection with human immunodeficiency virus type 1 (HIV-1) is characterized by a type I interferon (IFN) response, resulting in the induction of antiviral genes, including restriction factors (1–3). One such restriction factor is BST2 (also known as tetherin), which counteracts diverse enveloped viruses by tethering them to the host cell surface and preventing their release (4, 5). To overcome this restriction, many viruses encode countermeasures which, with the current exception of the Ebola envelope glycoprotein, act by surface downregulation and/or targeting of BST2 for degradation (reviewed in reference 6). The BST2 countermeasure encoded by HIV-1 is Vpu, which decreases the amount of BST2 on the plasma membrane (5) through the interaction between its transmembrane domain (TMD) and that of BST2 and which directs the degradation of BST2 through the interaction of its cytoplasmic domain with a β-TrCP containing SCF (Skp-Cullin-F-box)/CRL1 (Cullin1-RING ubiquitin ligase) E3 ubiquitin ligase complex (7–10). Neither the downmodulation of BST2 from the cell surface nor its degradation is strictly correlated with the ability of Vpu to enhance virion release from the cell surface (11). This lack of correlation might be explained in part by the recently described ability of Vpu to displace BST2 from sites of viral assembly (12).
The anti-BST2 function of Vpu has been proposed as a key adaptation enabling simian immunodeficiency virus (SIVcpz) to evolve into pandemic group M HIV-1 (13–16). Since Vpu's anti-BST2 activity was important for cross-species transmission of HIV-1, we hypothesized that it might be important for human-to-human transmission, particularly since BST2 is upregulated during the initial interferon response to HIV-1 infection (17) and a successful founder virus must presumably be able to counteract this.
Vpu modulates the expression of other cellular membrane proteins in addition to BST2, including CD4. Vpu acts on newly synthesized CD4 in the endoplasmic reticulum, targeting it for proteasomal degradation in a β-TrCP-dependent fashion (18, 19). Vpu is one of the three HIV-1 proteins (the other two are Env and Nef) that target CD4, underscoring the importance of this function for the virus. Following infection of a host cell, the presence of CD4 on the cell surface interferes with virion release and the packaging of Env into virions, substantially reduces infectivity of the released virions, and induces the exposure of epitopes within Env that facilitate the clearance of infected cells by antibody-dependent cytotoxicity (ADCC) (20–23). By enhancing virion infectivity and contributing to the evasion of ADCC through its downmodulation of CD4, Vpu might impact viral transmission by a mechanism other than the antagonism of BST2.
Transmission of HIV-1 to a new host has been shown to involve very few viruses or only one virus, the “transmitted/founder” (T/F) virus, which evolves into a diverse “quasispecies” under conditions of selection by the host immune response (24–28). Moreover, about half of transmission events occur during the early stages of infection when the transmitting partner is unaware of his or her HIV status and plasma viremia is highest (29). For these reasons, determining the phenotypic characteristics of T/F viruses that enable them to initiate an infection in a new host is of great interest. Because of its modulation of host factors involved in the immune response and its influence on virion infectivity, we hypothesized that Vpu might contribute to successful person-to-person transmission and the establishment of a new infection.
In the present study, vpu alleles from 10 sexually transmitted clade B group M HIV-1 transmitted/founder (T/F) infectious molecular clones (IMCs) (24–28) were epitope tagged and cloned into a Rev-dependent expression vector. For comparison, vpu alleles from laboratory-adapted chimeric virus NL4-3 (30) and pairs of plasma viruses from the acute and chronic phases of infection acquired longitudinally from four patients infected with clade B HIV-1 in the San Diego Primary Infection Cohort (SDPIC) (31) were cloned in a similar fashion. By evaluating the capacity of each allele for directing BST2 downregulation, enhancement of virion release, and CD4 downregulation, we found that the tested Vpu proteins from T/F viruses trended toward enhanced anti-CD4 function compared to Vpu proteins from NL4-3 and the chronic clade B isolates from the SDPIC. T/F Vpu proteins as a group were no better at BST2 downregulation than Vpu from NL4-3 or the chronic isolates. These data support the possibility that optimization of Vpu's anti-CD4 activity may play a role in transmission and the establishment of a productive infection in a new host.
Moreover, the data enabled us to identify a tryptophan residue at position 76 in the Vpu cytoplasmic tail that is conserved among clade B sequences (as well as clades D and G) and that is critical for Vpu-mediated enhancement of virion release but dispensable for the surface downregulation of BST2. This phenotypic decoupling by the Vpu W76 mutants supports the notion that the anti-BST2 activity of Vpu is not necessarily dependent on the downmodulation of cell surface BST2 and suggests an alternative mechanism for the ability of Vpu to stimulate virion release.
MATERIALS AND METHODS
Cell culture.
HeLa P4-R5 cells (32–34) were acquired from the NIH AIDS Research & Reference Reagent Program from Nathaniel Landau and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin, and 1 μg/ml puromycin. HEK293T cells were maintained in DMEM supplemented with 10% FBS and penicillin-streptomycin.
Plasmids and cloning.
The Rev-dependent expression vector pcDNA-RRE was constructed by cloning the NL4-3 Rev response element (RRE) into pcDNA3.1(-) (Invitrogen). A 375-bp PCR fragment encoding the RRE was amplified from pNL4-3 using primers NL43-RRE-XhoI-F (5′-TTGAGCTCGAGGTAGCACCCACCAAGG-3′) and NL43-RRE-EcoRI-R (5′-GGATCTGAATTCTAGCATTCCAAGGCACAG-3′). The PCR product was gel purified using a QIAquick gel extraction kit (Qiagen) and cloned into pcDNA3.1(-) after digestion of the PCR products and backbone with XhoI and EcoRI (NEB) to make pcDNA-RRE. The panel of 10 full-length transmitted/founder (T/F) HIV-1 infectious molecular clones (IMCs) was obtained through the NIH AIDS Research & Reference Reagent Program from John Kappes (24–27). vpu sequences were amplified from these IMCs and NL4-3 as a control by PCR with vpu sequence-specific primers designed to introduce a C-terminal FLAG tag (Table 1), gel purified using a QIAquick gel extraction kit (Qiagen), and then cloned into the pcDNA-RRE expression vector after digestion of the purified PCR products and backbone with NheI and XhoI (NEB). To clone vpu alleles from longitudinally acquired plasma samples from the San Diego Primary Infection Cohort (SDPIC) (31), HIV RNA was extracted from plasma samples using a High Pure viral RNA kit (Roche), reverse transcribed using a RETROscript kit (Ambion) with random decamer primers, and then amplified by nested PCR with primers targeting conserved sequences flanking the vpu coding region (Table 2). Nested PCR products were column purified using a QIAquick PCR purification kit (Qiagen) prior to bulk Sanger sequencing using the PCR 2 primers (Table 2). Clones with mixed peaks are noted in Fig. 1. Cloning primers which included a C-terminal FLAG tag were designed based on the bulk sequencing, with multiple primer sets designed for samples exhibiting variation in the 5′ and 3′ vpu coding sequences (Table 2). These primers were used to amplify FLAG-tagged vpu coding sequences from the nested PCR products. PCR products were cloned into the pcDNA-RRE vector using NheI and XhoI (NEB) and verified by sequencing as described above. Representative clones from acute and chronic pairs were initially chosen for further analysis, and where phenotypic discordance was noted, additional clones were selected and analyzed. The Rev expression vector pRev-IRES-GFP (IRES, internal ribosome entry site; GFP, green fluorescent protein) was constructed by subcloning rev from a NL4-3 rev expression vector cloned previously in our laboratory (pcRev-E2) following digestion with XbaI into the pIRES2-AcGFP1 vector (Clontech) digested with NheI. For site-directed mutagenesis, mutations were introduced into the relevant pcDNA-Vpu-FLAG-RRE constructs using a QuikChange II site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions and verified by sequencing. The proviral mutants pNL4-3 ΔVpu (pNL43/Udel [35]) and pNL4-3-Vpu-52/56N (36) have been described previously. Mutants pNL4-3-Vpu-W76G and pNL4-3-Vpu-52/56N-W76G were constructed by site-directed mutagenesis of pNL4-3 and pNL4-3-Vpu-52/56N, respectively, using a QuikChange II site-directed mutagenesis kit.
TABLE 1.
Primers used for cloning NL4-3 and T/F Vpu proteins
| Vpu clone | Primer name | Primer sequence (5′–3′) |
|---|---|---|
| NL4-3 | NL43-Vpu-NheI-F | AGATCCGCTAGCATGCAACCTATAATAGTAGC |
| NL4-3 | NL43-Vpu-FLAG-XhoI-R | TTGAGCTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATCAATATCCCAAG |
| CH040 | CH040-Vpu-F | CGATGAGCTAGCATGAACTCTTTACAAATATCAG |
| CH040 | CH040-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATTAATATCCCCAA |
| CH058 | CH058-Vpu-F | CGATGAGCTAGCATGCAGCCTTTAAATATAGCAAT |
| CH058 | CH058-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCTAGATCATTAACATCCCAAG |
| CH077 | CH077-Vpu-F | CGATGAGCTAGCATGCAATCTTTATATATATTAGGAAT |
| CH077 | CH077-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCTGATCATCAATAACCCAAG |
| CH106 | CH106-Vpu-F | CGATGAGCTAGCATGAACTCTTTACAAGTAGTAG |
| CH106 | CH106-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATCAACATTCCAAG |
| REJO | REJO-Vpu-F | CGATGAGCTAGCATGCAAACTTTACAAATCTTAGC |
| REJO | REJO-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATCAATATCCCAAG |
| RHPA | RHPA-Vpu-F | CGATGAGCTAGCATGCAATCTTTACAAATAGTAGC |
| RHPA | RHPA-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATTAATAACCAAAGG |
| SUMA | SUMA-Vpu-F | CGATGAGCTAGCATGCAAGCTTTATATATATTAGGA |
| SUMA | SUMA-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATTAACATCCCAAG |
| THRO | THRO-Vpu-F | CGATGAGCTAGCATGCAATCCTTAGAAATACTTG |
| THRO | THRO-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCATATCATCAATATCCCAAG |
| TRJO | TRJO-Vpu-F | CGATGAGCTAGCATGTCACCTTTAGTAATAGCAT |
| TRJO | TRJO-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATCAATATCCCAAG |
| WITO | WITO-Vpu-F | CGATGAGCTAGCATGCAACCTTTAGAAATATTAGC |
| WITO | WITO-Vpu-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCTCATCATTAACATCCCAAG |
TABLE 2.
Primers used for nested PCR and cloning of acute and chronic Vpu proteins
| PCR no. or Vpu clone(s) | Primer name | Primer sequence (5′–3′) |
|---|---|---|
| PCR 1 | 5′Tat-Vpu-F1 | CTTAGGCATCTCCTATGGCAGGAAG |
| PCR 1 | 3′Env-Vpu-R1 | CTACTGGCCTAATTCCATGTGTACATTGTA |
| PCR 2 (nested) | 5′Tat-Vpu-nested-F2 | AAGCGGAGACAGCGACGAAGAGCT |
| PCR 2 (nested) | 3′Env-Vpu-nested-R2 | TGGACAGGCCTGTGTAATGACTGAGGT |
| Q303 Acute and Chronic | Q303-AC-F | CGATGAGCTAGCATGCAACCTTTACAAATAGTAG |
| Q303 Acute and Chronic | Q303-AC-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATCAACATCCCAA |
| N528 Acute-1 and -2 | N528-A1&2-F | CGATGAGCTAGCATGCCACCTTTATACATATCA |
| N528 Acute-1 and -2 and Chronic and N988 Acute | N528-A1&2C-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATTAACATCCCAAG |
| N528 Chronic | N528-C-F | CGATGAGCTAGCATGCCACCTTTATACATAACA |
| R611 Acute and Chronic-1 and -2 | R611-AC1&2-F | CGATGAGCTAGCATGCAGATTTTAGATATATCAGT |
| R611 Acute | R611-A-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATTAATATCCCAAG |
| R611 Chronic-1 | R611-C1-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCGTGAATATCCCAAG |
| R611 Chronic-2 | R611-C2-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATTGTGAATATCCCAAG |
| N988 Acute | N988-A-F | CGATGAGCTAGCATGCAACCCCTAGAGATAG |
| N988 Chronic-1 | N988-C1-F | CGATGAGCTAGCATGCAATCCCTAAAGATAGC |
| N988 Chronic-1 and -2 | N988-C1&2-FLAG-R | CGATGACTCGAGTCACTTATCGTCGTCATCCTTGTAATCCAGATCATTAACATCCCCA |
| N988 Chronic-2 | N988-C2-F | CGATGAGCTAGCATGCAATCCCTAGGGATAG |
FIG 1.

Variability in vpu sequences from longitudinally acquired plasma from acute and chronic phases of infection. The predicted amino acid sequences from bulk Sanger sequencing of Vpu isolates from the indicated plasma samples are aligned with the DNA chromatograms provided for regions with multiple peaks encoding variable residues.
Transfections.
HeLa P4-R5 cells in 6-well plates were transfected with 2 μg of the Vpu expression construct (pcDNA-Vpu-FLAG-RRE) and 2 μg of Rev- and GFP-coexpressing plasmid pRev-IRES-GFP using Lipofectamine 2000 (Invitrogen). Cells were harvested the next day for immunoblot analysis and staining for flow cytometry. For virion release assays, HeLa P4-R5 cells in 12-well plates were transfected with 500 ng of the Vpu expression construct (pcDNA-Vpu-FLAG-RRE) and 600 ng of the proviral plasmid lacking Vpu, pNL4-3 ΔVpu (pNL43/Udel). Interferon-treated cells were cultured in 1,000 U/ml human alpha interferon (IFN-α) A/D (Sigma). Supernatants and cells were collected 24 h later for p24 enzyme-linked immunosorbent assay (ELISA) and immunoblot analysis, respectively. For the titration of Vpu-FLAG expression plasmids, HeLa P4-R5 cells in 12-well plates were transfected using Lipofectamine 2000 with 800 ng of Rev- and GFP-expressing vector pRev-IRES-GFP and up to 2,400 ng (for 3×) of the Vpu-FLAG plasmid. When less than 2,400 ng of the Vpu plasmid was used, pcDNA3.1(-) was added for a total of 3,200 ng of plasmid DNA [Rev plus Vpu-FLAG plus pcDNA3.1(-)] per well.
Flow cytometry.
Transfected HeLa P4-R5 cells were stained with allophycocyanin (APC) anti-human CD4 antibody (OKT4; Biolegend), APC mouse IgG2b, κ isotype control (Biolegend), Alexa Fluor 647 anti-human CD317 (BST2, tetherin; RS38E [Biolegend]), or Alexa Fluor 647 mouse IgG1, κ isotype control, according to manufacturer's instructions, and then fixed in 1.5% paraformaldehyde. Surface expression of CD4 and BST2 was analyzed using a BD Accuri C6 flow cytometer after gating on live GFP-positive (Rev-expressing) cells. For staining of intracellular p24, cells were fixed and permeabilized using a BD Cytofix/Cytoperm kit according to the manufacturer's instructions, followed by staining with fluorescein isothiocyanate (FITC)-conjugated anti-HIV-1 p24 antibody (KC57; Beckman Coulter).
Virion release assays.
Supernatants from NL4-3 ΔVpu provirus- and Vpu expression plasmid-cotransfected HeLa P4-R5 cells were collected 24 h after transfection as previously described (5). Virion-associated p24 was pelleted through a 20% sucrose cushion before measurement by p24 ELISA (PerkinElmer or Advanced Bioscience Laboratories).
Immunoblot analyses.
Immunoblot analyses for Vpu, BST2, Gag/p55, actin, and FLAG epitope were performed as previously described (10, 37, 38).
Infections.
For infection with site-directed viral mutants, 4 μg proviral plasmid DNA, pNL4-3 ΔVpu (pNL43/Udel [35]), pNL4-3, pNL4-3-Vpu-52/56N (36), pNL4-3-Vpu-W76G, or pNL4-3-Vpu-52/56N-W76G was complexed with Lipofectamine 2000 and used to transfect HEK293T cells plated in 10-cm-diameter dishes. After 36 h, viral supernatants were harvested, cellular debris was pelleted, and clarified supernatants were concentrated by centrifugation at 4,000 × g for 20 min at room temperature in Amicon Ultra-15 100K centrifugal filter tubes (EMD Millipore). Infectivity was determined by infecting 20,000 HeLa P4-R5 cells plated in a 48-well plate in triplicate with serial dilutions of the viral supernatants. Two days after infection, the cells were fixed and stained and the number of infectious centers was measured by computer-assisted image analysis as described previously (39). HeLa P4-R5 cells plated in triplicate in 12-well plates were infected at an multiplicity of infection (MOI) of 0.2. After 5 h, the viral supernatants were removed, the cells were washed twice with phosphate-buffered saline (PBS), and fresh culture medium was added. Thirty hours later, supernatants and cells were harvested for p24 ELISA and fluorescence-activated cell sorter (FACS) and immunoblot analyses as described above.
Immunofluorescence microscopy.
HeLa P4-R5 cells were plated at 100,000 per well on glass coverslips in a 24-well plate. The following day, they were transfected with 400 ng of Vpu-FLAG expression vector and 400 ng of the Rev expression vector (pcRev-E2) using Lipofectamine 2000. At 24 h posttransfection, the cells were fixed with 3% paraformaldehyde for 15 min at room temperature, washed once with phosphate-buffered saline (PBS), and then permeabilized with 0.2% NP-40 for 5 min at room temperature. Fixed and permeabilized cells were washed twice with PBS and then blocked with 5% normal goat serum and 3% bovine serum albumin (BSA) for 30 min at 4°C. Cells were stained with rabbit anti-Vpu antiserum (from the NIH AIDS Research and Reference Reagent Program and contributed by Klaus Strebel) and/or mouse anti-BST2/HM1.24/CD317 antibody (Chugai Pharmaceutical Co., Kanagawa, Japan) in 5% normal goat serum for 30 min at room temperature and then washed four times with PBS. Cells were then incubated with FITC goat anti-rabbit and/or Rhodamine Red-X goat anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) in 5% goat serum for 30 min at room temperature. The cells were washed 5 times with PBS, mounted, and imaged using a fluorescence microscope (Olympus) and SlideBook version 4.1 software (Intelligent Imaging Innovations, Denver, CO). For each field, images in a series along the z axis were collected, the images were deconvolved using the nearest-neighbor method, and a representative single image plane was chosen. Images were assembled using Adobe Photoshop software.
RESULTS
T/F Vpu proteins are optimized for CD4 downmodulation.
Transmitted/founder Vpu proteins (amino acid sequence alignment shown in Fig. 2A) were FLAG tagged and cloned into a Rev-dependent expression construct (Fig. 2B). To test for the downregulation of CD4 and BST2 from the cell surface, we used a flow cytometric assay in which HeLa P4-R5 cells were transfected with a bicistronic plasmid coexpressing Rev and GFP as well as a Rev-dependent vector expressing FLAG-tagged Vpu proteins (Fig. 2B) and then surface stained with fluorophore-conjugated anti-CD4 or anti-BST2 antibodies. Transfected cells that produce enough Rev to express Vpu are also GFP positive and, depending on the functionality of the tested Vpu protein, exhibit a decrease in surface CD4 and BST2. This is evident in the two-color plots in Fig. 2C; as GFP (and Rev) expression increases, CD4 and BST2 surface expression decreases. For T/F Vpu proteins CH040 (second column of Fig. 2C) and CH077 (third column of Fig. 2C), BST2 downregulation (bottom row) appears similar to that of VpuNL4-3 (first column), and all three of these appear more active than RHPA (fourth column of Fig. 2C; discussed further below). However, for CD4 downregulation (top row), CH077 appears more active than CH040 and VpuNL4-3, with the activity of RHPA most similar to that of CH077. These relationships are shown quantitatively in Fig. 2D, where the CD4 downregulation activity of each T/F Vpu is plotted as a ratio compared to the activity of VpuNL4-3. All T/F Vpu proteins, with the exception of CH040, exhibited relatively enhanced anti-CD4 activity, reducing the surface expression of CD4 25% to 60% more effectively than VpuNL4-3. The efficient counteraction with respect to CD4 exhibited by Vpu depends primarily, although not exclusively, on its cytoplasmic domain (CD) (36, 40), and the differences in both the predicted amino acid sequences and the anti-CD4 activities of these T/F Vpu proteins suggest that the tertiary structure of this domain, although apparently dynamic and tolerant of multiple polymorphisms, might be related to its functional efficiency (Fig. 2A).
FIG 2.

CD4 and BST2 downregulatory activity and expression of transmitted/founder Vpu proteins. (A) Amino acid sequence alignment of T/F Vpu proteins compared to NL4-3 and the clade B consensus sequence. The amino acid sequences of the T/F Vpu proteins are aligned to the reference clade B consensus Vpu sequence (LANL). Residues shaded dark gray are exactly the same as the reference sequence; residues shaded light gray are of the same class (basic, acidic, polar, or hydrophobic) as the corresponding clade B consensus residue; and unshaded residues are of a different class. (B) Construction of Vpu-FLAG expression vectors. Vpu alleles from transmitted/founder HIV-1 infectious molecular clones (IMCs) were PCR amplified using allele-specific primers, with a FLAG tag included in the 3′ (reverse) primer, and cloned into the multiple-cloning site of the pcDNA-RRE vector, which includes a downstream Rev response element (RRE). (C) Plasmids encoding Vpu-FLAG-RRE and Rev-IRES-GFP were used to cotransfect HeLa P4-R5 cells. The next day, the cells were stained for either CD4 or BST2 and analyzed by flow cytometry. Two-color plots show surface downregulation of CD4 (y axis, upper panels) or BST2 (y axis, lower panels) versus GFP (x axis) by NL4-3 Vpu and three selected T/F Vpu proteins (CH040, CH077, and RHPA). (D) Relative levels of CD4 downregulation by T/F Vpu alleles. Samples were gated on live GFP-positive (GFP+ [Rev+]) cells. The mean fluorescence intensity (MFI) for the isotype control was subtracted from the MFI for each anti-CD4-stained sample. The value for each T/F Vpu was then subtracted from that of the no-Vpu control, and this value was divided by the same calculation made for NL4-3 Vpu to derive the relative activity. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (E) Relative levels of BST2 downregulation by T/F Vpu alleles. Samples were gated on live GFP+ (Rev+) cells, and the activity relative to NL4-3 Vpu was calculated as described above for CD4. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (F) Immunoblot analysis of the expression of T/F Vpu alleles. Cell lysates were analyzed for FLAG (Vpu), BST2, and actin.
Most T/F Vpu proteins downregulate BST2 with the same efficiency as VpuNL4-3.
As BST2 is an interferon-induced antiviral restriction factor and the acute phase of HIV-1 infection is marked by a substantial type I interferon response, we hypothesized that T/F Vpu proteins might be optimized for BST2 downmodulation, since they were derived from viruses that successfully established infection under this selective pressure. To test this, HeLa P4-R5 cells were transfected with FLAG-tagged Vpu and Rev expression constructs and analyzed for surface expression of BST2. As seen in Fig. 2C and E, all T/F Vpu proteins except for RHPA were similar to VpuNL4-3 in their BST2 surface downregulation activity. The impaired RHPA protein notably differs from NL4-3 by an A15G polymorphism in the transmembrane domain; this is the second A in the AxxxAxxxAxxxW motif previously found to be important for the interaction between the TMDs of Vpu and BST2 (41, 42). RHPA also encodes a variant amino acid (leucine) instead of the conserved W76 in the cytoplasmic tail, which is studied further below. The expression levels of these Vpu proteins were detected by immunoblot analysis for the FLAG epitope tag (Fig. 2F). While expression levels of these Vpu proteins appear to be correlated only roughly with their CD4 downregulation activities, RHPA, which is impaired for BST2 surface downregulation, is the most poorly expressed among the T/F Vpu proteins.
T/F Vpu proteins enhance virion release with effectiveness similar to that of VpuNL4-3 even when the expression of BST2 is stimulated by IFN-α.
Treatment of cells with type I interferon upregulates BST2 and enhances the inhibition of virion release. To test whether T/F Vpu proteins are superior to VpuNL4-3 at counteracting BST2's inhibitory effect on virion release, we transfected HeLa P4-R5 cells with HIV-1 provirus that lacks Vpu (NL4-3 ΔVpu) along with the Vpu-FLAG-RRE expression constructs to provide Vpu in trans. Cells were cultured in media with or without 1,000 U/ml alpha IFN (IFN-α), and supernatants and cells were collected after 24 h. Viral supernatants were pelleted through 20% sucrose cushions, and p24 was measured by ELISA (Fig. 3A). Cells were analyzed for FLAG, BST2, actin, and Gag expression by immunoblotting (Fig. 3B). As a control, the pNL4-3 proviral plasmid was transfected, and cell lysates were analyzed by immunoblotting with rabbit antiserum to HIV-1 Vpu. This antiserum did not uniformly detect the FLAG-tagged primary Vpu proteins compared to the anti-FLAG antibody (data not shown). Although variation is evident among the T/F Vpu proteins, their phenotypes with respect to enhancement of virion release were similar to that of VpuNL4-3, except for CH040, which was substantially impaired. Despite being impaired for enhancement of virion release, CH040 Vpu was as potent as VpuNL4-3 at downregulating cell surface BST2 (Fig. 2E), suggesting that these two functions of Vpu are separable. As discussed further below, this phenotypic discordance appears largely due to a W76G polymorphism near the protein's C terminus. The expression of these Vpu proteins as detected by immunoblot analysis for FLAG (Fig. 3B) did not reveal a clear correlation with the virion release phenotype (as was similarly the case as shown in Fig. 2 with respect to the phenotypes of CD4 and BST2 downregulation).
FIG 3.

Enhancement of virion release by transmitted/founder Vpu proteins. (A) Virion release enhancement by T/F Vpu alleles in the presence or absence of IFN-α. Cells were transfected with the pNL4-3 plasmid encoding Vpu (NL4-3) or cotransfected with the indicated T/F Vpu expression plasmids (or a plasmid encoding the FLAG-tagged NL4-3 Vpu control) and with a provirus that does not express Vpu (NL4-3 ΔVpu; all other samples) and then cultured either without or with IFN-α (1,000 U/ml) overnight. The concentration of pelletable p24 antigen in the culture supernatants was measured by ELISA. Error bars represent the standard deviations of the results of quadruplicate experiments. (B) Immunoblot analysis of cell lysates showing the induction of BST2 following IFN-α treatment as well as actin, Gag (p55 and p24), FLAG, and Vpu (using a polyclonal rabbit antiserum to the cytoplasmic domain of NL4-3 Vpu) for the NL4-3 controls.
Treatment of the cells with IFN-α resulted in lower levels of virion release enhancement by all Vpu proteins tested, suggesting that, compared to NL4-3 Vpu, no T/F Vpu protein provides relatively increased resistance to the effects of interferon, which, as expected, induced the expression of BST2 as detected by immunoblot analysis (Fig. 3B). Unexpectedly, the analysis was complicated by reduced expression of all Vpu-FLAG proteins in the presence of IFN-α (Fig. 3B), correlating with the attenuated enhancement of virion release under these conditions. This reduction might be due to inhibitory effects of interferon on the pcDNA expression vector's cytomegalovirus (CMV) promoter, as previously described for type II interferon (43). In contrast, immunoblot analysis using Vpu-specific antibody (Fig. 3B, bottom panel) revealed that expression of VpuNL4-3 directed by the HIV promoter (in cis from the proviral plasmid) was increased following interferon treatment (shown in the first two lanes of the immunoblot in Fig. 3B). Nevertheless, the data suggest that Vpu proteins from T/F viruses do not enhance virion release more effectively than VpuNL4-3, even when the experimental conditions include exogenous IFN-α to mimic the antiviral state that occurs in response to acute infection in vivo.
The anti-CD4 and anti-BST2 activity of T/F Vpu proteins is not entirely explained by their expression levels.
Fig. 2 suggests a rough correlation between the expression levels of T/F Vpu proteins as assessed by immunoblot analysis (Fig. 2F) and their levels of activity against CD4 and/or BST2. For instance, the most highly expressed clone, WITO, is among those with the highest levels of both CD4 and BST2 surface downregulation. The most poorly expressed clones in this system, RHPA and TRJO, exhibited the weakest BST2 surface downregulation phenotypes. To test whether the differential activity levels of the T/F Vpu proteins could be explained solely by their expression levels, we titrated the expression of the control VpuNL4-3 in comparison with the most highly expressed and active clone, WITO, along with a poorly expressed clone, RHPA. HeLa P4-R5 cells were transfected with 800 ng of pRev-IRES-GFP and with from 200 ng (for 0.25×) to 2,400 ng (for 3×) of Vpu-FLAG expression plasmid. Where less than 2,400 ng of Vpu-FLAG plasmid was used, pcDNA3.1(-) was added for a total amount of 3,200 ng plasmid DNA per well. The next day, cells were harvested for FACS and immunoblot analysis as described above.
When the expression level of WITO Vpu was near that of 1× VpuNL4-3 (around 0.5× or 400 ng WITO plasmid), the CD4 and BST2 surface downregulatory activities were similar to those of VpuNL4-3 (Fig. 4). Comparison of mean fluorescence intensities (MFI) from three experiments for 1× NL4-3 and 0.5× WITO yields a P value of 0.495 for CD4 and a P value of 0.391 for BST2 (paired Student's t test, two tailed). This suggests that, at least in the case of WITO, superior function is related to robust expression in this system. For RHPA Vpu, however, an expression level similar to that of VpuNL4-3 did not yield equal activity. Despite a level of expression similar to that of VpuNL4-3 after transfection of 1× to 2× RHPA expression plasmid (Fig. 4C), RHPA Vpu was better than VpuNL4-3 at downregulating CD4 (Fig. 4A) but remained less active at downregulating BST2 (Fig. 4B). Comparison of the MFIs for 1× NL4-3 and 1× RHPA yielded a P value of 0.230 for CD4 and a P value of 0.006 for BST2 (paired Student's t test, two tailed). RHPA achieved VpuNL4-3-like levels of BST2 surface downregulation activity only when comparatively overexpressed (between 2× and 3× the amount of expression plasmid) (Fig. 4C). Comparison of the MFIs for 1× NL4-3 and 2× RHPA (which was more highly expressed than NL4-3) yielded a P value of 0.086 for BST2 (paired Student's t test, two tailed). These data support the notion that RHPA is impaired for BST2 surface downregulation relative to VpuNL4-3 even when its level of expression in the system is equalized.
FIG 4.
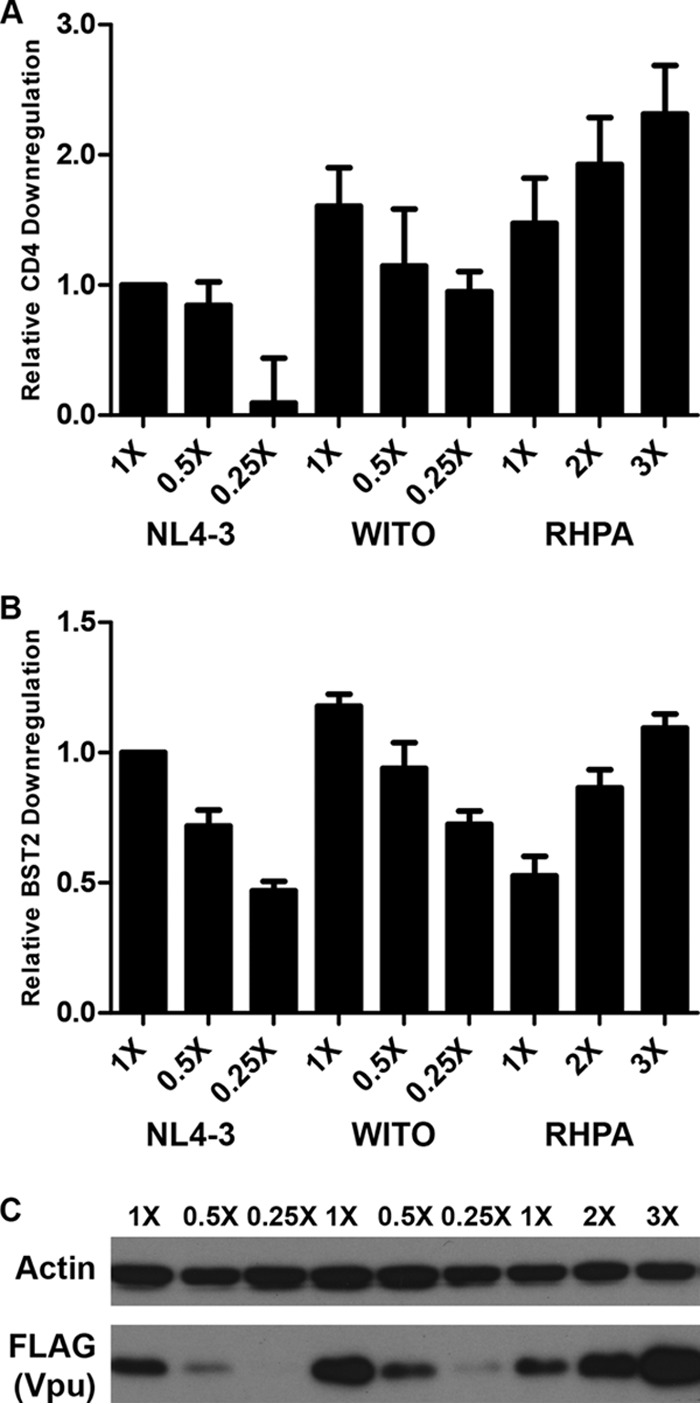
Titration of highly and poorly expressed T/F Vpu proteins. HeLa P4-R5 cells were transfected in 12-well plates with 800 ng of Rev- and GFP-expressing vector pRev-IRES-GFP and up to 2,400 ng (for 3×) of the Vpu-FLAG plasmid. When less than 2,400 ng of the Vpu plasmid was used, pcDNA3.1(-) was added for a total of 3,200 ng of plasmid DNA per well. (A) Relative levels of CD4 downregulation by titrated T/F Vpu alleles. CD4 surface downregulation compared to 1× NL4-3 Vpu was calculated as described for Fig. 2D. The values shown are means from three experiments. Error bars are the standard deviations. (B) Relative levels of BST2 downregulation by titrated T/F Vpu alleles. BST2 surface downregulation compared to 1× NL4-3 Vpu was calculated as described for Fig. 2E. The values shown are means from three experiments. Error bars are the standard deviations. (C) Immunoblot analysis of cell lysates showing the expression levels of titrated Vpu-FLAG with actin as a control.
Individual Vpu proteins isolated from chronically infected persons differ substantially in their anti-CD4 and anti-BST2 effects.
While the tested T/F Vpu proteins as a group appear modestly superior to VpuNL4-3 for CD4 downregulation, VpuNL4-3 is from a laboratory-adapted HIV-1 strain and might not be representative of Vpu proteins derived from primary chronic clade B isolates. To compare the function of T/F Vpu proteins to that of Vpu proteins isolated from chronically infected persons, we cloned vpu alleles from plasma samples acquired longitudinally from treatment-naive subjects in the San Diego Primary Infection Cohort (SDPIC). Viral RNA was extracted from pairs of plasma samples from the acute and chronic phases of infection for each of four subjects who remained untreated with antiretroviral therapy for the entirety of the sampled time period (Table 3). The isolates from the acute phase were collected 45 to 85 days after the estimated date of infection, so they were likely subjected to selection pressures in the host, in particular, the cytotoxic T lymphocyte response, that would presumably induce coding changes relative to the T/F virus in that individual. Therefore, studies of the acute Vpu proteins are most relevant in comparison to their matching chronic Vpu proteins, where differences in function can be attributed to specific amino acid changes that developed in vivo. Nonetheless, the chronic Vpu proteins also provide a comparison to the T/F Vpu proteins.
TABLE 3.
Acute/chronic sample information
| Sample name | Collection time (days from EDIa) | CD4 (no. of cells/μl) | Viral load (no. of copies/ml)b |
|---|---|---|---|
| Q303 Acute | 45 | 642 | 89,900 |
| Q303 Chronic | 545 | 522 | 3,910 |
| N528 Acute | 85 | 813 | 7,200 |
| N528 Chronic | 1,069 | 224 | 57,500 |
| R611 Acute | 85 | 418 | 17,600 |
| R611 Chronic | 456 | 704 | 13,100 |
| N988 Acute | 45 | 959 | 109,000 |
| N988 Chronic | 1,777 | 408 | 2,270 |
EDI, estimated date of infection; calculated as described in 46.
All subjects were antiretroviral therapy naive at all time points sampled.
Viral RNA extracted from plasma was reverse transcribed into cDNA, and then amplicons that included the vpu sequence were cloned by nested PCR and Sanger sequenced in bulk using the PCR 2 primers (Table 2). Based on the sequencing results, primers (listed in Table 2) were designed to FLAG tag and clone vpu into the Rev-dependent expression vector pcDNA-RRE as described above. The amino acid sequence alignment of the cloned acute and chronic Vpu proteins (shown in Fig. 5A) revealed that, as anticipated, the acute and chronic Vpu pairs are more similar to each other than to acute or chronic Vpu isolates from other patients. Analysis of CD4 downregulation by FACS as described above revealed that all the acute and chronic Vpu pairs were as good as or better than VpuNL4-3 at CD4 downregulation, except for the pair from subject N528 (Fig. 5B). Acute and chronic Vpu pairs were also similar to VpuNL4-3 in their ability to downregulate BST2 from the cell surface (Fig. 5C). While the N988 acute Vpu was the most active against CD4 among the tested isolates, it was the least active for BST2 downregulation.
FIG 5.

Activity and expression level of Vpu proteins isolated from the acute and chronic phases of infection. (A) Amino acid sequence alignment of acute and chronic Vpu proteins compared to NL4-3 and the clade B consensus sequence. Residues are shaded as described for Fig. 2A, with the clade B Vpu consensus as the reference sequence. (B) Relative levels of CD4 downregulation by acute and chronic Vpu alleles. The experiments were done and the samples were analyzed as described in the Fig. 2D legend. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (C) Relative levels of BST2 downregulation by acute and chronic Vpu alleles. The experiments were done and the samples were analyzed as described in the Fig. 2E legend. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (D) Virion release enhancement by acute and chronic Vpu alleles. The experiments were conducted and the samples analyzed as described in the Fig. 3 legend, except that IFN was not used. Error bars represent the standard deviations of the results of quadruplicate experiments. (E) Immunoblot analysis of the expression of acute and chronic Vpu alleles. Lysates from cells transfected for virion release analysis as described for panel D were analyzed for Gag/p55, actin, and FLAG (Vpu) by immunoblotting.
Acute and chronic Vpu proteins were also tested for their ability to enhance virion release as described above but in the absence of interferon treatment (Fig. 5D). Although the ability to enhance virion release was highly variable among these Vpu proteins, N528 acute-1 and N988 chronic-1 Vpu proteins were substantially impaired for this phenotype. Similar to the T/F Vpu proteins, the expression levels of the acute and chronic Vpu proteins did not clearly correlate with their relative anti-CD4 or anti-BST2 activity (Fig. 5E). For N528 acute-1 Vpu, which is also less active for BST2 surface downregulation, this impairment for virion release enhancement might be related to the presence of two hydrophilic residues at positions 7 and 8 (serine-threonine), whereas the N528 chronic clone with better activity has only the threonine. These hydrophilic substitutions could reduce the efficiency of interaction between the transmembrane domains (TMDs) of Vpu and BST2 by altering the tilt angle of the Vpu TMD helix in the lipid bilayer as a result of hydrophobic mismatch (41).
N988 chronic-1 Vpu was slightly impaired for BST2 surface downregulation but was nearly defective for enhancement of virion release. Notably, the N988 acute Vpu was not markedly impaired for enhancement of virion release, although it was similar to N988 chronic-1 Vpu in its ability to downregulate BST2 from the cell surface. These two paired proteins differ by only three amino acids: N988 acute Vpu has a proline at amino acid 3 whereas N988 chronic-1 has a serine; it has a glutamic acid at amino acid 5 whereas N988 chronic-1 has a lysine; and it has the conserved tryptophan at position 76 whereas N988 chronic-1 has a glycine. This W76G polymorphism near the C terminus of the Vpu cytoplasmic tail is, as noted above, also present in T/F Vpu CH040, a Vpu that is characterized by the same discordance between the phenotypes of BST2 downregulation and enhancement of virion release (Fig. 2 and 3). The role of this residue in enhancing virion release was studied in depth by site-directed mutagenesis as described below.
Although bulk sequencing of cDNA reverse transcribed from the subjects' plasma samples revealed few sequence variations (shown as sequence chromatograms in Fig. 1), in those cases where phenotypic discordance existed and the sequenced variants encoded amino acids with different properties, we tested additional clones to determine whether our findings were representative of the detected viruses. N528 acute-2 Vpu differs from N528 acute-1 Vpu by one amino acid: acute-2 has M at amino acid 67 instead of L (Fig. 5A). N528 acute-1 and acute-2 were phenotypically very similar: both had relatively poor CD4 downregulation activity (Fig. 6A), slightly impaired BST2 downregulation activity (Fig. 6B), and impaired virion release enhancement activity (Fig. 6C). R611 chronic-2 differs from R611 chronic-1 at position 40, where it has Q instead of K, and position 80, where it has N instead of D (Fig. 5A). R611 chronic Vpu proteins have similarly high levels of CD4 and BST2 downregulation activities (Fig. 6A and B), but virion release enhancement by R611 chronic-2 was higher than that by R611 chronic-1 (Fig. 6C). Differential expression levels as tested by immunoblot analysis did not explain this variation (Fig. 6D). N988 chronic-1 and N988 chronic-2 exhibited similar levels of activity despite differing at amino acids 5 (N988 chronic-2 has G instead of K) and 16 (N988 chronic-2 has T instead of A) (Fig. 5A). Both N988 chronic Vpu variants tested exhibited high levels of CD4 downregulation (Fig. 6A), had relatively low levels of BST2 downregulation (Fig. 6B), and were greatly impaired for virion release enhancement (Fig. 6C). Notably, both N988 chronic variants have G at position 76 instead of the conserved W. These data indicated that, in general, marked functional variation was not present among clones obtained from the same sample despite coding variations.
FIG 6.

Activity and expression of additional Vpu variants isolated from the acute and chronic phases of infection. (A) Relative levels of CD4 downregulation by additional acute and chronic Vpu variants. Alleles labeled “-1” are those initially evaluated in Fig. 5 and reevaluated alongside the variants labeled “-2” here. The experiments were done and the samples were analyzed as described in the Fig. 2D legend. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (B) Relative levels of BST2 downregulation by additional acute and chronic Vpu variants. The experiments were done and the samples were analyzed as described in the Fig. 2E legend. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (C) Virion release enhancement by additional acute and chronic Vpu variants. The experiments were conducted and the samples analyzed as described in the Fig. 3 legend, except that IFN was not used. Error bars represent the standard deviations of the results of quadruplicate experiments. (D) Immunoblot analysis of the expression of additional acute and chronic Vpu variants. Lysates from cells transfected for virion release analysis as described for panel C were analyzed for Gag/p55, actin, and FLAG (Vpu) by immunoblotting.
Site-directed mutation of tryptophan 76 reveals its specific importance for Vpu-mediated enhancement of virion release.
Among the primary Vpu proteins analyzed here, the three most impaired for enhancement of virion release, the T/F Vpu CH040 and the chronic Vpu proteins from subject N988, all have a tryptophan-76-to-glycine (W76G) polymorphism. Despite their impaired ability to counteract BST2-mediated restriction of virion release, these Vpu proteins were remarkably effective or impaired only mildly for downregulation of cell surface BST2 (Fig. 2, 3, 5, and 6). To investigate whether the W76G substitution is sufficient to impair the enhancement of virion release, site-directed mutations encoding it were introduced in NL4-3 vpu and the vpu of high-performance and well-expressed T/F clone CH077 (Fig. 7A to D). As noted above, T/F Vpu RHPA also has a polymorphism involving this residue: W76L. However, while RHPA Vpu is impaired only mildly for the enhancement of virion release compared to VpuNL4-3, it was the most impaired for BST2 surface downregulation among the T/F Vpu proteins—essentially the opposite of the phenotype of CH040 Vpu, which has the W76G polymorphism (Fig. 2 to 4). Site-directed mutagenesis was therefore also used to introduce the W76L substitution into VpuNL4-3. All of these mutants were tested for CD4 and BST2 surface downregulation (Fig. 7A and B) as well as for enhancement of virion release (Fig. 7C). Site-directed mutation of W76G appeared to have little effect on the expression of these Vpu proteins as determined by immunoblot analysis for FLAG (Fig. 7D). As expected (Fig. 7A), the W76G and W76L mutations had no effect on the downregulation of CD4 by NL4-3 or CH077 Vpu. BST2 downregulation (Fig. 7B) was likewise minimally perturbed or unaffected by the W76G or W76L substitutions. This suggests that the impaired downregulation of surface BST2 by RHPA Vpu (which has L76) and the N988 chronic-1 Vpu (with G76) is not due to this polymorphism.
FIG 7.

Site-directed mutagenesis of Vpu amino acid 76: effects on CD4 and BST2 downregulation and enhancement of virion release. (A to D) Function and expression of Vpu proteins with site-directed mutation of W76. (E to H) Test of whether mutation of amino acid 76 to W restores function of impaired alleles. (A and E) Relative levels of CD4 downregulation by site-directed Vpu mutants. Data are expressed relative to NL4-3 Vpu as described for Fig. 2D. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (B and F) Relative levels of BST2 downregulation by site-directed Vpu mutants. Data are expressed relative to NL4-3 Vpu as described for Fig. 2E. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (C and G) Virion release enhancement by the site-directed Vpu mutants. Pelletable p24 antigen was measured by ELISA in supernatants from cultures of cells transfected with the Vpu expression plasmid and NL4-3 ΔVpu as described for Fig. 5D. Error bars represent the standard deviations of the results of quadruplicate experiments. (D) Immunoblot analysis of the expression of site-directed Vpu mutants and controls. Lysates from cells transfected for FACS analysis as described for panels A and B were analyzed for actin and Vpu (FLAG) by immunoblotting. (H) Immunoblot analysis of the expression of site-directed mutants of functionally impaired Vpu proteins. Lysates from cells transfected for virion release analysis as described for panel G were analyzed for Gag/p55, actin, and FLAG (Vpu) by immunoblotting.
In contrast to their minimal impact on the downregulation of surface BST2, both the W76G and W76L substitutions substantially decreased the ability of NL4-3 Vpu to enhance virion release (Fig. 7C). The W76G substitution also impaired virion release enhancement by CH077 Vpu. Together, these data indicate that the substitution of W76 near the C-terminal end of the Vpu cytoplasmic tail with glycine or leucine substantially impairs Vpu's ability to enhance virus release without markedly affecting its ability to reduce the total amount of BST2 at the cell surface. This phenotypic discordance is especially evident in the case of the W76G substitution in the context of the T/F CH077 Vpu.
To further support the hypothesis of the role of W76 in virion release, we tested site-directed mutants of the primary Vpu proteins that were selectively impaired for virion release and that encoded residues other than tryptophan at this site, replacing those residues with tryptophan: plasmids expressing CH040 Vpu-G76W, RHPA Vpu-L76W, and N988-C1 Vpu-G76W were constructed. Changing residue 76 to the conserved tryptophan had no effect on the CD4 downregulation (Fig. 7E) or BST2 downregulation (Fig. 7F) activities of these Vpu proteins. However, these site-directed mutations had substantial effects on the virion release enhancement activity of these Vpu proteins. Mutation of G76 to tryptophan in CH040 and N988 chronic-1 Vpu proteins restored their virion release enhancement activity (Fig. 7G), confirming a role for W76 in virion release enhancement. Conversely, mutation of L76 of RHPA Vpu to W greatly reduced its virion release activity. The explanation for the effect of the L76W substitution is unclear, although the C terminus of RHPA is unique among tested alleles not only for L76 but also for the presence of D72, whereas the other Vpu proteins have a histidine residue. Conceivably, D72 and L76 of RHPA Vpu determine the tertiary structure of its C-terminal end that accounts for its virion release enhancement function, and mutation of L76 to W disrupts this. As shown by the immunoblot in Fig. 7H, the changes in the virion release enhancement phenotype were not due to differential expression of these site-directed mutants. Taking the data together, this gain-of-function and loss-of-function analysis indicates that W76 is a novel and specific determinant of Vpu-mediated enhancement of virion release, at least in certain genetic contexts.
Functional consequences of site-directed mutation of tryptophan 76 compared to mutation of β-TrCP-binding residues S52,56.
We examined whether the defect in anti-BST2 activity conferred by the W76G substitution relates to Vpu's ability to interact with β-TrCP, which recruits a SCF (Skp-Cullin 1-F box) E3 ubiquitin ligase to direct ubiquitination and degradation of BST2 (7, 8, 10, 44). Mutations of the serines in the β-TrCP-binding motif of Vpu (S52,56N) abrogate β-TrCP binding, not only abolishing Vpu-mediated downregulation of CD4 but also substantially impairing its ability to downregulate BST2 and enhance virion release (7, 19). To determine whether the W76G substitution affects Vpu's anti-BST2 activity via β-TrCP or instead by a different mechanism, mutations encoding the W76G and S52,56N substitutions were introduced separately and in combination into the highly functional T/F CH077 vpu allele. These mutants were tested for CD4 and BST2 surface downregulation and for enhancement of virion release. As expected, CH077-S52,56N was markedly impaired for CD4 downregulation while CH077-W76G was not (Fig. 8A). Similarly, CH077-W76G was not impaired for BST2 surface downregulation (Fig. 8B), whereas CH077-S52,56N was slightly impaired. The combination mutant CH077 S52,56N-W76G was most impaired for BST2 downregulation, despite the lack of effect of the W76G single mutation, although all of these effects were modest (Fig. 8B).
FIG 8.

Functional consequences of W76G site-directed mutation compared to mutation of β-TrCP-binding sites S52,56N in the background of a highly functional Vpu allele (CH077). (A) Effects of W76G and S52,56N mutations, alone and in combination, on surface downregulation of CD4 by CH077 Vpu. Data are expressed relative to NL4-3 Vpu as described for Fig. 2D. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (B) Effects of W76G and S52,56N mutations on surface downregulation of BST2. Data are expressed relative to NL4-3 Vpu as described for Fig. 2E. The values shown are means of the results of two experiments performed in duplicate. Error bars are the standard deviations. (C) Virion release enhancement by the Vpu proteins. Pelletable p24 antigen was measured by ELISA in supernatants from cultures of the cells analyzed as described for panel E which were transfected with the Vpu expression plasmid and NL4-3 ΔVpu as described for Fig. 5D. Error bars represent the standard deviations of the results of quadruplicate experiments. (D) Immunoblot analysis of the expression of site-directed Vpu mutants and controls. Lysates from cells transfected for FACS analysis as described for panels A and B were analyzed for actin and Vpu (FLAG) by immunoblotting. (E) Surface downregulation of BST2 in cells cotransfected with provirus lacking Vpu (NL4-3 ΔVpu) and the indicated Vpu-FLAG expression constructs. The day after transfection, the cells were analyzed by flow cytometry. The two-color plots show BST2 expression (y axis) versus intracellular p24 expression (x axis). Results are representative of two experiments performed in duplicate.
Importantly, substitution of W76G and mutation of the β-TrCP-binding site caused independent and additive impairments of Vpu's ability to enhance virion release (Fig. 8C): Vpu CH077-W76G was just as impaired for enhancement of virion release as Vpu CH077-S52,56N, and the Vpu with both S52,56N and W76G substitutions had no activity. To ensure a direct comparison between the phenotypes of BST2 downregulation and enhancement of virion release, we assessed surface BST2 on the cells used for the virion release assays (Fig. 8E), which confirmed the findings of experiments where Vpu was expressed in the absence of viral assembly (Fig. 8B). Immunoblot analysis of cell lysates (Fig. 8D) revealed that the changes in the virion release enhancement phenotype were not due to differential expression of the W76G and S52,56N mutants. The additive effects of W76G and S52,56N mutations on virion release enhancement suggest that these mutations are acting at different steps through which Vpu counteracts the restriction imposed by BST2. Moreover, the data support the hypothesis that W76 is important for an aspect of BST2 antagonism that is disassociated mechanistically from surface downregulation and degradation.
We further confirmed the effects of the W76G substitution by characterizing site-directed mutants in the NL4-3 proviral context. HeLa P4-R5 cells were infected with virus made by transfection of HEK293T cells with proviral plasmids encoding NL4-3 ΔVpu (pNL43/Udel), NL4-3, NL4-3-Vpu-S52,56N, NL4-3-Vpu-W76G, or NL4-3-Vpu-S52,56N-W76G. After 5 h of incubation, viral supernatants were removed from the HeLa cells, the cells were washed twice with PBS, and fresh cell culture medium was added. After 30 h, viral supernatants were taken for p24 ELISA and cells were harvested for immunoblot and FACS analysis for surface BST2 and intracellular p24. As with the CH077 mutant studied in trans, NL4-3-Vpu-W76G was not substantially impaired for BST2 surface downregulation (Fig. 9A and B). In the NL4-3 proviral context, the S52,56N mutation markedly impaired the downregulation of BST2. When the S52,56N mutation was combined with W76G (W76G-S52,56N), BST2 surface expression was, in fact, slightly upregulated (Fig. 9A and B). Virion release was impaired for both NL4-3-Vpu-W76G and NL4-3-Vpu-S52,56N (Fig. 9C).
FIG 9.

Functional consequences of W76G alone and in combination with S52,56N mutations in the context of NL4-3 virus. (A) Surface downregulation of BST2 in HeLa P4-R5 cells infected with NL4-3 virus or the indicated viral mutants at a multiplicity of infection of 0.2. Viral supernatants were added to the cells for 5 h, the cells were washed, and then fresh culture medium was added. At 30 h after infection, the cells were analyzed by flow cytometry. The two-color plots show BST2 expression (y axis) versus intracellular p24 expression (x axis). Results are representative of three experiments performed in triplicate. (B) Relative levels of BST2 downregulation by site-directed viral mutants as described for panel A. Data are expressed relative to NL4-3. The values shown are means of the results of the representative experiment described in the panel A legend and performed in triplicate. Error bars are the standard deviations. (C) Virion release by Vpu mutant provirus. Pelletable p24 antigen was measured by ELISA in supernatants from cultures of the cells analyzed as described for panels A and B which were infected with the indicated Vpu mutant viruses. Error bars are the standard deviations of triplicate experiments. (D) Immunoblot analysis of the expression of Vpu from cells infected with viral mutants. Cell lysates were analyzed for Gag, actin, BST2, and Vpu by immunoblotting.
Importantly, these mutations had additive effects on virion release: the combination proviral mutant NL4-3-Vpu-S52,56N-W76G was as impaired in virion release as virus lacking Vpu (Fig. 9C). Immunoblot analysis confirmed equal levels of expression of Gag/p55 in the provirus-transfected cells (Fig. 9D). Since a minority of the cells were infected, the BST2 degradation phenotype was not evident by immunoblot analysis (Fig. 9D). Less Vpu-W76G and Vpu-S52,56N-W76G protein was detected by immunoblot analysis performed with anti-Vpu antiserum than was detected in the case of the NL4-3 Vpu and Vpu-S52,56N proteins (Fig. 9D), suggesting that Vpu with the W76G mutation might be more poorly expressed in the NL4-3 proviral context. However, FLAG-tagged NL4-3 Vpu-W76G and CH077 Vpu-W76G mutants provided in trans and assessed by anti-FLAG immunoblot analysis (Fig. 7D) were expressed just as well as NL4-3 Vpu-FLAG and CH077 Vpu-FLAG, respectively. These data support the notion that the rabbit anti-Vpu antiserum does not detect W76G variants as well as the wild-type Vpu. Nonetheless, Vpu-W76G was characterized by robust BST2 downregulation activity, suggesting that this effect is not necessarily sufficient to yield enhanced virion release.
Tryptophan 76 is not required for colocalization of Vpu and BST2.
We determined the intracellular distribution of NL4-3 Vpu site-directed mutants W76G, S52,56N, and W76G-S52,56N as it related to BST2 to determine whether changes in the colocalization of these proteins accounted for the impaired virion release enhancement activity of the W76G and combination W76G-S52,56N mutants. HeLa P4-R5 cells were cotransfected with plasmids expressing NL4-3 Vpu, NL4-3 Vpu-W76G, NL4-3 Vpu-S52,56N, or NL4-3 Vpu-W76G-S52,56N and Rev (pcRev-E2). The next day, cells were fixed, permeabilized, and stained for Vpu and BST2 as described in Materials and Methods. As shown in Fig. 10, the intracellular distributions of the Vpu proteins (in green, left column) were not markedly different: each Vpu protein concentrated in a perinuclear region as well as in more peripheral endosomes. Likewise, the colocalizations of Vpu proteins and BST2 (in red) were similar among the mutants and indistinguishable from the wild-type results (merged images in the right column of Fig. 10). These data suggest that the impaired virion release enhancement activity of the W76G Vpu mutant is not the result of mislocalization away from BST2, a conclusion supported by the preserved ability of this protein to downregulate BST2.
FIG 10.

Subcellular distribution and colocalization of Vpu site-directed mutants with BST2. HeLa P4-R5 cells were cotransfected with plasmids expressing the indicated NL4-3 Vpu-FLAG site-directed mutants and Rev. The following day, Vpu and BST2 were visualized as described in Materials and Methods, using polyclonal rabbit anti-Vpu and monoclonal mouse anti-BST2 antibodies. The left column shows anti-Vpu staining, the middle column shows anti-BST2 staining, and the right column shows a merged image in which Vpu is green and BST2 is red, with the overlap in yellow. Scale bar, 20 μm.
DISCUSSION
Despite facing the robust type I interferon response of acute infection and the associated upregulation of BST2 (17), the transmitted/founder Vpu proteins tested here are no better at BST2 downregulation or enhancement of virion release than the “prototype” clade B VpuNL4-3 or the clade B Vpu proteins obtained from a cohort of chronically infected patients. These results suggest that the recent observation that clade B T/F viruses (some of which were the originating proviruses of the Vpu proteins studied here) are relatively more resistant to interferon than chronic clade B controls (45) is not likely related to an optimized anti-BST2 activity of Vpu. However, we observed a trend toward superior CD4 downregulation activity of the evaluated T/F Vpu proteins compared to VpuNL4-3 and the chronic Vpu proteins. This raises the possibility that the anti-CD4 activity of Vpu plays a role in transmission, possibly through consequent enhancement of the infectivity of newly produced virions. This hypothesis is consistent with recent results indicating that T/F viruses exhibit enhanced cell-free infectivity and greater virion incorporation of envelope glycoprotein (45).
Examination of the amino acid sequences of primary Vpu isolates with differential anti-BST2 activities suggested that the conserved tryptophan at position 76 near the protein's C terminus is important for the Vpu-mediated enhancement of virion release but is less critical for the downregulation of BST2 from the cell surface. This conclusion was directly supported by site-directed mutagenesis of both NL4-3 and a highly functional T/F Vpu protein (CH077). Specifically, the naturally occurring W76G polymorphism is associated with impaired enhancement of virion release but preserved BST2 surface downregulation. Site-directed mutants encoding W76G were similarly impaired whether provided in trans or in the context of a replication-competent virus. Moreover, reciprocal mutation of the impaired primary Vpu proteins from glycine to a tryptophan at position 76 rescued the virion release enhancement phenotype of the impaired primary Vpu proteins (CH040 and N988 chronic-1). We found that W76G alone or in combination with S52,56N had no effect on the colocalization of Vpu with BST2. Importantly, evaluation of the combination mutant encoding S52,56N (which abrogates binding to β-TrCP) and W76G indicated that these mutations are additive, and thus their effects are likely mechanistically distinct.
The mechanism by which W76 specifically contributes to the enhancement of virion release by Vpu is unclear, but it appears to be independent of the downregulation of BST2 from the cell surface. Also, given the additive nature of its contribution to the release enhancement phenotype of the β-TrCP-binding mutant S52,56N, the mechanism is also apparently independent of the degradation of BST2. The contribution of W76 might involve the recently described ability of Vpu to displace BST2 from virion budding sites in the plane of the plasma membrane (12). This phenotype mapped to the C-terminal α-helix in the Vpu cytoplasmic domain, which is adjacent to the unstructured extreme C terminus in which W76 resides. Interestingly, the displacement effect is phenotypically additive with the effect of β-TrCP, reminiscent of the additive effect of the W76G and S52,56N mutations described here. But rather than accounting for the residual activity of a Vpu mutant with substitutions of serines 52 and 56 that lacks the ability to bind β-TrCP and is essentially defective in downregulating BST2 (12), the phenotype of the W76G variants shown here suggests that the overall downregulation of cell surface BST2 is not sufficient for the antagonism of restricted virion release.
Currently, we hypothesize that despite the downregulation of total cell surface BST2 by Vpu W76G, sufficient BST2 likely remains at virion budding sites to preserve the restriction of release. How W76 would specifically enable Vpu to exclude BST2 from budding sites remains to be elucidated. Nonetheless, we speculate that Vpu might require such an activity due to the timing of its expression. We note that Vpu, like Gag, is a Rev-dependent protein that is expressed relatively late in the virus replication cycle. Vpu has no “head start” to remove BST2 from the cell surface before the onset of viral assembly, as might be the case for the SIV Nef proteins that antagonize BST2 (reviewed in reference 6). Thus, optimal Vpu activity with respect to the enhancement of virion release might require a mechanism that enables the antagonism of BST2 that is poised to associate with assembling virions, and we hypothesize that this mechanism requires W76. Notably, W76 is a conserved feature of clade B Vpu proteins such as those studied here, as well as clades D and G, but it is not conserved within one of the most prevalent clades of group M HIV-1, clade C. This lack of conservation might be explained by alternative sequences at the extreme C terminus of Vpu proteins that provide a functional contribution similar to that of W76. Alternatively, the downregulation of BST2 from the cell surface, an activity for which W76 is dispensable, might actually be a more robustly conserved phenotype than the enhancement of virion release.
In summary, the members of the subset of clade B T/F Vpu proteins studied here do not appear to be superior to Vpu proteins from a group of chronically infected persons with respect to their anti-BST2 activity, although they may be optimized for CD4 downregulation. Whether the enhanced anti-CD4 activity of T/F Vpu proteins generally plays a role in transmission requires further study. In addition, the study of Vpu proteins encoded by primary isolates led us to identify C-terminal residue W76 as important for enhancement of virion release by clade B Vpu by a mechanism that is not associated with the removal of BST2 from the cell surface.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI081668 to J.G. M.K.L. was supported in part by NIH training grant T32 AI007384. The San Diego Primary Infection Cohort is supported by NIH grant AI74621. The CFAR Translational Virology Core is supported by NIH grant AI036214.
We are grateful to Marissa Suarez for performing the p24 assays and Susan Little, Douglas Richman, Davey M. Smith, and Gabriel Wagner for providing plasma samples from the San Diego Primary Infection Cohort. We also thank the NIH AIDS Research and Reference Reagent Program and the UCSD CFAR.
Footnotes
Published ahead of print 26 February 2014
REFERENCES
- 1.Gaines H, von Sydow MA, von Stedingk LV, Biberfeld G, Bottiger B, Hansson LO, Lundbergh P, Sonnerborg AB, Wasserman J, Strannegaard OO. 1990. Immunological changes in primary HIV-1 infection. AIDS 4:995–999. 10.1097/00002030-199010000-00008 [DOI] [PubMed] [Google Scholar]
- 2.Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, Lebedeva M, DeCamp A, Li D, Grove D, Self SG, Borrow P. 2009. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 83:3719–3733. 10.1128/JVI.01844-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bitzegeio J, Sampias M, Bieniasz PD, Hatziioannou T. 2013. Adaptation to the interferon-induced antiviral state by human and simian immunodeficiency viruses. J. Virol. 87:3549–3560. 10.1128/JVI.03219-12 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 4.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430. 10.1038/nature06553 [DOI] [PubMed] [Google Scholar]
- 5.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252. 10.1016/j.chom.2008.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans DT, Serra-Moreno R, Singh RK, Guatelli JC. 2010. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol. 18:388–396. 10.1016/j.tim.2010.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell RS, Katsura C, Skasko MA, Fitzpatrick K, Lau D, Ruiz A, Stephens EB, Margottin-Goguet F, Benarous R, Guatelli JC. 2009. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 5:e1000450. 10.1371/journal.ppat.1000450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Fruh K, Moses AV. 2009. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J. Virol. 83:7931–7947. 10.1128/JVI.00242-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubé M, Roy BB, Guiot-Guillain P, Binette J, Mercier J, Chiasson A, Cohen EA. 2010. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 6:e1000856. 10.1371/journal.ppat.1000856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tokarev AA, Munguia J, Guatelli JC. 2011. Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J. Virol. 85:51–63. 10.1128/JVI.01795-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyagi E, Andrew AJ, Kao S, Strebel K. 2009. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. U. S. A. 106:2868–2873. 10.1073/pnas.0813223106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McNatt MW, Zang T, Bieniasz PD. 2013. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 9:e1003299. 10.1371/journal.ppat.1003299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, Takehisa J, Ogando Y, Ochsenbauer C, Kappes JC, Ayouba A, Peeters M, Learn GH, Shaw G, Sharp PM, Bieniasz P, Hahn BH, Hatziioannou T, Kirchhoff F. 2009. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6:409–421. 10.1016/j.chom.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT. 2009. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 5:e1000429. 10.1371/journal.ppat.1000429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T. 2009. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6:54–67. 10.1016/j.chom.2009.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang SJ, Lopez LA, Hauser H, Exline CM, Haworth KG, Cannon PM. 2010. Anti-tetherin activities in Vpu-expressing primate lentiviruses. Retrovirology 7:13. 10.1186/1742-4690-7-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homann S, Smith D, Little S, Richman D, Guatelli J. 2011. Upregulation of BST-2/tetherin by HIV infection in vivo. J. Virol. 85:10659–10668. 10.1128/JVI.05524-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Margottin F, Benichou S, Durand H, Richard V, Liu LX, Gomas E, Benarous R. 1996. Interaction between the cytoplasmic domains of HIV-1 Vpu and CD4: role of Vpu residues involved in CD4 interaction and in vitro CD4 degradation. Virology 223:381–386. 10.1006/viro.1996.0491 [DOI] [PubMed] [Google Scholar]
- 19.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. 1998. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1:565–574. 10.1016/S1097-2765(00)80056-8 [DOI] [PubMed] [Google Scholar]
- 20.Levesque K, Zhao YS, Cohen EA. 2003. Vpu exerts a positive effect on HIV-1 infectivity by down-modulating CD4 receptor molecules at the surface of HIV-1-producing cells. J. Biol. Chem. 278:28346–28353. 10.1074/jbc.M300327200 [DOI] [PubMed] [Google Scholar]
- 21.Bour S, Perrin C, Strebel K. 1999. Cell surface CD4 inhibits HIV-1 particle release by interfering with Vpu activity. J. Biol. Chem. 274:33800–33806. 10.1074/jbc.274.47.33800 [DOI] [PubMed] [Google Scholar]
- 22.Lama J, Mangasarian A, Trono D. 1999. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Biol. 9:622–631. 10.1016/S0960-9822(99)80284-X [DOI] [PubMed] [Google Scholar]
- 23.Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 88:2633–2644. 10.1128/JVI.03230-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552–7557. 10.1073/pnas.0802203105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee HY, Giorgi EE, Keele BF, Gaschen B, Athreya GS, Salazar-Gonzalez JF, Pham KT, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Hahn BH, Shaw GM, Korber BT, Bhattacharya T, Perelson AS. 2009. Modeling sequence evolution in acute HIV-1 infection. J. Theor. Biol. 261:341–360. 10.1016/j.jtbi.2009.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, Manigart O, Mulenga J, Anderson JA, Swanstrom R, Haynes BF, Athreya GS, Korber BT, Sharp PM, Shaw GM, Hahn BH. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 82:3952–3970. 10.1128/JVI.02660-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206:1273–1289. 10.1084/jem.20090378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, Hahn BH, Kappes JC. 2012. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J. Virol. 86:2715–2728. 10.1128/JVI.06157-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brenner BG, Roger M, Routy JP, Moisi D, Ntemgwa M, Matte C, Baril JG, Thomas R, Rouleau D, Bruneau J, Leblanc R, Legault M, Tremblay C, Charest H, Wainberg MA. 2007. High rates of forward transmission events after acute/early HIV-1 infection. J. Infect. Dis. 195:951–959. 10.1086/512088 [DOI] [PubMed] [Google Scholar]
- 30.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris SR, Little SJ, Cunningham T, Garfein RS, Richman DD, Smith DM. 2010. Evaluation of an HIV nucleic acid testing program with automated Internet and voicemail systems to deliver results. Ann. Intern. Med. 152:778–785. 10.7326/0003-4819-152-12-201006150-00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661–666. 10.1038/381661a0 [DOI] [PubMed] [Google Scholar]
- 33.Morgenstern JP, Land H. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18:3587–3596. 10.1093/nar/18.12.3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charneau P, Mirambeau G, Roux P, Paulous S, Buc H, Clavel F. 1994. HIV-1 reverse transcription. A termination step at the center of the genome. J. Mol. Biol. 241:651–662. 10.1006/jmbi.1994.1542 [DOI] [PubMed] [Google Scholar]
- 35.Klimkait T, Strebel K, Hoggan MD, Martin MA, Orenstein JM. 1990. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 64:621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schubert U, Bour S, Ferrer-Montiel AV, Montal M, Maldarell F, Strebel K. 1996. The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J. Virol. 70:809–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tokarev A, Suarez M, Kwan W, Fitzpatrick K, Singh R, Guatelli J. 2013. Stimulation of NF-kappaB activity by the HIV restriction factor BST2. J. Virol. 87:2046–2057. 10.1128/JVI.02272-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Day JR, Munk C, Guatelli JC. 2004. The membrane-proximal tyrosine-based sorting signal of human immunodeficiency virus type 1 gp41 is required for optimal viral infectivity. J. Virol. 78:1069–1079. 10.1128/JVI.78.3.1069-1079.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Day JR, Martinez LE, Sasik R, Hitchin DL, Dueck ME, Richman DD, Guatelli JC. 2006. A computer-based, image-analysis method to quantify HIV-1 infection in a single-cycle infectious center assay. J. Virol. Methods 137:125–133. 10.1016/j.jviromet.2006.06.019 [DOI] [PubMed] [Google Scholar]
- 40.Magadan JG, Bonifacino JS. 11 November 2011. Transmembrane domain determinants of CD4 downregulation by HIV-1 Vpu. J. Virol. 10.1128/JVI.05933-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skasko M, Wang Y, Tian Y, Tokarev A, Munguia J, Ruiz A, Stephens EB, Opella SJ, Guatelli J. 2012. HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J. Biol. Chem. 287:58–67. 10.1074/jbc.M111.296772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vigan R, Neil SJ. 2010. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J. Virol. 84:12958–12970. 10.1128/JVI.01699-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harms JS, Splitter GA. 1995. Interferon-gamma inhibits transgene expression driven by SV40 or CMV promoters but augments expression driven by the mammalian MHC I promoter. Hum. Gene Ther. 6:1291–1297. 10.1089/hum.1995.6.10-1291 [DOI] [PubMed] [Google Scholar]
- 44.Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. 2009. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 5:e1000574. 10.1371/journal.ppat.1000574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP, Jr, Bjorkman PJ, Wilen CB, Doms RW, O'Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH. 29 March 2013. Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. U. S. A. 10.1073/pnas.1304288110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le T, Wright EJ, Smith DM, He W, Catano G, Okulicz JF, Young JA, Clark RA, Richman DD, Little SJ, Ahuja SK. 2013. Enhanced CD4+ T-cell recovery with earlier HIV-1 antiretroviral therapy. N. Engl. J. Med. 368:218–230. 10.1056/NEJMoa1110187 [DOI] [PMC free article] [PubMed] [Google Scholar]