

ABSTRACT
Antivector immunity limits the response to homologous boosting for viral vector vaccines. Here, we describe a new, potent vaccine vector based on replication-competent vesicular stomatitis virus pseudotyped with the glycoprotein of the lymphocytic choriomeningitis virus (VSV-GP), which we previously showed to be safe in mice. In mice, VSV and VSV-GP encoding ovalbumin (OVA) as a model antigen (VSV-OVA and VSV-GP-OVA) induced equal levels of OVA-specific humoral and cellular immune responses upon a single immunization. However, boosting with the same vector was possible only for VSV-GP-OVA as neutralizing antibodies to VSV limited the immunogenicity of the VSV-OVA boost. OVA-specific cytotoxic T-lymphocyte (CTL) responses induced by VSV-GP-OVA were at least as potent as those induced by an adenoviral state-of-the-art vaccine vector and completely protected mice in a Listeria monocytogenes challenge model. VSV-GP is so far the only replication-competent vaccine vector that does not lose efficacy upon repeated application.
IMPORTANCE Although there has been great progress in treatment and prevention of infectious diseases in the past several years, effective vaccines against some of the most serious infections, e.g., AIDS, malaria, hepatitis C, or tuberculosis, are urgently needed. Here, several approaches based on viral vector vaccines are under development. However, for all viral vaccine vectors currently in clinical testing, repeated application is limited by neutralizing antibodies to the vector itself. Here, we have exploited the potential of vesicular stomatitis virus pseudotyped with the glycoprotein of the lymphocytic choriomeningitis virus (VSV-GP) as a vaccine platform. VSV-GP is the first replication-competent viral vector vaccine that does not induce vector-specific humoral immunity, i.e., neutralizing antibodies, and therefore can boost immune responses against a foreign antigen by repeated applications. The vector allows introduction of various antigens and therefore can serve as a platform technology for the development of novel vaccines against a broad spectrum of diseases.
INTRODUCTION
Despite the progress in treatment and prevention of many infectious diseases, there is still an urgent need for effective vaccines against diseases like AIDS, malaria, hepatitis C, or tuberculosis. Many successful vaccines are based on live-attenuated pathogens (e.g., those for polio, measles, mumps, and rubella) and usually confer strong and long-lasting immunity (1, 2). For those infectious agents that cannot be attenuated or where, despite attenuation, safety concerns remain (e.g., HIV), replication-competent viral vector vaccines can be used. These viral vectors express the antigen of choice with the expectation that the induced immunity will be as strong and lasting as the response to live-attenuated vaccines. An ideal viral vector vaccine must fulfill several criteria: it must be safe, it must induce strong and durable cellular and humoral immune responses, there should be no preexisting immunity in the human population, and ideally the vaccine should not lose its potency upon repeated application. All current replication-competent viral vector vaccines lose efficacy upon repeated application but are very effective in a heterologous prime-boost regimen. Preexisting or vector-induced neutralizing antibodies can limit replication of the vector vaccine as shown, e.g., for adenoviral and poxvirus-based vector vaccines (3–5).
Vesicular stomatitis virus (VSV), a negative-strand RNA virus of the rhabdovirus family, is a potent candidate vaccine vector. VSV-based vaccines can mount both strong humoral (6) and potent cellular immune responses against pathogens (7). Accordingly, VSV vectors have been shown to induce protective immunity against a large number of different pathogens in animal models, e.g., HIV, influenza virus, Marburg virus, and Ebola virus (8–10). VSV has several characteristics required for an ideal vaccine vector: the general population lacks antibodies against VSV, which can interfere with vaccination efficacy (11); infections in humans are rare and mostly asymptomatic (12); there is no risk of genotoxicity as the virus does not integrate into the host genome and replicates in the cytoplasm (13); due to the helical structure of the capsid, the viral genome can accommodate large antigenic transgenes (14); the packaging capacity of VSV can even be increased further by using a semi-replication-competent vector system, consisting of a pair of VSV variants with trans-complementing mutations (15); and finally, as VSV is an enveloped virus, viral glycoproteins are potentially presented in their natural, lipid envelope-associated conformation on the viral surface, thereby exposing conformational epitopes not always found in the recombinant protein. Such conformational epitopes on the surface of a viral pathogen can be potent inducers of neutralizing antibodies.
However, VSV has two major limitations. Neutralizing antibodies against the VSV glycoprotein G are already induced after the first application, so that homologous boosting with VSV-based vector vaccines is ineffective (6). Therefore, for VSV, boosting can be achieved only by using multiple serotypes (16). Additionally, VSV replicates in neurons and thereby can cause viral encephalitis (17, 18). Therefore, only attenuated VSV variants can be used in humans, but these, however, have been found to be less potent (6, 19).
Our group has recently shown that neurotropism (studied in mice and human neurons) and neurotoxicity (mice) are completely abolished by pseudotyping viruses with the glycoprotein (GP) of the lymphocytic choriomeningitis virus (LCMV) WE-HPI strain (20–22). In contrast to the LCMV Armstrong strain, the WE strain of LCMV is not neurotropic and is not significantly pathogenic (23). The LCMV-GP-WE pseudotype VSV-GP is not neurotoxic even at high intracranial doses in mice. Further extensive safety studies in mice have not shown any pathogenicity of VSV-GP even at extremely high systemic doses (A. Muik, L. J. Stubbert, R. Z. Jahedi, Y. Geiss, C. Dold, R. Tober, A. Volk, S. Klein, U. Dietrich, B. Yadollahi, T. Falls, H. Miletic, D. Stojdl, J. C. Bell, and D. von Laer, submitted for publication).
Here, we evaluated the immunogenicity of a VSV-GP-based vector vaccine using the model antigen ovalbumin (OVA) in mice. We found that VSV-GP was as immunogenic as VSV and adenoviral vector vaccines upon prime, but VSV-GP was the only vector that did not induce neutralizing antibodies to the vector itself and therefore did not lose immunogenicity upon repeated application.
MATERIALS AND METHODS
Ethics statement.
Animal experiments were performed in compliance with the Austrian national animal experimentation law (“Tierversuchsgesetz”), and animal trial permission was granted by Austrian national authorities (Bundesministerium für Wissenschaft und Forschung, no. 66.011/154-II/3b/2011).
Cell lines and bacteria.
BHK-21 cells (American Type Culture Collection, Manassas, VA) were cultured in Glasgow minimum essential medium (GMEM) (Gibco, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS; PAA Laboratories), 5% tryptose phosphate broth (Gibco, Carlsbad, CA), 100 units/ml penicillin (Gibco), and 0.1 mg/ml streptomycin (Gibco). Murine splenocytes were isolated from spleens of immunized C57BL/6 mice, treated with ACK buffer (Lonza, Basel, Switzerland) to lyse erythrocytes, washed with phosphate-buffered saline (PBS), and subsequently cultured in RPMI 1620 medium (Lonza, Basel, Switzerland) supplemented with 10% FCS.
A Listeria monocytogenes strain expressing OVA134–387 (Lm_OVA) (24) was kindly provided by Hao Shen, University of Pennsylvania, USA. Bacteria were grown in LB medium supplemented with 5 μg/ml erythromycin and 5 mg/liter glucose.
Viruses.
VSV, VSV-GP, VSV-gfp, and VSV-GP-gfp were described previously (25, 26) (Muik et al., submitted). VSV-OVA and VSV-GP-OVA were generated de novo. The OVA-enhanced green fluorescent protein (eGFP) fusion cassette was inserted on position 5 in the viral genomes between G/GP and L genes. For construction of the OVA-eGFP fusion cassette, the full-length OVA sequence was amplified (GenBank sequence accession number NM_205152) using primers 5′-GCA TGG ACG AGC TGT ACA AGA TGG GCT CCA TCG GCG CA-3′ and 5′-CAA ACA TGA AGA ATC TGG CTA GAT CAT CAA GGG GAA ACA CAT CTG CC-3′. The eGFP sequence starting with a unique NheI restriction site and the intergenic region between the VSV G and L genes was obtained from pVSV-gfp using primers 5′-AAA GTA ACT CAA ATC CTG CTA GG-3′ and 5′-TGC GCC GAT GGA GCC CAT CTT GTA CAG CTC GTC CAT GC-3′. The sequence spanning the L gene up to a unique HpaI restriction site was amplified with primers 5′-GGC AGA TGT GTT TCC CCT TGA TGA TCT AGC CAG ATT CTT CAT GTT TG-3′ and 5′-GTA AAA AAC TAT ACC CTT GAC TGG-3′ from pVSV-gfp. To obtain the full-length eGFP_OVA sequence, a fusion PCR with these three PCR products and the primers 5′-AAA GTA ACT CAA ATC CTG CTA GG-3′ and 5′-GTA AAA AAC TAT ACC CTT GAC TGG-3′ was performed. The resulting cassette was ligated via unique NheI/HpaI sites into pVSV-GP to obtain pVSV-GP-OVA. To create pVSV-OVA, LCMV-GP was replaced in pVSV-GP-OVA by VSV-G from pVSV-XN2 using the unique restriction sites MluI and XhoI.
ΔM51-GP-OVA was created by exchange of wild-type M in VSV-GP-OVA with an M gene containing the ΔM51 mutation (kindly provided by Oliver Ebert, Munich, Germany).
Recombinant VSVs were rescued and plaque purified as described elsewhere (27). Titers were determined on confluent BHK-21 monolayers via plaque assay.
The adenovirus vectors described here are replication-defective E1-deleted vectors based on human adenovirus type 5 (Ad5) and bear a human cytomegalovirus (hCMV) promoter-driven expression cassette for secreted, full-length ovalbumin or N-terminally truncated intracellular ovalbumin. Cloning details can be obtained upon request. Vectors were produced on N52.E6 cells (28) and purified by double CsCl gradients. Vector titration was performed by a DNA-based slot blot procedure (29).
Preparation of cell lysates and Western blot analysis.
BHK-21 cells were infected with VSV-OVA or VSV-GP-OVA at a multiplicity of infection (MOI) of 0.1, and cell lysates were prepared 24 h later. Uninfected BHK-21 cells were used as a control. Cells were lysed in ice-cold cell lysis buffer (50 mmol/liter HEPES, pH 7.5; 150 mmol/liter NaCl; 1% Triton X-100; 2% aprotinin; 2 mmol/liter EDTA, pH 8.0; 50 mmol/liter sodium fluoride; 10 mmol/liter sodium pyrophosphate; 10% glycerol; 1 mmol/liter sodium vanadate; and 2 mmol/liter Pefabloc SC) for 30 min. Subsequently, cell lysates were centrifuged (13,000 rpm) for 10 min to remove cell debris, and lysates were stored at −80°C until use.
SDS-PAGE of protein lysates was performed under standard reducing conditions on a 10% polyacrylamide gel. Proteins were electrophoretically transferred to 0.45-μm nitrocellulose membranes (Whatman, Dassel, Germany). Membranes were blocked with PBSTM (PBS containing 5% skim milk and 0.1% Tween 20) and stained overnight at 4°C with a GFP-specific mouse monoclonal antibody (B-2, sc-9996; Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:1,000 in PBSTM. Detection was performed with a peroxidase-conjugated mouse IgG-specific antibody from goat (Invitrogen, Carlsbad, CA), diluted 1:10,000 in PBSTM. Blots were developed with enhanced chemiluminescence (ECL) (30). For beta-actin staining, blots were stripped with NaOH (0.1%) for 10 min at room temperature and blocked with PBSTM. Actin was stained with a beta-actin-specific monoclonal antibody from mouse (A2228; Sigma, Munich, Germany) diluted 1:5,000 in PBSTM and a secondary horseradish peroxidase-conjugated mouse IgG-specific antibody from goat and development with ECL.
TCID50 assay.
For in vitro growth curves, virus titers were determined using a 50% tissue culture infective dose (TCID50) assay using the method of Spearman-Kärber as described previously (31). Briefly, 10-fold serial dilutions of virus were prepared. One hundred microliters of each dilution was added in quadruplicate to confluent BHK-21 cells in 96-well plates and incubated for 24 to 48 h at 37°C until a cytopathic effect was visible. Numbers of infected wells were counted, and TCID50 values were calculated.
Mouse experiments.
C57BL/6 wild-type mice were purchased from Harlan Laboratories (Rossdorf, Germany) and bred and maintained in the animal facilities of the Innsbruck Medical University. All experiments were performed in compliance with local animal experimentation guidelines and approved by local authorities (Bundesministerium für Wissenschaft und Forschung no. 66.011/154-II/3b/2011). Mice were immunized intramuscularly with 1 × 106 PFU of VSV vectors or 2 × 109 vector particles (vp) of recombinant adenovirus vectors diluted in 50 μl PBS, respectively. Control animals received immunizations with 50 μl PBS without virus.
Serum transfer experiment.
C57BL/6 mice were immunized on days 0 and 26 with VSV-OVA or VSV-GP-OVA. For generation of nonimmune sera, naive C57BL/6 mice were used. On day 7 post-boost immunization, mice were bled. Blood was allowed to coagulate for 1 h at room temperature, and subsequently, serum was obtained by centrifugation for 5 min at 8,000 rpm. Serum was stored at −80°C till use. Three hundred fifty to 500 μl of serum was injected intravenously into naive C57BL/6 mice. Mice were immunized 24 h later with the respective virus or PBS as control. On day 7 postimmunization, OVA-specific cytotoxic T-lymphocyte (CTL) responses were determined via tetramer and intracellular cytokine staining.
IFN-γ enzyme-linked immunosorbent spot (ELISpot) assay.
The mouse gamma interferon (IFN-γ) ELISpot Plus kit (Mabtech, Nacka Strand, Sweden) was used to quantify ovalbumin-specific CD8+ T cells. The assay was performed according to the manufacturer's protocol. Briefly, 2 × 105 splenocytes per well were incubated under standard cell culture conditions in 96-well plates precoated with a mouse IFN-γ-specific antibody. For stimulation of OVA-specific CTLs, 2.5 μg/ml of a peptide containing the ovalbumin-specific CTL epitope (SIINFEKL) was added. Anti-CD3/anti-CD28-coupled magnetic beads (32) were added to stimulate CTLs in positive-control wells, while negative-control wells remained unstimulated. After 20 h, the cells were removed. The plates were subsequently incubated with a biotinylated IFN-γ-specific antibody and a streptavidin-horseradish peroxidase (HRP)-conjugated secondary antibody. The plates were developed with tetramethylbenzidine (TMB) substrate, and spots were counted in a CTL-Immunospot plate reader (CTL, Cleveland, OH). The assay was performed in duplicate, and counts of unstimulated samples were subtracted from those of peptide-stimulated samples.
Intracellular cytokine staining.
Intracellular production of the cytokines IFN-γ, tumor necrosis factor alpha (TNF-α), and interleukin-2 (IL-2) was examined by flow cytometric analysis. Briefly, 106 splenocytes were incubated in the presence or absence of 2.5 μg/ml peptide (SIINFEKL) in standard medium supplemented with 1 μg/ml GolgiPlug (BD, Franklin Lakes, NJ, USA) for 6 h at 37°C in a humidified 6% CO2 incubator. Cells were washed once with fluorescence-activated cell sorting (FACS) buffer (PBS supplemented with 1% fetal calf serum and 0.05% sodium azide) and stained with a peridinin chlorophyll protein (PerCp)-Cy5-conjugated CD8+-specific antibody (BD, Franklin Lakes, NJ, USA). Intracellular staining with fluorescein isothiocyanate (FITC)-conjugated IFN-γ-specific, phycoerythrin (PE)-conjugated IL-2-specific, and allophycocyanin (APC)-conjugated TNF-α-specific antibodies (BD), respectively, was performed using the Cytofix/Cytoperm fixation/permeabilization solution kit (BD) according to the manufacturer's instructions. Samples were measured and analyzed using a FACSCanto II cytometer (BD) and DIVA software.
Tetramer staining.
SIINFEKL-specific CD8+ T cell counts were determined using APC-conjugated iTAg tetramers (iTAg tetramer/APC–H–2-kb OVA [SIINFEKL]; Beckman Coulter, catalog no. T03002). Splenocytes (106) were stained with PE-conjugated CD8+-specific, FITC-conjugated CD3+-specific antibodies (BD) and 2.5 μl of tetramer. After washing with FACS buffer, cells were fixed with 3.7% formaldehyde. The proportion of tetramer-positive CD8+ T cells was determined by flow cytometric analysis using a FACSCanto II cytometer and DIVA software.
Anti-OVA IgG enzyme-linked immunosorbent assay (ELISA).
Ninety-six-well microtiter plates were coated overnight at 4°C with 100 μl of ovalbumin protein (ICN Biomedicals Inc., Costa Mesa, CA) at a concentration of 100 μg/ml in PBS. Nonspecific binding sites were blocked with 100 μl PBSTM for 1 h at 37°C. All washing steps were performed with PBS containing 0.1% Tween 20 (PBST). Mouse plasma was diluted in PBST in 1:4 serial dilutions, starting with a 1:40 dilution. One hundred microliters of plasma dilution was added to the coated wells in duplicate and incubated for 1 h at 37°C. Detection was performed with a horseradish peroxidase-conjugated mouse IgG-specific antibody from goat (diluted 1:10,000; Invitrogen, Carlsbad, CA) and Sure Blue TMB detection reagent and TMB stop solution (KPL, Gaithersburg, MD). Plates were analyzed at 450 nm (signal) and 650 nm (background) on a model 680 microplate reader (Bio-Rad, Hercules, CA) using Microplate Manager 5.2.1 software (Bio-Rad, Hercules, CA). Endpoint titers were determined as the reciprocal maximum dilution at which the mean optical density at 450 nm (OD450) − OD650 of duplicates was greater than the mean OD450 − OD650 plus 2 standard deviations of naive sera.
For absolute quantification, a standard curve was generated using 2-fold serial dilutions (128 ng to 0.125 ng per well) of an ovalbumin-specific monoclonal antibody from mouse (A6075; Sigma).
LCMV-GP1 IgG ELISA.
ELISA plates (PS-Microplate, 96 wells; Greiner-Bio One) were coated overnight at 4°C with F(ab)2 fragments of anti-human IgG (Jackson ImmunoResearch) diluted 1:800 in 0.1 M sodium carbonate buffer (pH 9.6). After a 2-h blocking step at room temperature with 2% bovine serum albumin (BSA)-PBS, the F(ab)2 fragments were loaded with 100 μl/well of purified Fc-LCMV-GP1 (33) and incubated for 1 h at room temperature. On a parallel plate, plasma samples were prediluted 1:8 in 0.1% BSA-PBS and a 3-fold dilution series was performed. A total of 50 μl/well was applied to the Fc-LCMV-GP1-saturated plate and incubated for 1 h at room temperature. LCMV-GP1-specific IgG antibodies were detected with anti-mouse IgG-γ-HRP (Sigma) diluted 1:100 in 0.1% BSA-PBS. Plates were developed with 100 μl/well of a solution containing 0.2 mg/ml 2,2′-azino-di-(3-ethylbenzthiazoline sulfonic acid), 0.1 M NaH2PO4, 0.04% H2O2, pH 4. Plates were read at 405 nm in a Victor3 reader (Wallac 1420; PerkinElmer). Between each step, plates were washed five times with PBST. Titers were determined as the reciprocal maximum dilution at which the OD405 was greater than the 2-fold OD405 signal of the naive plasma.
Antiadenovirus ELISA.
Of a purified E1-deleted Ad5 vector without a transgene expression cassette, 6 × 108 vp were coated in 0.2 M Na2CO3-NaHCO3, pH 9.5, overnight in 96-well Maxisorp plates (Nunc). Blocking was performed for 1 h at room temperature with 3% (wt/vol) BSA in PBS. After blocking, serial dilutions of the mouse sera in blocking buffer were transferred into the wells and incubated for 2 h at room temperature. Detection was performed using a horseradish peroxidase-conjugated mouse IgG-specific antibody from goat (BD 554002) (incubation of a 1:2,000 dilution for 1 h at 37°C). Plates were analyzed at 491 nm (signal) and 620 nm (background).
VSV neutralization assay.
The VSV neutralization assay was performed as described elsewhere (34). Briefly, mouse plasma samples were diluted in 2-fold serial dilutions in PBS, starting with a 1:10 dilution. One hundred PFU of VSV-GFP or VSV-GP-GFP diluted in 50 μl serum-free GMEM was mixed with 50 μl of plasma dilution in duplicate samples, and mixtures were incubated at 37°C for 1 h. Then, samples were transferred to 96-well plates containing a monolayer of confluent BHK-21 cells in 100 μl complete GMEM. Plates were incubated at 37°C, 6% CO2, for 2 to 3 days and analyzed for cytopathic effect. Neutralizing titers are given as the highest plasma dilution which completely inhibited a VSV-GFP- or VSV-GP-GFP-induced cytopathic effect.
Lm_OVA challenge.
Immunized mice were intravenously challenged on day 7 postboost with 1 × 105 CFU of Listeria monocytogenes expressing OVA (Lm_OVA). On day 3 postchallenge, mice were sacrificed and splenocytes were isolated. Tenfold serial dilutions of splenocytes were performed in PBS. Dilutions were plated in duplicate on LB plates supplemented with 5 μg/ml erythromycin and 5 mg/liter glucose. After 2 days, colonies were counted and the number of CFU per spleen was calculated.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA).
RESULTS
Construction of VSV and VSV-GP vectors containing the model antigen ovalbumin.
We introduced the model antigen ovalbumin (OVA) fused to eGFP at position 5 into the VSV and VSV-GP genomes (Fig. 1A). After infection of BHK-21 cells with equal MOIs, similar amounts of OVA-eGFP fusion protein were expressed by the two viruses as determined by Western blotting (Fig. 1B). Both viruses replicated well in BHK-21 cells with a slightly delayed replication of VSV-GP-OVA compared to VSV-OVA in the initial phase. However, the two viruses reached comparable titers after 24 h (Fig. 1C).
FIG 1.
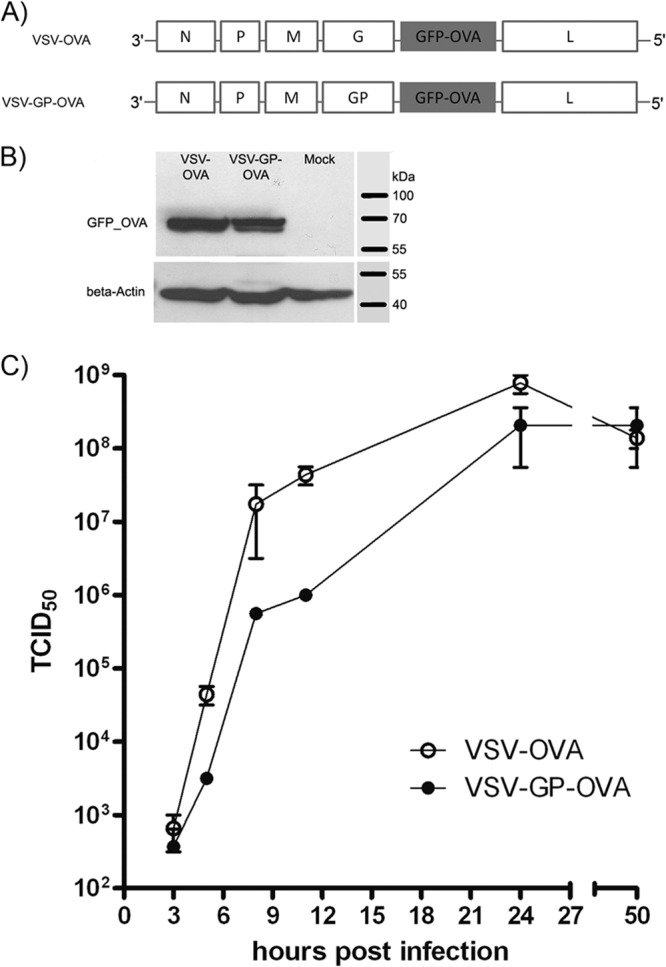
VSV vectors containing OVA antigen. (A) Genomes of VSV-OVA and GP-pseudotyped VSV-GP-OVA vectors containing ovalbumin (OVA). The sequence coding for full-length OVA, C terminally fused to eGFP, was introduced at position 5 into both vectors. (B) BHK-21 cells were infected with VSV-OVA or VSV-GP-OVA at an MOI of 0.1, and 24 h later, cell lysates were prepared. The eGFP_OVA fusion protein (69 kDa) was detected by Western blotting using a GFP-specific antibody. (C) BHK-21 cells were infected with VSV-OVA or VSV-GP-OVA at an MOI of 0.1 in duplicate. Virus production at the indicated time points was determined by TCID50 assay.
VSV-GP boosts immune response.
In a prime/boost immunization experiment, C57BL/6 mice were immunized intramuscularly with VSV-OVA, VSV-GP-OVA, or respective control vectors without the OVA transgene (VSV and VSV-GP) and cellular and humoral immune responses against OVA were analyzed at different time points. Only mice immunized with vectors containing the OVA transgene developed OVA-specific T cells and antibodies, whereas control mice immunized with vector VSV or VSV-GP did not (Fig. 2 and data not shown). Seven days post-prime immunization, VSV-OVA and VSV-GP-OVA induced comparable frequencies of IFN-γ-secreting OVA-specific cytotoxic T lymphocytes (CTLs) as determined in an enzyme-linked immunosorbent spot (ELISpot) assay with mean values of 363 and 273 IFN-γ+ cells per 106 splenocytes, respectively. CTL responses decreased over time for the two vectors with similar kinetics (Fig. 2A and data not shown). Prior to boosting (26 days postprime), fewer than 70 IFN-γ+ cells per 106 splenocytes were detected in both groups.
FIG 2.
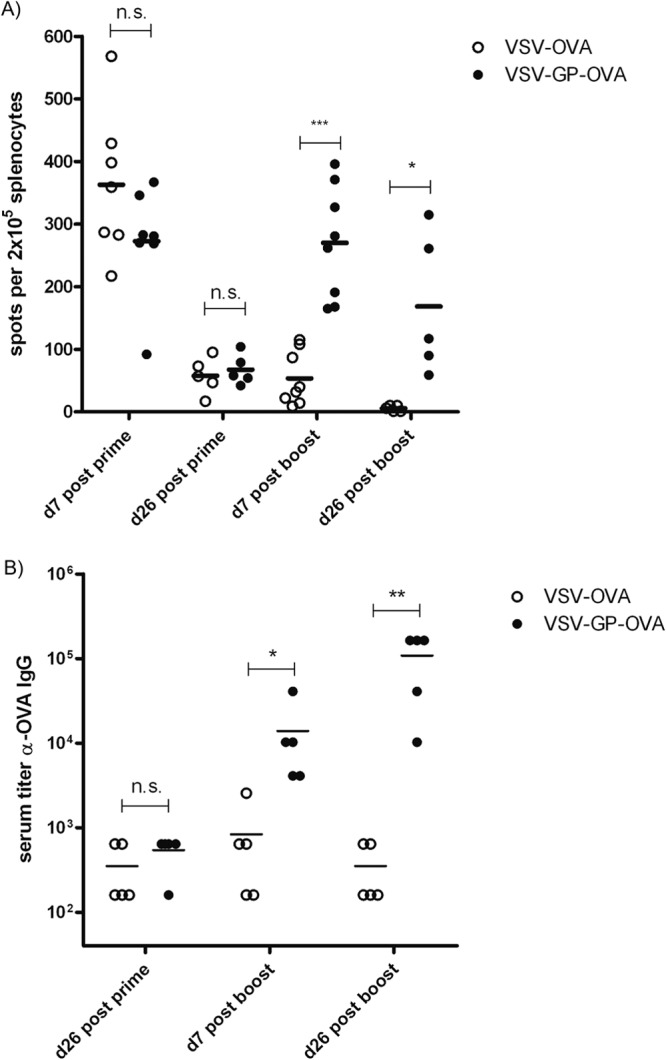
Cellular and humoral immune responses induced by VSV-OVA and VSV-GP-OVA. C57BL/6 mice were immunized intramuscularly on days 0 and 26 with 1 × 106 PFU of VSV-OVA or VSV-GP-OVA. Mice were sacrificed at the indicated time points, and splenocytes and plasma samples were collected. n was ≥5 mice per group and time point. (A) OVA-specific CD8+ T cell responses were determined in an IFN-γ–ELISpot assay after restimulation of splenocytes with OVA257–264 peptide (SIINFEKL). Values for unstimulated cells were subtracted as background. Statistical significances were determined in a one-way analysis of variance followed by Bonferroni's multiple comparison test (*, P ≤ 0.05; ***, P ≤ 0.001; n.s., not significant). (B) OVA-specific IgG titers in mouse plasma were determined by ELISA. Data were analyzed by nonparametric statistics (two-tailed, Mann-Whitney test), and statistically significant differences are marked (*, P ≤ 0.05; **, P ≤ 0.01).
A second administration of VSV-OVA did not boost CTL responses. In sharp contrast, boosting was highly efficient for the VSV-GP-OVA pseudotype, leading to CTL levels comparable to those after prime immunization. This secondary CTL response for VSV-GP-OVA lasted for at least 26 days after boost immunizations. In contrast, for VSV-OVA-immunized animals, OVA-specific CTLs further decreased over time and were undetectable 26 days after boost immunizations. Also, the humoral immune response against the OVA antigen was boosted only with VSV-GP-OVA (Fig. 2B). While the two vectors induced comparable titers of anti-OVA IgGs after prime immunization, VSV-OVA could not boost this response. In contrast, VSV-GP-OVA efficiently boosted antibody titers after the second immunization, and titers of up to 1:100,000 (corresponding to >25 μg/ml anti-OVA IgG) were reached in mouse plasma.
The attenuated VSV variant ΔM51 was shown to be less cytotoxic in vitro and less neurotoxic in animals than VSV expressing the wild-type M protein (35). Vaccination with such a less cytotoxic vector may lead to a prolonged antigen expression in vivo and consequently improve the immune response. To test this hypothesis, we compared OVA-specific CTL levels and antibody titers after prime/boost immunization with either VSV-GP-OVA or VSV-ΔM51-GP-OVA. Both vectors induced potent anti-OVA immune responses. However, reduction of cytotoxicity had no advantage for vaccination, as VSV-GP-OVA induced significantly higher levels of IFN-γ- and TNF-α-secreting CTLs and higher titers of anti-OVA IgGs than did VSV-ΔM51-GP-OVA (Fig. 3).
FIG 3.

VSV-GP-OVA containing the less cytopathic M mutant (ΔM51) induces reduced anti-OVA immune responses. C57BL/6 mice (n = 5) were immunized in a prime/boost schedule intramuscularly with either VSV-GP-OVA or VSV-ΔM51-GP-OVA. Seven days after the second boost, mice were sacrificed and splenocytes were harvested. Cells were stimulated with the OVA-specific CTL peptide SIINFEKL and analyzed via intracellular cytokine staining for production of IFN-γ and TNF-α. (A and B) Means ± standard deviations for IFN-γ+ (A) and IFN-γ+/TNF-α+ cells within CD8+ T cells (B) are depicted. Statistical significances were determined using a two-tailed t test, and asterisks indicate statistical significance (**, P = 0.01). (C) Plasma was collected at indicated time points, and the titer of anti-OVA antibodies was determined by ELISA. Statistical significances were determined by nonparametric statistics (two-tailed, Mann-Whitney test).
VSV-GP does not induce neutralizing antibodies against the vector.
Previous studies demonstrated that high titers of neutralizing antibodies are induced as early as 1 week after infection with VSV, whereas neutralizing antibodies against LCMV do not readily develop (36, 37). Therefore, we next determined whether this difference in neutralizing antibody responses to the viral vector itself could explain the difference in boosting capacity observed between VSV-OVA and VSV-GP-OVA. Titers of neutralizing antibodies were measured in an in vitro neutralization assay on BHK-21 cells infected with either VSV-G or VSV-GP vectors expressing eGFP. All VSV-OVA-immunized mice had high titers of neutralizing antibodies against VSV-G already after prime immunization (mean, 2 × 103), which did not significantly increase upon boost immunization (Fig. 4A). In contrast, none of the sera of the VSV-GP-OVA-immunized mice was able to neutralize either VSV or the VSV-GP pseudotype (Fig. 4B), although we found antibodies binding to LCMV-GP (Fig. 4C).
FIG 4.

Vaccination with VSV-GP does not induce neutralizing antibodies against the vector. Mice were immunized intramuscularly on days 0 and 26 with 1 × 106 PFU of VSV-OVA or VSV-GP-OVA in a prime/boost regimen, and plasma samples collected at the indicated time points were analyzed for the titer of neutralizing antibodies against VSV (A) or VSV-GP (B). Titers of neutralizing antibodies are given as the highest dilution which completely inhibited the cytopathic effect induced by 100 PFU of VSV-GFP or VSV-GP-GFP. At least 5 mice were analyzed per time point and virus. Plasma samples from VSV- or VSV-GP-immunized mice were collected at the indicated time points and were analyzed for the titer of LCMV-GP1 binding antibodies by ELISA using recombinant LCMV-GP1 (C). Plasma samples from 4 mice per group were pooled, and data show means ± standard deviations from 4 independent experiments. Statistical significances were determined using nonparametric statistics (two-tailed, Mann-Whitney test; *, ≤0.05). For serum transfer, naive mice received either immune serum from VSV-OVA- or VSV-GP-OVA-immunized mice or nonimmune serum from naive mice as a control. Subsequently, mice were immunized intramuscularly with 1 × 106 PFU of VSV-OVA or VSV-GP-OVA, and OVA-specific CTL responses were determined via tetramer (D) and intracellular cytokine (E) staining on day 7 postimmunization. Four mice per group were analyzed. Means ± standard deviations are shown. Statistical significances were determined using an unpaired, two-tailed t test (*, P ≤ 0.05; ***, P ≤ 0.01; ns, not significant).
To confirm that neutralizing antibodies against the VSV-G glycoprotein were indeed the reason for the inability of VSV-OVA to reinduce/boost immune responses after second immunizations, we performed a serum transfer experiment. Naive C57BL/6 mice received either immune serum from VSV-OVA- or VSV-GP-OVA-vaccinated mice or nonimmune serum from naive mice as a negative control. Subsequently, mice were vaccinated with VSV-OVA, VSV-GP-OVA, or PBS. OVA-specific CTL responses were determined via tetramer staining and intracellular cytokine staining for IFN-γ-secreting T cells. After transfer of nonimmune serum, both VSV-OVA and VSV-GP-OVA induced potent CTL responses, whereas immunization with PBS did not raise any OVA-specific response (Fig. 4D and E). However, after transfer of VSV-OVA immune serum, vaccination with VSV-OVA induced only a minor OVA-specific CD8+ T cell response, while transfer of VSV-GP-OVA immune serum had no influence on vaccination with VSV-GP-OVA.
VSV-GP-OVA induces immune responses as strong as those induced by adenovirus-OVA, a state-of-the-art vaccine vector.
In a next step, we compared our VSV-GP-OVA pseudotype to an adenoviral vector expressing OVA as a state-of-the-art vaccine vector. Mice were immunized on day 0 (prime), day 26 (first boost), and day 52 (second boost) intramuscularly with either 106 PFU of VSV-GP-OVA or 2 × 109 viral particles of an adenoviral vector expressing intracellular or secreted OVA, AdiOVA and AdsOVA, respectively. Seven days after the second boost, OVA-specific CTL responses were determined. While the levels of IFN-γ-producing CTLs were not significantly different between the VSV-GP-OVA pseudotype and the adenoviral vector AdiOVA, VSV-GP-OVA induced significantly higher frequencies of multifunctional OVA-specific T cells (Fig. 5).
FIG 5.

VSV-GP-OVA is at least as potent as a state-of-the-art adenoviral vaccine vector. Mice (n = 5) were immunized with VSV-GP-OVA, AdiOVA (expressing intracellular OVA), or AdsOva (expressing secreted OVA) in a prime/boost schedule with two boost immunizations. (A) Seven days after the second boost immunization, mice were sacrificed and splenocytes were harvested. Cells were stimulated with the OVA-specific CTL epitope SIINFEKL and analyzed by intracellular cytokine staining for production of IFN-γ, TNF-α, and IL-2. Means ± standard deviations for IFN-γ+, IFN-γ+/TNF-α+, and IFN-γ+/TNF-α+/IL-2+ cells within CD8+ T cell population are shown. Statistical significances were determined using an unpaired, two-tailed t test (**, P ≤ 0.01; *, P ≤ 0.05; n.s., not significant). (B) Plasma was collected at the indicated time points, and the titer of OVA-specific antibodies was determined by ELISA. Statistical significances were determined using nonparametric statistics (two-tailed, Mann-Whitney test; *, P ≤ 0.05). (C) Mice (n ≥ 4) were immunized with AdiOVA in a prime/boost schedule with two boost immunizations. Plasma was collected at indicated time points, and the titer of antiadenovirus antibodies was determined via ELISA.
Mice immunized with VSV-GP had high titers of anti-OVA IgGs in the plasma already after the first immunization, which were boosted by subsequent immunizations (Fig. 5). Mice immunized with AdiOVA, however, only occasionally developed OVA-specific antibodies, while mice immunized with AdsOVA exhibited high titers of anti-OVA antibodies. After prime immunization, these were comparable to those of VSV-GP-OVA-immunized animals. The titers of anti-OVA antibodies increased for both groups (AdsOVA and VSV-GP-OVA) after the first boost. However, boosting was more pronounced for VSV-GP-OVA, where OVA-specific IgG titers rose even further after the second boost. These data are consistent with the antivector antibody titers. Whereas for VSV-GP-OVA no neutralizing antibodies were induced even after multiple immunizations (Fig. 4 and data not shown), adenovirus-immunized mice had high titers of adenovirus-specific antibodies after the first boost immunization (Fig. 5C), a time point after which further boosting with the AdsOVA became inefficient. For adenovirus-specific antibodies, titers of binding antibodies typically correlate with neutralizing capacity when mice are immunized intramuscularly with Ad5 (38, 39).
VSV-GP-OVA vaccination protects mice from Lm_OVA challenge.
Finally, we tested the efficacy of VSV-GP-OVA vaccination in a challenge model using Listeria monocytogenes expressing OVA (Lm_OVA). Mice were immunized in a prime/boost schedule with VSV-GP-OVA, VSV-OVA, AdiOVA, VSV-GP control vector, or PBS and challenged 7 days postboost with Lm_OVA. Three days after challenge, mice were sacrificed and bacterial load in the spleen was measured (Fig. 6). In all PBS and VSV-GP control mice, high-level Lm_OVA replication was detected in the spleen (5 × 106 to 9 × 106 CFU per spleen). After vaccination with VSV-GP-OVA or AdiOVA, however, bacterial load in the spleen was below quantification limit (<102 CFU per spleen) for all mice. For VSV-OVA, however, only a partial protection from Lm_OVA challenge was observed; 5 out of 8 mice were completely protected, but for 3 animals a significant replication of Lm_OVA was observed (2 × 103, 9 × 103, and 2 × 106 CFU per spleen). Consistently, mean SIINFEKL-specific CD8+ T cell frequencies were lower for VSV-OVA-vaccinated mice than for the two protected groups, VSV-GP-OVA and AdiOVA. This corresponds well to the lower frequencies of OVA-specific IFN-γ-secreting CTLs after prime/boost immunization for VSV-OVA compared to VSV-GP-OVA observed in the preceding experiment (Fig. 2).
FIG 6.
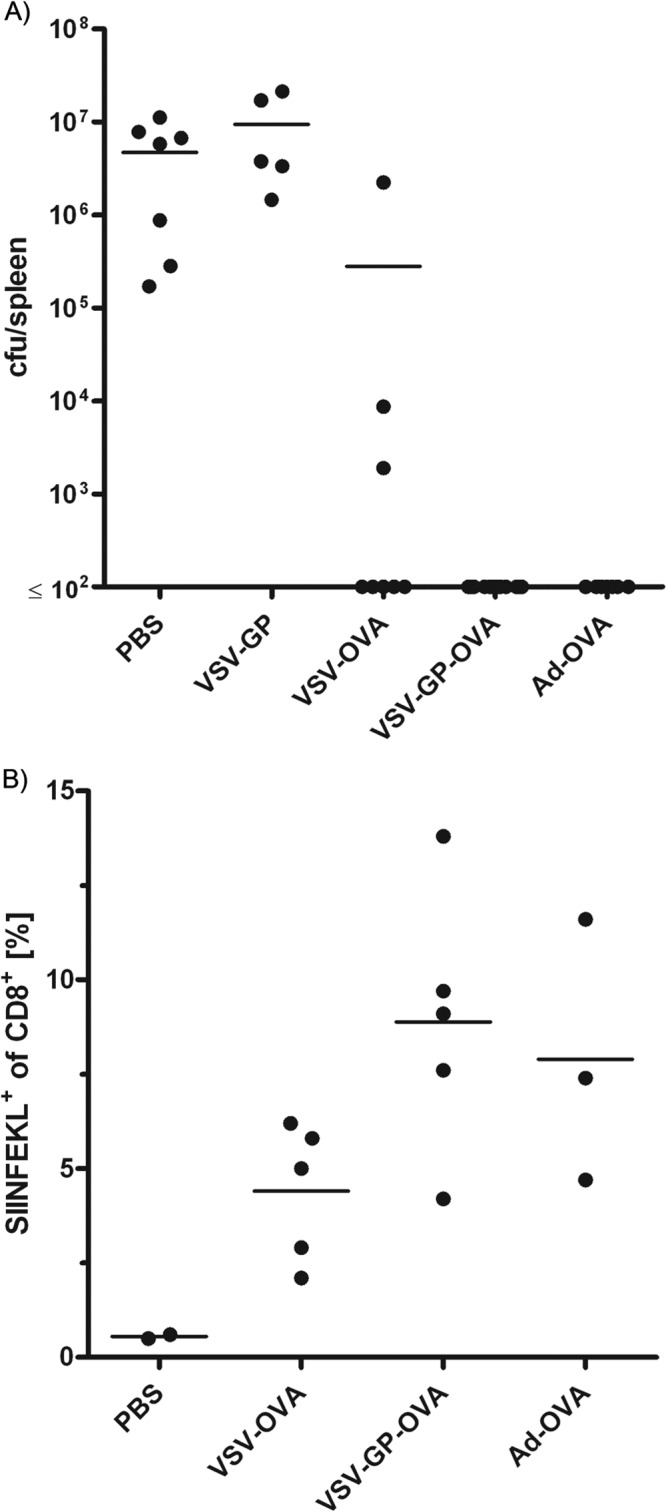
VSV-GP-OVA-immunized mice were protected from challenge with Listeria monocytogenes expressing OVA. Mice were immunized in a prime/boost regimen with the indicated vectors or PBS as a control and challenged 7 days later with Lm_OVA. Three days after challenge, mice were sacrificed. (A) Bacterial load in the spleen was determined (n ≥ 5). Data from two independent experiments were combined in the graph. (B) Frequency of OVA-specific CTLs was determined by tetramer staining in the second experiment (n ≥ 2).
DISCUSSION
In this study, we describe a replication-competent vesicular stomatitis virus pseudotyped with the glycoprotein of an LCMV WE strain (WE-HPI), VSV-GP, which was found to be a highly potent vaccine vector.
VSV has previously been shown to be a highly efficient vaccine vector; however, its inherent neurotoxicity has limited clinical application. Several groups have engineered VSV mutants with reduced neurotoxicity (18, 19), and the level of toxicity/safety in mice of an attenuated VSV variant has been highly predictive of the safety level in nonhuman primates (40, 41). Initial studies in humans with attenuated VSV strains have confirmed this prediction ability of mouse experiments [J. Fuchs, I. Frank, N. Kochar, M. Elizaga, M. Allen, D. K. Carter, N. Frahm, S. Kalams, M. Mulligan, R. Sheets, M. Pensiero, D. Clarke, and J. Eldridge, First-in-human phase I clinical trial of a recombinant vesicular stomatitis virus (rVSV)-based preventive HIV-1 vaccine, presented at AIDS Vaccine 2012, Boston, MA, 9 to 12 September 2012; http://www.epostersonline.com/aidsvax2012/?q=node/2820]. VSV-GP is the only attenuated VSV variant that has completely lost detectable neurotoxicity in mice while preserving replication fitness (22) (Muik et al., submitted). This lack of toxicity in mice should be predictive of an excellent safety profile for VSV-GP in humans.
The required level of safety for a therapeutic vaccine, e.g., for severe chronic infections or cancer, is already high but could well be met by the VSV-GP vaccine vector described here. However, safety requirements for a prophylactic vaccine used in a large number of healthy individuals are even higher. This maximal safety level is most likely difficult to achieve by any of the replication-competent vector vaccines under investigation (vaccinia virus, canarypox virus, CMV, and VSV pseudotyped with Ebola virus envelope). However, as VSV-GP has a small genome of limited complexity, several additional modifications could easily be introduced to maximize vaccine safety, such as splitting the genome to generate a semireplicative virus system as described by us previously for VSV (15).
Among all replication-competent vector vaccines, VSV-GP was found to be unique in the ability to boost the vaccine response upon repeated applications. Accordingly, VSV-GP-OVA reinduced OVA-specific CTL responses and boosted the titers of OVA-specific antibodies. The lack of vector-neutralizing antibodies in VSV-GP-OVA-immunized mice was found to be the reason for VSV-GP-OVA's ability to boost immune responses upon repeated applications. Accordingly, serum transfer from VSV-immunized mice reduced the subsequent T cell response to OVA after VSV-OVA vaccination, while serum transfer from VSV-GP-immunized mice had no effect on the response to VSV-GP-OVA vaccination. Indeed, VSV is known to rapidly induce neutralizing antibodies (11), while neutralizing antibodies to LCMV are, if at all, induced only after chronic infection of 50 to 150 days (42). By exchanging the glycoproteins between VSV and LCMV, Pinschewer and colleagues showed that the ability to induce neutralizing antibodies is determined solely by the viral glycoprotein and not the backbone (43).
Antivector immunity is an important issue for viral vector vaccines. For those, currently in clinical testing (poxviruses, adenoviruses, VSV, etc.), repeated application is limited by antivector immunity (44). There are several other candidates in preclinical development. But among those, only a replication-deficient LCMV vector allows homologous boosting (45, 46). To circumvent this problem, heterologous prime-boost regimes or different serotypes of the same vector may be used.
Interestingly, we were able to boost anti-OVA antibody titers once in mice immunized with AdsOVA (the OVA protein is secreted). These data correlated with titers of adenovirus-specific antibodies in the plasma of the mice, which were low after prime. It is known that the induction of adenovirus-specific antibodies in mice by immunization with human Ad5 vectors is strongly dependent on both the applied dose and the route of delivery (39) and can be boosted by repeated vector administration. Consistently, only after the first low-dose boost were adenovirus-specific antibody titers high in all Ad-immunized animals. Importantly, a large fraction of the human population—in particular in sub-Saharan Africa—is already immune to several adenovirus serotypes (47). While clinical data have demonstrated that preexisting immunity restricts the efficacy of Ad5-based vaccine vectors, the underlying mechanisms may well go beyond simple neutralization but still have to be elucidated. In contrast, the seroprevalence to LCMV is extremely low (48), and this can be considered a significant advantage over vaccine vectors whose wild-type counterparts have high seroprevalence.
Adenovirus vectors based on the human adenovirus serotype 5 were chosen for comparison with VSV-GP as a “gold standard” since they have been shown by various groups to be the most immunogenic viral vector vaccines currently available (44, 49). Additionally, adenoviral vector vaccines showed efficacy in animal models for several diseases, e.g., HIV infection and malaria. Here, VSV-GP-OVA induced specific CTL responses as potent as those induced by an adenoviral vector. The high frequencies of OVA-specific CTLs also translated into protection in a Listeria monocytogenes challenge model, where control of the bacteria is mainly achieved by CTLs. All VSV-GP-OVA- and AdiOVA-immunized mice were able to completely control Listeria monocytogenes infection. In contrast, for VSV-OVA-immunized mice, which showed lower OVA-specific CTL frequencies, only part of the animals controlled the infection. Potential differences between AdiOVA- and VSV-GP-OVA-induced immune protection could not be detected as both vectors provided full protection against the dose of Listeria used in the described experiment.
While T cell responses induced by adenoviral vectors and VSV-GP were comparable, we found drastic differences in OVA-specific antibody titers. In contrast to VSV infection, adenovirus infection is not cytopathic. Therefore, the antibody response to adenovirus immunization is weak for intracellular antigens but potent for secreted antigens. For example, in the STEP trial, an adenoviral vector containing HIV Gag, Pol, and Nef induced only T cell responses whereas similar vectors encoding HIV Env also elicited high titers of Env-specific antibodies (50, 51). In line with these findings, we found that when expressed from an adenoviral vector, only the secreted but not the intracellular OVA antigen induced a significant antibody response. The same is true for other vaccines. Fusion of an IgG signal sequence to HIV Nef or Chlamydia pneumoniae Omp2 in a DNA vaccine enhanced secretion of antigens and titers of antigen-specific antibodies (52). Boyle and colleagues showed that a DNA vaccine encoding OVA induced higher titers of OVA-specific antibodies after intramuscular immunization of mice when the OVA protein was secreted or membrane bound than when it was in a cytoplasmic form (53). In contrast, OVA, when expressed from VSV, induced high titers of antibodies even in the intracellular form. Accordingly, for VSV, the localization of the antigen seems to be less important than it is for other delivery systems. VSV encoding either wild-type hepatitis B virus middle envelope surface glycoprotein or a secretion-deficient variant induced identical T cell and antibody responses (54). The most likely explanation is that the intracellular antigen is set free by virus-induced cell lysis.
We found that the noncytopathic ΔM51 variant was less immunogenic than the more cytopathic vector (VSV-GP or VSV) containing the wild-type M protein. Consistently, after intranasal or intramuscular immunization the HIV Env-specific CTL response was lower for a VSV-ΔM51 variant than for VSV with wild-type M protein (55). Likewise, a recent study showed that a VSV variant containing the ΔM51 protein induced lower titers of antibodies against hepatitis B virus middle surface protein than the corresponding virus with wild-type M protein (54). The release of antigen from cells killed by virus infection again seems to be important for the induction of a potent humoral immune response.
VSV-GP is the first replicating viral vector vaccine that does not readily induce neutralizing antibodies to the vector itself and thus can repeatedly boost immune responses against a vaccine antigen. In future, this unique feature could potentially be conferred also on other enveloped viral vaccine vectors simply by pseudotyping these with LCMV GP protein.
ACKNOWLEDGMENTS
We kindly thank Hao Shen and Dirk Busch for providing Listeria monocytogenes expressing OVA and protocols. We are grateful to Katja Kotsch and Cornelia Fabritius for their practical support. We thank Manuela Lunardon and Bettina Grosslercher for excellent technical support.
This work has been supported by the intramural funding program of Innsbruck Medical University for young scientists, MUI-Start, project 2012032006.
D.V.L. is an inventor of VSV-GP and a participator in the biotech company ViraTherapeutics GmbH, which holds intellectual property rights for VSV-GP. D.V.L. is listed as inventor on a patent application related to VSV-GP. For the other authors, no competing financial interests exist.
Footnotes
Published ahead of print 19 February 2014
REFERENCES
- 1.Robert-Guroff M. 2007. Replicating and non-replicating viral vectors for vaccine development. Curr. Opin. Biotechnol. 18:546–556. 10.1016/j.copbio.2007.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manrique J, Piatak M, Lauer W, Johnson W, Mansfield K, Lifson J, Desrosiers R. 2013. Influence of mismatch of Env sequences on vaccine protection by live attenuated simian immunodeficiency virus. J. Virol. 87:7246–7254. 10.1128/JVI.00798-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barouch DH, Pau MG, Custers JH, Koudstaal W, Kostense S, Havenga MJ, Truitt DM, Sumida SM, Kishko MG, Arthur JC, Korioth-Schmitz B, Newberg MH, Gorgone DA, Lifton MA, Panicali DL, Nabel GJ, Letvin NL, Goudsmit J. 2004. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J. Immunol. 172:6290–6297 [DOI] [PubMed] [Google Scholar]
- 4.Casimiro DR, Chen L, Fu TM, Evans RK, Caulfield MJ, Davies ME, Tang A, Chen M, Huang L, Harris V, Freed DC, Wilson KA, Dubey S, Zhu DM, Nawrocki D, Mach H, Troutman R, Isopi L, Williams D, Hurni W, Xu Z, Smith JG, Wang S, Liu X, Guan L, Long R, Trigona W, Heidecker GJ, Perry HC, Persaud N, Toner TJ, Su Q, Liang X, Youil R, Chastain M, Bett AJ, Volkin DB, Emini EA, Shiver JW. 2003. Comparative immunogenicity in rhesus monkeys of DNA plasmid, recombinant vaccinia virus, and replication-defective adenovirus vectors expressing a human immunodeficiency virus type 1 gag gene. J. Virol. 77:6305–6313. 10.1128/JVI.77.11.6305-6313.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santra S, Sun Y, Parvani JG, Philippon V, Wyand MS, Manson K, Gomez-Yafal A, Mazzara G, Panicali D, Markham PD, Montefiori DC, Letvin NL. 2007. Heterologous prime/boost immunization of rhesus monkeys by using diverse poxvirus vectors. J. Virol. 81:8563–8570. 10.1128/JVI.00744-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts A, Buonocore L, Price R, Forman J, Rose JK. 1999. Attenuated vesicular stomatitis viruses as vaccine vectors. J. Virol. 73:3723–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haglund K, Leiner I, Kerksiek K, Buonocore L, Pamer E, Rose JK. 2002. High-level primary CD8(+) T-cell response to human immunodeficiency virus type 1 gag and env generated by vaccination with recombinant vesicular stomatitis viruses. J. Virol. 76:2730–2738. 10.1128/JVI.76.6.2730-2738.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geisbert TW, Feldmann H. 2011. Recombinant vesicular stomatitis virus-based vaccines against Ebola and Marburg virus infections. J. Infect. Dis. 204(Suppl 3):S1075–S1081. 10.1093/infdis/jir349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rose NF, Marx PA, Luckay A, Nixon DF, Moretto WJ, Donahoe SM, Montefiori D, Roberts A, Buonocore L, Rose JK. 2001. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell 106:539–549. 10.1016/S0092-8674(01)00482-2 [DOI] [PubMed] [Google Scholar]
- 10.Schwartz JA, Buonocore L, Suguitan AL, Jr, Silaghi A, Kobasa D, Kobinger G, Feldmann H, Subbarao K, Rose JK. 2010. Potent vesicular stomatitis virus-based avian influenza vaccines provide long-term sterilizing immunity against heterologous challenge. J. Virol. 84:4611–4618. 10.1128/JVI.02637-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner RR, Rose JK. 1996. Rhabdoviridae: the viruses and their replication, p 1121–1136 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology. Lippincott-Raven, Philadelphia, PA [Google Scholar]
- 12.Stallknecht DE. 2000. VSV-NJ on Ossabaw Island, Georgia. The truth is out there. Ann. N. Y. Acad. Sci. 916:431–436. 10.1111/j.1749-6632.2000.tb05322.x [DOI] [PubMed] [Google Scholar]
- 13.Lichty BD, Power AT, Stojdl DF, Bell JC. 2004. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol. Med. 10:210–216. 10.1016/j.molmed.2004.03.003 [DOI] [PubMed] [Google Scholar]
- 14.Haglund K, Forman J, Krausslich HG, Rose JK. 2000. Expression of human immunodeficiency virus type 1 Gag protein precursor and envelope proteins from a vesicular stomatitis virus recombinant: high-level production of virus-like particles containing HIV envelope. Virology 268:112–121. 10.1006/viro.1999.0120 [DOI] [PubMed] [Google Scholar]
- 15.Muik A, Dold C, Geiss Y, Volk A, Werbizki M, Dietrich U, von Laer D. 2012. Semireplication-competent vesicular stomatitis virus as a novel platform for oncolytic virotherapy. J. Mol. Med. 90:959–970. 10.1007/s00109-012-0863-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rose NF, Roberts A, Buonocore L, Rose JK. 2000. Glycoprotein exchange vectors based on vesicular stomatitis virus allow effective boosting and generation of neutralizing antibodies to a primary isolate of human immunodeficiency virus type 1. J. Virol. 74:10903–10910. 10.1128/JVI.74.23.10903-10910.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson JE, Nasar F, Coleman JW, Price RE, Javadian A, Draper K, Lee M, Reilly PA, Clarke DK, Hendry RM, Udem SA. 2007. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology 360:36–49. 10.1016/j.virol.2006.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper D, Wright KJ, Calderon PC, Guo M, Nasar F, Johnson JE, Coleman JW, Lee M, Kotash C, Yurgelonis I, Natuk RJ, Hendry RM, Udem SA, Clarke DK. 2008. Attenuation of recombinant vesicular stomatitis virus-human immunodeficiency virus type 1 vaccine vectors by gene translocations and g gene truncation reduces neurovirulence and enhances immunogenicity in mice. J. Virol. 82:207–219. 10.1128/JVI.01515-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke DK, Cooper D, Egan MA, Hendry RM, Parks CL, Udem SA. 2006. Recombinant vesicular stomatitis virus as an HIV-1 vaccine vector. Springer Semin. Immunopathol. 28:239–253. 10.1007/s00281-006-0042-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miletic H, Fischer YH, Neumann H, Hans V, Stenzel W, Giroglou T, Hermann M, Deckert M, Von Laer D. 2004. Selective transduction of malignant glioma by lentiviral vectors pseudotyped with lymphocytic choriomeningitis virus glycoproteins. Hum. Gene Ther. 15:1091–1100. 10.1089/hum.2004.15.1091 [DOI] [PubMed] [Google Scholar]
- 21.Beyer WR, Miletic H, Ostertag W, von Laer D. 2001. Recombinant expression of lymphocytic choriomeningitis virus strain WE glycoproteins: a single amino acid makes the difference. J. Virol. 75:1061–1064. 10.1128/JVI.75.2.1061-1064.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muik A, Kneiske I, Werbizki M, Wilflingseder D, Giroglou T, Ebert O, Kraft A, Dietrich U, Zimmer G, Momma S, von Laer D. 2011. Pseudotyping vesicular stomatitis virus with lymphocytic choriomeningitis virus glycoproteins enhances infectivity for glioma cells and minimizes neurotropism. J. Virol. 85:5679–5684. 10.1128/JVI.02511-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moskophidis D, Battegay M, van den Broek M, Laine E, Hoffmann-Rohrer U, Zinkernagel RM. 1995. Role of virus and host variables in virus persistence or immunopathological disease caused by a non-cytolytic virus. J. Gen. Virol. 76:381–391. 10.1099/0022-1317-76-2-381 [DOI] [PubMed] [Google Scholar]
- 24.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. 2002. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J. Immunol. 168:1528–1532 [DOI] [PubMed] [Google Scholar]
- 25.Boritz E, Gerlach J, Johnson JE, Rose JK. 1999. Replication-competent rhabdoviruses with human immunodeficiency virus type 1 coats and green fluorescent protein: entry by a pH-independent pathway. J. Virol. 73:6937–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schnell MJ, Buonocore L, Whitt MA, Rose JK. 1996. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J. Virol. 70:2318–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebert O, Shinozaki K, Kournioti C, Park MS, Garcia-Sastre A, Woo SL. 2004. Syncytia induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Cancer Res. 64:3265–3270. 10.1158/0008-5472.CAN-03-3753 [DOI] [PubMed] [Google Scholar]
- 28.Schiedner G, Hertel S, Kochanek S. 2000. Efficient transformation of primary human amniocytes by E1 functions of Ad5: generation of new cell lines for adenoviral vector production. Hum. Gene Ther. 11:2105–2116. 10.1089/104303400750001417 [DOI] [PubMed] [Google Scholar]
- 29.Kreppel F, Biermann V, Kochanek S, Schiedner G. 2002. A DNA-based method to assay total and infectious particle contents and helper virus contamination in high-capacity adenoviral vector preparations. Hum. Gene Ther. 13:1151–1156. 10.1089/104303402320138934 [DOI] [PubMed] [Google Scholar]
- 30.Mruk DD, Cheng CY. 2011. Enhanced chemiluminescence (ECL) for routine immunoblotting: an inexpensive alternative to commercially available kits. Spermatogenesis 1:121–122. 10.4161/spmg.1.2.16606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaerber G. 1931. 50% end-point calculation. Arch. Exp. Pathol. Pharmakol. 162:480–483. 10.1007/BF01863914 [DOI] [Google Scholar]
- 32.Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M, Meyer J, Hartmann S, Hansmann ML, Fehse B, von Laer D. 2008. Resistance of mature T cells to oncogene transformation. Blood 112:2278–2286. 10.1182/blood-2007-12-128751 [DOI] [PubMed] [Google Scholar]
- 33.Eschli B, Zellweger RM, Wepf A, Lang KS, Quirin K, Weber J, Zinkernagel RM, Hengartner H. 2007. Early antibodies specific for the neutralizing epitope on the receptor binding subunit of the lymphocytic choriomeningitis virus glycoprotein fail to neutralize the virus. J. Virol. 81:11650–11657. 10.1128/JVI.00955-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altfeld M, Rosenberg ES, Shankarappa R, Mukherjee JS, Hecht FM, Eldridge RL, Addo MM, Poon SH, Phillips MN, Robbins GK, Sax PE, Boswell S, Kahn JO, Brander C, Goulder PJ, Levy JA, Mullins JI, Walker BD. 2001. Cellular immune responses and viral diversity in individuals treated during acute and early HIV-1 infection. J. Exp. Med. 193:169–180. 10.1084/jem.193.2.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jayakar HR, Whitt MA. 2002. Identification of two additional translation products from the matrix (M) gene that contribute to vesicular stomatitis virus cytopathology. J. Virol. 76:8011–8018. 10.1128/JVI.76.16.8011-8018.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diallo JS, Vaha-Koskela M, Le Boeuf F, Bell J. 2012. Propagation, purification, and in vivo testing of oncolytic vesicular stomatitis virus strains. Methods Mol. Biol. 797:127–140. 10.1007/978-1-61779-340-0_10 [DOI] [PubMed] [Google Scholar]
- 37.Hangartner L, Zinkernagel RM, Hengartner H. 2006. Antiviral antibody responses: the two extremes of a wide spectrum. Nat. Rev. Immunol. 6:231–243. 10.1038/nri1783 [DOI] [PubMed] [Google Scholar]
- 38.Wortmann A, Vohringer S, Engler T, Corjon S, Schirmbeck R, Reimann J, Kochanek S, Kreppel F. 2008. Fully detargeted polyethylene glycol-coated adenovirus vectors are potent genetic vaccines and escape from pre-existing anti-adenovirus antibodies. Mol. Ther. 16:154–162. 10.1038/sj.mt.6300306 [DOI] [PubMed] [Google Scholar]
- 39.Croyle MA, Patel A, Tran KN, Gray M, Zhang Y, Strong JE, Feldmann H, Kobinger GP. 2008. Nasal delivery of an adenovirus-based vaccine bypasses pre-existing immunity to the vaccine carrier and improves the immune response in mice. PLoS One 3:e3548. 10.1371/journal.pone.0003548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mire CE, Miller AD, Carville A, Westmoreland SV, Geisbert JB, Mansfield KG, Feldmann H, Hensley LE, Geisbert TW. 2012. Recombinant vesicular stomatitis virus vaccine vectors expressing filovirus glycoproteins lack neurovirulence in nonhuman primates. PLoS Negl. Trop. Dis. 6:e1567. 10.1371/journal.pntd.0001567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jenks N, Myers R, Greiner SM, Thompson J, Mader EK, Greenslade A, Griesmann GE, Federspiel MJ, Rakela J, Borad MJ, Vile RG, Barber GN, Meier TR, Blanco MC, Carlson SK, Russell SJ, Peng KW. 2010. Safety studies on intrahepatic or intratumoral injection of oncolytic vesicular stomatitis virus expressing interferon-beta in rodents and nonhuman primates. Hum. Gene Ther. 21:451–462. 10.1089/hum.2009.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Planz O, Seiler P, Hengartner H, Zinkernagel RM. 1996. Specific cytotoxic T cells eliminate B cells producing virus-neutralizing antibodies. Nature 382:726–729. 10.1038/382726a0 [DOI] [PubMed] [Google Scholar]
- 43.Pinschewer DD, Perez M, Jeetendra E, Bachi T, Horvath E, Hengartner H, Whitt MA, de la Torre JC, Zinkernagel RM. 2004. Kinetics of protective antibodies are determined by the viral surface antigen. J. Clin. Invest. 114:988–993. 10.1172/JCI22374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Small JC, Ertl HC. 2011. Viruses—from pathogens to vaccine carriers. Curr. Opin. Virol. 1:241–245. 10.1016/j.coviro.2011.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flatz L, Hegazy AN, Bergthaler A, Verschoor A, Claus C, Fernandez M, Gattinoni L, Johnson S, Kreppel F, Kochanek S, Broek M, Radbruch A, Levy F, Lambert PH, Siegrist CA, Restifo NP, Lohning M, Ochsenbein AF, Nabel GJ, Pinschewer DD. 2010. Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat. Med. 16:339–345. 10.1038/nm.2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halbherr SJ, Brostoff T, Tippenhauer M, Locher S, Berger Rentsch M, Zimmer G. 2013. Vaccination with recombinant RNA replicon particles protects chickens from H5N1 highly pathogenic avian influenza virus. PLoS One 8:e66059. 10.1371/journal.pone.0066059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiang Z, Li Y, Cun A, Yang W, Ellenberg S, Switzer WM, Kalish ML, Ertl HC. 2006. Chimpanzee adenovirus antibodies in humans, sub-Saharan Africa. Emerg. Infect. Dis. 12:1596–1599. 10.3201/eid1210.060078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lledo L, Gegundez MI, Saz JV, Bahamontes N, Beltran M. 2003. Lymphocytic choriomeningitis virus infection in a province of Spain: analysis of sera from the general population and wild rodents. J. Med. Virol. 70:273–275. 10.1002/jmv.10389 [DOI] [PubMed] [Google Scholar]
- 49.Chen H, Xiang ZQ, Li Y, Kurupati RK, Jia B, Bian A, Zhou DM, Hutnick N, Yuan S, Gray C, Serwanga J, Auma B, Kaleebu P, Zhou X, Betts MR, Ertl HC. 2010. Adenovirus-based vaccines: comparison of vectors from three species of adenoviridae. J. Virol. 84:10522–10532. 10.1128/JVI.00450-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, Gilbert PB, Lama JR, Marmor M, Del Rio C, McElrath MJ, Casimiro DR, Gottesdiener KM, Chodakewitz JA, Corey L, Robertson MN. 2008. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 372:1881–1893. 10.1016/S0140-6736(08)61591-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McElrath MJ, De Rosa SC, Moodie Z, Dubey S, Kierstead L, Janes H, Defawe OD, Carter DK, Hural J, Akondy R, Buchbinder SP, Robertson MN, Mehrotra DV, Self SG, Corey L, Shiver JW, Casimiro DR. 2008. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet 372:1894–1905. 10.1016/S0140-6736(08)61592-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Svanholm C, Bandholtz L, Lobell A, Wigzell H. 1999. Enhancement of antibody responses by DNA immunization using expression vectors mediating efficient antigen secretion. J. Immunol. Methods 228:121–130. 10.1016/S0022-1759(99)00086-1 [DOI] [PubMed] [Google Scholar]
- 53.Boyle JS, Koniaras C, Lew AM. 1997. Influence of cellular location of expressed antigen on the efficacy of DNA vaccination: cytotoxic T lymphocyte and antibody responses are suboptimal when antigen is cytoplasmic after intramuscular DNA immunization. Int. Immunol. 9:1897–1906. 10.1093/intimm/9.12.1897 [DOI] [PubMed] [Google Scholar]
- 54.Cobleigh MA, Bradfield C, Liu Y, Mehta A, Robek MD. 2012. The immune response to a vesicular stomatitis virus vaccine vector is independent of particulate antigen secretion and protein turnover rate. J. Virol. 86:4253–4261. 10.1128/JVI.05991-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Publicover J, Ramsburg E, Robek M, Rose JK. 2006. Rapid pathogenesis induced by a vesicular stomatitis virus matrix protein mutant: viral pathogenesis is linked to induction of tumor necrosis factor alpha. J. Virol. 80:7028–7036. 10.1128/JVI.00478-06 [DOI] [PMC free article] [PubMed] [Google Scholar]