

ABSTRACT
HLA-B*57:01 and HLA-B*57:03, the most prevalent HLA-B*57 subtypes in Caucasian and African populations, respectively, are the HLA alleles most protective against HIV disease progression. Understanding the mechanisms underlying this immune control is of critical importance, yet they remain unclear. Unexplained differences are observed in the impact of the dominant cytotoxic T lymphocyte (CTL) response restricted by HLA-B*57:01 and HLA-B*57:03 in chronic infection on the Gag epitope KAFSPEVIPMF (KF11; Gag 162 to 172). We previously showed that the HLA-B*57:03-KF11 response is associated with a >1-log-lower viral setpoint in C clade virus infection and that this response selects escape mutants within the epitope. We first examined the relationship of KF11 responses in B clade virus-infected subjects with HLA-B*57:01 to immune control and observed that a detectable KF11 response was associated with a >1-log-higher viral load (P = 0.02). No evidence of HLA-B*57:01-KF11-associated selection pressure was identified in previous comprehensive analyses of >1,800 B clade virus-infected subjects. We then studied a B clade virus-infected cohort in Barbados, where HLA-B*57:03 is highly prevalent. In contrast to findings for B clade virus-infected subjects expressing HLA-B*57:01, we observed strong selection pressure driven by the HLA-B*57:03-KF11 response for the escape mutation S173T. This mutation reduces recognition of virus-infected cells by HLA-B*57:03-KF11 CTLs and is associated with a >1-log increase in viral load in HLA-B*57:03-positive subjects (P = 0.009). We demonstrate functional constraints imposed by HIV clade relating to the residue at Gag 173 that explain the differential clade-specific escape patterns in HLA-B*57:03 subjects. Further studies are needed to evaluate the role of the KF11 response in HLA-B*57:01-associated HIV disease protection.
IMPORTANCE HLA-B*57 is the HLA class I molecule that affords the greatest protection against disease progression in HIV infection. Understanding the key mechanism(s) underlying immunosuppression of HIV is of importance in guiding therapeutic and vaccine-related approaches to improve the levels of HIV control occurring in nature. Numerous mechanisms have been proposed to explain the HLA associations with differential HIV disease outcome, but no consensus exists. These studies focus on two subtypes of HLA-B*57 prevalent in Caucasian and African populations, HLA-B*57:01 and HLA-B*57:03, respectively. These alleles appear equally protective against HIV disease progression. The CTL epitopes presented are in many cases identical, and the dominant response in chronic infection in each case is to the Gag epitope KF11. However, there the similarity ends. This study sought to better understand the reasons for these differences and what they teach us about which immune responses contribute to immune control of HIV infection.
INTRODUCTION
HLA polymorphism has a substantial impact on HIV disease outcome (1–5), yet the principal mechanisms underlying these effects remain unresolved (5). The most protective HLA class I molecule is HLA-B*57. HLA-B*57:01 is the most prevalent subtype in Caucasian populations, apparently conferring a level of protection against HIV disease progression similar to that conferred by HLA-B*57:03, the most prevalent subtype in African populations (6). One proposal is that the HLA-B*57-mediated protection is at least in part due to the breadth of the Gag-specific CD8+ T-cell response and that HLA-associated immune control of HIV is related to the ability of the cytotoxic T lymphocyte (CTL) response to drive selection pressure on the virus, such that escape can be achieved only at significant cost to viral replicative capacity (5, 7–10). However, although HLA-B*57:01 and HLA-B*57:03 appear to present identical Gag epitopes, previous studies suggest that significant differences exist in the impact of these responses on immune control.
The dominant HIV-specific CD8+ T-cell response in each case is directed toward the Gag epitope KAFSPEVIPMF (KF11; Gag 162 to 172). Published studies with HLA-B*57:01-positive subjects have been almost exclusively conducted in the context of B clade virus infection, and these have suggested that a response to KF11 is not associated with immune control (2, 11) and that the magnitude of HLA-B*57:01-KF11 responses may even be higher in progressors. Evaluation of full-length viral sequences in 1,888 B clade virus-infected subjects failed to identify any sequence polymorphisms within the KF11 epitope or flanking it that were directly associated with HLA-B*57:01 (12) and that would have suggested strong selection pressure imposed on the virus by this response. By comparison, 20 HIV amino acid polymorphisms were identified elsewhere in the HIV proteome that were directly associated with HLA-B*57:01. In contrast, in studies undertaken with C clade virus-infected African subjects expressing HLA-B*57:03, a KF11 response is associated with a >1-log-lower viral load (13), and there is strong evidence of selection pressure within this epitope, with approximately 70% of subjects carrying mutations at Ala-163 and/or Ser-165, positions 2 and 4 in the epitope (P2 and P4) (9, 10, 14).
The escape mutants that are selected within KF11 in C clade virus-infected HLA-B*57:03-positive subjects are typically A163G and S165N. The A163G mutation is selected first, reducing CTL recognition but also significantly lowering viral replicative capacity (14). The S165N mutant is then selected, substantially restoring viral replicative capacity at the same time as entirely abrogating recognition, an ideal result for the virus (14) that has been associated with higher viral loads (15). Thus, the impact of the HLA-B*57:03-KF11 response on the virus is consistent with the mechanism of HLA-mediated immune control described above, with CTL activity forcing the selection of viral escape mutants that reduce viral replicative capacity, whereas that of the HLA-B*57:01-KF11 response is not consistent with this mechanistic model.
Initial studies proposed that the HLA-B*57:01-KF11 T-cell receptors (TCRs) are highly conserved, with a dominant or exclusively expressed Vα5/Vβ19 TCR in 60 to 100% of subjects (15, 16). These HLA-B*57:01-KF11 TCRs were more likely than the HLA-B*57:03-KF11 TCRs to recognize epitopes containing A163G and S165N; hence, these mutations would not be selected in individuals expressing HLA-B*57:01 (15, 17). However, subsequent studies of HLA-B*57:01-KF11 TCR usage have been contradictory, with the Vα5/Vβ19 TCRs identified as the dominant receptor in 0/6 and 2/10 HLA-B*57:01-positive subjects, respectively (11, 18). Thus, it appears unlikely that HLA B*57 subtype-specific effects, namely, a public TCR clonotype with high functional avidity for HLA-B*57:01-KF11 (19), fully explain the observed differences in selection of KF11 escape mutants in HLA-B*57:01 and HLA-B*57:03.
An alternative hypothesis is that escape mutations within KF11 are tolerated in the context of the C clade, but not in the B clade, Gag sequence and that this may contribute to the high frequency of escape mutations within the KF11 epitope in HLA-B*57:03-positive individuals infected with C clade virus. To address this possibility, we here compared a cohort of B clade virus-infected subjects in Barbados, in which the HLA-B*57:03-subtype predominates, with a cohort of C clade virus-infected subjects in Botswana, where the HLA-B*57:03-subtype also predominates. We identified clade-specific sequence differences that influence the dynamics of viral escape within the HLA-B*57:03-restricted KF11 epitope. These differences were confirmed in a large multicohort data set featuring 3,298 subjects (including the Barbados and Botswana cohorts), including 1,732 C clade virus-infected Africans and 1,566 B clade virus-infected North Americans.
MATERIALS AND METHODS
Study cohorts and subjects.
HIV-1 B clade virus-infected subjects expressing HLA-B*57:01 were studied from antiretroviral therapy (ART)-naive cohorts in Oxford, United Kingdom (the Thames Valley Cohort, as previously described [20]), and in Barcelona, Spain (21). Additional study cohorts for evaluation of HLA-B*57:03 in the context of B clade and C clade virus infection in Barbados and Botswana, respectively, were (i) Bridgetown, Barbados (B clade; n = 246; median age, 38 years [interquartile range, 31 to 47]; female/male ratio, 60:40; samples collected between 2008 and 2010), where study subjects were attendees at the Ladymeade Reference Clinic, and (ii) Gaborone, Botswana (C clade; n = 514; median age, 27 years [interquartile range, 23 to 32]; female/male ratio, 100:0; samples collected between 2007 and 2008), where study subjects were antenatal women from the Mma Bana Study, as previously described (8, 13, 22, 23).
Ethics approval was given by the Health Research Development Committee, Botswana Ministry of Health, by the Barbados Ministry of Health, the Hospital Germans Trias i Pujol Ethics Committee, and by the Oxford Research Ethics Committee. Subjects received voluntary testing and counseling, and written informed consent was obtained from all individuals. Viral load in chronic infection was measured using the Roche Amplicor, version 1.5, assay; CD4+ T-cell counts were measured by flow cytometry. Viral load and absolute CD4 count measurements were obtained at study entry (baseline) for all individuals. All study subjects were ART naive.
Four-digit HLA typing of the class I locus was performed from genomic DNA as previously described (24) by sequence-based typing at the American Society for Histocompatability and Immunogenetics (ASHI)-accredited HLA typing laboratory, University of Oklahoma Health Sciences Center, USA. Exons 2 and 3 of HLA class I were amplified by locus-specific PCR and then sequenced. Resolution of ambiguities was undertaken according to the ASHI committee recommendations.
Additional viral sequence analyses were performed on two previously described multicenter cohorts: (i) the International HIV Adaptation Collaborative (IHAC), consisting of 1,443 clade B Gag sequences (12), and (ii) 1,470 African clade C Gag sequences from cohorts based in Durban (8), Bloemfontein (25), and Kimberley (20), South Africa, Zambia, and the Thames Valley area of the United Kingdom (20). Where high-resolution HLA typing was unavailable, we employed a published machine learning algorithm trained on a data set of high-resolution HLA class I types from >13,000 individuals with known ethnicity to complete these data to high resolution (26).
IFN-γ ELISPOT assays.
IFN-γ enzyme-linked immunospot (ELISPOT) assays were performed as previously described (13, 27), using optimally defined epitopes and 18-mer overlapping peptides (OLP) with input cells/well ranging from 30,000 to 100,000. The number of specific spot-forming cells (SFC) was calculated by subtracting the mean number of spots in the negative-control wells from the number of spots counted in each well. The magnitude of epitope-specific responses was calculated as SFC per million cells.
Site-directed mutagenesis of NL43.
The mutation S173T (serine to threonine at Gag HXB2 position 173) was introduced by site-directed mutagenesis (QuikChange I; Stratagene, United Kingdom) into wild-type NL43 plasmid DNA, as well as NL43 containing the mutations A163G and/or S165N (14). Whole plasmid DNA p83-2 (the 5′ half of strain HIV-1NL43) was PCR amplified in a mutagenesis reaction with two overlapping primers containing the target mutation. Primers used for the mutagenesis reaction were F (5′-CCCAGAAGTAATACCCATGTTTACGGCATTATCAGAAGGAGC-3′) and R (5′-GCTCCTCTGATAATGCCGTAAACATGGGTATTACTTCTGGG-3) (the mutagenesis site is underlined).The presence of mutations was verified by DNA Gag sequencing in newly generated plasmid clones. The DNA fragment ranging from the SapI to ApaI restriction sites was then subcloned into a new p83-2 vector to avoid potential carryover of additional mutations during the mutagenesis, and the coding region sequence was verified again as previously described (28).
Virus production and replication kinetics.
Viral stocks were produced by cotransfection of the different site-directed mutant plasmids (5′ half of strain HIV-1NL43) with p83-10eGFP (3′ half of strain HIV-1NL43) into MT4 cells (29). Viral stocks were harvested and viral RNA was extracted (Qiagen, United Kingdom). The gag p24/p17 coding region was PCR amplified and sequenced to confirm the presence of the mutations in the viral RNA and the absence of any other potential polymorphisms. The 50% tissue culture infective dose (TCID50) for each viral stock was determined in MT4 cells using the Reed and Muench method (30). For replication experiments, Jurkat, MT4, and H9 T cells were infected in triplicate at a multiplicity of infection (MOI) of 0.005 in a total volume of 3 ml with wild-type or mutant HIV-1 NL43 virus and incubated at 37°C for 2 h. Pellets were washed twice with phosphate-buffered saline (PBS) and cultured at 37°C and 5% CO2. After infection, around 50,000 cells were harvested daily in order to measure infectivity by percentage of enhanced green fluorescent protein (eGFP)-positive cells by fluorescence-activated cell sorting (FACS). Replication kinetics were determined by calculating the mean viral slope using the LOGEST function (Microsoft Excel) and converted to natural logs. Variation in replication slopes was assessed using Student's t test. All statistical calculations were performed in Prism 5.0 (GraphPad).
Amplification and sequencing of proviral DNA.
Gag p17/p24 sequences (from the Barbados cohort, n = 125; from the Botswana cohort, n = 322) were generated from genomic DNA extracted from peripheral blood mononuclear cells (PBMCs) where available and amplified by nested PCR using previously published primers to obtain population sequences, as previously described (31). Sequencing was undertaken using the BigDye Ready Reaction Terminator Mix (v3.1) (Applied Biosystems, United Kingdom). Sequences were analyzed using Sequencher v4.8 (Gene Codes Corporation) and aligned by SeAl to the HXB2 B clade reference strain.
Identification of HLA-associated viral polymorphisms from proviral DNA.
HLA-associated viral polymorphisms were identified from proviral DNA using a previously described method that corrects for phylogeny, HLA linkage disequilibrium, and codon covariation (8, 32). A Q value statistic, representing the P value analogue of the false discovery rate (FDR), was computed for each association. The FDR is the expected proportion of false positives among the associations identified at a given P value threshold; for example, among associations for which the Q value is ≤0.2, we expect 20% to be false positives. The phylogenetically corrected methods rely on an inferred phylogeny. We constructed two phylogenies for this study: (i) a phylogeny consisting of clade B and C sequences from Barbados and Botswana constructed using Phyml v2.4.5, under the general time reversible (GTR) model (33), and (ii) a phylogeny consisting of 3,298 p17/p24 sequences from all cohorts described for this study. This phylogeny was too large for Phyml, so we employed a 3-stage process to infer the phylogeny. In the first stage, a combined alignment was created, then sites with >10% missing data were removed, after which sequences with missing data in >10% of remaining sites were removed (resulting in the above-noted total). In the second stage, a phylogeny was inferred separately for clade B and C alignments, using Phyml v2.4.5 under the GTR model. Finally, in the third stage, the resulting phylogenies were joined by adding a single common ancestor to the two clade trees, and the branch lengths were optimized using hyphy, under the GTR model (34).
Phylogenetically corrected odds ratio.
Identification of HLA-associated polymorphisms and assessment of differential escape between viral clades and/or closely related HLA alleles were performed as previously described (12, 32, 35). Briefly, a maximum likelihood phylogenetic tree was constructed for each gene, and a model of conditional adaptation was inferred for each observed amino acid at each codon (32). In this model, the amino acid is assumed to evolve independently along the phylogeny until it reaches the observed hosts (tree tips). In each host, the HLA-mediated selection pressure is modeled using a weighted logistic regression, in which the individual's HLA repertoire is used as a predictor and the bias is determined by the transmitted sequence (35). Because the transmitted sequence is not observed, we average over the possible transmitted sequences, and all possible phylogenetic histories, as inferred from the phylogeny. Similarly, where high-resolution HLA types are not available, we perform a weighted average over possible completions (12).
To test for differential escape between HLA-B*57:01 and B*57:03, or to test for clade-specific effects on selection, interaction variables were added to the phylogenetically corrected logistic regression model and significance was determined via a likelihood ratio test, as previously described (35).
Effect of S173T mutation on epitope recognition by KF11-specific CD8+ T cells.
CD4+ T cells were enriched from PBMCs from healthy donors expressing HLA-B*57:03 using negative selection (Dynabeads) and activated for 3 to 6 days using interleukin 2 (IL-2) (50 U/ml; Roche) and phytohemagglutinin (PHA) (3 μg/ml). KF11-specific CD8+ T cells (<98% specificity) were enriched from PBMCs from HIV-infected donors using tetramers as previously described (36). B*57:03-positive CD4+ T cells were infected with NL43GFP or NL43GFP containing the S173T mutation as described above. To test for epitope recognition, epitope-specific CD8+ T cells (<98% specificity) were cocultured with the HIV-infected CD4+ T cells in the presence of CD107a antibodies (phycoerythrin [PE]-Cy5), 10 μg/ml of brefeldin A, Golgi stop (BD), CD49d, and CD28 for 6.5 h at 37°C in a 5% CO2 incubator. Cells were stained for surface and intracellular antibodies against CD4 (allophycocyanin [APC]), CD8 (Alexa Fluor 700), MIP1B (fluorescein isothiocyanate [FITC]), p24 (PE), gamma interferon (IFN-γ) (PE-Cy7), and LIVE/DEAD marker (Pacific blue) and then immediately acquired by FACS (BD LSRII).
Nucleotide sequence accession numbers.
Sequences obtained in this study were submitted to GenBank under accession numbers FJ497801 to FJ497875, FJ497885 to FJ497899, FJ497901 to FJ497905, FJ497907 to FJ497916, and FJ497918 to FJ497950.
RESULTS
B clade HLA-B*57:01-KF11 responders have higher viral setpoints than nonresponders.
Previous studies of B clade virus-infected subjects using peptide-major histocompatibility complex (MHC) class I tetramers have suggested that a detectable HLA-B*57:01 response is more frequently observed in progressors (including those with viral loads of >90,000) than in elite controllers or long-term nonprogressors (2, 11). These studies, however, were not sufficiently powered to demonstrate a statistically significant result. We therefore started by comparing responses to KF11 in B clade virus-infected, ART-naive individuals expressing HLA-B*57:01 whose viral setpoints ranged from undetectable to 500,000 copies/ml (Fig. 1). Here the association between KF11 responders and high viral setpoint reaches statistical significance (P = 0.02, Mann-Whitney test). These findings are consistent with the earlier studies cited of B clade virus-infected subjects expressing HLA-B*57:01 and provide a result opposite that obtained in HLA-B*57:03-positive individuals infected with C clade virus (13), using the identical approach of measuring IFN-γ ELISPOT responses to KF11, where a response was associated with a >10-fold-lower viral setpoint. Equivalent studies of KF11 responses in 17 HLA-B*57:03-positive subjects infected with B clade virus similarly showed substantially lower median viral loads in KF11 responders than in nonresponders (median viral loads, 1,629 and 6,127 copies/ml, respectively), although here this difference did not reach statistical significance (P = 0.28 [data not shown]).
FIG 1.
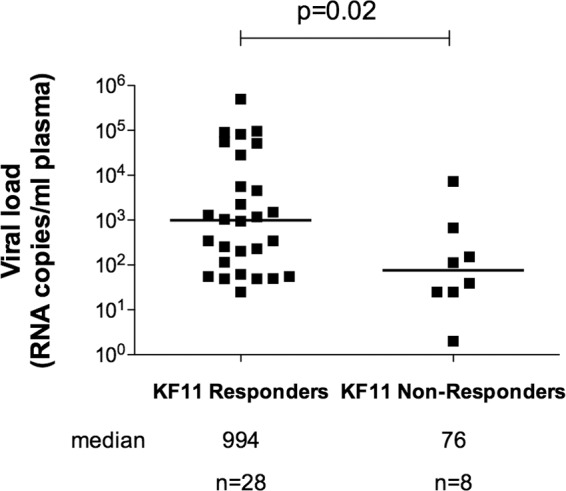
Median viral loads of KF11 responders versus nonresponders in B clade virus-infected individuals expressing HLA-B*57:01. PBMCs from B clade virus-infected, ART-naive individuals expressing HLA-B*57:01 were analyzed by IFN-γ ELISPOT assay for responses to the KF11 epitope. Viral loads of responders and nonresponders were compared. Mann-Whitney U tests were performed.
Differential escape in the B*57:03-KF11 epitope in B clade versus C clade virus infection.
In order to evaluate further the potential differences between HLA-B*57:01 and HLA-B*57:03, we investigated a B clade virus-infected, ART-naive study cohort in Barbados, where HLA-B*57:03 is highly prevalent. It has been noted in several other studies that HLA-B*57:03 is associated with immune control of HIV in B clade and C clade virus infection (1–5). Consistent with these studies, HLA-B*57:03-positive subjects in Barbados exhibited significantly lower median viral loads than HLA-B*57:03-negative subjects (median, 3,450 versus 13,350 [P = 0.015, Mann-Whitney test]) and significantly higher CD4+ counts (median, 565 versus 398 [P = 0.003, Mann-Whitney test]) (Fig. 2).
FIG 2.
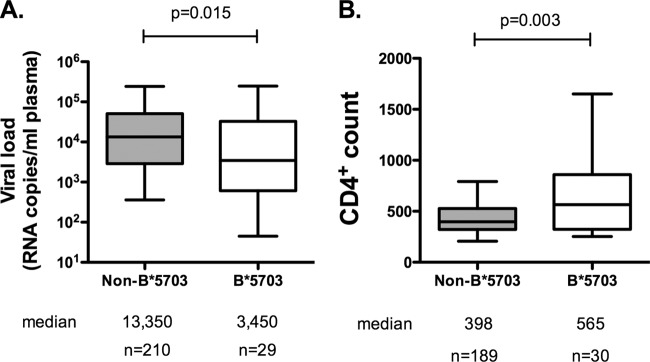
Median viral load and CD4 count of B*57:03-positive versus B*57:03-negative subjects in the Barbados cohort (B clade). B*57:03-positive subjects were compared to B*57:03-negative subjects for viral load (A) and CD4 count (B). Medians and 5th to 95th percentiles are shown. Mann-Whitney U tests were performed.
To determine the nature of any selection pressure imposed on the B clade virus through the HLA-B*57:03 KF11 response, we analyzed viral sequences in gag in the Barbados cohort in order to identify associations between HLA-B*57:03 and viral polymorphisms in the region of the KF11 epitope. This revealed that HLA-B*57:03 expression was associated with the previously described escape mutations T242N, in epitope TW10 (TSTLQEQIGW; Gag HXB2 240 to 249) (7, 37), and I147X, in the epitope ISW9 (ISPRTLNAW; Gag HXB2 147 to 155) (Table 1) (38, 39). However, the intraepitope escape mutations within KF11 (KAFSPEVIPMF; Gag HXB2 162 to 172), namely, A163G and S165N, selected in approximately 70% of C clade virus-infected HLA-B*57:03-positive subjects (3, 14), were not associated with HLA-B*57:03 in this Barbadian study cohort (Tables 1 and 2).
TABLE 1.
HLA-B*57:03-associated polymorphisms in Gag p17 and p24 (Barbados cohort)
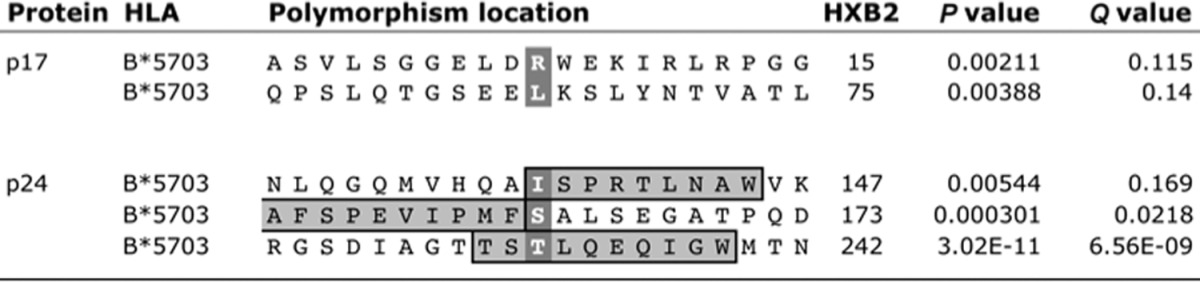
TABLE 2.
HXB2 sequences in B*5703-positive and -negative subjects (Barbados cohort)
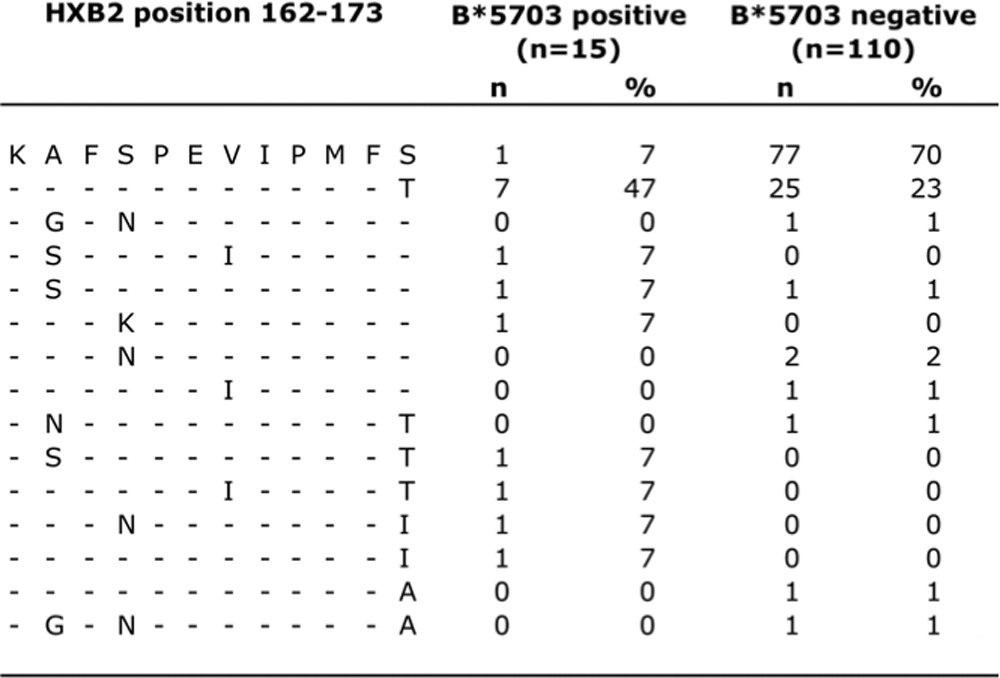
However, in the Barbados cohort, we identified an HLA-B*57:03-associated viral polymorphism located at Gag HXB2 position 173, which immediately flanks the C terminus of the KF11 epitope. This mutation has not been observed in association with HLA-B*57:03 in studies of C clade virus-infected cohorts, which in any case have threonine as the consensus residue at position 173 (14, 40, 41). The high frequency of selection of S173T by HLA-B*57:03-positive subjects (61% versus 24% in HLA-B*57:03-positive versus HLA-B*57:03-negative subjects) together with the lack of any selection of intraepitope KF11 mutations led to the hypothesis that selection of S173T in B clade virus may mitigate against further selection of KF11 escape mutations A163G and/or S165N (Table 2) (see below).
We performed a further analysis using a phylogenetically corrected method (12) to compare the impact of HLA-B*57:03 on the selection of Gag escape mutants in B clade versus C clade HIV, using data from the study cohorts in Barbados (B clade) and in Gaborone, Botswana (C clade). We found no statistical difference between odds of HLA-B*57:03-mediated escape in the two cohorts for T242N (P = 0.82) or I147L (P = 0.29). In contrast, we observed substantial clade differences for all three KF11 escape mutations: the strength of selection for A163G and S165N was significantly greater in the C clade cohort (P = 0.006 and P = 0.08, respectively), whereas Thr-173 was selected only in the B clade cohort (P = 0.0006). In fact, Gag Thr-173, the consensus in the C clade, arises at a significantly lower frequency in HLA-B*57:03-positive subjects in Botswana (P = 0.0062; discussed further below) (Table 3). These data demonstrate clade-specific differences in the impact of HLA-B*57:03 on Gag escape mutant selection, with differential effects at Gag 163, 165, and 173, within or immediately flanking the dominant KF11 epitope.
TABLE 3.
Differences in PhyloDOR of B*57:03-associated mutations in Barbados and Botswana
| X (HXB2) | Country | Phylogenetically corrected odds ratio | P value | Difference in effecta | P value |
|---|---|---|---|---|---|
| Asn-242 | Botswana | 4.9 | 3.26E−07 | N | 0.815 |
| Barbados | 11.67 | 6.15E−14 | |||
| Leu-147 | Botswana | 10.5 | 4.99E−05 | N | 0.2937 |
| Barbados | 13.26 | 4.00E−03 | |||
| Gly-163 | Botswana | 13.22 | 0.0003 | Y | 0.0067 |
| Barbados | 9.343 | 0.2006 | |||
| Asn-165 | Botswana | 4.213 | 0.0025 | N | 0.0818 |
| Barbados | 11.88 | 0.0887 | |||
| Thr-173 | Botswana | −2.798 | 0.0062 | Y | 0.0006 |
| Barbados | 13.52 | 0.0033 |
Presence of difference in B*5703's effect on X in Botswana versus Barbados. N, no; y, yes.
Impact of S173T on recognition of virus-infected target cells and on viral setpoint.
The location of the HLA-B*57:03-associated mutation immediately downstream of the KF11 epitope suggests that the S173T mutation reduces processing of the epitope. To test whether the HLA-B*57:03-associated S173T polymorphism reduces recognition of virus-infected target cells, CD4+ T cells from HLA-B*57:03+ healthy subjects were infected with NL43 HIV that was either wild type, expressing Ser-173, or engineered to express the S173T viral polymorphism. Infected cells were incubated with HLA-B*57:03-KF11-specific CD8+ T cells (>98% specific), and the level of CD8+ T-cell activation was monitored by CD107a and MIP1β expression. We observed that the S173T mutant indeed significantly reduced recognition by the KF11-specific CD8+ T cells (Fig. 3) (P = 0.0038, Student's t test). In the same assay, using CD8+ T cells specific for the HLA-B*57:03-restricted Pol-specific epitope IATESIVIW (IAW9), no difference were observed in the level of stimulation by the two viruses on the HLA-B*57:03-restricted IAW9-specific CD8+ T cells (Fig. 3). These data support the hypothesis that S173T specifically reduces presentation of the KF11 epitope by HLA-B*57:03. Furthermore, mismatched CD4+ T cells induced consistently low levels of stimulation, confirming that activation of the KF11- and IAW9-specific CD8+ T cells was HLA-B*57:03 dependent.
FIG 3.

Effect of viral mutation S173T on epitope recognition of HIV-infected cells by KF11-specific CD8+ T cells. Ex vivo CD4+ T cells from B*57:03+ and B*57:03− donors were infected with wild-type NL43 virus or NL43 virus harboring the S173T viral mutation. Infected CD4+ T cells were then cultured with KF11-specific CD8+ T cells (A) or IAW9-specific CD8+ T cells (B), and the level of CD8+ T-cell activation was monitored by expression of CD107 and Mip1β. Data from both experiments were standardized relative to percent recognition by wild-type virus (C). Experiments were performed in triplicate; means and standard deviations are shown. Student's t test was performed. *, P < 0.01; **, P < 0.001; ***, P < 0.0001. NS, not significant.
We next examined the viral setpoints and CD4 counts in HLA-B*57:03-positive subjects with and without the S173T mutation. Viral loads in HLA-B*57:03-positive subjects with the B clade wild type, serine at Gag 173, were more than 10-fold lower than in B*57:03-positive subjects with the S173T polymorphism (median viral loads, 520 and 6,905, respectively [P = 0.009, Mann-Whitney test]). Furthermore, Ser-173 was associated with a substantially higher CD4 count in HLA-B*57:03-positive subjects than Thr-173 (median CD4 counts, 787 and 375, respectively [P = 0.036, Mann-Whitney test]) (Fig. 4). However, no differences in median viral load or CD4 counts were observed in B*57:03-negative subjects with serine versus threonine at Gag 173 (median viral loads, 14,450 and 10,600, respectively [P = 0.949], and median CD4 counts, 358 and 374, respectively [P = 0.522, Mann-Whitney test]). These data together support the conclusion that HLA-B*57:03-KF11 responses drive the selection of the S173T mutation in B clade virus-infected individuals expressing HLA-B*57:03 and that this is an escape mutation in that it reduces recognition of virally infected targets. These findings are consistent with the hypothesis that this response contributes to HLA-B*57:03-associated control of HIV, since viral loads are significantly higher in those with the S173T escape mutation.
FIG 4.
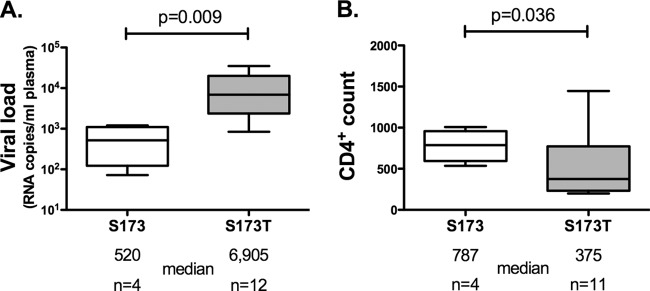
Median viral load and CD4 count of B*57:03-positive HIV-infected subjects with viral polymorphisms Ser-173 and Thr-173. Proviral DNA sequences from B*57:03-positive subjects from the Barbados cohort (B clade) were analyzed for the presence of the viral polymorphisms Ser-173 and Thr-173. Viral loads (A) and CD4 counts (B) were compared. Medians and 5th to 95th percentiles are shown. Mann-Whitney t tests were performed.
S173T with A163G and S165N significantly reduces viral replicative capacity.
The observations described above prompted the following question: if A163G and S165N are escape mutations frequently selected in HLA-B*57:03-positive subjects infected with C clade virus, why are they not selected in HLA-B*57:03-positive subjects with B clade virus infection? To assess the functional significance of the HLA-B*57:03-associated S173T mutation and the possible impact of this polymorphism on the selection of A163G and S165N, the viral polymorphisms S173T, A163G, and S165N were introduced by site-directed mutagenesis into the B clade virus backbone of NL43GFP. Infectious viral stocks were generated by transfecting MT4 T cells with the relevant DNA constructs. H9, MT4, or Jurkat T cells were then infected, and the rate of viral growth was determined by monitoring the percentage of NL43GFP-infected cells over 14 days.
Analysis of the rate of viral growth in MT4, H9, and Jurkat T cells showed, first, that the S173T polymorphism had no significant effect on viral fitness in this in vitro system in any of the three cells lines used (Fig. 5 and data not shown). We previously showed that the introduction of A163G or A163G/S165N into the NL43 backbone significantly reduced viral replicative capacity, with S165N acting as a partial compensatory mutant for A163G that also completely abrogated recognition of KF11 (14, 15). In this study, we observed that the introduction of either A163G or S165N into the NL43 backbone in combination with S173T also significantly reduces viral spread but substantially more than in the absence of S173T. Furthermore, the combination of S173T and both of the KF11 mutations, A163G and S165N, dramatically reduced viral spread even further, indicating a significant cost to viral fitness of this combination of viral mutations in a B clade virus (Fig. 5). These data together suggest that the KF11 escape mutant S173T is more commonly selected in B clade virus-infected subjects expressing HLA-B*57:03 because the cost to replicative capacity is negligible, less than that resulting from A163G or S165N. Subsequent mutations in addition to S173T result in such a substantial reduction in replicative capacity, without any apparent amelioration from S165N to reduce these fitness costs, that these arise very rarely (Table 2).
FIG 5.

Viral replication capacity of NL43GFP virus with multiple B*57:03-associated viral mutations. NL43GFP virus was engineered to contain combinations of the viral mutations Thr-173, Gly-163, and Asn-165. MT4 cells were infected and monitored for GFP-positive cells over 14 days (A). The slope of the curve was calculated from the exponential growth phase using the LOGEST function and converted to natural logs (B). Experiments were performed in triplicate; mean and standard deviations are shown. Dunnett's multiple-comparison tests were performed. *, P < 0.01; **, P < 0.001; ***, P < 0.0001.
As mentioned above, Gag Thr-173, the consensus in C clade viruses, arises at a significantly lower frequency in HLA-B*57:03-positive than in HLA-B*57:03-negative subjects in Botswana (P = 0.0062). A larger analysis of the KF11 epitope region of 1,899 C clade sequences confirmed that the presence of A163G, S165N, or both in combination was significantly associated with serine at position 173 and that this was the case for both HLA-B*57:03-positive and HLA-B*57:03-negative individuals (Fig. 6). Thus, although Thr-173 is the consensus in C clade viruses, it appears unfavorable in the context of the KF11 intraepitope escape mutations, supporting the findings in B clade viruses suggesting that this combination of mutations has a detrimental impact on viral fitness.
FIG 6.
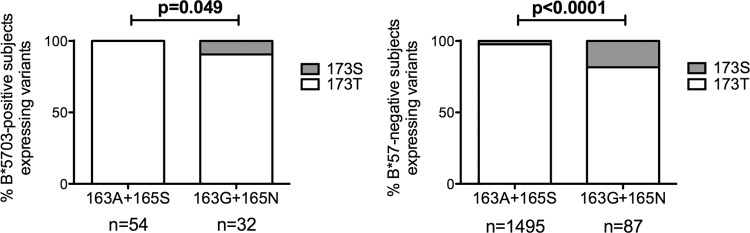
Frequency of KF11 mutations (A163G and S165N) and S173T in C clade virus Gag sequences (n = 1,899). HIV-1 p24 Gag sequences were analyzed for the presence of the KF11 mutations, A163G and S165N, in the presence of Thr-173 and Ser-173. B*57:03-positive subjects (A) and B*57-negative subjects (B) were analyzed. Fisher's exact tests were performed.
DISCUSSION
HLA-B*57:01 and HLA-B*57:03 are the two most protective HLA molecules against HIV disease progression in both B and C clade virus infection (5). These molecules differ by only two amino acids (D114N and S116Y), and the peptide binding motifs are almost indistinguishable (42, 43). In chronic infection, the dominant HIV-specific CD8+ T-cell response in subjects expressing HLA-B*57:01 or HLA-B*57:03 is to the Gag epitope KAFSPEVIPMF (KF11; Gag HXB2 162 to 172) (2, 10, 13). Studies of HLA-B*57:03-positive subjects infected with C clade virus indicate that this KF11 response makes an important contribution to immune control (3, 15) and contributes to the superiority of HLA-B*57:03 as a protective HLA class I molecule over the closely related HLA-B*57:02 and HLA-B*58:01 (10).
This study set out to investigate the following observations: first, that while the HLA-B*57:03-KF11 response is associated with significantly lower viral setpoints in C clade virus infection (13), studies of HLA-B*57:01-KF11 responses had suggested the opposite in B clade virus-infected individuals (2, 11); second, whereas the HLA-B*57:03-KF11 response frequently drives escape mutations within KF11 in C clade virus infection (A163G and S165N) (8), these are not selected in response to HLA-B*57:01 responses in B clade virus infection (12).
We first confirmed a statistically significant association between response to the KF11 epitope in B clade virus-infected subjects expressing HLA-B*57:01 and a >1-log-higher viral load. This result arose from the identical assays that were used in the studies that showed that a KF11 response was associated with a >1-log-lower viral load in C clade virus-infected subjects expressing HLA-B*57:03 (13). We next showed that HLA-B*57:03 has similar impacts in B and C clade virus infections in terms of the escape mutations selected in the Gag epitopes ISW9 and TW10 but a differential impact in the KF11 epitope. The S173T mutation selected in B clade virus infection reduces recognition of virus-infected targets and is associated with a >1-log increase in viral setpoint. This is consistent with studies of C clade virus infection (15), suggesting that this HLA-B*57:03 KF11 response contributes to HLA-B*57:03-associated immune control of HIV infection. The clade-specific differences in the selection of KF11-driven escape mutants observed in Barbados (B clade virus-infected cohort) and Botswana (C clade virus-infected cohort) were corroborated in analyses of larger data sets.
The position of S173T one residue downstream (P1′) of the KF11 epitope suggests that it may be a processing mutation, since this residue would be involved in the cleavage site of the proteasome (44). Previous studies of peptide cleavage motifs have suggested that the constitutive proteasome and immunoproteasome have a strong preference for alanine at P1′ but prefer serine over threonine; thus, the mutation S173T could affect efficient cleavage of the C-terminal end of the KF11 epitope by the proteasome (44).
We show that the HLA-B*57:03-associated S173T mutation effectively precludes further selection of the KF11 intraepitope viral mutations, A163G and S165N, since the combination of these three mutations in a B clade virus backbone results in a virus with a severely reduced replicative capacity. Indeed, the close proximity of the amino acid positions 173, 163, and 165 between helix 1 and helix 2 of the Gag p24 structure suggests that structural constraints prevent selection of A163G and S165N if S173T has already been selected. Previous work has shown that, using a B clade virus backbone, and in the presence of S173, the mutation A163G reduces replicative capacity but that the further addition of S165N, as observed in vivo, partially restores replicative capacity (14, 45). This fits with the order of selection of A163G and S165N, with S165N apparently always arising subsequent to A163G (14). However, in B clade virus infection, it appears that the selection of S173T prevents the selection of further mutants within the epitope because the fitness cost is too high. S173T appears to be the preferred choice of viral escape from the KF11-specific response, since it has minimal effect on viral fitness.
Our inference from the data described above and from previous studies (23) is that the HLA-B*57:03-KF11 response contributes to immune control of B and C clade HIV infection. The reduced recognition of S173T virus-infected cells by KF11-specific CTLs together with the lack of cost to viral replicative capacity resulting from S173T is consistent with the observation that viral loads are higher and CD4 counts lower in B clade virus-infected subjects expressing HLA-B*57:03.
In view of the substantial reduction in viral replicative capacity resulting from the A163G/S165N/S173T combination in B clade virus infection, it is perhaps surprising to observe the selection of A163G/S165N at high frequency in HLA-B*57:03-positive subjects infected with C clade virus in which the vast majority of sequences carry Thr at Gag 173. It may be inferred from this that the presence of consensus Thr-173 in the context of C clade virus Gag does not have the same prohibitive effect on viral fitness, as it does not prevent the selection of A163G and S165N. Nevertheless, in C clade virus infection, both in HLA-B*57:03-positive and in HLA-B*57:03-negative individuals, A163G/S165N are significantly associated with Ser at Gag 173 (Fig. 6), as opposed to the consensus Thr at this position, suggesting that the combination of A163G/S165N/S173T is not favored in either B or C clade virus infection.
Gag 173 has been well studied in relation to HLA-B*27, another protective HLA molecule, because of the S173A mutation that accompanies the R264K escape variant within the dominant HLA-B*27-restricted epitope KRWIILGLNK (KK10) (46–48). It is noteworthy that in C clade virus infection, R264K escape in HLA-B*27-positive subjects is typically accompanied by compensatory mutations not at Gag 173 but at Gag 165 (Brener et al., unpublished data). These data underline the tight constraints on amino acid substitutions in the capsid protein, the interdependence of residues at certain key positions in the structure, including Gag 163, Gag 165, and Gag 173, and therefore the impact that clade can have on the escape options for the virus.
These data help to explain why HLA-B*57:03 is not associated with the “usual” KF11 intraepitope mutations A163G/S165N in clade B, but they do not explain why HLA-B*57:01 is not associated either with the S173T flanking mutation or with any KF11 intraepitope mutations. Previous studies have suggested that TCR usage for the HLA-B*57:01-KF11 response allows recognition of the KF11 variants (15), but these initial TCR studies indicating conservation of a “public” HLA-B*57:01-KF11 TCR have not been borne out by subsequent studies (11, 18). One possible explanation is that the potency of the HLA-B*57:01-KF11 response is so great that a moderate reduction in processed epitope would not affect killing sufficiently to be selected; however, preliminary data suggest that the HLA-B*57:03 response is, if anything, the more potent. Further studies with a large number of KF11-specific clones would be needed to establish whether clear-cut differences between the responses restricted by HLA-B*57:03 and HLA-B*57:01 exist in terms of potency and the relevance of this to viral escape patterns. A recent study comparing the impact of individual HLA class I molecules on immune control (viral load < 2,000 copies/ml) versus noncontrol (viral load > 10,000 copies/ml) of B clade virus infection showed the identical odds ratio for protection via HLA-B*57:01 in a European American cohort and for that via HLA-B*57:03 in an African American cohort (6).
These studies therefore provide an explanation for the distinct clade-specific selection of escape mutants by the HLA-B*57:03-KF11 response but do not resolve the question of why the HLA-B*57:01-KF11 response does not select escape mutants. Insufficient studies have been undertaken with C clade virus-infected subjects who express HLA-B*57:01 to be certain of whether this response selects no escape mutants in C clade as well as in B clade virus infection. The absence of the KF11 response in elite controllers with HLA-B*57:01 does not necessarily mean that these responses have not contributed to immune control in these subjects, since it is possible that the period of detectability may be transient. It is clear that many responses that are undetectable in elite controllers can become detectable after peptide stimulation (49). However, if the KF11-specific CTL response contributes to immune control of HIV in HLA-B*57:01-positive subjects in B clade virus infection, it would be unique in failing to select escape mutants in the process and the mechanism would be invaluable for directing successful vaccine targets.
ACKNOWLEDGMENTS
P.J.R.G. is supported by the Wellcome Trust and the NIH (RO1 AI046995). Z.L.B. is the recipient of a New Investigator Award from the Canadian Institutes of Health Research and a Scholar Award from the Michael Smith Foundation for Health Research.
Footnotes
Published ahead of print 5 February 2014
REFERENCES
- 1.Kaslow RA, Carrington M, Apple R, Park L, Munoz A, Saah AJ, Goedert JJ, Winkler C, O'Brien SJ, Rinaldo C, Detels R, Blattner W, Phair J, Erlich H, Mann DL. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2:405–411. 10.1038/nm0496-405 [DOI] [PubMed] [Google Scholar]
- 2.Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, Hallahan CW, Selig SM, Schwartz D, Sullivan J, Connors M. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. U. S. A. 97:2709–2714. 10.1073/pnas.050567397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L, Zimbwa P, Moore S, Allen T, Brander C, Addo MM, Altfeld M, James I, Mallal S, Bunce M, Barber LD, Szinger J, Day C, Klenerman P, Mullins J, Korber B, Coovadia HM, Walker BD, Goulder PJ. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432:769–775. 10.1038/nature03113 [DOI] [PubMed] [Google Scholar]
- 4.Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Gao X, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O'Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DM, Vine S, Addo MM, Allen TM, et al. 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557. 10.1126/science.1195271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goulder PJ, Walker BD. 2012. HIV and HLA class I: an evolving relationship. Immunity 37:426–440. 10.1016/j.immuni.2012.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Apps R, Qi Y, Carlson JM, Chen H, Gao X, Thomas R, Yuki Y, Del Prete GQ, Goulder P, Brumme ZL, Brumme CJ, John M, Mallal S, Nelson G, Bosch R, Heckerman D, Stein JL, Soderberg KA, Moody MA, Denny TN, Zeng X, Fang J, Moffett A, Lifson JD, Goedert JJ, Buchbinder S, Kirk GD, Fellay J, McLaren P, Deeks SG, Pereyra F, Walker B, Michael NL, Weintrob A, Wolinsky S, Liao W, Carrington M. 2013. Influence of HLA-C expression level on HIV control. Science 340:87–91. 10.1126/science.1232685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282–289. 10.1038/nm992 [DOI] [PubMed] [Google Scholar]
- 8.Matthews PC, Prendergast A, Leslie A, Crawford H, Payne R, Rousseau C, Rolland M, Honeyborne I, Carlson J, Kadie C, Brander C, Bishop K, Mlotshwa N, Mullins JI, Coovadia H, Ndung'u T, Walker BD, Heckerman D, Goulder PJ. 2008. Central role of reverting mutations in HLA associations with human immunodeficiency virus set point. J. Virol. 82:8548–8559. 10.1128/JVI.00580-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crawford H, Lumm W, Leslie A, Schaefer M, Boeras D, Prado JG, Tang J, Farmer P, Ndung'u T, Lakhi S, Gilmour J, Goepfert P, Walker BD, Kaslow R, Mulenga J, Allen S, Goulder PJ, Hunter E. 2009. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J. Exp. Med. 206:909–921. 10.1084/jem.20081984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kloverpris HN, Stryhn A, Harndahl M, van der Stok M, Payne RP, Matthews PC, Chen F, Riddell L, Walker BD, Ndung'u T, Buus S, Goulder P. 2012. HLA-B*57 micropolymorphism shapes HLA allele-specific epitope immunogenicity, selection pressure, and HIV immune control. J. Virol. 86:919–929. 10.1128/JVI.06150-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendoza D, Royce C, Ruff LE, Ambrozak DR, Quigley MF, Dang T, Venturi V, Price DA, Douek DC, Migueles SA, Connors M. 2012. HLA B*5701-positive long-term nonprogressors/elite controllers are not distinguished from progressors by the clonal composition of HIV-specific CD8+ T cells. J. Virol. 86:4014–4018. 10.1128/JVI.06982-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, Chui CK, Cotton LA, Knapp DJ, Riddler SA, Haubrich R, Nelson G, Pfeifer N, Deziel CE, Heckerman D, Apps R, Carrington M, Mallal S, Harrigan PR, John M, Brumme ZL. 2012. Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J. Virol. 86:13202–13216. 10.1128/JVI.01998-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53. 10.1038/nm1520 [DOI] [PubMed] [Google Scholar]
- 14.Crawford H, Prado JG, Leslie A, Hue S, Honeyborne I, Reddy S, van der Stok M, Mncube Z, Brander C, Rousseau C, Mullins JI, Kaslow R, Goepfert P, Allen S, Hunter E, Mulenga J, Kiepiela P, Walker BD, Goulder PJ. 2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 81:8346–8351. 10.1128/JVI.00465-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu XG, Lichterfeld M, Chetty S, Williams KL, Mui SK, Miura T, Frahm N, Feeney ME, Tang Y, Pereyra F, Labute MX, Pfafferott K, Leslie A, Crawford H, Allgaier R, Hildebrand W, Kaslow R, Brander C, Allen TM, Rosenberg ES, Kiepiela P, Vajpayee M, Goepfert PA, Altfeld M, Goulder PJ, Walker BD. 2007. Mutually exclusive T-cell receptor induction and differential susceptibility to human immunodeficiency virus type 1 mutational escape associated with a two-amino-acid difference between HLA class I subtypes. J. Virol. 81:1619–1631. 10.1128/JVI.01580-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillespie GM, Stewart-Jones G, Rengasamy J, Beattie T, Bwayo JJ, Plummer FA, Kaul R, McMichael AJ, Easterbrook P, Dong T, Jones EY, Rowland-Jones SL. 2006. Strong TCR conservation and altered T cell cross-reactivity characterize a B*57-restricted immune response in HIV-1 infection. J. Immunol. 177:3893–3902 [DOI] [PubMed] [Google Scholar]
- 17.Stewart-Jones GB, Simpson P, van der Merwe PA, Easterbrook P, McMichael AJ, Rowland-Jones SL, Jones EY, Gillespie GM. 2012. Structural features underlying T-cell receptor sensitivity to concealed MHC class I micropolymorphisms. Proc. Natl. Acad. Sci. U. S. A. 109:E3483–E3492. 10.1073/pnas.1207896109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simons BC, Vancompernolle SE, Smith RM, Wei J, Barnett L, Lorey SL, Meyer-Olson D, Kalams SA. 2008. Despite biased TRBV gene usage against a dominant HLA B57-restricted epitope, TCR diversity can provide recognition of circulating epitope variants. J. Immunol. 181:5137–5146 [DOI] [PubMed] [Google Scholar]
- 19.Berger CT, Frahm N, Price DA, Mothe B, Ghebremichael M, Hartman KL, Henry LM, Brenchley JM, Ruff LE, Venturi V, Pereyra F, Sidney J, Sette A, Douek DC, Walker BD, Kaufmann DE, Brander C. 2011. High-functional-avidity cytotoxic T lymphocyte responses to HLA-B-restricted Gag-derived epitopes associated with relative HIV control. J. Virol. 85:9334–9345. 10.1128/JVI.00460-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthews PC, Adland E, Listgarten J, Leslie A, Mkhwanazi N, Carlson JM, Harndahl M, Stryhn A, Payne RP, Ogwu A, Huang KH, Frater J, Paioni P, Kloverpris H, Jooste P, Goedhals D, van Vuuren C, Steyn D, Riddell L, Chen F, Luzzi G, Balachandran T, Ndung'u T, Buus S, Carrington M, Shapiro R, Heckerman D, Goulder PJ. 2011. HLA-A*7401-mediated control of HIV viremia is independent of its linkage disequilibrium with HLA-B*5703. J. Immunol. 186:5675–5686. 10.4049/jimmunol.1003711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mothe B, Llano A, Ibarrondo J, Zamarreno J, Schiaulini M, Miranda C, Ruiz-Riol M, Berger CT, Herrero MJ, Palou E, Plana M, Rolland M, Khatri A, Heckerman D, Pereyra F, Walker BD, Weiner D, Paredes R, Clotet B, Felber BK, Pavlakis GN, Mullins JI, Brander C. 2012. CTL responses of high functional avidity and broad variant cross-reactivity are associated with HIV control. PLoS One 7:e29717. 10.1371/journal.pone.0029717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shapiro RL, Hughes MD, Ogwu A, Kitch D, Lockman S, Moffat C, Makhema J, Moyo S, Thior I, McIntosh K, van Widenfelt E, Leidner J, Powis K, Asmelash A, Tumbare E, Zwerski S, Sharma U, Handelsman E, Mburu K, Jayeoba O, Moko E, Souda S, Lubega E, Akhtar M, Wester C, Tuomola R, Snowden W, Martinez-Tristani M, Mazhani L, Essex M. 2010. Antiretroviral regimens in pregnancy and breast-feeding in Botswana. N. Engl. J. Med. 362:2282–2294. 10.1056/NEJMoa0907736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright JK, Brumme ZL, Carlson JM, Heckerman D, Kadie CM, Brumme CJ, Wang B, Losina E, Miura T, Chonco F, van der Stok M, Mncube Z, Bishop K, Goulder PJ, Walker BD, Brockman MA, Ndung'u T. 2010. Gag-protease-mediated replication capacity in HIV-1 subtype C chronic infection: associations with HLA type and clinical parameters. J. Virol. 84:10820–10831. 10.1128/JVI.01084-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bunce M, O'Neill CM, Barnardo MC, Krausa P, Browning MJ, Morris PJ, Welsh KI. 1995. Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (PCR-SSP). Tissue Antigens 46:355–367. 10.1111/j.1399-0039.1995.tb03127.x [DOI] [PubMed] [Google Scholar]
- 25.Huang KH, Goedhals D, Fryer H, van Vuuren C, Katzourakis A, De Oliveira T, Brown H, Cassol S, Seebregts C, McLean A, Klenerman P, Phillips R, Frater J. 2009. Prevalence of HIV type-1 drug-associated mutations in pre-therapy patients in the Free State, South Africa. Antivir. Ther. 14:975–984. 10.3851/IMP1416 [DOI] [PubMed] [Google Scholar]
- 26.Listgarten J, Brumme Z, Kadie C, Xiaojiang G, Walker B, Carrington M, Goulder P, Heckerman D. 2008. Statistical resolution of ambiguous HLA typing data. PLoS Comput. Biol. 4:e1000016. 10.1371/journal.pcbi.1000016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altfeld MA, Trocha A, Eldridge RL, Rosenberg ES, Phillips MN, Addo MM, Sekaly RP, Kalams SA, Burchett SA, McIntosh K, Walker BD, Goulder PJ. 2000. Identification of dominant optimal HLA-B60- and HLA-B61-restricted cytotoxic T-lymphocyte (CTL) epitopes: rapid characterization of CTL responses by enzyme-linked immunospot assay. J. Virol. 74:8541–8549. 10.1128/JVI.74.18.8541-8549.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prado JG, Honeyborne I, Brierley I, Puertas MC, Martinez-Picado J, Goulder PJ. 2009. Functional consequences of human immunodeficiency virus escape from an HLA-B*13-restricted CD8+ T-cell epitope in p1 Gag protein. J. Virol. 83:1018–1025. 10.1128/JVI.01882-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber J, Weberova J, Carobene M, Mirza M, Martinez-Picado J, Kazanjian P, Quinones-Mateu ME. 2006. Use of a novel assay based on intact recombinant viruses expressing green (EGFP) or red (DsRed2) fluorescent proteins to examine the contribution of pol and env genes to overall HIV-1 replicative fitness. J. Virol. Methods 136:102–117. 10.1016/j.jviromet.2006.04.004 [DOI] [PubMed] [Google Scholar]
- 30.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 31.Leslie A, Kavanagh D, Honeyborne I, Pfafferott K, Edwards C, Pillay T, Hilton L, Thobakgale C, Ramduth D, Draenert R, Le Gall S, Luzzi G, Edwards A, Brander C, Sewell AK, Moore S, Mullins J, Moore C, Mallal S, Bhardwaj N, Yusim K, Phillips R, Klenerman P, Korber B, Kiepiela P, Walker B, Goulder P. 2005. Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA. J. Exp. Med. 201:891–902. 10.1084/jem.20041455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carlson JM, Brumme ZL, Rousseau CM, Brumme CJ, Matthews P, Kadie C, Mullins JI, Walker BD, Harrigan PR, Goulder PJ, Heckerman D. 2008. Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLoS Comput. Biol. 4:e1000225. 10.1371/journal.pcbi.1000225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704. 10.1080/10635150390235520 [DOI] [PubMed] [Google Scholar]
- 34.Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. 10.1093/bioinformatics/bti079 [DOI] [PubMed] [Google Scholar]
- 35.Carlson JM, Listgarten J, Pfeifer N, Tan V, Kadie C, Walker BD, Ndung'u T, Shapiro R, Frater J, Brumme ZL, Goulder PJ, Heckerman D. 2012. Widespread impact of HLA restriction on immune control and escape pathways of HIV-1. J. Virol. 86:5230–5243. 10.1128/JVI.06728-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Payne RP, Kløverpris H, Sacha JB, Brumme Z, Brumme C, Buus S, Sims S, Hickling S, Riddell L, Chen F, Luzzi G, Edwards A, Phillips R, Prado JG, Goulder PJR. 2010. Efficacious early antiviral activity of HIV Gag- and Pol-specific HLA-B*2705-restricted CD8+ T cells. J. Virol. 84:10543–10557. 10.1128/JVI.00793-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feeney ME, Tang Y, Roosevelt KA, Leslie AJ, McIntosh K, Karthas N, Walker BD, Goulder PJ. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 78:8927–8930. 10.1128/JVI.78.16.8927-8930.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Draenert R, Le Gall S, Pfafferott KJ, Leslie AJ, Chetty P, Brander C, Holmes EC, Chang SC, Feeney ME, Addo MM, Ruiz L, Ramduth D, Jeena P, Altfeld M, Thomas S, Tang Y, Verrill CL, Dixon C, Prado JG, Kiepiela P, Martinez-Picado J, Walker BD, Goulder PJ. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J. Exp. Med. 199:905–915. 10.1084/jem.20031982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boutwell CL, Rowley CF, Essex M. 2009. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J. Virol. 83:2460–2468. 10.1128/JVI.01970-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chopera DR, Woodman Z, Mlisana K, Mlotshwa M, Martin DP, Seoighe C, Treurnicht F, de Rosa DA, Hide W, Karim SA, Gray CM, Williamson C. 2008. Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog. 4:e1000033. 10.1371/journal.ppat.1000033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang KH, Goedhals D, Carlson JM, Brockman MA, Mishra S, Brumme ZL, Hickling S, Tang CS, Miura T, Seebregts C, Heckerman D, Ndung'u T, Walker B, Klenerman P, Steyn D, Goulder P, Phillips R, van Vuuren C, Frater J. 2011. Progression to AIDS in South Africa is associated with both reverting and compensatory viral mutations. PLoS One 6:e19018. 10.1371/journal.pone.0019018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barber LD, Percival L, Arnett KL, Gumperz JE, Chen L, Parham P. 1997. Polymorphism in the alpha 1 helix of the HLA-B heavy chain can have an overriding influence on peptide-binding specificity. J. Immunol. 158:1660–1669 [PubMed] [Google Scholar]
- 43.Ostrov DA, Grant BJ, Pompeu YA, Sidney J, Harndahl M, Southwood S, Oseroff C, Lu S, Jakoncic J, de Oliveira CA, Yang L, Mei H, Shi L, Shabanowitz J, English AM, Wriston A, Lucas A, Phillips E, Mallal S, Grey HM, Sette A, Hunt DF, Buus S, Peters B. 2012. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc. Natl. Acad. Sci. U. S. A. 109:9959–9964. 10.1073/pnas.1207934109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toes RE, Nussbaum AK, Degermann S, Schirle M, Emmerich NP, Kraft M, Laplace C, Zwinderman A, Dick TP, Muller J, Schonfisch B, Schmid C, Fehling HJ, Stevanovic S, Rammensee HG, Schild H. 2001. Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. J. Exp. Med. 194:1–12. 10.1084/jem.194.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boutwell CL, Carlson JM, Lin TH, Seese A, Power KA, Peng J, Tang Y, Brumme ZL, Heckerman D, Schneidewind A, Allen TM. 2013. Frequent and variable cytotoxic-T-lymphocyte escape-associated fitness costs in the human immunodeficiency virus type 1 subtype B Gag proteins. J. Virol. 87:3952–3965. 10.1128/JVI.03233-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, Conlon C, Workman C, Shaunak S, Olson K, Goulder P, Brander C, Ogg G, Sullivan JS, Dyer W, Jones I, McMichael AJ, Rowland-Jones S, Phillips RE. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375–386. 10.1084/jem.193.3.375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, Rinaldo CR, Craggs SL, Allgaier RL, Power KA, Kuntzen T, Tung CS, LaBute MX, Mueller SM, Harrer T, McMichael AJ, Goulder PJ, Aiken C, Brander C, Kelleher AD, Allen TM. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 81:12382–12393. 10.1128/JVI.01543-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneidewind A, Brockman MA, Sidney J, Wang YE, Chen H, Suscovich TJ, Li B, Adam RI, Allgaier RL, Mothe BR, Kuntzen T, Oniangue-Ndza C, Trocha A, Yu XG, Brander C, Sette A, Walker BD, Allen TM. 2008. Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. J. Virol. 82:5594–5605. 10.1128/JVI.02356-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ndhlovu ZM, Proudfoot J, Cesa K, Alvino DM, McMullen A, Vine S, Stampouloglou E, Piechocka-Trocha A, Walker BD, Pereyra F. 2012. Elite controllers with low to absent effector CD8+ T cell responses maintain highly functional, broadly directed central memory responses. J. Virol. 86:6959–6969. 10.1128/JVI.00531-12 [DOI] [PMC free article] [PubMed] [Google Scholar]