

ABSTRACT
Vif is a lentiviral accessory protein that regulates viral infectivity in part by inducing proteasomal degradation of APOBEC3G (A3G). Recently, CBFβ was found to facilitate Vif-dependent degradation of A3G. However, the exact role of CBFβ remains unclear. Several studies noted reduced Vif expression in CBFβ knockdown cells while others saw no significant impact of CBFβ on Vif stability. Here, we confirmed that CBFβ increases Vif steady-state levels. CBFβ affected expression of neither viral Gag nor Vpu protein, indicating that CBFβ regulates Vif expression posttranscriptionally. Kinetic studies revealed effects of CBFβ on both metabolic stability and the rate of Vif biosynthesis. These effects were dependent on the ability of CBFβ to interact with Vif. Importantly, at comparable Vif levels, CBFβ further enhanced A3G degradation, suggesting that CBFβ facilitates A3G degradation by increasing the levels of Vif and by independently augmenting the ability of Vif to target A3G for degradation. CBFβ also increased expression of RUNX1 by enhancing RUNX1 biosynthesis. Unlike Vif, however, CBFβ had no detectable effect on RUNX1 metabolic stability. We propose that CBFβ acts as a chaperone to stabilize Vif during and after synthesis and to facilitate interaction of Vif with cellular cofactors required for the efficient degradation of A3G.
IMPORTANCE In this study, we show that CBFβ has a profound effect on the expression of the HIV-1 infectivity factor Vif and the cellular transcription factor RUNX1, two proteins that physically interact with CBFβ. Kinetic studies revealed that CBFβ increases the rate of Vif and RUNX1 biosynthesis at the level of translation. Mutants of Vif unable to physically interact with CBFβ were nonresponsive to CBFβ. Our data suggest that CBFβ exerts a chaperone-like activity (i) to minimize the production of defective ribosomal products (DRiPs) by binding to nascent protein to prevent premature termination and (ii) to stabilize mature protein conformation to ensure proper function of Vif and RUNX1. Thus, we identified a novel mechanism of protein regulation that affects both viral and cellular factors and thus has broad implications beyond the immediate HIV field.
INTRODUCTION
The human immunodeficiency virus type 1 (HIV-1) accessory protein Vif plays an important role in regulating virus infectivity. The primary function of Vif is to counteract the antiviral activity of several human cytidine deaminases, including APOBEC3G (A3G), APOBEC3F, APOBEC3DE, and APOBEC3H (reviewed in references 1 and 2). A3G and the other members of the APOBEC3 (A3) family are cytidine deaminases that when packaged into HIV-1 virions can severely inhibit viral infectivity by editing the viral genome during cDNA synthesis. Deamination of deoxycytidine on single-stranded DNA produces deoxyuridine that leads to guanidine-to-adenosine changes upon second-strand synthesis (reviewed in reference 3). The presence of deoxyuridine in single-stranded DNA may also lead to activation of a cellular DNA excision repair machinery that in the case of single-stranded viral cDNA can cause fragmentation and degradation of the viral genome (4).
The antiviral activity of A3G is dependent on its encapsidation into the core of retroviral particles (reviewed in reference 1). This is accomplished by the association of A3G with viral or cellular RNAs (5–15). Vif inhibits encapsidation of A3G in part by inducing its ubiquitination and subsequent degradation by the cellular proteasome machinery (16–22). The mechanism of Vif-induced A3G degradation has been extensively studied (for a review, see reference 23). Vif does not have enzymatic or catalytic activity but functions as a molecular adapter that connects A3G to a Cullin 5 (Cul5)-dependent E3 ubiquitin ligase complex, resulting in ubiquitination and subsequent degradation of A3G. The interaction domains of Vif and A3G have been mapped to the N-terminal region in Vif as well as to regions in the N-terminal half of A3G (24–32). A conserved HCCH motif in the C-terminal half of Vif has been shown to be critical for the interaction with Cul5 (33–36).
Using coimmunoprecipitation (co-IP) assays in combination with mass spectrometry, CBFβ was recently identified as a novel Vif-interacting protein (37, 38). CBFβ has previously been described in the literature as a transcriptional cofactor of RUNX. The RUNX/CBFβ complex can either activate or repress the expression of genes important for cell growth, differentiation, and cancer development (reviewed in reference 39). CBFβ itself does not bind DNA but increases the DNA binding affinity of RUNX proteins and stabilizes RUNX, presumably by forming a RUNX/CBFβ complex. CBFβ is ubiquitously expressed and exists in two isoforms, both of which can bind to Vif (40, 41). Binding of CBFβ to Vif is required for efficient A3G degradation and production of infectious virus (37, 38, 40, 42). Mutational analyses identified regions in the N-terminal halves of Vif and CBFβ that are important for the interaction of the two proteins (38, 42–45). However, the mechanism by which CBFβ facilitates Vif-induced degradation of A3G is not fully understood. CBFβ by itself interacts with neither A3G nor Cul5 (37, 38). Instead, CBFβ binding to Vif was found to be important for the assembly of a Vif-Cul5 (Cul5) complex possibly by stabilizing a favorable conformation of Vif (38, 46). This is supported by the observation that under certain in vitro assembly conditions, the Vif/EloB/EloC complex was found to interact with Cul5/Rbx even in the absence of CBFβ (43). In vitro, CBFβ binding to Vif reduces Vif sensitivity to chymotrypsin digestion, further suggesting a conformational stabilization of Vif by CBFβ (43).
Several reports found that knockdown of CBFβ resulted in reduced steady-state levels of Vif (37, 40) while others found that CBFβ could facilitate Vif-induced A3G degradation without apparent effect on Vif stability (38, 44). Neither the cause underlying the reduced steady-state levels of Vif in the absence of CBFβ nor the reason for the discrepant observations regarding the effects of CBFβ on Vif expression is currently understood. Here, we report that CBFβ indeed affects the steady-state expression of Vif. Knockdown of CBFβ in HeLa cells through a short hairpin RNA (shRNA) resulted in the reduction of Vif levels. Vif expression was restored by ectopic expression of CBFβ. Kinetic studies revealed that CBFβ significantly enhanced the rate of Vif de novo biosynthesis and, to a more limited extent, increased the metabolic stability of Vif. Deletions in the N-terminal region of Vif resulted in loss of CBFβ responsiveness. Independent from its effects on Vif expression, CBFβ also enhanced the Vif-mediated degradation of A3G, in agreement with reports that observed CBFβ-enhanced degradation of A3G without an effect on Vif expression (38, 44). Taken together, our data suggest that CBFβ functions as a molecular chaperone to enhance Vif biosynthesis, to stabilize mature Vif protein, and to facilitate the assembly of an A3G-Vif-Cul5 E3 ligase complex that, overall, results in more efficient degradation of A3G.
MATERIALS AND METHODS
Transfections.
For transfection, cells were grown in 25-cm2 flasks to about 80% confluence (∼3 × 106 cells). Cells were transfected using Lipofectamine Plus (Invitrogen Corp., Carlsbad, CA) or TransIT (Mirus Bio LLC, Madison, WI) following the manufacturer's recommendations. Where appropriate, empty vector DNA was used to adjust total DNA amounts.
Generation of stable CBFβ knockdown HeLa cells.
A HeLa CBFβ knockdown cell line (HeLa-KD) was established using a GIPZ Lentiviral shRNA system (catalog number VGH5526; Thermo Fisher Scientific, Rockford, IL) according to the supplier's instructions. A cell line transduced with nonspecific shRNA (catalog number RHS4348; Thermo Fisher Scientific, Rockford, IL) was created as a reference. Stable cell lines expressing the shRNAs were selected by addition of 3 μg/ml puromycin. Successful knockdown of CBFβ was assessed by immunoblotting. Cells were then maintained in Dulbecco's modified Eagle's medium (DMEM) without puromycin.
Antibodies.
A rabbit polyclonal antibody to CBFβ (catalog number PA1-317; Thermo Fisher Scientific, Rockford, IL) was used for immunoblot analyses. A mouse monoclonal antibody to Vif (antibody 319) was a generous gift of Michael Malim and was used for immunoblot analyses. For immunoprecipitation of Vif, a rabbit polyclonal antibody raised against recombinant Vif (Vif93) was used (47). Production of an APOBEC3G-specific peptide antibody (ApoC17) was reported previously (48). This antibody is also available through the NIH Research and Reference Reagent Program (catalog number 10082). HIV-1 Gag was identified using pooled HIV Ig (catalog number 3957; NIH Research and Reference Reagent Program). A Vpu-specific antibody was produced by immunizing rabbits with purified recombinant protein corresponding to the Vpu cytoplasmic domain. This antibody is freely available through the NIH Research and Reference Reagent Program (catalog number 969). A mouse monoclonal antibody to alpha-tubulin, polyclonal antibody to actin, and a polyclonal antibody to RUNX1 were purchased from Sigma-Aldrich, Inc. (catalog numbers T9026, A5060, and R9528, respectively; St. Louis, MO). For coimmunoprecipitation studies, c-Myc agarose affinity gel was used (catalog number A7470; Sigma-Aldrich, Inc., St. Louis, MO). For immunoblot analysis of RUNX1-hemagglutinin (HA), mouse anti-HA antibodies were used (H3663; Sigma-Aldrich, Inc., St. Louis, MO). RUNX1 and enhanced green fluorescent protein (eGFP) were immunoprecipitated using HA antibody-conjugated agarose beads (A2095; Sigma-Aldrich, Inc., St. Louis, MO). A polyclonal antibody to GFP (catalog number 632592; Clontech Laboratories, Inc., Mountain View, CA) was used to detect eGFP expression.
Plasmids.
Human CBFβ (GenBank NM_001755.2) was cloned by reverse transcription-PCR (RT-PCR) from HeLa cell poly(A)+ mRNA using primers 5′-AATACTCGAGAAGATGCCGCGCGTCGTGCCCGACCA and 5′-AATAGGTACCCTAGGGTCTTGTTGTCTTCTTGCC. The RT-PCR was performed using a SuperScript III One-step RT-PCR kit (catalog number 12574-026; Invitrogen Corp., Carlsbad, CA) according to the manufacturer's instructions. PCR products were digested with XhoI and KpnI and cloned into pcDNA3.1(−) (Invitrogen Corp., Carlsbad, CA). Vif was expressed either from a cytomegalovirus (CMV) promoter-driven codon-optimized vector, pcDNA-human Vif (hVif) (49), or from the full-length molecular clone pNL4-3 (50). The Vif-HA expression vector for the expression of C-terminally HA-tagged HXB2 Vif was a gift of Xiao-Fang Yu (17). A vector for the expression of untagged human APOBEC3G has been reported previously (16). Construction of Vif deletion mutants was previously reported (51). A vif-defective variant of the full-length molecular clone NL4-3, NL4-3/Vif(−), was reported elsewhere (47). N-terminally HA-tagged RUNX1 was constructed by PCR amplification of RUNX1 (52) using the forward primer 5′-GGCCATGGAGGCCATGGCTTCAGACAGCATATTTGAG and reverse primer 5′-CTCGAGTCAGTAGGGCCTCCACACGGCCT and TaKaRa LA Taq polymerase (Clontech, Mountain View, CA) as per the manufacturer's recommendation. Resulting PCR fragments were cloned into the SfiI and XhoI sites of pCMV-HA (Clontech, Mountain View, CA). The HA-eGFP vector was obtained from Genecopoeia, Inc. (catalog number Ex-eGFP-M06; Rockville, MD).
Immunoblotting.
For immunoblot analysis of cell-associated proteins, whole-cell lysates were prepared as follows. Cells were washed once with phosphate-buffered saline (PBS), suspended in PBS, and mixed with an equal volume of sample buffer (4% sodium dodecyl sulfate, 125 mM Tris-HCl, pH 6.8, 10% glycerol, and 0.002% bromophenol blue). Proteins were solubilized by heating 10 to 15 min at 95°C with occasional vortexing. Cell lysates were subjected to SDS-PAGE; proteins were transferred to polyvinylidene difluoride (PVDF) membranes and reacted with appropriate antibodies as described in the text. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Piscataway, NJ), and proteins were visualized by enhanced chemiluminescence (GE Healthcare, Piscataway, NJ).
Northern blot analysis.
Total RNA was prepared using RNeasy Mini Kits (Qiagen, Valencia, CA). RNA samples were electrophoresed on denaturing 1% agarose gels and transferred to nylon membranes by capillary blotting using a Turbo blotter (Schleicher & Schuell, Inc., Keene, NH). After UV cross-linking, the membranes were prehybridized with 10 ml of QuickHyb Hybridization Solution (Stratagene, La Jolla, CA) for 1 h at 68°C. Probes were labeled with [α-32P]dTTP using a Ladderman random primer labeling kit (PanVera, Madison, WI). Labeled probes were mixed with sonicated salmon sperm DNA (Stratagene, La Jolla, CA), heated at 94°C for 3 min, and then chilled on ice. Membranes were incubated for 5 h at 68°C with 32P-radiolabeled DNA probes (1 × 107 cpm) specific for CBFβ, hVif, or beta-actin. Following hybridization, membranes were washed twice with wash buffer (2× SSPE, 0.1% SDS) (1× SSPE is 0.18 M NaCl, 10 mM NaH2PO4, and 1 mM EDTA [pH 7.7]) for 15 min at room temperature, followed by one wash in 0.2× SSPE with 0.1% SDS for 15 min at 65°C. To reprobe the blots, membranes were stripped by a 15-min incubation in 1% SDS at 100°C.
Metabolic labeling and immunoprecipitation.
Cells were suspended in 5 ml of RPMI medium lacking methionine and cysteine (MP Biomedical, Solon, OH) and incubated for 20 min at 37°C to deplete the intracellular methionine/cysteine pool. Labeling was done at 37°C in 200 μl of methionine-free RPMI medium containing 5% fetal calf serum (FCS) supplemented with 150 μCi of the 35S protein labeling mix Expre35S35S (NEG072; PerkinElmer, Waltham, MA). For pulse-chase analyses, cells were pulsed for 10 min (Vif) or 20 min (RUNX1) at 37°C with radioisotope. Cells were then pelleted to remove unincorporated isotope, suspended in 1 ml of complete RPMI medium, and distributed into equal aliquots, one sample for each chase time point. Samples were collected at the specified time points; cells were pelleted and stored on dry ice. For pulse-labeling experiments, the total volume of the reaction mixture was 280 μl. Also, MG132 (catalog number 474791; EMD Millipore, Billerica, MA) was added during starvation and labeling (10 μM) to inhibit proteasomal degradation. After the initial starvation period, isotope was added at room temperature, and the sample was immediately divided into five equal aliquots (50 μl each). Samples were incubated at 37°C for the times indicated in the text and then quick-frozen on dry ice to stop the reaction. Cell lysates were prepared by detergent lysis followed by reextraction of the detergent-insoluble material with sample buffer, as reported previously (53). Soluble and detergent-insoluble extracts were pooled prior to immunoprecipitation.
Coimmunoprecipitation analysis.
HeLa CBFβ knockdown (KD) cells were transfected with 1 μg of expression vectors for myc-tagged CBFβ or empty vector and 4 μg of Vif expression vectors as indicated in the text. Cells were harvested at 24 h posttransfection and washed twice with cold PBS. Cells were lysed in 500 μl of lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% [vol/vol] Triton X-100, and complete protease inhibitor cocktail) at 4°C for 30 min and then clarified by centrifugation at 10,000 × g for 2 min. Five percent of the lysate was used as the input control, and the remaining lysate was used for immunoprecipitation of Myc-tagged antigens. Precleared cell lysates were mixed with Myc antibody-conjugated agarose beads (Sigma-Aldrich, Inc., St. Louis, MO) and incubated at 4°C for 1 h. Samples were then washed three times with wash buffer (20 mM Tris-HCl [pH 7.5], 100 mM NaCl, 0.1 mM EDTA, 0.05% [vol/vol] Tween 20). Proteins were eluted by boiling beads in sample buffer and subjected to immunoblot analysis using antibodies to Vif and CBFβ. Coimmunoprecipitation of RUNX1 and CBFβ was done essentially the same way except that HA-tagged RUNX1 was used for the precipitation of untagged CBFβ.
Viral infectivity assay.
Virus-containing supernatants were harvested at 24 h after transfection of cells. Cellular debris was removed by centrifugation (3 min at 1,500 rpm), and clarified supernatants were filtered (0.45-μm pore size) to remove residual cellular debris. A total of 150 μl of viral stock was used to infect 5 × 104 TZM-bl cells in a 24-well plate in a total volume of 1.1 ml. Typically, infections were done in triplicate. Infection was allowed to proceed for 48 h at 37°C. Medium was removed, and cells were lysed in 300 μl of Promega 1× reporter lysis buffer (Promega Corp., Madison, WI) and frozen at −80°C for a minimum of 30 min. To determine the luciferase activity in the lysates, 5 μl of each lysate was combined with 20 μl of luciferase substrate (Steady-Glo; Promega Corp., Madison, WI), and light emission was measured using a Modulus ii microplate reader (Turner Biosystems Inc., Sunnyvale, CA).
RESULTS
CBFβ facilitates Vif-induced A3G degradation and increases steady-state levels of Vif.
We created a stable CBFβ knockdown cell line (HeLa-KD) as described in Materials and Methods. To assess the effective down-modulation of CBFβ in HeLa-KD cells and to validate the reported effect of CBFβ on Vif expression and A3G expression, we transfected HeLa-KD cells with increasing amounts of pcDNA-hVif in the presence of constant amounts of pcDNA-A3G and in the presence or absence of CBFβ (Fig. 1A). Mock-transfected HeLa-KD cells revealed undetectable levels of A3G, CBFβ, and Vif (Fig. 1A, lane 1). Expressing increasing amounts of CBFβ in the absence of Vif had no effect on A3G stability (Fig. 1A, lanes 2 to 6). In contrast, CBFβ significantly increased the steady-state levels of Vif in transfected HeLa-KD cells (Fig. 1A, lanes 7 to 12, and B). Similar effects of CBFβ on Vif stability were seen when Vif was expressed from the full-length molecular clone NL4-3 (see Fig. 3B below). However, the effect of CBFβ on Vif expression was most pronounced at low levels of Vif (Fig. 1B, compare bars 7 and 10 to 9 and 12). In the absence of CBFβ, Vif only modestly reduced A3G levels (Fig. 1A, lanes 7 to 9). In contrast, in the presence of CBFβ, A3G was virtually undetectable even at the lowest level of Vif (Fig. 1A, lanes 10 to 12). These results are in agreement with published reports demonstrating that CBFβ facilitates the Vif-induced degradation of A3G and increases Vif expression (37, 38, 40, 42).
FIG 1.

CBFβ increases steady-state levels of Vif via a posttranscriptional mechanism. (A) HeLa-KD cells were transfected with 1 μg of pcDNA-A3G (lanes 2 to 12) together with 0.1, 0.5, 1, or 2 μg of pcDNA-CBFβ (lanes 3 to 6) or 0.5, 1.5, or 3 μg of pcDNA-hVif in the absence (lanes 7 to 9) or presence (lanes 10 to 12) of 1 μg of pcDNA-CBFβ. Total amounts of transfected DNA in each sample were adjusted to 5 μg using empty vector DNA as appropriate. Lane 1 represents a mock-transfected sample. Cells were harvested 24 h after transfection, and total cell extracts were prepared for immunoblot analysis. Samples were separated by SDS-PAGE and analyzed with antibodies to A3G, CBFβ, Vif (319), or alpha-tubulin (tub) as indicated. (B) Vif-specific bands from the experiment shown in panel A were quantified by densitometric scanning of the immunoblot, and results are expressed as relative units, with the Vif signal in lane 7 defined as 1. (C) HeLa-KD cells were either mock transfected (lane 1), transfected with 4 μg of pcDNA-hVif (lane 2) or 1 μg of pcDNA-CBFβ (lane 3), or cotransfected with 4 μg of pcDNA-hVif and 1 μg of pcDNA-CBFβ (lane 4). Total amounts of transfected DNA in all samples were adjusted to 5 μg with empty vector DNA. A portion of the samples was used for total RNA extraction and Northern blot analysis (mRNA). For that purpose, 20 μg of total RNA was separated by denaturing 1% agarose gel electrophoresis and transferred to a nylon membrane. The membranes were probed with 32P-labeled probes specific for CBFβ, Vif, or actin as indicated. The mRNAs were visualized by autoradiography. The remaining samples were used for protein analysis (protein). Total cell extracts were subjected to immunoblot analysis using a Vif-specific polyclonal antibody. The same blot was then reprobed with a CBFβ-specific polyclonal antibody, followed by probing with an actin-specific polyclonal antibody as a loading control.
FIG 3.

CBFβ independently affects Vif stability and Vif function. (A) HeLa-KD cells were transfected with pcDNA-A3G (1 μg) in the absence (lanes 2 to 6) or presence (lanes 7 to 12) of pcDNA-CBFβ (0.5 μg). The amount of pcDNA-hVif varied from 0.1 μg to 5.0 μg as indicated. All DNA amounts were adjusted to 6 μg of total transfected DNA using empty vector DNA. Cells were harvested at 24 h posttransfection, and total cell lysates were analyzed by immunoblotting with antibodies to A3G, Vif (319), CBFβ, or alpha-tubulin (tub). (B) HeLa-KD cells were transfected with 4 μg of pNL4-3 or pNL4-3/Vif(−) DNA in the presence or absence of pcDNA-A3G (1 μg) and pcDNA-CBFβ (1 μg) as indicated at the top. Cells and virus-containing supernatants were harvested 24 h later, and total cell extracts were prepared for immunoblot analysis (cell). A fraction of the cell-free virus (80%) was pelleted through a 20% sucrose cushion and suspended in sample buffer for immunoblot analysis (virus). Samples were separated by SDS-PAGE and analyzed with antibodies to A3G, CBFβ, Vif (319), Vpu, or HIV Ig (Gag) as indicated. (C) A portion of the remaining filtered culture supernatant was normalized for equal reverse transcriptase activity and used for the infection of TZM-bl cells. HIV-induced activation of luciferase was measured 48 h after infection. Results are shown as a scatter blot of two independent experiments performed in triplicate infections each. Each dot represents one experimental data point. The mean of the values obtained for NL4-3 in the absence of A3G and CBFβ was defined as 100%.
The effect of CBFβ on Vif expression is posttranscriptional.
To gain insights into the mechanism that leads to reduced Vif expression in the absence of CBFβ, we compared the Vif mRNA levels in transfected HeLa-KD cells in the presence or absence of exogenous CBFβ (Fig. 1C). We found that CBFβ had little or no effect on the levels of Vif mRNA, nor did Vif expression affect CBFβ mRNA levels (Fig. 1C, mRNA). In contrast, Vif protein levels were significantly lower in the absence of CBFβ (Fig. 1C, protein), indicating that the effect of CBFβ on Vif expression occurs at a posttranscriptional level. We also performed a polysome analysis to identify potential differences in the polysome loading of vif mRNA in the presence or absence of CBFβ. The polysome profile of actin mRNA as well as those of 28S and 18S rRNAs were included for reference. All RNA profiles shown in Fig. 2 derived from the same gradient. Thus, differences in the relative profiles of Vif mRNA and actin mRNA or ribosomal RNAs are real and not due to differences in the gradient centrifugation. The results as shown in Fig. 2 therefore suggest that CBFβ also had no effect on the translational activity on vif mRNAs (Fig. 2, top).
FIG 2.
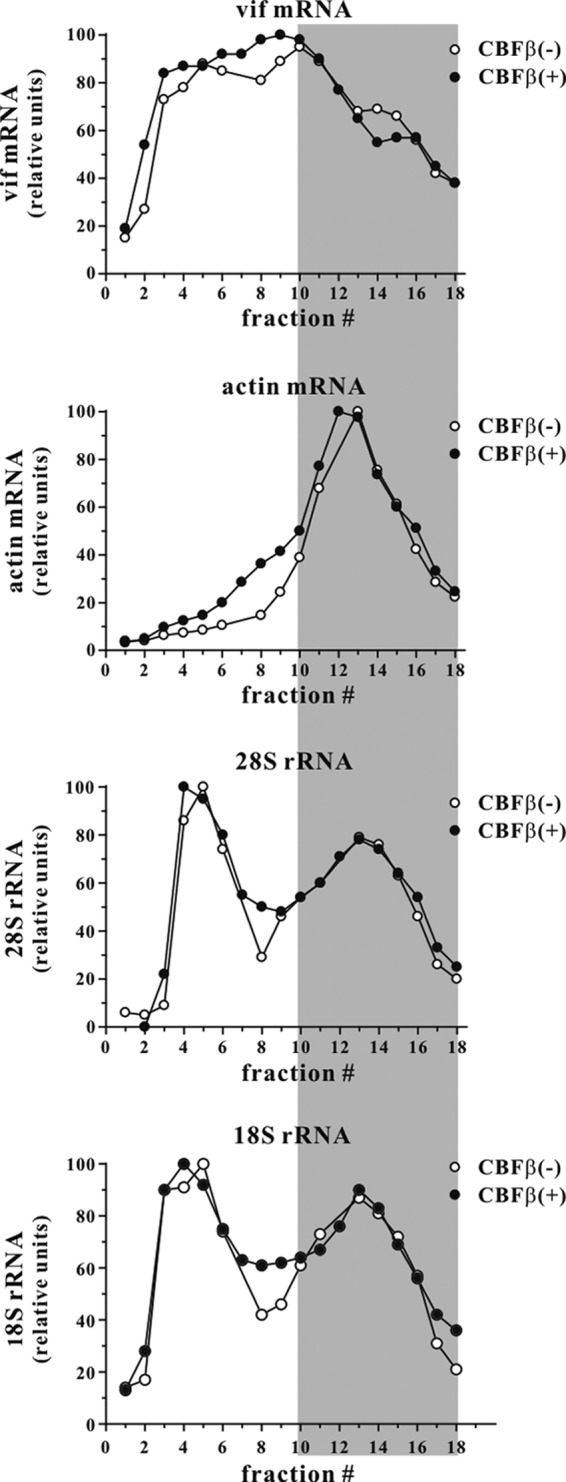
CBFβ does not affect translational activity on Vif mRNA. Polysome analysis was done using a modification of a previously described protocol (63). Briefly, HeLa-KD cells (9 × 106 in a 75-cm2 flask) were transfected with 12 μg of pcDNA-hVif together with either 3 μg of pcDNA-CBFβ or 3 μg of empty vector DNA. After 24 h, transfected cells were treated with 100 μg/ml of cycloheximide for 15 min at 37°C. Cells were then washed twice with ice-cold PBS containing cycloheximide (100 μg/ml), followed by two washes with solution A (150 mM KCl, 10 mM MgCl2, and 50 mM Tris-HCl, pH 7.6). Cells were suspended in 500 μl of solution A containing 0.5% NP-40 and lysed on ice for 10 min. Nuclei were removed by low-speed centrifugation (2,800 × g for 10 min at 4°C). Supernatants were layered on linear 15% to 50% sucrose gradients in solution A. The gradients were centrifuged in a SW55 rotor (Beckman Coulter Inc., Indianapolis, IN) at 36,000 rpm for 90 min at 4°C. Eighteen fractions (250 μl each) were collected from top to bottom. Total RNA was extracted from each fraction using TRIzol LS reagent (Invitrogen Corp., Carlsbad, CA) according to the manufacturer's directions and loaded on an agarose gel for Northern blot analysis. Prior to Northern blot analysis, membranes were stained with methylene blue to visualize ribosomal RNAs. The signal intensity of 18S and 28S ribosomal RNAs in each fraction was determined by optical scanning. Northern blotting was done on the same gradient samples using Vif- or actin-specific 32P-labeled probes. Vif- or actin-specific signals were quantified by PhosphorImager analysis. To allow direct comparison, the highest signal intensity in each gradient was defined as 100 relative units. The shaded area represents the polysome fractions.
CBFβ independently affects Vif stability and function.
To test if the enhanced degradation of A3G by Vif in the presence of CBFβ is simply a consequence of the elevated levels of Vif, we modulated Vif expression in HeLa-KD cells in the presence of constant amounts of A3G and in the presence or absence of CBFβ (Fig. 3A). To achieve comparable expression of Vif, the amounts of transfected Vif vector ranged from 0.5 to 5 μg in the absence of CBFβ and from 0.1 to 1 μg in the presence of CBFβ, as indicated in Fig. 3A. We found that transfecting 3 μg of pcDNA-hVif in the absence of CBFβ yielded levels of Vif comparable to 0.3 to 0.4 μg of pcDNA-hVif in the presence of CBFβ (Fig. 3A, compare lane 5 to lane 9 or 10). As observed in Fig. 1, Vif induced only modest degradation of A3G in the absence of CBFβ, while in the presence of CBFβ, comparable levels of Vif resulted in almost quantitative depletion of A3G. These results indicate that CBFβ exerts both a quantitative and a qualitative effect on Vif and Vif-induced A3G degradation.
CBFβ knockdown affects the ability of Vif to regulate viral infectivity.
We previously reported that the steady-state levels of Vif expressed from the proviral genome are low and regulated not only at the level of RNA splicing but also at the level of protein turnover (53). Indeed, even modest overexpression of Vif was found to inhibit viral infectivity due to a Vif-induced assembly defect (54). Thus, while it appears that relatively low levels of Vif are desired and sufficient to ensure the production of fully infectious virus from A3G-expressing cells, we wondered how the reduced levels of Vif expressed in virus-producing CBFβ knockdown cells, combined with the reduced ability of Vif to target A3G, would affect viral infectivity.
To that end we transfected HeLa-KD cells with wild-type NL4-3 or its vif-defective NL4-3/Vif(−) variant in the presence or absence of A3G and exogenously supplied CBFβ (Fig. 3B). Protein expression in the transfected cells and the presence of virus-associated proteins in the culture supernatants were assessed by immunoblotting. As expected, endogenous CBFβ was undetectable, and exogenously provided CBFβ was efficiently expressed in the transfected cells. Of note, CBFβ was effectively excluded from cell-free virions. CBFβ had no effect on viral Gag protein synthesis or on the production of cell-free virions. Also, CBFβ had no effect on the expression of Vpu, which—like Vif—is expressed from a partially spliced mRNA. In contrast, CBFβ significantly increased the steady-state level of Vif. Thus, of the HIV-1 proteins analyzed, only Vif expression was affected by CBFβ, further arguing against possible effects of CBFβ on HIV-1 transcription, RNA splicing, or RNA transport. Surprisingly, the increased level of Vif in CBFβ-expressing cells was not paralleled by increased packaging of Vif into cell-free virions (Fig. 3B, lanes 2 and 3 of cell versus virus). The reason for this is unclear; however, it is conceivable that association of Vif with CBFβ leads to intracellular sequestration of Vif. Such a scenario is consistent with our observation that expression of Vif can induce cellular redistribution of CBFβ (65) as well as the recent finding that Vif-CBFβ complexes are stable enough to interfere with the formation of heterodimeric RUNX-CBFβ complexes (43). As to A3G, the presence of CBFβ resulted in enhanced degradation of A3G and more efficient exclusion from virions by Vif. As expected, CBFβ had no significant effect on expression or encapsidation of A3G in the absence of Vif.
The infectivity of the virions produced in the experiment shown in Fig. 3B was measured by infection of TZM-bl cells. Resulting luciferase activity was normalized to wild-type NL4-3 produced in the absence of A3G and CBFβ, which was defined as 100% (Fig. 3C). As expected, expression of A3G reduced the infectivity of vif-defective virus by about 10- to 20-fold, irrespective of the presence or absence of CBFβ (Fig. 3C, columns 5 and 6). In contrast, NL4-3 was almost fully infectious when produced in the presence of CBFβ (Fig. 3C, column 3). Surprisingly, even in the absence of CBFβ, the infectivity of wild-type NL4-3 was inhibited only about 3-fold (Fig. 3C, column 2). These results suggest that the low levels of Vif produced in CBFβ-depleted cells are sufficient for the production of viruses with only modestly reduced infectivity.
The N-terminal region in Vif is important for interaction with and responsiveness to CBFβ.
CBFβ was previously shown to interact with Vif through N-terminal motifs (38, 42–45). In particular, the mutations W21A, L64S, and I66S in Vif were found to reduce or abolish interaction with CBFβ (38, 43). We tested these mutants and found that in our hands all of them still interacted with CBFβ. The reason for the discrepant results is not clear. However, previous studies employed C-terminally tagged Vif mutants while we used untagged Vif proteins in our co-IP studies. Since we were unable to detect interference of point mutants with CBFβ binding, we tested a series of in-frame deletion mutants of Vif that we had previously created to characterize dominant negative Vif variants (51). For this experiment, C-terminally myc-tagged CBFβ was used. The Vif mutants employed in the current analysis are schematically shown in Fig. 4A, and their expression in the presence or absence of CBFβ is shown in the top panel of Fig. 4B. As expected, expression of wild-type Vif (Fig. 4B, WT) was significantly increased in the presence of CBFβ. In contrast, expression of Vif-ΔK, Vif-ΔD, Vif-ΔL, and Vif-ΔG was unaffected by CBFβ. Vif-ΔH was partially CBFβ responsive while Vif-ΔC retained full responsiveness to CBFβ (Fig. 4B, top panel).
FIG 4.

The N-terminal region in Vif is important for interaction with and responsiveness to CBFβ. (A) Vif deletion mutants used in this analysis were constructed on the backbone of pcDNA-hVif and are schematically shown. The positioning of the deletions is roughly to scale. Numbers above the deletions indicate the first and last residues that are deleted. (B) HeLa-KD cells were transfected with 4 μg each of the indicated Vif expression vector together with 1 μg of empty vector (−) or 1 μg of pcDNA-CBFβ-Myc. Detergent extracts were prepared 24 h later. A portion of the extract was analyzed directly by sequential immunoblotting using antibodies to Vif, CBFβ, or tubulin (Input). The remaining samples were immunoprecipitated with c-Myc agarose gel. Precipitated samples were separated by SDS-PAGE and probed with antibodies to CBFβ (IP) or Vif (co-IP). WB, Western blotting; α, anti.
To address whether CBFβ responsiveness is correlated with the ability of Vif variants to interact with CBFβ, coimmunoprecipitation analyses were performed. Aliquots of the samples indicated in the top panel of Fig. 4B were immunoprecipitated with Myc-specific antibody beads. The precipitates were eluted from the beads, separated by SDS-PAGE, and probed with antibodies to CBFβ or Vif (Fig. 4B, lower panels). Wild-type Vif and the CBFβ-responsive Vif-ΔH and Vif-ΔC variants were able to efficiently interact with CBFβ in this coimmunoprecipitation assay. Vif-ΔK did not interact with CBFβ, and only a very weak signal was detectable for Vif-ΔD. Expression levels of Vif-ΔL and Vif-ΔG were too low to allow for definitive conclusions regarding CBFβ interaction. Nevertheless, our results suggest a correlation of CBFβ responsiveness of Vif with the ability of Vif to interact with CBFβ, which is in agreement with previous studies (37, 38, 40, 42).
CBFβ affects the turnover of Vif.
Vif levels in HIV-infected cells are lower than those of other viral proteins, presumably due to the short Vif half-life (53). To see if the reduced expression of wild-type Vif in the absence of CBFβ is due to more rapid turnover of Vif, pulse-chase analyses were performed in transfected HeLa-KD cells in the presence or absence of exogenous CBFβ. For comparison, the CBFβ nonresponsive Vif-ΔD variant was analyzed in parallel (Fig. 5A and B). Transfected cells were pulse-labeled for 10 min and, after removal of unincorporated isotope, chased for up to 60 min. Cell extracts were immunoprecipitated with a Vif-specific polyclonal antibody, separated by SDS-PAGE, and visualized by fluorography (Fig. 5A). Vif-specific bands were quantified by PhosphorImager analysis, and the results were plotted as a function of time (Fig. 5B). We found that wild-type Vif was indeed more rapidly degraded in the absence of CBFβ than Vif produced in the presence of CBFβ (Fig. 5B). In contrast, the presence or absence of CBFβ had no significant impact on the turnover of Vif-ΔD. The half-life of Vif-ΔD in the presence or absence of CBFβ was similar to that of wild-type Vif expressed in the absence of CBFβ, indicating that binding of CBFβ to Vif stabilizes the protein in HeLa cells. Curiously, the absolute amount of wild-type Vif recovered at the pulse time point (0 min) was higher in the presence of CBFβ. This could indicate a lower rate of Vif biosynthesis or cotranslational degradation of Vif in the absence of CBFβ. Importantly, this phenomenon was not observed for Vif-ΔD, where the levels of Vif recovered at the pulse time point were comparable in the presence and absence of CBFβ (Fig. 5A, compare 0-min time points).
FIG 5.

CBFβ stabilizes Vif and increases Vif biosynthesis. (A) HeLa-KD cells were transfected with 5 μg of pcDNA-hVif (Vif wt) or pcDNA-hVif-ΔD (Vif-ΔD) in the presence of 1 μg of empty vector [CBFβ(−)] or 1 μg of pcDNA-CBFβ [CBFβ(+)]. One day later, cells were labeled for 10 min with [35S]methionine-cysteine and chased for up to 60 min, as described in Materials and Methods. Cell lysates were immunoprecipitated with a Vif-specific polyclonal antibody (Vif93) and separated by SDS-PAGE. Proteins were visualized by fluorography. A representative experiment is shown. (B) Vif-specific protein bands as shown in panel A were quantified by PhosphorImager analysis. The time-zero value for each sample was defined as 100%, and results are plotted as a function of time. Data represent means ± standard errors of the means from three independent experiments. (C) HeLa-KD cells were transfected with 5 μg of pcDNA-hVif (Vif wt) or pcDNA-hVif-ΔD (Vif-ΔD) in the presence of 1 μg of empty vector [CBFβ(−)] or 1 μg of pcDNA-CBFβ [CBFβ(+)]. One day later, cells were labeled with [35S]methionine-cysteine for 2 to 10 min as detailed in Materials and Methods. Cell lysates were immunoprecipitated with a Vif polyclonal antibody (Vif93) and separated by SDS-PAGE. Proteins were visualized by fluorography. (D) Vif-specific protein bands from the experiment shown in panel C were quantified by PhosphorImager analysis, and results were plotted as a function of time. Time zero was not experimentally determined but was defined as zero for graphical representation. Amounts of wild-type Vif recovered at the last time point in the absence of CBFβ (open circles) were defined as 100%. Data represent the means ± standard errors of the means from three independent experiments. (E) CBFβ increases de novo biosynthesis of CBFβ binding-competent Vif variants. HeLa-KD cells were transfected with 5 μg of pcDNA-hVif-ΔK (Vif-ΔK), pcDNA-hVif-ΔH (Vif-ΔH), or pcDNA-hVif-ΔC (Vif-ΔC) in the presence of 1 μg of empty vector (open circles) or 1 μg of pcDNA-CBFβ (filled circles). One day later, cells were labeled with [35S]methionine-cysteine for 2 to 10 min. Cell lysates were immunoprecipitated with a Vif polyclonal antibody and separated by SDS-PAGE. Vif-specific protein bands were quantified by PhosphorImager analysis, and results were plotted as a function of time. Amounts of Vif recovered at the last time point in the absence of CBFβ were defined as 100%.
CBFβ enhances the rate of Vif biosynthesis.
To test if the increased level of wild-type Vif shown in Fig. 5A at the pulse time point was due to a difference in the rate of Vif biosynthesis or caused by cotranslational degradation of Vif in the absence of CBFβ, we measured the kinetics of Vif biosynthesis as described in Materials and Methods. To prevent cotranslational protein degradation, the experiment was performed in the presence of MG132 (10 μM). Vif biosynthesis was monitored for 10 min with samples collected at 2-min intervals. Cell extracts were immunoprecipitated with a Vif-specific polyclonal antibody; samples were separated by SDS-PAGE and visualized by fluorography (Fig. 5C). Vif-specific bands were quantified by PhosphorImager analysis, and the results were plotted as a function of time (Fig. 5D). We found that wild-type Vif was synthesized at a higher rate when CBFβ was present. In contrast, the rate of Vif-ΔD biosynthesis was unaffected by the presence or absence of CBFβ. Indeed, Vif-ΔD synthesis with or without CBFβ occurred at a similar rate as wild-type Vif in the absence of CBFβ. These results suggest that CBFβ, in addition to its effect on Vif degradation, also increased the rate of de novo synthesis of wild-type Vif but not of Vif-ΔD. We also tested Vif mutants Vif-ΔK, Vif-ΔH, and Vif-ΔC and found that Vif-ΔH and Vif-ΔC, both of which interact with CBFβ, showed increased de novo protein biosynthesis in the presence of CBFβ (Fig. 5E). These results are consistent with the increased steady-state levels observed in the experiment shown in Fig. 4B. In contrast, Vif-ΔK, which does not interact with CBFβ, behaved like Vif-ΔD and revealed no change in the rate of protein biosynthesis in response to CBFβ expression (Fig. 5E). Thus, the effect of CBFβ on Vif biosynthesis is specific and dependent on the ability of CBFβ to interact with Vif.
It is conceivable that interaction of CBFβ with Vif affects folding of Vif, which might affect the relative affinity of our polyclonal Vif antibody used for immunoprecipitation and thus could be misconstrued as differences in Vif protein biosynthesis. To rule out that CBFβ-induced protein folding affected the Vif-antibody interaction, we repeated the experiment shown in Fig. 5C using pVif-HA (17) in place of pcDNA-hVif (Fig. 6). Of note, pcDNA-hVif expresses NL4-3 Vif from a codon-optimized unspliced transcript while pVif-HA expresses HA-tagged HXB2 Vif from a non-codon-optimized spliced transcript (17). Thus, Vif protein produced from pVif-HA differed from hVif not only in the presence of a C-terminal HA tag but also in its primary amino acid sequence (10% amino acid variation) (Fig. 6A) and in the mode of expression (spliced versus unspliced transcripts). In addition to the Vif antibody, we employed a rat monoclonal anti-HA antibody for immunoprecipitation (Fig. 6B). Irrespective of the antibody used for immunoprecipitation, CBFβ increased the rate of Vif-HA biosynthesis (Fig. 6C and D). These results indicate that the observed differences in the pulse-labeling experiment shown in Fig. 5D and E reflect true differences in the rate of Vif biosynthesis and are not due to CBFβ-induced differences in antibody recognition.
FIG 6.
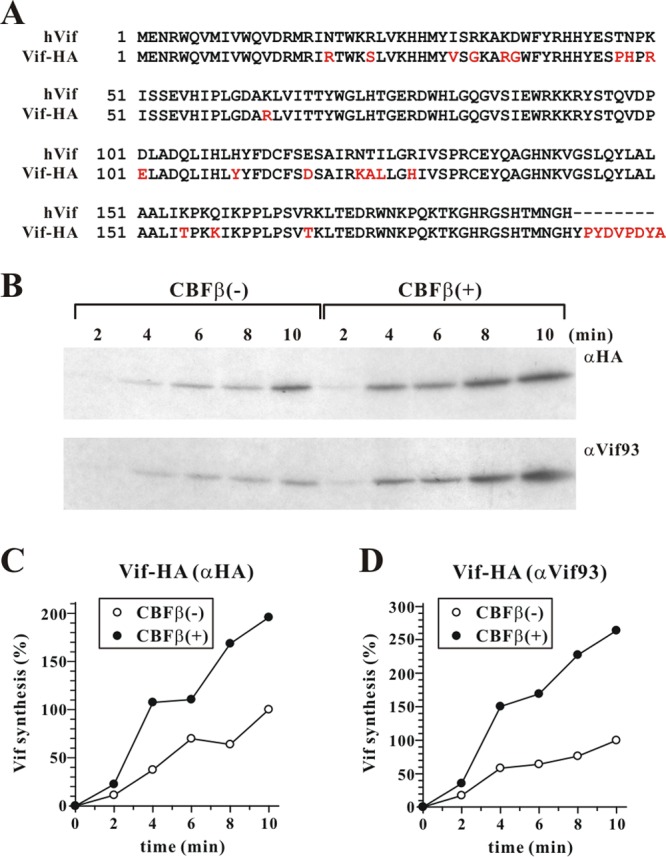
The increased rate of Vif synthesis in CBFβ-positive cells is not due to differential antibody recognition. (A) Amino acid sequence alignment of hVif (based on the NL4-3 isolate) and Vif-HA (based on the HXB2 isolate). Amino acid differences are shown in red. (B) HeLa-KD cells were transfected with 5 μg of pVif-HA in the presence of 1 μg of empty vector [CBFβ(−)] or 1 μg of pcDNA-CBFβ [CBFβ(+)]. Cells were pulse-labeled in 2-min intervals for up to 10 min as described in the legend of Fig. 5C. Cell lysates were precipitated with an HA-specific rat monoclonal antibody (C) or the Vif93 polyclonal antibody (D). Samples were quantified by PhosphorImager analysis, and Vif levels were plotted as described for Fig. 5D.
CBFβ increases RUNX steady-state levels through a posttranscriptional mechanism.
It was previously reported that heterodimerization of CBFβ with the transcription factor RUNX1 results in stabilization of RUNX1 (55). We verified this observation by expressing human RUNX1 in HeLa-KD cells without or with exogenously expressed CBFβ (Fig. 7A). As a control, we included RUNX G108R, a mutant previously reported to lack the ability of forming heterodimers with CBFβ (55, 56). Indeed, expression of CBFβ increased the steady-state levels of RUNX1 (Fig. 7A, protein, compare lanes 6 and 7). In contrast, expression of the CBFβ binding-defective RUNX1 G108R (Fig. 7A, protein, lanes 8 and 9) or an irrelevant control, eGFP (Fig. 7A, protein, lanes 4 and 5), was not affected by the coexpression of CBFβ. Northern blot analysis (Fig. 7A, mRNA) did not reveal any CBFβ-induced changes in RUNX1 or eGFP mRNA level, indicating that consistent with our observations on Vif (Fig. 1C), CBFβ affects RUNX1 stability at a posttranscriptional level.
FIG 7.

CBFβ increases RUNX steady-state levels through a posttranscriptional mechanism. (A) HeLa-KD cells were either mock transfected (lane 1), or transfected with 0.3 μg each of pCMV-HA (lanes 2 and 3), eGFP-HA (lanes 4 and 5), RUNX1-HA (lanes 6 and 7), or RUNX-HA G108R (lanes 8 and 9) either in the absence of CBFβ (lanes 2, 4, 6, and 8) or the presence of 1 μg of pcDNA-CBFβ (lanes 3, 5, 7, and 9). Transfected DNA in all samples was adjusted to 5 μg of total DNA using empty vector DNA. A portion of the samples was used for total RNA extraction and Northern blot analysis (mRNA). For that purpose, 15 μg of total RNA was separated by denaturing 1% agarose gel electrophoresis and transferred to a nylon membrane. The membranes were probed with 32P-labeled probe specific to RUNX1, eGFP, CBFβ, or actin, as indicated. The mRNAs were visualized by autoradiography. The remaining samples were used for protein analysis (protein). Total cell extracts were subjected to immunoblot analysis using an HA-specific polyclonal antibody (RUNX1 and eGFP). The same blot was then reprobed with a CBFβ-specific polyclonal antibody (CBFβ), followed by probing with an actin-specific polyclonal antibody as a loading control (actin). (B) eGFP or RUNX G108R does not interact with CBFβ. HeLa-KD cells were transfected as described in panel A. Detergent extracts were prepared 24 h later. A portion of the extracts was analyzed directly by immunoblotting using polyclonal antibodies to RUNX1, GFP, CFβ, or actin as indicated (input). The remaining samples were immunoprecipitated with HA agarose beads (IP). Samples were separated by SDS-PAGE and probed with polyclonal antibodies to CBFβ (IP).
Coimmunoprecipitation studies using HA-tagged eGFP (Fig. 7B, lanes 4 and 5) HA-tagged RUNX1 (Fig. 7B, lanes 6 and 7), or RUNX1 G108R (lanes 8 and 9) verified that CBFβ interacts with wild-type RUNX1 (Fig. 7B, lane 7, IP) but not with the G108R mutant (Fig. 7B, lane 9, IP) or the irrelevant eGFP control (Fig. 7B, lane 5, IP).
CBFβ increases RUNX1 biosynthesis.
To test if CBFβ affects RUNX1 expression the same way it affects Vif, pulse-chase and pulse-labeling analyses were done as described for Fig. 5. HeLa-KD cells were transfected with RUNX1 or RUNX G108R in the presence or absence of CBFβ. For determination of RUNX1 stability, cells were subjected to pulse-chase analysis using a 20-min pulse and a 4-h chase protocol (Fig. 8A). For determination of differences in the rate of RUNX1 biosynthesis, cells were subjected to a 20-min pulse-labeling protocol (Fig. 8B). Interestingly, in contrast to a previous report (55), we could not detect a significant impact of CBFβ on the half-life of RUNX1 in our 4-h assay. In fact, the decay profiles of RUNX1 as well as of RUNX G108R were essentially superimposable (Fig. 8A, graph). Instead, we saw a clear enhancement of RUNX1 de novo synthesis by CBFβ (Fig. 8B, filled circles). This effect was not observed for the CBFβ binding-defective RUNX G108R mutant (Fig. 8B, filled triangles), indicating a dependence on the physical interaction of RUNX1 and CBFβ and attesting to the specificity of the effect. Taken together, these results suggest that the enhancement of protein biosynthesis by CBFβ is not limited to Vif but is seen for the other known CBFβ binding partner as well.
FIG 8.

(A) HeLa-KD cells were transfected with 0.3 μg of HA-tagged RUNX1 or RUNX G108R in the absence (−) or presence (+) of 1 μg of CBFβ expression vector. Total transfected DNA was adjusted to 5 μg in all samples. Cells were harvested 24 h after transfection. Pulse-chase analysis of RUNX1 was performed as described for Fig. 5A except that the labeling time was 20 min, and the chase time was extended to 4 h. Data represent the means ± standard errors of the means from three independent experiments. (B) For pulse-labeling cells were transfected as described for panel A. To prevent protein degradation, all steps of the experiment were performed in the presence of MG132 (10 μM). Labeling was done as described for Fig. 5C except that the labeling time was extended to 20 min. RUNX1 was immunoprecipitated from total cell extracts with HA-specific antibody-conjugated agarose beads, separated by 12.5% PAGE, and visualized by fluorography. Quantitation of the gels was performed by PhosphorImager analysis as described for Fig. 5B and D. Data represent the means ± standard errors of the means from three independent experiments.
DISCUSSION
Regulation of protein expression in mammalian cells occurs at multiple levels. There is regulation at the level of de novo protein biosynthesis that includes transcriptional activation and mRNA splicing. At the other end of the spectrum is the turnover of proteins by proteolytic processes, the rate of which determines a protein's half-life. Proteolysis is not limited to mature proteins. In fact, protein synthesis in itself is imperfect, and up to 50% of nascent polypeptide chains in a cell may be degraded by processive proteolysis before reaching full size (57, 58). Thus, the overall steady-state level of a protein in a cell is derived from a combination of multiple parameters affecting protein synthesis and proteolytic degradation.
In the case of HIV, all proteins are encoded by a single mRNA precursor. Consequently, all HIV-encoded proteins are subject to the same transcriptional control. However, regulation of gene expression is achieved by differential splicing of the mRNA precursor, which regulates the relative levels of mRNAs encoding individual HIV proteins. In fact, only Gag and Gag-Pol precursor proteins are produced from full-length unspliced HIV-1 mRNAs while all other HIV-encoded proteins are produced from partially or fully spliced transcripts (59, 60). Vif is expressed from a partially spliced 5.3-kb mRNA that is produced in HIV-infected cells at relatively low abundance (59, 60). In addition, the inherent half-life of Vif is low due to rapid proteasome-mediated degradation (53). Thus, steady-state levels of Vif in HIV-infected cells are low compared to those of other viral proteins such as Gag, Env, and even Vpu. It is likely that HIV-1 has evolved to operate with low levels of Vif to avoid the negative impact of high levels of Vif on viral infectivity caused by interference of Vif with the maturation of the Gag precursor during virus assembly (54). Yet levels of Vif have to be sufficiently high to prevent the packaging of A3G into nascent virions. Thus, there is a relatively narrow range of Vif at which optimal support for virus replication can be achieved.
The recent identification of CBFβ as a Vif-interacting cellular protein exposed a new level of Vif regulation. Indeed, two studies reported reduced expression of Vif upon depletion of CBFβ (37, 40), a phenomenon that, curiously, was not observed in other studies (38, 44). Our own results clearly support the observation that CBFβ affects Vif expression. The mechanism of how CBFβ regulates Vif expression has thus far been obscure. CBFβ was previously known as a transcriptional cofactor regulating the DNA binding capacity of RUNX1 (61). In addition, RUNX1 and CBFβ were found to synergistically inhibit Tat-mediated HIV-1 long terminal repeat (LTR) transactivation (52). However, our data rule out a role of CBFβ in the transcriptional regulation of Vif expression. First, the effect of CBFβ is promoter independent and was observed for Vif expressed under the control of a CMV promoter in the absence of Tat (pcDNA-hVif and pVif-HA) as well as the Tat-dependent HIV-1 LTR (pNL4-3). The effect of CBFβ is also independent of the RNA sequence since codon-optimized Vif differs from non-codon-optimized Vif in 68 of 576 residues (11.8%). CBFβ also did not affect splicing since expression of Vif from partially (pNL4-3), fully (pVif-HA), and unspliced (pcDNA-hVif) vectors was equally sensitive to CBFβ. Finally, polysome analyses did not reveal any effects of CBFβ on activation of translation of Vif from pcDNA-hVif, suggesting that CBFβ affects steps downstream of translational initiation. Indeed, we demonstrate for the first time that Vif expression is regulated not only at the level of proteasomal degradation but also at the level of de novo protein biosynthesis. It is difficult to judge which of the two mechanisms contributes more to the changes in Vif steady-state levels. However, our data strongly indicate that CBFβ plays an important role in the regulation of Vif steady-state levels in HIV-infected cells. The effect of CBFβ on protein biosynthesis is not limited to Vif since we noted a similar effect on the synthesis of RUNX1. CBFβ was previously found to stabilize RUNX1 by inhibiting its proteasomal degradation (55). Our own data indicate that CBFβ increases the steady-state expression of RUNX1 but has only little effect on the half-life of RUNX1 in a 4-h observation period. The reason for this discrepancy is unclear. Adjusting our experimental conditions (i.e., labeling, lysis buffer, etc.) to those employed by Huang et al. (55) did not affect our results (data not shown). Alternatively, our studies were performed in CBFβ-knockdown HeLa cells while the studies by Huang et al. (55) involved mouse P19 cells. It is thus possible that cell-type- or species-specific differences account for the observed differences. Importantly, however, CBFβ enhanced the de novo protein biosynthesis of RUNX1, similar to its effect on Vif. Thus, the effect of CBFβ on protein biosynthesis is not limited to Vif.
Our data support the view that CBFβ, in addition to its effect on Vif expression, potentiates the ability of Vif to target A3G for degradation. Thus, at comparable levels of Vif, Vif-induced A3G degradation was significantly more effective when CBFβ was coexpressed. This observation is consistent with studies reporting CBFβ-facilitated A3G degradation in the absence of measurable effects on Vif expression (38, 44). Surprisingly, CBFβ did not have a similarly dramatic effect on the production of infectious virus. In fact, infectivity of virions produced in the absence of CBFβ was only approximately three times lower in wild-type NL4-3 than in CBFβ-expressing cells (Fig. 3C). The reason for this apparent disconnect between enhancing Vif expression and regulation of viral infectivity is not completely clear. However, the finding is consistent with our previous observation that the ability of Vif to produce infectious virus is not directly linked to its ability to efficiently induce A3G degradation, and the current data further support this notion (62).
How can we explain the seemingly unconnected effects of CBFβ on Vif biosynthesis, Vif stability, and Vif-induced A3G degradation? In fact, all of the effects described here and reported previously indicate that CBFβ has a chaperone-like function. For instance, a role of CBFβ in preventing premature termination of nascent Vif or RUNX1 polypeptide chains could explain the increased rate of Vif biosynthesis observed in our studies (Fig. 9A, step 1). This effect would require physical interaction of CBFβ with the nascent polypeptide chains and would therefore not be observed for CBFβ binding-defective mutants of Vif, consistent with our pulse-labeling results. Binding of CBFβ to Vif protein may also stabilize its mature conformation and thus reduce intrinsic proteolytic degradation (Fig. 9A, step 2). Finally, stabilizing a mature conformation of Vif would also explain its increased potential to induce degradation of A3G (Fig. 9B), as suggested previously (37, 38, 40, 43). In the absence of CBFβ, Vif can still bind to A3G. However, only in the presence of CBFβ will Vif assume a conformation that allows for the efficient assembly of a Cul5-based E3 ubiquitin ligase complex capable of ubiquitinating A3G and thus trigger its proteasomal degradation (Fig. 9B, step 3). In conclusion, all of the effects of CBFβ described in this study and reported previously can be explained by a chaperone-like activity of CBFβ that involves binding to nascent polypeptide chains of Vif or RUNX1 to prevent premature chain termination and/or to stabilize mature protein conformations.
FIG 9.

Model of CBFβ-mediated enhancement of Vif biosynthesis, stabilization of Vif protein, and enhancement of A3G degradation. (A) During Vif biosynthesis, CBFβ binds to nascent Vif polypeptide chains and inhibits their premature termination and degradation as defective ribosomal products (64) (step 1). This results in increased levels of newly synthesized Vif. CBFβ bound to Vif further stabilizes Vif and reduces intrinsic degradation of Vif by cellular proteasomes (step 2). The result is an increased half-life of Vif. (B) Vif binding to A3G is CBFβ independent. However, only in the presence of CBFβ can Vif effectively assemble a Cul5-based E3 ubiquitin (Ub) ligase complex (step 3) required for ubiquitination and subsequent proteolytic degradation of A3G. Other components of the Cul5 E3 ubiquitin ligase complex were omitted for simplicity.
ACKNOWLEDGMENTS
We thank Takeshi Yoshida, Sarah Welbourn, Haruka Yoshii-Kamiyama, Chia-Yen Chen, and Amy Andrew for helpful discussions and critical reading of the manuscript. We also thank Alicia Buckler-White and Ronald Plishka for conducting nucleotide sequence analyses. Finally, we thank Michael Malim for the Vif monoclonal antibody and Xiao-Fang Yu for the pVif-HA vector.
This work was supported by the Intramural Research Program of the NIH, NIAID, to K.S.
Footnotes
Published ahead of print 12 February 2014
REFERENCES
- 1.Goila-Gaur R, Strebel K. 2008. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 5:51. 10.1186/1742-4690-5-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albin JS, Harris RS. 2010. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev. Mol. Med. 12:e4. 10.1017/S1462399409001343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris RS, Liddament MT. 2004. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 4:868–877. 10.1038/nri1489 [DOI] [PubMed] [Google Scholar]
- 4.Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G. 2002. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 277:39926–39936. 10.1074/jbc.M207107200 [DOI] [PubMed] [Google Scholar]
- 5.Khan MA, Goila-Gaur R, Kao S, Miyagi E, Walker RC, Jr, Strebel K. 2009. Encapsidation of APOBEC3G into HIV-1 virions involves lipid raft association and does not correlate with APOBEC3G oligomerization. Retrovirology 6:99. 10.1186/1742-4690-6-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khan MA, Kao S, Miyagi E, Takeuchi H, Goila-Gaur R, Opi S, Gipson CL, Parslow TG, Ly H, Strebel K. 2005. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 79:5870–5874. 10.1128/JVI.79.9.5870-5874.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Svarovskaia ES, Xu H, Mbisa JL, Barr R, Gorelick RJ, Ono A, Freed EO, Hu WS, Pathak VK. 2004. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J. Biol. Chem. 279:35822–35828. 10.1074/jbc.M405761200 [DOI] [PubMed] [Google Scholar]
- 8.Burnett A, Spearman P. 2007. APOBEC3G multimers are recruited to the plasma membrane for packaging into human immunodeficiency virus type 1 virus-like particles in an RNA-dependent process requiring the NC basic linker. J. Virol. 81:5000–5013. 10.1128/JVI.02237-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cen S, Guo F, Niu M, Saadatmand J, Deflassieux J, Kleiman L. 2004. The interaction between HIV-1 Gag and APOBEC3G. J. Biol. Chem. 279:33177–33184. 10.1074/jbc.M402062200 [DOI] [PubMed] [Google Scholar]
- 10.Douaisi M, Dussart S, Courcoul M, Bessou G, Vigne R, Decroly E. 2004. HIV-1 and MLV Gag proteins are sufficient to recruit APOBEC3G into virus-like particles. Biochem. Biophys. Res. Commun. 321:566–573. 10.1016/j.bbrc.2004.07.005 [DOI] [PubMed] [Google Scholar]
- 11.Luo K, Liu B, Xiao Z, Yu Y, Yu X, Gorelick R, Yu XF. 2004. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J. Virol. 78:11841–11852. 10.1128/JVI.78.21.11841-11852.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navarro F, Bollman B, Chen H, Konig R, Yu Q, Chiles K, Landau NR. 2005. Complementary function of the two catalytic domains of APOBEC3G. Virology 333:374–386. 10.1016/j.virol.2005.01.011 [DOI] [PubMed] [Google Scholar]
- 13.Schafer A, Bogerd HP, Cullen BR. 2004. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology 328:163–168. 10.1016/j.virol.2004.08.006 [DOI] [PubMed] [Google Scholar]
- 14.Zennou V, Perez-Caballero D, Gottlinger H, Bieniasz PD. 2004. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 78:12058–12061. 10.1128/JVI.78.21.12058-12061.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alce TM, Popik W. 2004. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J. Biol. Chem. 279:34083–34086. 10.1074/jbc.C400235200 [DOI] [PubMed] [Google Scholar]
- 16.Kao S, Khan MA, Miyagi E, Plishka R, Buckler-White A, Strebel K. 2003. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J. Virol. 77:11398–11407. 10.1128/JVI.77.21.11398-11407.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060. 10.1126/science.1089591 [DOI] [PubMed] [Google Scholar]
- 18.Marin M, Rose KM, Kozak SL, Kabat D. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9:1398–1403. 10.1038/nm946 [DOI] [PubMed] [Google Scholar]
- 19.Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404–1407. 10.1038/nm945 [DOI] [PubMed] [Google Scholar]
- 20.Stopak K, de Noronha C, Yonemoto W, Greene WC. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12:591–601. 10.1016/S1097-2765(03)00353-8 [DOI] [PubMed] [Google Scholar]
- 21.Conticello SG, Harris RS, Neuberger MS. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 13:2009–2013. 10.1016/j.cub.2003.10.034 [DOI] [PubMed] [Google Scholar]
- 22.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. 2004. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 279:7792–7798. 10.1074/jbc.M313093200 [DOI] [PubMed] [Google Scholar]
- 23.Niewiadomska AM, Yu XF. 2009. Host restriction of HIV-1 by APOBEC3 and viral evasion through Vif. Curr. Top. Microbiol. Immunol. 339:1–25. 10.1007/978-3-642-02175-6_1 [DOI] [PubMed] [Google Scholar]
- 24.Chen G, He Z, Wang T, Xu R, Yu XF. 2009. A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J. Virol. 83:8674–8682. 10.1128/JVI.00653-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dang Y, Davis RW, York IA, Zheng YH. 2010. Identification of 81LGxGxxIxW89 and 171EDRW174 domains from human immunodeficiency virus type 1 Vif that regulate APOBEC3G and APOBEC3F neutralizing activity. J. Virol. 84:5741–5750. 10.1128/JVI.00079-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang Y, Wang X, York IA, Zheng YH. 2010. Identification of a critical T(Q/D/E)x5ADx2(I/L) motif from primate lentivirus Vif proteins that regulate APOBEC3G and APOBEC3F neutralizing activity. J. Virol. 84:8561–8570. 10.1128/JVI.00960-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang Y, Wang X, Zhou T, York IA, Zheng YH. 2009. Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization. J. Virol. 83:8544–8552. 10.1128/JVI.00651-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He Z, Zhang W, Chen G, Xu R, Yu XF. 2008. Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J. Mol. Biol. 381:1000–1011. 10.1016/j.jmb.2008.06.061 [DOI] [PubMed] [Google Scholar]
- 29.Pery E, Rajendran KS, Brazier AJ, Gabuzda D. 2009. Regulation of APOBEC3 proteins by a novel YXXL motif in human immunodeficiency virus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J. Virol. 83:2374–2381. 10.1128/JVI.01898-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell RA, Pathak VK. 2007. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 81:8201–8210. 10.1128/JVI.00395-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huthoff H, Malim MH. 2007. Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and virion encapsidation. J. Virol. 81:3807–3815. 10.1128/JVI.02795-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell RA, Smith J, Barr R, Bhattacharyya D, Pathak VK. 2009. Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J. Virol. 83:1992–2003. 10.1128/JVI.01621-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. U. S. A. 102:11444–11449. 10.1073/pnas.0502440102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehle A, Thomas ER, Rajendran KS, Gabuzda D. 2006. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J. Biol. Chem. 281:17259–17265. 10.1074/jbc.M602413200 [DOI] [PubMed] [Google Scholar]
- 35.Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu XF. 2006. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology 349:290–299. 10.1016/j.virol.2006.02.002 [DOI] [PubMed] [Google Scholar]
- 36.Xiao Z, Xiong Y, Zhang W, Tan L, Ehrlich E, Guo D, Yu XF. 2007. Characterization of a novel Cullin5 binding domain in HIV-1 Vif. J. Mol. Biol. 373:541–550. 10.1016/j.jmb.2007.07.029 [DOI] [PubMed] [Google Scholar]
- 37.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. 2011. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481:371–375. 10.1038/nature10693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang W, Du J, Evans SL, Yu Y, Yu XF. 2011. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481:376–379. 10.1038/nature10718 [DOI] [PubMed] [Google Scholar]
- 39.Blyth K, Cameron ER, Neil JC. 2005. The RUNX genes: gain or loss of function in cancer. Nat. Rev. Cancer 5:376–387. 10.1038/nrc1607 [DOI] [PubMed] [Google Scholar]
- 40.Hultquist JF, Binka M, LaRue RS, Simon V, Harris RS. 2012. Vif proteins of human and simian immunodeficiency viruses require cellular CBFβ to degrade APOBEC3 restriction factors. J. Virol. 86:2874–2877. 10.1128/JVI.06950-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou X, Evans SL, Han X, Liu Y, Yu X-F. 2012. Characterization of the interaction of full-length HIV-1 Vif protein with its key regulator CBFβ and CRL5 E3 ubiquitin ligase components. PLoS One 7:e33495. 10.1371/journal.pone.0033495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hultquist JF, McDougle RM, Anderson BD, Harris RS. 2012. HIV type 1 viral infectivity factor and the RUNX transcription factors interact with core binding factor β on genetically distinct surfaces. AIDS Res. Hum. Retroviruses 28:1543–1551. 10.1089/aid.2012.0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DY, Kwon E, Hartley PD, Crosby DC, Mann S, Krogan NJ, Gross JD. 2013. CBFβ stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol. Cell 49:632–644. 10.1016/j.molcel.2012.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du J, Zhao K, Rui Y, Li P, Zhou X, Zhang W, Yu X-F. 2013. Differential requirements for HIV-1 Vif-mediated APOBEC3G degradation and RUNX1-mediated transcription by core binding factor beta. J. Virol. 87:1906–1911. 10.1128/JVI.02199-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou J, Zhang Z, Mi Z, Wang X, Zhang Q, Li X, Liang C, Cen S. 2012. Characterization of the interface of the bone marrow stromal cell antigen 2-Vpu protein complex via computational chemistry. Biochemistry 51:1288–1296. 10.1021/bi2015986 [DOI] [PubMed] [Google Scholar]
- 46.Salter JD, Lippa GM, Belashov IA, Wedekind JE. 2012. Core-binding factor β increases the affinity between human Cullin 5 and HIV-1 Vif within an E3 ligase complex. Biochemistry 51:8702–8704. 10.1021/bi301244z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karczewski MK, Strebel K. 1996. Cytoskeleton association and virion incorporation of the human immunodeficiency virus type 1 Vif protein. J. Virol. 70:494–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kao S, Miyagi E, Khan MA, Takeuchi H, Opi S, Goila-Gaur R, Strebel K. 2004. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology 1:27. 10.1186/1742-4690-1-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen KL, Llano M, Akari H, Miyagi E, Poeschla EM, Strebel K, Bour S. 2004. Codon optimization of the HIV-1 vpu and vif genes stabilizes their mRNA and allows for highly efficient Rev-independent expression. Virology 319:163–175. 10.1016/j.virol.2003.11.021 [DOI] [PubMed] [Google Scholar]
- 50.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walker RC, Jr, Khan MA, Kao S, Goila-Gaur R, Miyagi E, Strebel K. 2010. Identification of dominant negative human immunodeficiency virus type 1 Vif mutants that interfere with the functional inactivation of APOBEC3G by virus-encoded Vif. J. Virol. 84:5201–5211. 10.1128/JVI.02318-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cunningham L, Finckbeiner S, Hyde RK, Southall N, Marugan J, Yedavalli VRK, Dehdashti SJ, Reinhold WC, Alemu L, Zhao L, Yeh J-RJ, Sood R, Pommier Y, Austin CP, Jeang K-T, Zheng W, Liu P. 2012. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBFβ interaction. Proc. Natl. Acad. Sci. U. S. A. 109:14592–14597. 10.1073/pnas.1200037109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fujita M, Akari H, Sakurai A, Yoshida A, Chiba T, Tanaka K, Strebel K, Adachi A. 2004. Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 6:791–798. 10.1016/j.micinf.2004.04.011 [DOI] [PubMed] [Google Scholar]
- 54.Akari H, Fujita M, Kao S, Khan MA, Shehu-Xhilaga M, Adachi A, Strebel K. 2004. High level expression of human immunodeficiency virus type-1 Vif inhibits viral infectivity by modulating proteolytic processing of the Gag precursor at the p2/nucleocapsid processing site. J. Biol. Chem. 279:12355–12362. 10.1074/jbc.M312426200 [DOI] [PubMed] [Google Scholar]
- 55.Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y. 2001. Dimerization with PEBP2β protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J. 20:723–733. 10.1093/emboj/20.4.723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akamatsu Y, Tsukumo S, Kagoshima H, Tsurushita N, Shigesada K. 1997. A simple screening for mutant DNA binding proteins: application to murine transcription factor PEBP2α subunit, a founding member of the Runt domain protein family. Gene 185:111–117. 10.1016/S0378-1119(96)00644-0 [DOI] [PubMed] [Google Scholar]
- 57.Wheatley DN, Giddings MR, Inglis MS. 1980. Kinetics of degradation of “short-” and “long-lived” proteins in cultured mammalian cells. Cell Biol. Int. Rep. 4:1081–1090. 10.1016/0309-1651(80)90045-4 [DOI] [PubMed] [Google Scholar]
- 58.Turner GC, Varshavsky A. 2000. Detecting and measuring cotranslational protein degradation in vivo. Science 289:2117–2120. 10.1126/science.289.5487.2117 [DOI] [PubMed] [Google Scholar]
- 59.Rabson A, Daugherty D, Venkatesan S, Boulukos KE, Benn S, Folks TM, Feorino P, Martin MA. 1985. Transcription of novel open reading frames of AIDS retrovirus during infection of lymphocytes. Science 229:1388–1390. 10.1126/science.2994220 [DOI] [PubMed] [Google Scholar]
- 60.Karn J, Stoltzfus CM. 2012. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2:a006916. 10.1101/cshperspect.a006916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, Shiina M, Sato K, Kumasaka T, Yamamoto M, Ishii S, Ogata K. 2001. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFβ. Cell 104:755–767. 10.1016/S0092-8674(01)00271-9 [DOI] [PubMed] [Google Scholar]
- 62.Kao S, Goila-Gaur R, Miyagi E, Khan MA, Opi S, Takeuchi H, Strebel K. 2007. Production of infectious virus and degradation of APOBEC3G are separable functional properties of human immunodeficiency virus type 1 Vif. Virology 369:329–339. 10.1016/j.virol.2007.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tscherne JS, Pestka S. 1975. Inhibition of protein synthesis in intact HeLa cells. Antimicrob. Agents Chemother. 8:479–487. 10.1128/AAC.8.4.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. 2000. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404:770–774. 10.1038/35008096 [DOI] [PubMed] [Google Scholar]
- 65.Klase Z, Yedavalli VSRK, Houzet L, Perkins M, Maldarelli F, Brenchley J, Strebel K, Liu P, Jeang KT. Activation of HIV-1 from latent infection via synergy of RUNX1 inhibitor Ro5-3335 and SAHA. PLoS Pathog., in press [DOI] [PMC free article] [PubMed] [Google Scholar]