

ABSTRACT
There are currently no U.S. Food and Drug Administration (FDA)-approved vaccines or therapeutics to prevent or treat Argentine hemorrhagic fever (AHF). The causative agent of AHF is Junin virus (JUNV); a New World arenavirus classified as a National Institute of Allergy and Infectious Disease/Centers for Disease Control and Prevention category A priority pathogen. The PTAP late (L) domain motif within JUNV Z protein facilitates virion egress and transmission by recruiting host Tsg101 and other ESCRT complex proteins to promote scission of the virus particle from the plasma membrane. Here, we describe a novel compound (compound 0013) that blocks the JUNV Z-Tsg101 interaction and inhibits budding of virus-like particles (VLPs) driven by ectopic expression of the Z protein and live-attenuated JUNV Candid-1 strain in cell culture. Since inhibition of the PTAP-Tsg101 interaction inhibits JUNV egress, compound 0013 serves as a prototype therapeutic that could reduce virus dissemination and disease progression in infected individuals. Moreover, since PTAP l-domain-mediated Tsg101 recruitment is utilized by other RNA virus pathogens (e.g., Ebola virus and HIV-1), PTAP inhibitors such as compound 0013 have the potential to function as potent broad-spectrum, host-oriented antiviral drugs.
IMPORTANCE There are currently no FDA-approved vaccines or therapeutics to prevent or treat Argentine hemorrhagic fever (AHF). The causative agent of AHF is Junin virus (JUNV); a New World arenavirus classified as an NIAID/CDC category A priority pathogen. Here, we describe a prototype therapeutic that blocks budding of JUNV and has the potential to function as a broad-spectrum antiviral drug.
INTRODUCTION
Junin virus (JUNV) is a New World arenavirus endemic in Argentina that causes severe Argentine hemorrhagic fever and significant mortality in humans (1). The virus is present in excreta from infected rodents and is typically spread to humans via aerosolization (1). JUNV is important because it is a biosafety level 4 pathogen with potential use as a bioterror agent, for which there are no U.S. Food and Drug Administration (FDA)-approved vaccines or therapeutics. JUNV is an enveloped, bisegmented negative-sense RNA virus that encodes four proteins: surface glycoprotein precursor (GPC), RNA polymerase (L), nucleoprotein (NP), and small RING finger matrix protein (Z). The Z protein of arenaviruses plays a key role in promoting virus egress and spread and is capable of budding independently from mammalian cells in the form of virus-like particles (VLPs) (2–6). Efficient separation of arenavirus Z VLPs from the plasma membrane is promoted by viral L domains within the Z protein (for a review, see reference 7). L domains function by recruiting host proteins that facilitate efficient virus-cell separation (“pinching-off”) (7). JUNV Z protein contains a single PTAP type L domain which interacts with host Tsg101, a component of the endosomal sorting complexes required for transport (ESCRT) machinery (5–7). L-domain-mediated recruitment of host proteins by virus represents a potential target for the development of novel antiviral inhibitors to disrupt virus egress from infected cells to reduce virus dissemination and disease progression (3, 6, 8–13).
Based upon the known nuclear magnetic resonance structure of the Tsg101-PTAP interaction site, we conducted an in silico screen for competitive binding inhibitors. We identified several compounds, and the most potent (compound 0013) blocks both Z VLP and live JUNV egress at nanomolar concentrations. Moreover, compound 0013 specifically blocks PTAP-Tsg101 interactions. Intriguingly, since L-domain-containing matrix proteins are generally required for efficient virus-cell separation of emerging RNA viruses (filoviruses, arenaviruses, henipaviruses, rhabdoviruses, paramyxoviruses, and retroviruses), we speculate that inhibitors of this virus-host interaction could represent broad-spectrum antiviral drugs against a range of enveloped RNA viruses.
MATERIALS AND METHODS
Cell lines, virus, and antibodies.
VeroE6 and HEK293T cells were grown in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum. The Candid-1 vaccine strain of JUNV was kindly provided by Robert B. Tesh (University of Texas Medical Branch, Galveston, TX) via Susan R. Ross (University of Pennsylvania, Philadelphia, PA) and was propagated in VeroE6 cells as described previously (14). The antisera used included a monoclonal anti-Flag antiserum (Sigma-Aldrich, St. Louis, MO), a monoclonal anti-JUNV-GP antiserum QC03-BF11 (BEI Resources, Manassas, VA), a monoclonal anti-Tsg101 antiserum (Clone C2; Santa Cruz Biotechnology, Santa Cruz, CA), a rabbit polyclonal anti-EBOV VP40 antiserum described previously (15), and a monoclonal anti-actin antiserum (Sigma-Aldrich).
Compound 4816-0013.
Compound 4816-0013 (>95% purity) was purchased from ChemDiv (San Diego, CA), and a 100 mM stock solution was prepared in dimethyl sulfoxide (DMSO) and stored at −20°C.
Plasmids.
A JUNV Z-WT expression plasmid was kindly provided by Cybele Garcia (Ciudad Universitaria, Buenos Aires, Argentina). A Flag-tagged copy of full-length JUNV Z-WT was PCR amplified and cloned into the pCAGGS vector. A PTAP deletion mutant of JUNV Z was generated in pCAGGS by standard PCR and cloning techniques. JUNV-Z-CYFP was constructed by joining the C-terminal half of EYFP to the C terminus of JUNV-Z in pCAGGS using standard PCR and cloning techniques. Plasmids expressing EBOV VP40, MARV VP40, and NYFP-Tsg101 have been described previously (12).
BiMC assay.
Bimolecular complementation (BiMC) in HEK293T cells was performed as described previously (12). Briefly, HEK293T cells were cotransfected for 3 h, and the cells were then treated with the indicated concentrations of compound 0013 for an additional 24 h. The cells were examined by fluorescence microscopy.
Virus infection and focus-forming assay.
For the experiment described in Fig. 5, VeroE6 cells were infected with JUNV (Candid-1) at a multiplicity of infection of 0.02 for 42 h at 37°C. Supernatants were removed, and the cells were washed three times with 1× phosphate-buffered saline (PBS). The cells were then treated with DMSO alone or the indicated concentration of compound 0013 for an additional 30 h. Virions were harvested from the supernatant samples as described below for VLPs and then used to infect fresh monolayers of VeroE6 cells for 48 h for the quantification of foci using fluorescence microscopy (Fig. 5).
FIG 5.

The antibudding activity of compound 0013 is reversible. Western blot of JUNV Z in VLPs from HEK293T cells treated with DMSO alone (lane 1) or 400 nM compound 0013 (lane 2) for 20 h. After 20 h, the cells were washed once with PBS and then overlaid with fresh medium lacking the drug for an additional 16 (lane 3) or 24 h (lane 4). JUNV Z VLP budding was restored to levels comparable to control (VLPs; compare lane 1 to lanes 3 and 4), indicating that the antibudding activity of compound 0013 is reversible.
VLP budding assay and Western blotting.
VLP budding assays and Western blotting were performed as described previously (15–17).
Immunoprecipitation/Western blot assay.
VeroE6 cells in a 100-mm dish were transfected with 2.0 μg of JUNV-Z-WT using Lipofectamine reagent (Invitrogen) and the protocol of the supplier. DMSO or the indicated concentrations of compound 0013 were added at 6 h posttransfection. Cells were harvested and lysed in nondenaturing buffer (20 mM Tris-HCl [pH 8.0], 137 mM NaCl, 1.0% Nonidet P-40, 2.0 mM EDTA, 10% glycerol) at 30 h posttransfection. The cell lysates were clarified for 10 min and then incubated with anti-Flag or preimmune control (Invitrogen) antisera overnight at 4°C. Protein G-agarose beads (Invitrogen) were added to the samples, which were then incubated with agitation for 7 h at 4°C. The beads were washed three times in nondenaturing lysis buffer, suspended in loading buffer with boiling, and then fractionated by SDS-PAGE. Endogenous Tsg101 was detected in samples by Western blotting with anti-Tsg101 antiserum.
MTT cell viability assay.
MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assays on HEK293T and VeroE6 cells were performed as recommended by the supplier (Amresco, Solon, OH). Briefly, cells were seeded onto collagen-coated 96-well plates at a density of 5 × 104 cells/well. HEK293T cells were transfected with empty vector (pCAGGS) and incubated under appropriate conditions with the indicated concentrations of compound 0013. MTT (5 mg/ml in PBS) was added to the media for 3 h, and the absorbance at 590 nm was measured and recorded.
RESULTS
Identification of host-oriented inhibitors targeting L domains.
We and others have established that efficient budding of hemorrhagic syndrome viruses (e.g., arenaviruses and filoviruses) is dependent upon subversion of host proteins (e.g., Tsg101) by viral L domains (e.g., PTAP L domain) (Fig. 1A). These viral L-domain–host interactions represent viable targets for broad-spectrum, host-oriented antiviral therapeutics. Since disruption of virus budding would reduce virus dissemination, small-molecule inhibitors that block these virus-host interactions should effectively block virion egress and thus disease progression and transmission (Fig. 1A). We used an in silico screen to identify candidate small molecule compounds that would interfere with the interaction between the viral PTAP l-domain and the UEV domain of host Tsg101 (Fig. 1B) (18, 19). Briefly, the in silico screen involved computational docking with AutoDock 4.0, energy minimization using CHARMM with the MMFF force field, and ranking with Accelrys LigScore2 of more than two million drug-like compounds from the ZINC database. After experimentally testing the top-10-scoring compounds, we identified compound 5539-0062 (Fig. 1C), which achieved low micromolar inhibition of EBOV VP40 budding, as described previously (12). Next, we identified and tested numerous structural analogs of compound 5539-0062, including compound 0013 (Fig. 1C). Based upon VLP budding assays and studies with live virus, we report that compound 0013 is a more potent PTAP-specific inhibitor and antiviral compound than 5539-0062.
FIG 1.
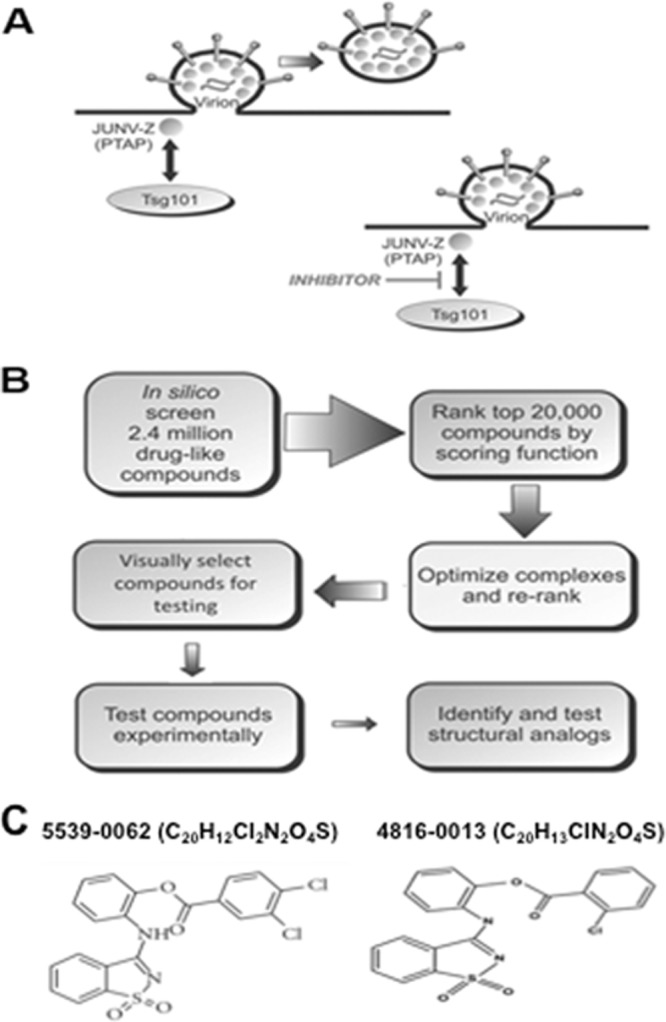
Mechanism of budding inhibition and in silico strategy. (A) Schematic diagram of JUNV budding at the plasma membrane showing the PTAP-dependent interaction between JUNV Z and host Tsg101 in the absence or presence of a PTAP budding inhibitor. (B) Flow chart of the in silico screening strategy used to identify PTAP-Tsg101 inhibitors. (C) Chemical formula and structure of PTAP-Tsg101 inhibitors 5539-0062 and 4816-0013.
Compound 0013 blocks JUNV Z-Tsg101 interactions.
We sought to determine whether compound 0013 specifically blocks the JUNV Z-Tsg101 interaction first by using a BiMC approach to evaluate interactions between NYFP-Tsg101 and JUNV-Z-CYFP. Briefly, the mechanistic basis of the BiMC assay involves the splitting of yellow fluorescent protein (YFP) into N-terminal (NYFP) and C-terminal (CYFP) halves that are then joined to two proteins of interest (Tsg101 and JUNV Z) (Fig. 2A). If Tsg101 and JUNV Z interact when coexpressed in mammalian cells, then the N- and C-terminal halves of YFP will come together to reconstitute a functionally fluorescent YFP signal (12). Expression and efficient budding of JUNV-Z-CYFP was confirmed (Fig. 2B). NYFP-Tsg101 and JUNV-Z-CYFP were coexpressed in human HEK293T cells for 3 h, and then the cells were treated with the indicated concentrations of compound 0013 (Fig. 2C). At 24 h after transfection, the cells were examined for YFP fluorescence (Fig. 2C). YFP-positive cells were abundant in the presence of vehicle alone, indicating that this assay is capable of detecting JUNV Z interactions with host Tsg101. Importantly, the extent of complementation (YFP-positive cells) decreased with a dose dependence on compound 0013 concentration (Fig. 2C), indicating that compound 0013 inhibits PTAP-mediated interactions between JUNV Z and host Tsg101. Indeed, quantification of fluorescent cells revealed a significant difference between samples in the absence or presence of the indicated concentrations of compound 0013 (Fig. 2D).
FIG 2.

BiMC assay showing that compound 0013 inhibits JUNV Z/Tsg101 interaction. (A) Schematic diagrams of YFP(Venus), NYFP-Tsg101, and JUNV-Z-CYFP (Flag-tagged) fusion proteins showing amino acid and Flag epitope positions. (B) Western blots with anti-Flag antiserum to detect JUNV-Z-CYFP fusion protein expressed in cells and VLPs (lane 1) compared to cells receiving vector (pCAGGS) alone (lane 2). (C) BiMC assay and representative confocal images of HEK293T cells coexpressing NYFP-Tsg101 and JUNV-Z-CYFP fusion proteins in the absence (DMSO alone) or in the presence of the indicated concentrations of compound 0013. Bar, 200 μm. (D) Quantification and statistical analysis of positively fluorescing cells in the BiMC assay. *, P < 0.5; **, P < 0.01 (determined by a one-way analysis of variance [ANOVA] and Student t test).
To further verify that compound 0013 inhibits JUNV Z-Tsg101 interactions, we used an immunoprecipitation/Western blot approach (Fig. 3). VeroE6 cells were transfected with vector alone or a plasmid expressing FLAG-tagged JUNV Z in the absence (DMSO alone) or presence of the indicated concentration of compound 0013 (Fig. 3). Proteins were extracted from cells under nondenaturing conditions at 30 h posttransfection, and cell proteins were immunoprecipitated with either preimmune serum (control antibody [Ab]) or anti-FLAG antiserum to pulldown Flag-tagged JUNV Z. Levels of endogenous Tsg101 in the precipitates were quantified by Western analysis using anti-Tsg101 antiserum (Fig. 3, top panel). Tsg101 was detected in JUNV Z precipitates but not in precipitates using preimmune serum (Fig. 3, compare lanes 1 and 2). Moreover, the levels of endogenous Tsg101 in JUNV Z precipitates diminished as the concentration of compound 0013 increased (Fig. 3, top panel, compare lanes 3, 5, and 7). The expression levels of endogenous Tsg101, actin, and JUNV Z are shown in the bottom three panels (Fig. 3). Together with results from BiMC assays, these data indicate that compound 0013 specifically blocks PTAP-mediated recruitment of host Tsg101 by JUNV Z.
FIG 3.
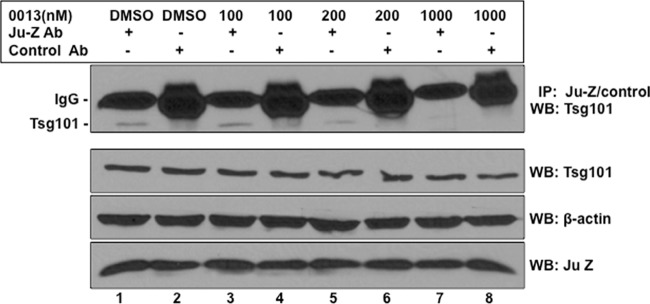
Immunoprecipitation/Western blot assay showing that compound 0013 inhibits JUNV Z/Tsg101 interaction. Cell proteins from JUNV Z (Flag-tagged)-transfected VeroE6 cells treated with DMSO alone or the indicated concentrations of compound 0013 were first immunoprecipitated with either mouse preimmune antiserum (control Ab; lanes 2, 4, 6, and 8) or mouse anti-Flag antiserum (lanes 1, 3, 5, and 7), and endogenous Tsg101 was detected in the precipitated samples by Western analysis with anti-Tsg101 antiserum (top panel). Western blot controls (bottom panels) for the expression of endogenous Tsg101, β-actin, and Flag-tagged JUNV Z are shown.
Compound 0013 inhibits JUNV Z VLP egress.
We next used a VLP budding assay to determine whether the ability of compound 0013 to disrupt the physical PTAP-Tsg101 interaction produces functional inhibition of JUNV Z VLP budding. First, we confirmed that efficient JUNV Z VLP egress from human HEK293T cells is dependent on the sole PTAP L-domain motif at the JUNV Z C terminus (Fig. 4A). Indeed, deletion of the PTAP motif from JUNV Z resulted in a marked decrease in JUNV Z VLP egress compared to JUNV Z-WT (Fig. 4A). Human HEK293T cells were then treated with vehicle (DMSO) alone or with increasing concentrations of compound 0013, and then budding of JUNV Z-WT VLPs was examined (Fig. 4B). Compound 0013 produced a dose-dependent inhibition of VLP formation and inhibited JUNV Z VLP egress by >90% at concentrations >100 nM (Fig. 4B, compare lane 2 to lanes 4 and 5). Compound 0013 had no effect on expression levels of cellular JUNV Z and actin at all of the drug concentrations tested (Fig. 4B).
FIG 4.
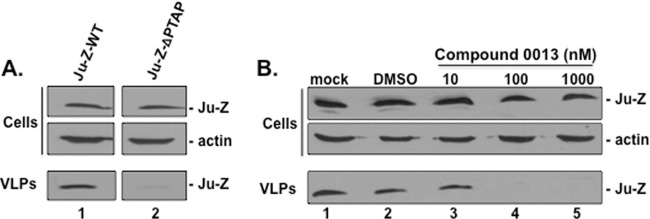
Compound 0013 inhibits egress of JUNV Z VLPs. (A) HEK293T cells were transfected with either JUNV Z-WT or JUNV-Z-ΔPTAP plasmids, and cell extracts and VLPs were harvested at 24 h posttransfection. JUNV Z-WT and JUNV-Z-ΔPTAP were detected by Western blotting in cells and VLPs. A Western blot control for β-actin in cells is shown. (B) HEK293T cells transfected with JUNV Z-WT were mock treated (lane 1), treated with DMSO alone (lane 2), or treated with the indicated concentrations of compound 0013 (lanes 3 to 5). Cells and VLPs were harvested at 24 h posttransfection, and JUNV Z was detected by Western blotting. A Western blot control for β-actin in cells is shown.
Interestingly, the inhibitory effect of compound 0013 on JUNV Z VLP egress was reversible (Fig. 5). Briefly, human HEK293T cells were treated with vehicle (DMSO) alone or with 400 nM concentration of compound 0013 (Fig. 5). As expected, budding of JUNV Z-WT VLPs was markedly reduced in the presence of compound 0013 (Fig. 5, compare lanes 1 and 2); however, budding of JUNV Z VLPs was restored to control levels 16 or 24 h after the compound was removed from the media (Fig. 5, lanes 3 and 4).
We next tested the PTAP specificity of compound 0013 by assessing its ability to inhibit production of EBOV or MARV VP40 VLPs (Fig. 6). We observed that compound 0013 blocked PTAP-dependent egress of EBOV VP40 VLPs by >50% at a concentration of 40 nM (Fig. 6A) but, as expected, it had little to no effect on MARV VP40 VLP production, which occurs in a PTAP-independent manner (Fig. 6B) (20). Lastly, we found that compound 0013 also blocked HIV-1 Gag VLP formation by >50% at similar concentrations, as expected from the fact that HIV-1 Gag VLP formation is PTAP dependent (18, 19, 21–26). Taken together, these data demonstrated that compound 0013 inhibits the PTAP-dependent egress of arenavirus, filovirus, and retrovirus VLPs at nanomolar concentrations.
FIG 6.
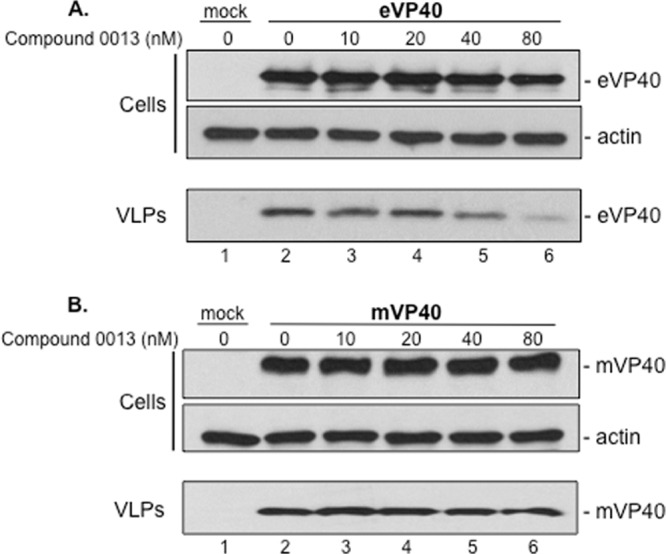
Effect of compound 0013 on filovirus VLP egress. VLP budding assay and Western blots for EBOV VP40 (eVP40) (A) and MARV VP40 (mVP40) (B) in the absence (DMSO alone, lane 2) or in the presence of the indicated concentrations of compound 0013. (A) The levels of eVP40 and actin in cells were equivalent in all samples (cells, lanes 2 to 6), whereas the level of eVP40 in VLPs was reduced in a dose-dependent manner in the presence of increasing amounts of compound 0013 (VLPs, lanes 2 to 6). (B) The levels of mVP40 and actin in cells were equivalent in all samples (cells, lanes 2 to 6), and the level of mVP40 in VLPs remained essentially unchanged in the presence of increasing amounts of compound 0013 (VLPs, lanes 2 to 6).
Compound 0013 inhibits egress of live JUNV virus (Candid-1 strain).
We next sought to determine whether compound 0013 could inhibit egress of live JUNV in a cell culture model of infection. Briefly, VeroE6 cells were infected with live-attenuated Candid-1 vaccine strain of JUNV in the presence of 10, 100, or 1,000 nM compound 0013. Cell extracts and supernatants were harvested, and virions purified from supernatant samples were then used to infect fresh VeroE6 cell monolayers for 48 h in the absence of drug (Fig. 7). JUNV foci were detected using indirect immunofluorescence with anti-GP antiserum to obtain a relative measure of the number of virions produced from infected cells in the absence or presence of compound 0013 (Fig. 7A). Qualitative and quantitative analyses revealed that the number of viral foci were decreased significantly in a dose-dependent manner by compound 0013 (Fig. 7A and B). These results agree well with those described above for JUNV Z VLP budding and suggest that compound 0013 reduces egress of live JUNV from infected VeroE6 cells. Importantly, compound 0013 did not have a detrimental effect on the synthesis of either JUNV GP or cellular actin in infected VeroE6 cells at all of the concentrations tested (Fig. 7C).
FIG 7.

Compound 0013 inhibits egress of live JUNV (Candid-1 strain) from VeroE6 cells. (A) Representative confocal microscopy images of viral foci (red) visualized with anti-JUNV GP antiserum 48 h postinfection. Bar, 1,000 μm. An enlarged viral focus is shown for clarity. Bar, 100 μm. (B) Bar graph showing quantification of JUNV foci observed in the absence or presence of compound 0013 and represented as a percent relative to the DMSO control (average from three independent experiments). An asterisk (*) indicates a P value of <0.01, as determined by a Student t test and one-way ANOVA. (C) Western blots to detect levels of JUNV GP and β-actin in JUNV (Candid-1)-infected VeroE6 cells in the absence (DMSO alone) or presence of the indicated concentrations of compound 0013.
Host Tsg101 is packaged into JUNV virions.
Since the JUNV Z PTAP-Tsg101 interaction is important for budding, we sought to determine whether endogenous Tsg101 was packaged into budding virions. VeroE6 cells were infected with JUNV (Candid-1) for 72 h, and both cell extracts and supernatants were harvested (Fig. 8). As expected, JUNV GP was detected only in virus-infected cells, whereas endogenous Tsg101 was detected in both JUNV-infected and mock-infected cells (Fig. 8, cells, top panels). Both JUNV GP and endogenous Tsg101 were detected in supernatant samples from JUNV-infected cells but not in supernatant samples from mock-infected cells (Fig. 8, media, bottom panels). These results indicate that endogenous Tsg101 is packaged into Junin virions released into the supernatant.
FIG 8.
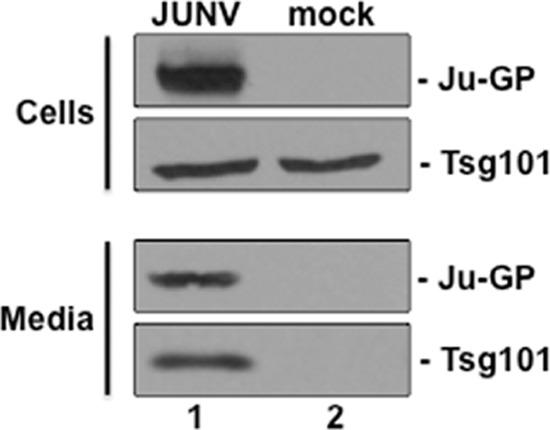
Host Tsg101 is packaged into JUNV virions. Vero cells were infected with JUNV (Candid-1) for 72 h, and both cell extracts and supernatants (media) were harvested. As expected, JUNV GP was detected only in virus-infected cells (cells, lane 1), whereas endogenous Tsg101 was detected in both JUNV-infected and mock-infected cells (cells, lanes 1 and 2). Both JUNV GP and endogenous Tsg101 were detected in supernatant (media) samples from JUNV-infected cells (media, lane 1) but not in supernatant samples from mock-infected cells (media, lane 2).
Compound 0013 is not cytotoxic.
Next, we wanted to determine whether compound 0013 was cytotoxic to HEK293T cells under conditions used for transfection experiments and to VeroE6 cells under conditions used for JUNV infection experiments. MTT cell viability assays were performed on HEK293T (Fig. 9A) or VeroE6 (Fig. 9B) cells that were treated with DMSO alone or the indicated concentrations of compound 0013 under conditions that mimicked those used for VLP (Fig. 9A) or JUNV (Fig. 9B) budding experiments. We found that compound 0013 was not cytotoxic to HEK293T or VeroE6 cells at the concentrations tested, nor did compound 0013 inhibit the expression of endogenous Tsg101 in HEK293T cells, as determined by Western blotting (Fig. 9C).
FIG 9.
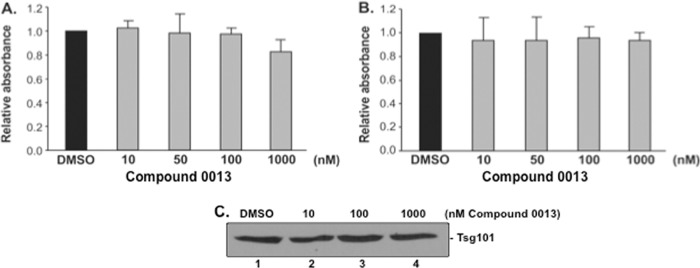
MTT cell viability assays. MTT cell viability assays were performed on HEK293T cells (A) and VeroE6 cells (B) that were treated with DMSO or the indicated concentrations of compound 0013 under conditions that mimicked those used for JUNV Z VLP transfection (A) or Junin virus (Candid-1) infection (B). (C) Western blot showing that levels of endogenous Tsg101 remain unchanged in HEK293T cells treated with DMSO or the indicated concentrations of compound 0013.
DISCUSSION
JUNV is classified as category A priority pathogen and potential agent of bioterrorism. JUNV is endemic in regions of Argentina and West Africa and is the cause of severe episodes of hemorrhagic fever with high mortality rates. The search for effective therapeutics to treat arenavirus infections, particularly those caused by Junin and Lassa viruses, has been an active area of research for many years (8, 27–42). More recent efforts in the field of antiviral therapeutics for emerging RNA viruses have focused on identifying host factors or virus-host interactions, rather than solely viral proteins, as targets for novel antivirals (6, 8–13, 39, 40, 43, 44). Host Tsg101 represents one such target (3, 12, 19, 22, 45). As described here and elsewhere, budding of arenaviruses is critically dependent on the subversion of host proteins, such as Tsg101, and that PTAP late (L) budding domains expressed arenaviruses are critical for interaction with these host proteins (3, 6). Not surprisingly, we and others have detected host proteins such as Tsg101 packaged into mature virions, likely as a result of their function in promoting virus-cell separation (Fig. 8) (46–48). Our rationale for targeting this virus-host interaction is that disruption of virus budding would prevent virus dissemination, and the identification of small molecule inhibitors that block these critical virus-host interactions should effectively block disease progression and transmission. Moreover, targeting a virus-host interaction rather than a viral target should greatly diminish or eliminate the high frequency of drug-resistant mutations. Importantly, L-domain-containing matrix proteins are required for efficient virus-cell separation of numerous emerging RNA viruses, including arenaviruses, filoviruses, henipaviruses, rhabdoviruses, paramyxoviruses, and retroviruses. We predict that targeting this interaction domain will serve as the basis for the development of powerful broad-spectrum antiviral drugs that will serve as a countermeasure against multiple biodefense pathogens. Indeed, our prediction is supported by findings presented here showing that compound 0013 inhibits egress of both JUNV and EBOV particles. It should be noted that compound 0013 had no virucidal effect and did not inhibit the entry or transcription of JUNV Candid-1 (J. Lu and R. N. Harty, unpublished data).
We describe here the identification and validation of a host-oriented PTAP-specific budding inhibitor of JUNV Z VLPs and infectious virions. Both immunoprecipitation/Western blot and BiMC assays demonstrated that compound 0013 blocked the interaction between JUNV Z and host Tsg101. The ability of compound 0013 to physically disrupt this virus-host interaction correlated with its functional inhibition of JUNV Z mediated the budding of both VLPs and live-attenuated JUNV (Candid-1). The 50% effective concentration (EC50) of compound 0013 for both the VLP and the live virus budding assays is in the nanomolar range, and its antibudding activity appears to be reversible. Importantly, compound 0013 likely possesses broad-spectrum antibudding activity, since we found that it blocks PTAP-mediated egress of both EBOV VP40 (Fig. 6) and HIV-1 Gag VLPs. The specificity of compound 0013 for inhibition of PTAP-mediated egress was demonstrated in the finding that PTAP-independent egress of MARV VP40 VLPs was not inhibited (Fig. 6).
In conclusion, compound 0013 possesses a number of properties desirable for further development as an antiviral therapeutic, including (i) many sites and functional group handles for modification to further improve its potency, selectivity, metabolic profile, and physiochemical properties, (ii) low cytotoxicity in cell culture, (iii) a known mechanism of action, and (iv) a current EC50 of <1.0 μM in vitro. Our findings with compound 0013 are encouraging, since its antiviral properties displayed improved potency over those of previously identified PTAP inhibitor 5539-0062 (12). Future studies include using medicinal chemistry approaches to improve both potency and drug-like properties, as well as proof-of-concept efficacy studies in an animal model of infection.
ACKNOWLEDGMENTS
We thank Cybele Garcia, Susan Ross, and Robert B. Tesh for generously providing reagents. We thank Su Chiang, Jay Wrobel, and Allen Reitz for medicinal chemistry support. We also thank Deborah Argento for assistance in the preparation of the manuscript and figures, Leslie King for critical reading of the manuscript, Claude Krummenacher for helpful suggestions, and members of the Harty lab for fruitful discussions and suggestions.
This study was supported in part by National Institutes of Health/National Institute of Allergy and Infectious Diseases grants AI102104 and U54-AI057168 to R.N.H.
Footnotes
Published ahead of print 12 February 2014
REFERENCES
- 1.Grant A, Seregin A, Huang C, Kolokoltsova O, Brasier A, Peters C, Paessler S. 2012. Junin virus pathogenesis and virus replication. Viruses 4:2317–2339. 10.3390/v4102317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fehling SK, Lennartz F, Strecker T. 2012. Multifunctional nature of the arenavirus RING finger protein Z. Viruses 4:2973–3011. 10.3390/v4112973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perez M, Craven RC, de la Torre JC. 2003. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc. Natl. Acad. Sci. U. S. A. 100:12978–12983. 10.1073/pnas.2133782100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perez M, Greenwald DL, de la Torre JC. 2004. Myristoylation of the RING finger Z protein is essential for arenavirus budding. J. Virol. 78:11443–11448. 10.1128/JVI.78.20.11443-11448.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urata S, Yasuda J. 2012. Molecular mechanism of arenavirus assembly and budding. Viruses 4:2049–2079. 10.3390/v4102049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urata S, de la Torre JC. 2011. Arenavirus budding. Adv. Virol. 2011:180326. 10.1155/2011/180326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen BJ, Lamb RA. 2008. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 372:221–232. 10.1016/j.virol.2007.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linero FN, Sepulveda CS, Giovannoni F, Castilla V, Garcia CC, Scolaro LA, Damonte EB. 2012. Host cell factors as antiviral targets in arenavirus infection. Viruses 4:1569–1591. 10.3390/v4091569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harty RN. 2009. No exit: targeting the budding process to inhibit filovirus replication. Antiviral Res. 81:189–197. 10.1016/j.antiviral.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harty RN, Schmitt AP, Bouamr F, Lopez CB, Krummenacher C. 2011. Virus budding/host interactions. Adv. Virol. 2011:963192. 10.1155/2011/963192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Harty RN. 2010. Viral and host proteins that modulate filovirus budding. Future Virol. 5:481–491. 10.2217/fvl.10.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Lee MS, Olson MA, Harty RN. 2011. Bimolecular complementation to visualize filovirus VP40-host complexes in live mammalian cells: toward the identification of budding inhibitors. Adv. Virol. 2011:341816. 10.1155/2011/341816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunz S, de la Torre JC. 2005. Novel antiviral strategies to combat human arenavirus infections. Curr. Mol. Med. 5:735–751. 10.2174/156652405774962353 [DOI] [PubMed] [Google Scholar]
- 14.Cuevas CD, Lavanya M, Wang E, Ross SR. 2011. Junin virus infects mouse cells and induces innate immune responses. J. Virol. 85:11058–11068. 10.1128/JVI.05304-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarthy SE, Johnson RF, Zhang YA, Sunyer JO, Harty RN. 2007. Role for amino acids 212KLR214 of Ebola virus VP40 in assembly and budding. J. Virol. 81:11452–11460. 10.1128/JVI.00853-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Licata JM, Simpson-Holley M, Wright NT, Han Z, Paragas J, Harty RN. 2003. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77:1812–1819. 10.1128/JVI.77.3.1812-1819.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Cocka L, Okumura A, Zhang YA, Sunyer JO, Harty RN. 2010. Conserved motifs within Ebola and Marburg virus VP40 proteins are important for stability, localization, and subsequent budding of virus-like particles. J. Virol. 84:2294–2303. 10.1128/JVI.02034-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pornillos O, Alam SL, Davis DR, Sundquist WI. 2002. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat. Struct. Biol. 9:812–817. 10.1038/nsb856 [DOI] [PubMed] [Google Scholar]
- 19.Pornillos O, Alam SL, Rich RL, Myszka DG, Davis DR, Sundquist WI. 2002. Structure and functional interactions of the Tsg101 UEV domain. EMBO J. 21:2397–2406. 10.1093/emboj/21.10.2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolnik O, Kolesnikova L, Stevermann L, Becker S. 2010. Tsg101 is recruited by a late domain of the nucleocapsid protein to support budding of Marburg virus-like particles. J. Virol. 84:7847–7856. 10.1128/JVI.00476-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55–65. 10.1016/S0092-8674(01)00506-2 [DOI] [PubMed] [Google Scholar]
- 22.Freed EO. 2003. The HIV-TSG101 interface: recent advances in a budding field. Trends Microbiol. 11:56–59. 10.1016/S0966-842X(02)00013-6 [DOI] [PubMed] [Google Scholar]
- 23.Martin-Serrano J, Zang T, Bieniasz PD. 2003. Role of ESCRT-I in retroviral budding. J. Virol. 77:4794–4804. 10.1128/JVI.77.8.4794-4804.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yasuda J. 2005. HIV budding and Tsg101. Uirusu 55:281–286 (In Japanese.). 10.2222/jsv.55.281 [DOI] [PubMed] [Google Scholar]
- 25.Palencia A, Martinez JC, Mateo PL, Luque I, Camara-Artigas A. 2006. Structure of human TSG101 UEV domain. Acta Crystallogr. Sect. D Biol. Crystallogr. 62:458–464. 10.1107/S0907444906005221 [DOI] [PubMed] [Google Scholar]
- 26.Usami Y, Popov S, Popova E, Inoue M, Weissenhorn W, H GG. 2009. The ESCRT pathway and HIV-1 budding. Biochem. Soc. Trans. 37:181–184. 10.1042/BST0370181 [DOI] [PubMed] [Google Scholar]
- 27.Barradas JS, Errea MI, D'Accorso NB, Sepulveda CS, Talarico LB, Damonte EB. 2008. Synthesis and antiviral activity of azoles obtained from carbohydrates. Carbohydr. Res. 343:2468–2474. 10.1016/j.carres.2008.06.028 [DOI] [PubMed] [Google Scholar]
- 28.Candurra NA, Maskin L, Damonte EB. 1996. Inhibition of arenavirus multiplication in vitro by phenotiazines. Antiviral Res. 31:149–158. 10.1016/0166-3542(96)06956-2 [DOI] [PubMed] [Google Scholar]
- 29.Damonte EB, Coto CE. 2002. Treatment of arenavirus infections: from basic studies to the challenge of antiviral therapy. Adv. Virus Res. 58:125–155. 10.1016/S0065-3527(02)58004-0 [DOI] [PubMed] [Google Scholar]
- 30.Garcia CC, Candurra NA, Damonte EB. 2000. Antiviral and virucidal activities against arenaviruses of zinc-finger active compounds. Antiviral Chem. Chemother. 11:231–237 [DOI] [PubMed] [Google Scholar]
- 31.Garcia CC, Candurra NA, Damonte EB. 2002. Mode of inactivation of arenaviruses by disulfide-based compounds. Antiviral Res. 55:437–446. 10.1016/S0166-3542(02)00076-1 [DOI] [PubMed] [Google Scholar]
- 32.Garcia CC, Djavani M, Topisirovic I, Borden KL, Salvato MS, Damonte EB. 2006. Arenavirus Z protein as an antiviral target: virus inactivation and protein oligomerization by zinc finger-reactive compounds. J. Gen. Virol. 87:1217–1228. 10.1099/vir.0.81667-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia CC, Topisirovic I, Djavani M, Borden KL, Damonte EB, Salvato MS. 2010. An antiviral disulfide compound blocks interaction between arenavirus Z protein and cellular promyelocytic leukemia protein. Biochem. Biophys. Res. Commun. 393:625–630. 10.1016/j.bbrc.2010.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sepulveda CS, Garcia CC, Damonte EB. 2010. Inhibition of arenavirus infection by thiuram and aromatic disulfides. Antiviral Res. 87:329–337. 10.1016/j.antiviral.2010.06.005 [DOI] [PubMed] [Google Scholar]
- 35.Sepulveda CS, Garcia CC, Fascio ML, D'Accorso NB, Docampo Palacios ML, Pellon RF, Damonte EB. 2012. Inhibition of Junin virus RNA synthesis by an antiviral acridone derivative. Antiviral Res. 93:16–22. 10.1016/j.antiviral.2011.10.007 [DOI] [PubMed] [Google Scholar]
- 36.Madrid PB, Chopra S, Manger ID, Gilfillan L, Keepers TR, Shurtleff AC, Green CE, Iyer LV, Dilks HH, Davey RA, Kolokoltsov AA, Carrion R, Jr, Patterson JL, Bavari S, Panchal RG, Warren TK, Wells JB, Moos WH, Burke RL, Tanga MJ. 2013. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS One 8:e60579. 10.1371/journal.pone.0060579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLay L, Ansari A, Liang Y, Ly H. 2013. Targeting virulence mechanisms for the prevention and therapy of arenaviral hemorrhagic fever. Antiviral Res. 97:81–92. 10.1016/j.antiviral.2012.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vela E. 2012. Animal models, prophylaxis, and therapeutics for arenavirus infections. Viruses 4:1802–1829. 10.3390/v4091802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radoshitzky SR, Kuhn JH, de Kok-Mercado F, Jahrling PB, Bavari S. 2012. Drug discovery technologies and strategies for Machupo virus and other New World arenaviruses. Expert Opin. Drug Discov. 7:613–632. 10.1517/17460441.2012.687719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panchal RG, Reid SP, Tran JP, Bergeron AA, Wells J, Kota KP, Aman J, Bavari S. 2012. Identification of an antioxidant small-molecule with broad-spectrum antiviral activity. Antiviral Res. 93:23–29. 10.1016/j.antiviral.2011.10.011 [DOI] [PubMed] [Google Scholar]
- 41.Gowen BB, Bray M. 2011. Progress in the experimental therapy of severe arenaviral infections. Future Microbiol. 6:1429–1441. 10.2217/fmb.11.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emonet SE, Urata S, de la Torre JC. 2011. Arenavirus reverse genetics: new approaches for the investigation of arenavirus biology and development of antiviral strategies. Virology 411:416–425. 10.1016/j.virol.2011.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolokoltsov AA, Adhikary S, Garver J, Johnson L, Davey RA, Vela EM. 2012. Inhibition of Lassa virus and Ebola virus infection in host cells treated with the kinase inhibitors genistein and tyrphostin. Arch. Virol. 157:121–127. 10.1007/s00705-011-1115-8 [DOI] [PubMed] [Google Scholar]
- 44.Radoshitzky SR, Dong L, Chi X, Clester JC, Retterer C, Spurgers K, Kuhn JH, Sandwick S, Ruthel G, Kota K, Boltz D, Warren T, Kranzusch PJ, Whelan SP, Bavari S. 2010. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J. Virol. 84:10569–10580. 10.1128/JVI.00103-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H, Liu X, Li Z, Zhan P, De Clercq E. 2010. TSG101: a novel anti-HIV-1 drug target. Curr. Med. Chem. 17:750–758. 10.2174/092986710790514444 [DOI] [PubMed] [Google Scholar]
- 46.Irie T, Licata JM, Harty RN. 2005. Functional characterization of Ebola virus L-domains using VSV recombinants. Virology 336:291–298. 10.1016/j.virol.2005.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luttge BG, Freed EO. 2010. FIV Gag: virus assembly and host-cell interactions. Vet. Immunol. Immunopathol. 134:3–13. 10.1016/j.vetimm.2009.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gottwein E, Bodem J, Muller B, Schmechel A, Zentgraf H, Krausslich HG. 2003. The Mason-Pfizer monkey virus PPPY and PSAP motifs both contribute to virus release. J. Virol. 77:9474–9485. 10.1128/JVI.77.17.9474-9485.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]