

ABSTRACT
Application of manure from antibiotic-treated animals to crops facilitates the dissemination of antibiotic resistance determinants into the environment. However, our knowledge of the identity, diversity, and patterns of distribution of these antibiotic resistance determinants remains limited. We used a new combination of methods to examine the resistome of dairy cow manure, a common soil amendment. Metagenomic libraries constructed with DNA extracted from manure were screened for resistance to beta-lactams, phenicols, aminoglycosides, and tetracyclines. Functional screening of fosmid and small-insert libraries identified 80 different antibiotic resistance genes whose deduced protein sequences were on average 50 to 60% identical to sequences deposited in GenBank. The resistance genes were frequently found in clusters and originated from a taxonomically diverse set of species, suggesting that some microorganisms in manure harbor multiple resistance genes. Furthermore, amid the great genetic diversity in manure, we discovered a novel clade of chloramphenicol acetyltransferases. Our study combined functional metagenomics with third-generation PacBio sequencing to significantly extend the roster of functional antibiotic resistance genes found in animal gut bacteria, providing a particularly broad resource for understanding the origins and dispersal of antibiotic resistance genes in agriculture and clinical settings.
IMPORTANCE
The increasing prevalence of antibiotic resistance among bacteria is one of the most intractable challenges in 21st-century public health. The origins of resistance are complex, and a better understanding of the impacts of antibiotics used on farms would produce a more robust platform for public policy. Microbiomes of farm animals are reservoirs of antibiotic resistance genes, which may affect distribution of antibiotic resistance genes in human pathogens. Previous studies have focused on antibiotic resistance genes in manures of animals subjected to intensive antibiotic use, such as pigs and chickens. Cow manure has received less attention, although it is commonly used in crop production. Here, we report the discovery of novel and diverse antibiotic resistance genes in the cow microbiome, demonstrating that it is a significant reservoir of antibiotic resistance genes. The genomic resource presented here lays the groundwork for understanding the dispersal of antibiotic resistance from the agroecosystem to other settings.
INTRODUCTION
The accumulation of antibiotic resistance (AR) determinants in human pathogens is currently one of the most significant threats to public health (reviewed in reference 1). The extensive use of antibiotics in animal agriculture is thought have created a selection pressure for resistant organisms and made farms an important source of antibiotic-resistant bacteria and AR genes (2–5), including in human commensal bacteria (6). Antibiotic-resistant bacteria can be transferred directly from animals to humans (7, 8) and can spread to soil, food, and groundwater through the application of manure to agricultural fields (reviewed in 9, 10). Manure bacteria that do not persist in the environment may transfer resistance genes to resident soil microorganisms, including pathogens, via horizontal gene transfer (11, 12). Therefore, it is essential to understand the contribution of animal agricultural practices to the environmental AR gene pool in more detail.
Cows in the United States generate between 1.9 and 14.2 billion pounds of manure each day (13), which is applied to fields as fertilizer and may serve as reservoirs of zoonotic pathogens and AR genes (reviewed in 14, 15). Valuable insight into the manure resistomes of pigs, cattle, and chickens has been gained from direct culturing of antibiotic-resistant bacteria (10, 16, 17), quantitative PCR (qPCR) amplification of AR genes (5, 18–20), and the exogenous isolation of plasmids carrying AR genes (21, 22). These approaches have shown that animal microbiomes are a substantial source of AR genes, especially of those conferring resistance to tetracycline and sulfonamide, which are often carried on plasmids (18, 21–26). Some of these approaches are limited by the recalcitrance of certain bacteria to growth in culture and lack of AR gene expression under the culture conditions used. For example, among the culturable bacteria, which represent nearly half of the species in the gut microbiota of animals and humans, most are restricted to growth under anaerobic conditions (27). However, most AR screens rely on aerobic culturing conditions. Similarly, qPCR is limited by the sequences of primers used to amplify target AR genes. More recently, sequence-based metagenomics has been used to explore the resistome in diverse environments, including cattle manure (28). However, sequence-based discovery is limited to those genes with similarity to known AR genes and therefore cannot identify novel AR genes. In addition, the short read length of many novel sequencing platforms prevents characterization of the genomic context of the putative AR genes.
A complementary strategy, functional metagenomics, provides a culture-independent tool that relies on expression of resistance in a surrogate host, enabling characterization and discovery of AR genes (29–31). Functional screens of metagenomic libraries constructed with DNA from human and avian feces have found that the majority of AR genes in gut microbial communities share only 40 to 60% identity with genes previously deposited in public databases, and as such might not have been recognized as resistance genes based on sequence alone (3, 32–34).
In this study, we used functional metagenomics to investigate the dairy cow microbiome as a reservoir for AR determinants to four antibiotic classes: beta-lactams, phenicols, aminoglycosides, and tetracyclines. We discovered a new clade of chloramphenicol resistance genes and a great diversity of resistance determinants originating from a diverse set of phyla. The study combined a novel sequencing technology, PacBio, with a classical functional metagenomics approach, which afforded more insight than was possible with either method alone.
RESULTS
Resistant clones in metagenomic libraries.
Dairy cows are treated with antibiotics such as beta-lactams to treat and prevent mastitis (35). To assess the dairy cow microbiome as a potential source of AR genes, we constructed fosmid and small-insert metagenomic libraries from each of five manure samples (Table 1). Libraries containing a total of 25.9 Gb of DNA were screened for resistance to beta-lactam antibiotics (carbenicillin, representing the penicillin class of beta-lactam antibiotics, and ceftazidime, representing the cephalosporin class) and kanamycin (an aminoglycoside). Fosmid libraries were also screened for resistance to tetracycline, and small-insert libraries were also screened for chloramphenicol resistance. The frequency of carbenicillin-, chloramphenicol-, kanamycin-, and tetracycline-resistant clones was consistent across libraries constructed from different cows and between small-insert and fosmid libraries (see Fig. S1 in the supplemental material). In contrast, the frequency of ceftazidime resistance was variable and detected in five of the ten libraries close to the detection limit (10−7). Four of the five libraries containing ceftazidime-resistant clones were derived from manure of cows that had been treated with beta-lactam antibiotics prior to the initiation of this study (Table 1).
TABLE 1 .
Functional metagenomic libraries constructed with DNA from dairy cow manure
| Cow no. | Library name | Library type | No. of clones | Average insert size (kb) | Gb of cloned DNA | Antibiotic treatment history between September 2011 and March 2012 |
|---|---|---|---|---|---|---|
| 01 | Cow01_fos | Fosmid | 90,000 | 24.1 | 2.1 | No treatment |
| Cow01_si | Small insert | 480,000 | 3.2 | 2.5 | ||
| 02 | Cow02_fos | Fosmid | 80,000 | 29.1 | 2.3 | No treatment |
| Cow02_si | Small insert | 1,044,780 | 2.5 | 2.6 | ||
| 03 | Cow03_fos | Fosmid | 60,000 | 28.1 | 1.6 | Repeatedly treated prior to sampling: January 2012, 125 mg ceftiofur, 5×; February 2012, 200 mg cephapirin, 20× |
| Cow03_si | Small insert | 1,427,800 | 3.1 | 4.3 | ||
| 04 | Cow04_before_fos | Fosmid | 60,000 | 32.3 | 1.9 | No antibiotic treatment history; sample obtained before antibiotic treatment |
| Cow04_before_si | Small insert | 1,304,000 | 2.1 | 2.8 | ||
| Cow04_after_fos | Fosmid | 142,800 | 31.3 | 4.4 | Sample obtained after antibiotic treatment in March 2012: 125 mg ceftiofur, 5× |
|
| Cow04_after_si | Small insert | 1,428,000 | 2.3 | 1.4 | ||
| Total | 10 libraries | 6,117,380 | 25.9 |
Identification of resistance genes.
Of 87 clones with genes conferring resistance to chloramphenicol, kanamycin, tetracycline, or beta-lactam antibiotics, 80 carried unique AR genes (see Table S1 in the supplemental material). Further screening tetracycline and beta-lactam resistance rediscovered the same genes, but each new round for kanamycin and chloramphenicol resistance identified unique AR genes. This suggests a high diversity of kanamycin and chloramphenicol resistance genes in the libraries, which was corroborated by the finding that the majority of the 80 unique AR genes conferred resistance to either chloramphenicol or kanamycin (35% and 53%, respectively) (Fig. 1a). Chloramphenicol and kanamycin resistance was highly represented in all libraries (see Table S1 in the supplemental material).
FIG 1 .
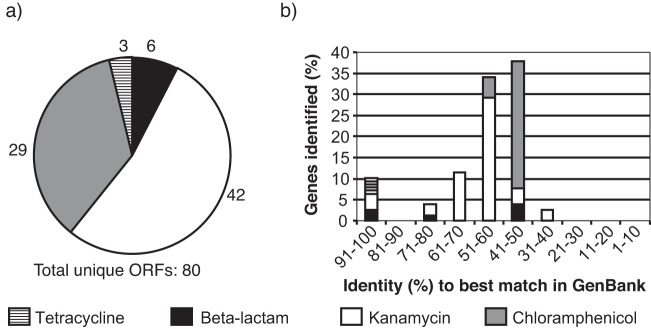
Antibiotic resistance genes from dairy cow manure. (a) Distribution of 80 unique antibiotic resistance (AR) genes among four classes of antibiotics. (b) Distribution of similarity of 80 unique AR genes from manure compared to homologues in GenBank.
The predicted protein sequences of the AR genes shared between 32% and 100% amino acid sequence identity with proteins deposited in public databases (Fig. 1b), and the average sequence identity differed among the types of AR genes. For example, all of the tetracycline resistance genes contained high (97% and 100%), and all of the chloramphenicol resistance genes shared low amino acid sequence identity (46% to 52%) (Fig. 1b) with sequences previously deposited in public databases. In contrast, the beta-lactamases ranged from 41 to 100% sequence identity with sequences previously deposited in the databases. Notably, all sequences were either very similar to (>96% identity) or very different from (<52% identity) sequences deposited in GenBank. This might indicate that the cow microbiome is a reservoir of AR genes which have not been identified in other environments yet.
We identified approximately the same number (21 to 26) of unique AR genes from each of the four cows, demonstrating that manure samples from different cows harbor similar-sized pools of resistance genes despite different prior exposure to antibiotics. This was reinforced by qPCR in total DNA of six of the AR genes reported here in cow manure from the pens of antibiotic-treated and untreated cows, which contained the same number of copies compared with 16S rRNA genes as the internal standard (Fig. 2). 16 rRNA copy numbers were very high, yielding low but highly sensitive detection of the AR genes.
FIG 2 .
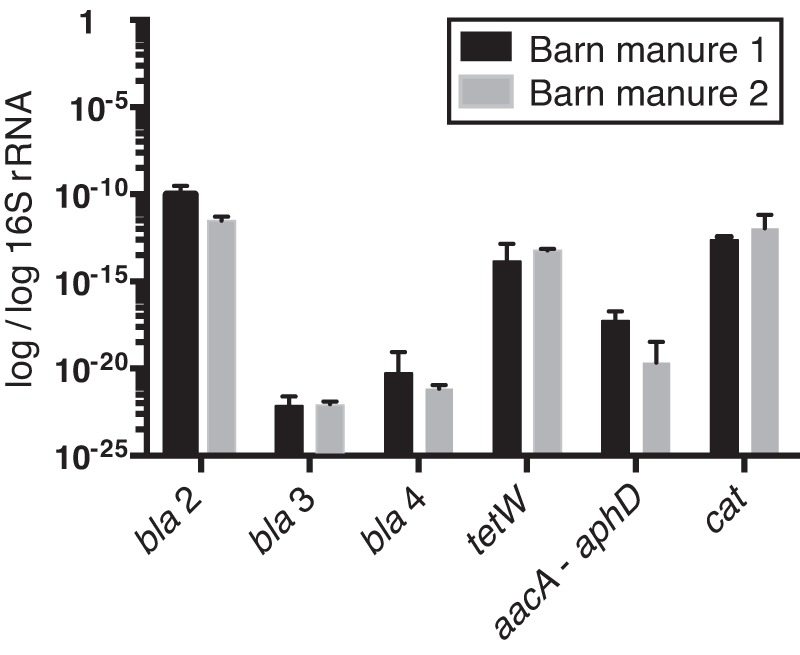
Quantitative PCR detection of the abundance of antibiotic resistance genes in manure. Values are expressed as the ratio of resistance gene copy number to 16S rRNA gene copy number. Genes: bla, beta-lactamases; tetW, tetracycline resistance; aacA-aphD, bifunctional aminoglycoside-modifying enzyme; cat, chloramphenicol acetyltransferase. Barn manure 1 is a sample from a pool of untreated cows, and barn manure 2 originated from a pool of antibiotic-treated cows.
Antibiotic resistance genes in clusters on fosmid clones.
To assess the genomic context of genes conferring resistance to kanamycin, tetracycline, and beta-lactam antibiotics, we used the Pacific Biosciences (PacBio) RS platform to sequence 12 fosmids. After error correction using the hierarchical genome assembly process (HGAP) pipeline (36), we obtained six completely assembled inserts ranging in size from 22.6 to 37.9 kb (Fig. 3). The other six inserts were only partially assembled, and two scaffolds smaller than 10 kb were excluded from further analyses. BLASTX analysis of the genes flanking the antibiotic resistance genes indicated that they originated from phyla that are characteristic of the cow microbiome (28, 37). Firmicutes were predominant (50%), followed by Bacteroidetes (23%), Proteobacteria (14%), and Actinobacteria (6%). Less abundant phyla included Spirochaeta, Planctomyces, Thermotogae, Fusobacteria, Chlorobi, Verrucomicrobia, and Cyanobacteria. One gene on one scaffold most closely matched (47% identity) an archaeal sequence affiliated with the Euryarchaeota (Fig. 3). Two of the clones (DCM001Kan16 and DCM001Kan17) harboring kanamycin resistance genes contained eight and five open reading frames (ORFs), respectively, with predicted functions related to AR or mobile genetic elements. One gene revealed high sequence identity (99%) to a gene encoding a bifunctional aminoglycoside-modifying enzyme, aacA-aphD from Ruminococcus flavefaciens. Although previously reported homologues in this family structurally modify a wide spectrum of aminoglycosides, including kanamycin, gentamicin, and amikacin (38, 39), the gene reported here confers resistance to kanamycin (MIC, 256 µg/ml) and amikacin (MIC, 32 µg/ml) and none of six other aminoglycosides tested. This clone also carries a beta-lactamase homologue but does not confer resistance to beta-lactam antibiotics. The abundance and arrangement of genes on these fosmid clones suggest that some manure bacteria harbor clusters of AR genes that may be transferred across species via mobile genetic elements. The genes putatively encoding mobile genetic elements were also diverse. These included insertion sequence (IS) elements belonging to different families (e.g., IS3 and IS116) and genes encoding other mechanisms of DNA mobility, such as excisionases, phage integrases, site-specific recombinases, and a plasmid-partitioning protein (parB).
FIG 3 .

Organization of genes on ten metagenomic fosmid clones conferring resistance to kanamycin (KM), tetracycline (TC), carbenicillin (CAR), or ceftazidime (CTZ) assessed using Pacific Biosciences RS sequencing technology. Genes are as follows: nat, N-acetyltransferases; aph, aminoglycoside phosphotransferases; aacA-aphD, bifunctional aminoglycoside-modifying enzyme; tetW and tetO, tetracycline resistance genes; cat, chloramphenicol acetyltransferase; bla, beta-lactamases. Genes with annotations related to mobile genetic elements are labeled as IS (insertion sequence elements/transposases) or parB (plasmid-partitioning protein). Identical genes are connected with gray lines. Antibiotic resistance genes for which we demonstrated a function by subcloning are indicated with gray boxes and asterisks. All other labels were assigned based on sequence homology.
Narrow- and extended-spectrum beta-lactamases.
Dairy cows are often treated with beta-lactam antibiotics, making resistance to them of special interest in the cow microbiome. Of the four beta-lactamases that we identified, one (beta-lactamase 2 [bla 2]) was found in two different clones from the same metagenomic library and showed high sequence identity (99%) to a beta-lactamase previously found only in Firmicutes (Fig. 3; also, see Tables S2, S3, and S4 in the supplemental material). Although both bla 2 genes were flanked by the same genes downstream, the two clones appear to have originated from different phyla, most likely Bacteroidetes and Spirochaeta, based on other genes on the fosmids (Fig. 3). This suggests that horizontal gene transfer distributed these beta-lactamase genes among various phyla within the cow microbiome.
To determine the relationships among all four unique beta-lactamase sequences, we constructed a phylogenetic tree (Fig. 4a). The range of similarity between the beta-lactamases from manure and previously identified sequences is striking—beta-lactamase 1 was highly identical (99%) to a beta-lactamase from Bacillus, whereas the highly divergent beta-lactamase 3 was 41% identical to a beta-lactamase from the genus Bacteroides. Beta-lactamase 4 clustered most closely with a cephalosporinase from a Parabacteroides sp. and provided high-level resistance against both penicillins and third-generation cephalosporins (see Table S5 in the supplemental material). All four identified beta-lactamases belong to Ambler class A (40).
FIG 4 .

(a) Phylogenetic tree inferred with maximum likelihood for the predicted amino acid sequences of four different beta-lactamases (bla) identified in metagenomic libraries constructed from the dairy cow microbiome and related sequences retrieved from GenBank. Numbers represent bootstrap values for 100 replications. Bootstrap values of >80 are shown. (b) Phylogenetic affiliations and genomic organization of chloramphenicol acetyltransferases (cat) identified in dairy cow manure small-insert libraries. The phylogenetic tree was inferred with maximum likelihood based on the predicted amino acid sequences of 29 chloramphenicol acetyltransferases and the most similar sequences retrieved from GenBank; one sequence was from the MG-RAST cow fecal metagenome data set 4444130.3 and is underlined (27). The aligned sequence was 148 amino acids. The genomic organization was assessed by a BLASTX search of the available flanking DNA. Dashed parts of arrows indicate regions for which only partial DNA sequences were available. Node tips with no arrows indicate that an insufficient amount of flanking DNA was present or that there were no significant BLASTX matches found. Percentage identity between cat sequences is indicated by a black/gray color gradation.
Novel clade of chloramphenicol acetyltransferases in dairy cow manure.
The low sequence identity with sequences in GenBank of the chloramphenicol acetyltransferases and N-acetyltransferases prompted us to explore the phylogenetic affiliations of these genes. A total of 29 unique chloramphenicol resistance genes and 33 unique N-acetyltransferases were identified in the manure libraries (see Table S1 in the supplemental material). Phylogenetic trees based on protein sequences (Fig. 4b; also, see Fig. S2 in the supplemental material) revealed a high diversity and divergence of the manure-derived genes. Furthermore, the phylogenetic tree strongly supported a divergent clade of chloramphenicol acetyltransferases originating from the manure samples from the four cows. The results show that dairy cow manure contains novel chloramphenicol resistance determinants that are only distantly related to previously known genes.
In order to determine whether these divergent chloramphenicol resistance genes are generally found in feces or are instead specific to the manure of cows raised at this particular farm, we interrogated metagenomic data sets of cow and buffalo rumens, animal and human fecal samples, and farm soils accessible through MG-RAST. A local TBLASTN search for the 29 newly discovered chloramphenicol resistance genes revealed significant matches to three reads in the cattle fecal data set (28) (MGRAST 4448367.3) (FQ16D9T05FSHBS, 85% identity; FPZANON03DEQ8T, 85% identity; and FLU3MGX162UVCV, 79% identity). No significant hits were found in the other MG-RAST data sets that we examined. From the cattle fecal data set, we included the nearly complete ORF on one of the reads, FQ16D9T05FSHBS, in the phylogenetic tree (Fig. 4b). This sequence clustered very closely with the 29 predicted protein sequences from the dairy cow manure metagenomic analysis, demonstrating that this type of chloramphenicol resistance is also found in cow microbiomes from another locations.
Based on sequence alignment with CLUSTALW, the chloramphenicol resistance genes shared between 71% and 99% sequence identity. Analysis of the flanking regions suggests that these AR genes likely originated from different genomic contexts (Fig. 4b). In addition, some chloramphenicol resistance genes were flanked by phage integrases or transposases, indicating that these genes may also be highly mobile within bacterial populations, which could contribute to the substantial diversity among these genes.
DISCUSSION
Using a combination of functional metagenomics and third-generation PacBio sequencing, we characterized the resistome of cow manure. The study provides a comprehensive profile of genes conferring resistance to various classes of antibiotics, including a novel clade of chloramphenicol resistance genes specific to the cow microbiome of cow manure. The resistance genes originated from diverse phyla, including Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria, indicating that the resistome resides in phylogenetically diverse organisms. We also found that the same AR genes are present in different taxa, and many resistance genes are flanked by mobile genetic elements such as transposases and IS elements (reviewed in reference 41). That resistance genes are present on mobile genetic elements in cow manure has been discovered previously using other approaches, including the exogenous isolation of plasmids and the use of conjugative transposons (42, 43). Together, these data indicate that these AR genes are likely horizontally transferred between bacterial species in the cow microbiome and therefore are candidates for dissemination to other bacteria in the environment, including pathogens.
Functional metagenomics in Eschericha coli captured diverse AR genes, some of which differed in expression, substrate specificity, and their identity as AR genes from predictions based on homology. First, although all of the genes were discovered based on their functional expression in E. coli, analysis of sequence alone would not have predicted their expression, as they most closely match (e.g., 58% identity) alleles from Gram-positive bacteria (in particular, the genera Streptococcus and Clostridium). Second, the spectrum of activity (against both penicillins and third-generation cephalosporins but not first-generation cephalosporins) of beta-lactamase 4 was surprising, because it shares significant sequence identity (79%) with a broad-spectrum cephalosporinase. Similarly, the aacA-aphD gene on the DCM001Kan17 clone shares high similarity (99%) with a bifunctional aminoglycoside-modifying enzyme, which would predict a broad activity spectrum (38), but in fact, this gene conferred resistance to a narrow spectrum of aminoglycosides. This limited activity could be due to the specificity of different domains in bifunctional proteins that are not always active, as demonstrated previously (29). Finally, many of the genes may not have been identified as AR genes based on their sequence alone, because they deeply diverge from AR genes with demonstrated function and their closest matches are frequently annotated as hypothetical proteins. Each of these examples demonstrates the power of functional metagenomics to assign functions to new genes.
Based on the amount of DNA and diversity of antibiotics screened, this work is the most comprehensive functional metagenomic screen performed from farm microbiomes to date (3, 33, 44). We found resistance genes in cow manure at a lower frequency (3 AR genes/Gb DNA) than was observed in a previous study of chicken (14 AR genes/Gb DNA). Both studies used a replicated experimental design, but such calculations must be treated cautiously, because many factors might skew these frequencies. However, the results are consistent with animal husbandry practice in the United States, in which dairy cows are treated with antibiotics less frequently and usually to treat and prevent diseases, whereas chickens are fed antibiotics to promote growth, resulting in approximately 4-fold more antibiotics being used in chickens than cows (45).
A key finding from this work is that different cows contain similar AR genes and gene families. This result indicates that we screened to completeness and isolated the full complement of manure-derived genes that are active in E. coli, contrasting with studies in soil, which contains a far more diverse set of AR genes based on the observation that every soil metagenomic library appears to contain AR genes not found in other soils (29, 46). Although only five cows were analyzed, the widespread distribution of the same genes is indicated by their detection in two additional samples of pooled manure from the barns housing the cows in this study (Fig. 2) and in the cattle fecal-pool data set available at MG-RAST (MGRAST 4448367.3). Therefore, the sequences and primers found and developed in this study may be used in the future to investigate factors driving abundance, diversity, and transferability of AR genes in cow manure from additional farms administering different antibiotics or feeding different diets. To understand in more detail how agricultural antibiotic use can affect the abundance, diversity, evolution, and horizontal gene transfer of novel AR genes identified in functional metagenomic screens, larger-scale studies with manures of animals raised with heavy usage of antibiotics (e.g., pigs) are necessary.
In conclusion, this study deepens our understanding of the diversity of AR gene classes in an important farm animal whose manure is often used as fertilizer. The combination of functional metagenomics and PacBio sequencing, which produces much longer reads than 454 pyrosequencing or Illumina, generated sufficient information without too-high sequence coverage about the genomic context of AR genes to infer the taxonomic affiliation of the origin of the DNA. This is critical for drawing conclusions about horizontal transfer of AR genes in bacterial populations. Further quantitative analysis is needed to assess the movement of AR genes between agriculture and hospitals or the food system. Such studies will provide a robust basis for management of antibiotic resistance in the future.
MATERIALS AND METHODS
Manure and fecal samples.
Cows were housed at the Kellogg Dairy Unit of the University of Connecticut, and sample collection was approved by their Institutional Animal Care and Use Committee (IACUC; approval no. A11-041). Fecal samples were obtained by rectal grabbing of approximately 70 g of fecal material from four different lactating dairy cows in March 2012. A fresh palpation sleeve was used for each sample, which was immediately inverted and sealed. The cows were selected by age and antibiotic treatment history to represent a broad spectrum of manure samples (Table 1). From one cow, we obtained two samples, one before antibiotic treatment and one 4 days after treatment (Table 1). The antibiotic concentration was not quantified in the fecal samples, since this cow was given intramammary treatment for mastitis. Therefore, the antibiotic concentration is likely low, and if antibiotic is present at all in the cow gut, it is likely degraded quickly due to the omnipresence of beta-lactamases in manure (47). All animals were fed the same lactating-cow diet consisting of 71% corn silage, 14% hay silage, 3% alfalfa hay, 3% beet pulp, 4% crude protein concentrate, 4% crude protein supplement, and 1% rumen-protected fat. Cows ranged from two to 11 years old.
Cows recently treated with antibiotics for mastitis prevention were housed separately from healthy cows. To compare AR gene abundance present in manure of pens, we collected random manure samples from the ground of each of the pens occupied by untreated and antibiotic-treated cows. Both rectal grab and pen manure samples were transported on ice to Yale University and stored at −80°C until DNA was extracted.
DNA isolation and library construction.
Fosmid libraries in the pCC2Fos vector (Epicentre, Madison, WI), which contains a chloramphenicol selection marker, were constructed using methods previously described for soil (46, 48), with a phenol-chloroform extraction followed by an ethanol-sodium acetate precipitation after end repair.
Manure DNA for small-insert libraries was extracted using the ZR fecal DNA miniprep kit (Zymo Research Corp., Irvine, CA) according to the manufacturer’s recommendations and cloned into pCF430, which encodes a tetracycline resistance marker (49). Insert DNA was digested with PstI and ligated into the linearized vector. The reaction mix was dialyzed using 0.2-µm filter membranes (Millipore, Billerica, MA), and the plasmids were electroporated into E. coli DH10β (Invitrogen, Carlsbad, CA) cells using a Micropulser (Bio-Rad, Hercules, CA). Cells were allowed phenotypic expression in super optimal broth with catabolite repression (SOC) medium for 1 h and plated on LB plates supplemented with tetracycline (5 µg/ml).
To estimate the average insert size for each library, we isolated DNA from ten randomly picked clones using QIAprep miniprep buffers (Qiagen, Valencia, CA) and chloroform extraction followed by isopropanol precipitation. For fosmids, fragments resulting from digestion with NotI (New England Biolabs, Beverly, MA) were separated on a 1% pulsed-field agarose gel (Bio-Rad) in 0.5× Tris-acetate-EDTA (TAE). For clones from small-insert libraries, fragments resulting from digestion with BamHI were separated on a 1% agarose gel in Tris-borate-EDTA (TBE) buffer. Clones were pooled and stored at −80°C in LB broth supplemented with 30% glycerol and chloramphenicol (20 µg/ml) or tetracycline (5 µg/ml).
Identification of antibiotic-resistant clones.
Metagenomic libraries (100 µl of the pooled clones) were grown in 50 ml of LB supplemented with the appropriate marker antibiotic (either chloramphenicol or tetracycline) for 2 h at 37°C and 200 rpm. Library mixtures were serially diluted and cultured on LB plates containing an antibiotic of interest: carbenicillin (50 µg/ml), ceftazidime (4 µg/ml), chloramphenicol (12.5 µg/ml), kanamycin (50 µg/ml), or tetracycline (10 µg/ml). Fosmid and small-insert libraries were not screened on the antibiotic used to select for the vector (chloramphenicol and tetracycline, respectively). The average frequency of resistant clones was determined by culturing each library on three replicate plates of each antibiotic. For subcloning, the resistant clones were scraped from the plates, pooled, and stored at −80°C in LB supplemented with 30% glycerol.
Subcloning or direct sequencing using primers designed to the pCF430 vector was used to investigate active genes, depending on the library and the antibiotic used for the screen (see Table S6 in the supplemental material). For subcloning, we followed previously published protocols using the pBluescript II KS(+) or the pET28b vector (46). Primers used for sequencing are listed in Table S7 in the supplemental material.
When subcloning from pooled clones as described in Table S6 in the supplemental material, twenty-four antibiotic-resistant subclones were randomly chosen and grown in 96-well square well plates. Plasmid DNA from these antibiotic-resistant subclones was purified using the Nucleospin 96 plasmid kit (Macherey-Nagel, Bethlehem, PA) and sequenced using T7 promoter and T3 primers at the DNA Analysis Facility at Yale University (http://dna-analysis.research.yale.edu). Antibiotic-resistant subclones containing the pET28b vector were sequenced using the T7 promoter and terminator primers.
Determination of MICs.
MIC assays were conducted in Mueller-Hinton Broth (DIFCO, Franklin Lakes, NJ) according to previously published protocols (46). pCC2Fos in Epi300 and pCF430 in DH10β were used as empty-vector controls. The MIC was defined as the antibiotic concentration that inhibited growth of 105 cells after 16 h at 37°C. The optical density at 600 nm (OD600) was measured using a Synergy HT plate reader (Bio-Tek, Winooski, VT). All MIC assays were performed in triplicate, and the MIC was reported as the concentration at which the average OD600 indicated reduced visible growth (OD630 < 0.1).
Quantitative PCR.
To obtain sufficiently pure DNA for quantitative PCR (qPCR), DNA was extracted from manure samples as described above for small-insert libraries and further purified using the nucleic acid purification instrument Aurora (Boreal Genomics, Los Altos, CA) (50). The primers used are listed in Table S7 in the supplemental material. Fourteen nanograms of purified DNA was used for qPCR amplification with the SsoFast EvaGreen Supermix (Bio-Rad) in a total reaction volume of 20 µl. Thermal-cycling conditions were as follows: initial denaturation step at 98°C for 3 min and then 45 cycles of 5 s at 98°C and 10 s at 60°C. The specificities of primer pairs were verified by melting curve analysis.
Sequencing of fosmids carrying AR genes.
To better understand the genomic context of twelve distinct clones conferring AR, we sequenced the entire fosmid in each using Pacific Biosciences (PacBio) RS technology. DNA was prepared using the QIAprep kit (Qiagen) from 10 ml of bacterial culture in LB supplemented with chloramphenicol and 1× CopyControl induction solution (Epicentre). From each fosmid clone, 300 ng of DNA was pooled into a single reaction mixture and submitted to the Yale high-throughput sequencing center (http://medicine.yale.edu/keck/ycga). Assembly was performed using Celera (51) and trimmed for fosmid vector sequence using the ContaminationTrimmer.py command (https://github.com/PacificBiosciences/HBAR-DTK). The resulting scaffolds were assigned to the fosmids by performing a BLASTN search for the resistance genes and end sequences generated by Sanger sequencing of the fosmid vector using the primers recommended by the manufacturer. The assembly of 5,037 high-quality reads resulted in 45 scaffolds with an average coverage of 7.88×. Twelve of these scaffolds contained the expected antibiotic resistance genes.
Phylogenetic analyses and sequence comparisons to metagenomic data sets.
The Geneious software (version 6.0.5) was used for sequence comparisons and phylogenetic analyses. For sequence alignments, we used CLUSTALW (52), and the phylogenetic trees were inferred using maximum likelihood. Bootstrap values were calculated based on 100 replications. To compare our sequences with other data sets, we retrieved metagenomic sequence data sets accessible through the MG-RAST server. These included the following data sets: pooled cattle feces (28) (MGRAST number 4448367.3), chicken cecum (53) (MGRAST number 4440283.3), cow and buffalo rumens (54) (MGRAST numbers 4441679.3, 4441680.3, 4441682.3, and 4446901.3), human feces (MGRAST numbers 4440946.3 and 4444130.3), and farm soil (MGRAST number 4441091.3).
Nucleotide sequence accession numbers.
All nucleotide sequences are publicly available in GenBank and shown in Table S1 in the supplemental material. Sequences were deposited in GenBank under the following accession numbers: KJ512900, KJ512901, KJ512902, KJ512903, KJ512904, KJ512905, KJ512906, KJ512907, KJ512908, KJ512909, KJ512910, KJ512911, KJ512912, KJ512913, KJ512914, KJ512915, KJ512916 KJ512917, KJ512918, KJ512919, KJ512920, KJ512921, KJ512922, KJ512923, KJ512924, KJ512925, KJ512926, KJ512927, KJ512928, KJ512929, KJ512930, KJ512931, KJ512932, KJ512933, KJ512934, KJ512935, KJ512936, KJ512937, KJ512938, KJ512939, KJ512940, KJ512941, KJ512942, KJ512943, KJ512944, KJ512945, KJ512946, KJ512947, KJ512948, KJ512949, KJ512950, KJ512951, KJ512952, KJ512953, KJ512954, KJ512955, KJ512956, KJ512957, KJ512958, KJ512959, KJ512960, KJ512961, KJ512962, KJ512963, KJ512964, KJ512965, KJ512966, KJ512967, KJ512968, KJ512969, KJ512970, KJ512971, KJ512972, KJ512973, KJ512974, KJ512975, KJ512976, KJ512977, KJ512978, KJ512979, KJ512980, KJ512981, KJ512982, KJ512983, KJ512984, KJ512985, KJ512986, KJ512987, KJ512988, KJ512989, KJ512990, and KJ512991.
SUPPLEMENTAL MATERIAL
Frequency of manure-derived clones resistant to five antibiotics. CAR, carbenicillin; CTZ, ceftazidime; CM, chloramphenicol; KM, kanamycin; TC, tetracycline. Fosmid libraries (gray bars) were not screened for resistance to chloramphenicol because the fosmid contains a determinant of chloramphenicol resistance, and small-insert libraries (black bars) were not screened for resistance to tetracycline for the same reason, as indicated with the letters NA. The detection limit is indicated with a black line at a frequency of 10−7. Download
Phylogenetic affiliations of N-acetyltransferases (nat) identified in the cow microbiome. The phylogenetic tree was inferred with maximum likelihood based on the predicted amino acid sequences of 31 N-acetyltransferases and the sequences with the highest identity retrieved from GenBank. The length of the alignment was 108 amino acids. Numbers represent bootstrap values for 100 replications. Two very short N-acetyltransferases were not included in the phylogenetic analyses. Bootstrap values of >80 are shown. Download
Resistance genes on metagenomic clones from dairy cow manure and their homologues. The clones in bold letters were chosen for sequencing using Pacific Biosciences (PacBio) technology.
Closest matches (BLASTX) of the genes in the clone DCM005Tet05 harboring beta-lactamase 2.
Closest matches (BLASTX) of the genes in the clone DCM005Carb03 harboring beta-lactamase 2.
Closest matches (BLASTX) of the genes in the clone DCM005Cefta01 harboring beta-lactamase 4, which exhibited very high resistance to penicillins and third-generation cephalosporins.
Resistance of metagenomic clones to beta-lactam antibiotics. Piperacillin, penicillin G, carbenicillin, and ampicillin represented the penicillin class. Cephalothin and cephalexin represented the first-generation cephalosporins, and ceftiofur and ceftazidime represented the third-generation cephalosporins. The libraries with clones containing these genes are indicated in the first row. Clavulanic acid (2 µg/ml) is an inhibitor of certain beta-lactamases.
Methods used to identify antibiotic resistance genes in metagenomic libraries.
Primers used in this study for sequencing and quantitative PCR.
ACKNOWLEDGMENTS
This research was funded by the Swiss National Science Foundation SNF Project (PBZHP3-138800) and the postdoctoral Kirschstein-National Research Service Award T32 training grant for Genomics and Proteomics.
We thank Guilin Wang for assembling the PacBio data, Colleen Chapman for collecting dairy cow manure samples, and Ashley Ferguson and Nicole Price for contributions to isolation of resistance genes. We also thank Nichole Broderick, Ashley Shade, Philipp Engel, and Jesse Morin for review of the manuscript.
Footnotes
Citation Wichmann F, Udikovic-Kolic N, Andrew S, Handelsman J. 2014. Diverse antibiotic resistance genes in dairy cow manure. mBio 5(2):e01017-13. doi:10.1128/mBio.01017-13.
REFERENCES
- 1. Septimus EJ, Owens RC., Jr. 2011. Need and potential of antimicrobial stewardship in community hospitals. Clin. Infect. Dis. 53(Suppl 1):S8–S14. 10.1093/cid/cir363 [DOI] [PubMed] [Google Scholar]
- 2. Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO, Dantas G. 2012. The shared antibiotic resistome of soil bacteria and human pathogens. Science 337:1107–1111. 10.1126/science.1220761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou W, Wang Y, Lin J. 2012. Functional cloning and characterization of antibiotic resistance genes from the chicken gut microbiome. Appl. Environ. Microbiol. 78:3028–3032. 10.1128/AEM.06920-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levy SB. 1978. Emergence of antibiotic-resistant bacteria in the intestinal flora of farm inhabitants. J. Infect. Dis. 137:689–690 [PubMed] [Google Scholar]
- 5. Zhu YG, Johnson TA, Su JQ, Qiao M, Guo GX, Stedtfeld RD, Hashsham SA, Tiedje JM. 2013. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. U. S. A. 110:3435–3440. 10.1073/pnas.1302581110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forslund K, Sunagawa S, Kultima JR, Mende DR, Arumugam M, Typas A, Bork P. 2013. Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 23:1163–1169. 10.1101/gr.155465.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Price LB, Stegger M, Hasman H, Aziz M, Larsen J, Andersen PS, Pearson T, Waters AE, Foster JT, Schupp J, Gillece J, Driebe E, Liu CM, Springer B, Zdovc I, Battisti A, Franco A, Zmudzki J, Schwarz S, Butaye P, Jouy E, Pomba C, Concepcion Porrero M, Ruimy R, Smith TC, Robinson DA, Weese JS, Sofia Arriola C, Yu F, Laurent F, Keim P, Skov R, Aarestrup FM. 2012. Staphylococcus aureus CC398: host adaptation and emergence of methicillin resistance in livestock. mBio 3:e00305-11. 10.1128/mBio.00305-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Novais C, Freitas AR, Silveira E, Antunes P, Silva R, Coque TM, Peixe L. 2013. Spread of multidrug-resistant Enterococcus to animals and humans: an underestimated role for the pig farm environment. J. Antimicrob. Chemother. 68:2746–2754. 10.1093/jac/dld289 [DOI] [PubMed] [Google Scholar]
- 9. Heuer H, Schmitt H, Smalla K. 2011. Antibiotic resistance gene spread due to manure application on agricultural fields. Curr. Opin. Microbiol. 14:236–243. 10.1016/j.mib.2011.04.009 [DOI] [PubMed] [Google Scholar]
- 10. Brichta-Harhay DM, Arthur TM, Bosilevac JM, Kalchayanand N, Shackelford SD, Wheeler TL, Koohmaraie M. 2011. Diversity of multidrug-resistant Salmonella enterica strains associated with cattle at harvest in the United States. Appl. Environ. Microbiol. 77:1783–1796. 10.1128/AEM.01885-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shoemaker NB, Vlamakis H, Hayes K, Salyers AA. 2001. Evidence for extensive resistance gene transfer among Bacteroides spp. and among Bacteroides and other genera in the human colon. Appl. Environ. Microbiol. 67:561–568. 10.1128/AEM.67.2.561-568.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karami N, Martner A, Enne VI, Swerkersson S, Adlerberth I, Wold AE. 2007. Transfer of an ampicillin resistance gene between two Escherichia coli strains in the bowel microbiota of an infant treated with antibiotics. J. Antimicrob. Chemother. 60:1142–1145. 10.1093/jac/dkm327 [DOI] [PubMed] [Google Scholar]
- 13. NASS 2010. Cattle on feed. In USDA economics, statistics and market information system. Cornell University, Ithaca, NY, and USDA, Washington, DC [Google Scholar]
- 14. Ferens WA, Hovde CJ. 2011. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog. Dis. 8:465–487. 10.1089/fpd.2010.0673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith KE, Stenzel SA, Bender JB, Wagstrom E, Soderlund D, Leano FT, Taylor CM, Belle-Isle PA, Danila R. 2004. Outbreaks of enteric infections caused by multiple pathogens associated with calves at a farm day camp. Pediatr. Infect. Dis. J. 23:1098–1104. 10.1097/01.inf.0000145409.74116.e5 [DOI] [PubMed] [Google Scholar]
- 16. Walczak JJ, Xu S. 2011. Manure as a source of antibiotic-resistant Escherichia coli and enterococci: a case study of a Wisconsin, USA family dairy farm. Water Air Soil Pollut. 219:579–589. 10.1007/s11270-010-0729-x [DOI] [Google Scholar]
- 17. Sawant AA, Hegde NV, Straley BA, Donaldson SC, Love BC, Knabel SJ, Jayarao BM. 2007. Antimicrobial-resistant enteric bacteria from dairy cattle. Appl. Environ. Microbiol. 73:156–163. 10.1128/AEM.01551-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koike S, Krapac IG, Oliver HD, Yannarell AC, Chee-Sanford JC, Aminov RI, Mackie RI. 2007. Monitoring and source tracking of tetracycline resistance genes in lagoons and groundwater adjacent to swine production facilities over a 3-year period. Appl. Environ. Microbiol. 73:4813–4823. 10.1128/AEM.00665-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walsh F, Ingenfeld A, Zampicolli M, Hilber-Bodmer M, Frey JE, Duffy B. 2011. Real-time PCR methods for quantitative monitoring of streptomycin and tetracycline resistance genes in agricultural ecosystems. J. Microbiol. Methods 86:150–155. 10.1016/j.mimet.2011.04.011 [DOI] [PubMed] [Google Scholar]
- 20. Looft T, Johnson TA, Allen HK, Bayles DO, Alt DP, Stedtfeld RD, Sul WJ, Stedtfeld TM, Chai B, Cole JR, Hashsham SA, Tiedje JM, Stanton TB. 2012. In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. U. S. A. 109:1691–1696. 10.1073/pnas.1120238109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heuer H, Kopmann C, Binh CT, Top EM, Smalla K. 2009. Spreading antibiotic resistance through spread manure: characteristics of a novel plasmid type with low % G+C content. Environ. Microbiol. 11:937–949. 10.1111/j.1462-2920.2008.01819.x [DOI] [PubMed] [Google Scholar]
- 22. Heuer H, Binh CT, Jechalke S, Kopmann C, Zimmerling U, Krogerrecklenfort E, Ledger T, Gonzalez B, Top E, Smalla K. 2012. IncP-1epsilon plasmids are important vectors of antibiotic resistance genes in agricultural systems: diversification driven by class 1 integron gene cassettes. Front. Microbiol. 3:2. 10.3389/fmicb.2012.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aslam M, Stanford K, McAllister TA. 2010. Characterization of antimicrobial resistance and seasonal prevalence of Escherichia coli O157:H7 recovered from commercial feedlots in Alberta, Canada. Lett. Appl. Microbiol. 50:320–326. 10.1111/j.1472-765X.2010.02798.x [DOI] [PubMed] [Google Scholar]
- 24. Cessna AJ, Larney FJ, Kuchta SL, Hao X, Entz T, Topp E, McAllister TA. 2011. Veterinary antimicrobials in feedlot manure: dissipation during composting and effects on composting processes. J. Environ. Qual. 40:188–198. 10.2134/jeq2010.0079 [DOI] [PubMed] [Google Scholar]
- 25. Chee-Sanford JC, Aminov RI, Krapac IJ, Garrigues-Jeanjean N, Mackie RI. 2001. Occurrence and diversity of tetracycline resistance genes in lagoons and groundwater underlying two swine production facilities. Appl. Environ. Microbiol. 67:1494–1502. 10.1128/AEM.67.4.1494-1502.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hölzel CS, Harms KS, Küchenhoff H, Kunz A, Müller C, Meyer K, Schwaiger K, Bauer J. 2010. Phenotypic and genotypic bacterial antimicrobial resistance in liquid pig manure is variously associated with contents of tetracyclines and sulfonamides. J. Appl. Microbiol. 108:1642–1656. 10.1111/j.1365-2672.2009.04570.x [DOI] [PubMed] [Google Scholar]
- 27. Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. 2011. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. U. S. A. 108:6252–6257. 10.1073/pnas.1102938108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Durso LM, Harhay GP, Bono JL, Smith TP. 2011. Virulence-associated and antibiotic resistance genes of microbial populations in cattle feces analyzed using a metagenomic approach. J. Microbiol. Methods 84:278–282. 10.1016/j.mimet.2010.12.008 [DOI] [PubMed] [Google Scholar]
- 29. Allen HK, Moe LA, Rodbumrer J, Gaarder A, Handelsman J. 2009. Functional metagenomics reveals diverse beta-lactamases in a remote Alaskan soil. ISME J. 3:243–251. 10.1038/ismej.2008.86 [DOI] [PubMed] [Google Scholar]
- 30. Riesenfeld CS, Goodman RM, Handelsman J. 2004. Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ. Microbiol. 6:981–989. 10.1111/j.1462-2920.2004.00664.x [DOI] [PubMed] [Google Scholar]
- 31. Parsley LC, Consuegra EJ, Kakirde KS, Land AM, Harper WF, Liles MR. 2010. Identification of diverse antimicrobial resistance determinants carried on bacterial, plasmid, or viral metagenomes from an activated sludge microbial assemblage. Appl. Environ. Microbiol. 76:3753–3757. 10.1128/AEM.03080-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sommer MO, Church GM, Dantas G. 2010. The human microbiome harbors a diverse reservoir of antibiotic resistance genes. Virulence 1:299–303. 10.4161/viru.1.4.12010 [DOI] [PubMed] [Google Scholar]
- 33. Kazimierczak KA, Scott KP, Kelly D, Aminov RI. 2009. Tetracycline resistome of the organic pig gut. Appl. Environ. Microbiol. 75:1717–1722. 10.1128/AEM.02206-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martiny AC, Martiny JBH, Weihe C, Field A, Ellis JC. 2011. Functional metagenomics reveals previously unrecognized diversity of antibiotic resistance genes in gulls. Front. Microbiol. 2:238. 10.3389/fmicb.2011.00238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barlow J. 2011. Mastitis therapy and antimicrobial susceptibility: a multispecies review with a focus on antibiotic treatment of mastitis in dairy cattle. J. Mammary Gland Biol. Neoplasia 16:383–407. 10.1007/s10911-011-9235-z [DOI] [PubMed] [Google Scholar]
- 36. Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10:563–569. 10.1038/nmeth.2474 [DOI] [PubMed] [Google Scholar]
- 37. Durso LM, Harhay GP, Smith TP, Bono JL, Desantis TZ, Harhay DM, Andersen GL, Keen JE, Laegreid WW, Clawson ML. 2010. Animal-to-animal variation in fecal microbial diversity among beef cattle. Appl. Environ. Microbiol. 76:4858–4862. 10.1128/AEM.00207-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rouch DA, Byrne ME, Kong YC, Skurray RA. 1987. The aacA-aphD gentamycin and kanamycin resistance determinant of Tn4001 from Staphylococcus aureus—expression and nucleotide analysis. J. Gen. Microbiol. 133:3039–3052 [DOI] [PubMed] [Google Scholar]
- 39. Tenorio C, Zarazaga M, Martinez C, Torres C. 2001. Bifunctional enzyme 6′-N-aminoglycoside acetyltransferase-2′′-O-aminoglycoside phosphotransferase in Lactobacillus and Pediococcus isolates of animal origin. J. Clin. Microbiol. 39:824–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ambler RP. 1980. The structure of beta-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 289:321–331. 10.1098/rstb.1980.0049 [DOI] [PubMed] [Google Scholar]
- 41. O’Brien TF. 2002. Emergence, spread, and environmental effect of antimicrobial resistance: how use of an antimicrobial anywhere can increase resistance to any antimicrobial anywhere else. Clin. Infect. Dis. 34(Suppl 3):S78–S84. 10.1086/340244 [DOI] [PubMed] [Google Scholar]
- 42. Heuer H, Krögerrecklenfort E, Wellington EM, Egan S, van Elsas JD, van Overbeek L, Collard JM, Guillaume G, Karagouni AD, Nikolakopoulou TL, Smalla K. 2002. Gentamicin resistance genes in environmental bacteria: prevalence and transfer. FEMS Microbiol. Ecol. 42:289–302. 10.1111/j.1574-6941.2002.tb01019.x [DOI] [PubMed] [Google Scholar]
- 43. van Overbeek LS, Wellington EM, Egan S, Smalla K, Heuer H, Collard JM, Guillaume G, Karagouni AD, Nikolakopoulou TL, van Elsas JD. 2002. Prevalence of streptomycin-resistance genes in bacterial populations in European habitats. FEMS Microbiol. Ecol. 42:277–288. 10.1111/j.1574-6941.2002.tb01018.x [DOI] [PubMed] [Google Scholar]
- 44. Tian B, Fadhil NH, Powell JE, Kwong WK, Moran NA. 2012. Long-term exposure to antibiotics has caused accumulation of resistance determinants in the gut microbiota of honeybees. mBio 3:e00377-12. 10.1128/mBio.00377-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim KR, Owens G, Kwon SI, So KH, Lee DB, Ok YS. 2011. Occurrence and environmental fate of veterinary antibiotics in the terrestrial environment. Water Air Soil Pollut. 214:163–174. 10.1007/s11270-010-0412-2 [DOI] [Google Scholar]
- 46. Donato JJ, Moe LA, Converse BJ, Smart KD, Berklein FC, McManus PS, Handelsman J. 2010. Metagenomic analysis of apple orchard soil reveals antibiotic resistance genes encoding predicted bifunctional proteins. Appl. Environ. Microbiol. 76:4396–4401. 10.1128/AEM.01763-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Binh CT, Heuer H, Gomes NC, Kotzerke A, Fulle M, Wilke BM, Schloter M, Smalla K. 2007. Short-term effects of amoxicillin on bacterial communities in manured soil. FEMS Microbiol. Ecol. 62:290–302. 10.1111/j.1574-6941.2007.00393.x [DOI] [PubMed] [Google Scholar]
- 48. Liles MR, Williamson LL, Rodbumrer J, Torsvik V, Goodman RM, Handelsman J. 2008. Recovery, purification, and cloning of high-molecular-weight DNA from soil microorganisms. Appl. Environ. Microbiol. 74:3302–3305. 10.1128/AEM.02630-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Newman JR, Fuqua C. 1999. Broad-host-range expression vectors that carry the L-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197–203. 10.1016/S0378-1119(98)00601-5 [DOI] [PubMed] [Google Scholar]
- 50. Engel K, Pinnell L, Cheng J, Charles TC, Neufeld JD. 2012. Nonlinear electrophoresis for purification of soil DNA for metagenomics. J. Microbiol. Methods 88:35–40. 10.1016/j.mimet.2011.10.007 [DOI] [PubMed] [Google Scholar]
- 51. Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, Anson EL, Bolanos RA, Chou HH, Jordan CM, Halpern AL, Lonardi S, Beasley EM, Brandon RC, Chen L, Dunn PJ, Lai Z, Liang Y, Nusskern DR, Zhan M, Zhang Q, Zheng X, Rubin GM, Adams MD, Venter JC. 2000. A whole-genome assembly of Drosophila. Science 287:2196–2204. 10.1126/science.287.5461.2196 [DOI] [PubMed] [Google Scholar]
- 52. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2. Bioinformatics 23:2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 53. Qu A, Brulc JM, Wilson MK, Law BF, Theoret JR, Joens LA, Konkel ME, Angly F, Dinsdale EA, Edwards RA, Nelson KE, White BA. 2008. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS One 3:e2945. 10.1371/journal.pone.0002945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ross EM, Moate PJ, Bath CR, Davidson SE, Sawbridge TI, Guthridge KM, Cocks BG, Hayes BJ. 2012. High throughput whole rumen metagenome profiling using untargeted massively parallel sequencing. BMC Genet. 13:53. 10.1286/1471-2156-13-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Frequency of manure-derived clones resistant to five antibiotics. CAR, carbenicillin; CTZ, ceftazidime; CM, chloramphenicol; KM, kanamycin; TC, tetracycline. Fosmid libraries (gray bars) were not screened for resistance to chloramphenicol because the fosmid contains a determinant of chloramphenicol resistance, and small-insert libraries (black bars) were not screened for resistance to tetracycline for the same reason, as indicated with the letters NA. The detection limit is indicated with a black line at a frequency of 10−7. Download
Phylogenetic affiliations of N-acetyltransferases (nat) identified in the cow microbiome. The phylogenetic tree was inferred with maximum likelihood based on the predicted amino acid sequences of 31 N-acetyltransferases and the sequences with the highest identity retrieved from GenBank. The length of the alignment was 108 amino acids. Numbers represent bootstrap values for 100 replications. Two very short N-acetyltransferases were not included in the phylogenetic analyses. Bootstrap values of >80 are shown. Download
Resistance genes on metagenomic clones from dairy cow manure and their homologues. The clones in bold letters were chosen for sequencing using Pacific Biosciences (PacBio) technology.
Closest matches (BLASTX) of the genes in the clone DCM005Tet05 harboring beta-lactamase 2.
Closest matches (BLASTX) of the genes in the clone DCM005Carb03 harboring beta-lactamase 2.
Closest matches (BLASTX) of the genes in the clone DCM005Cefta01 harboring beta-lactamase 4, which exhibited very high resistance to penicillins and third-generation cephalosporins.
Resistance of metagenomic clones to beta-lactam antibiotics. Piperacillin, penicillin G, carbenicillin, and ampicillin represented the penicillin class. Cephalothin and cephalexin represented the first-generation cephalosporins, and ceftiofur and ceftazidime represented the third-generation cephalosporins. The libraries with clones containing these genes are indicated in the first row. Clavulanic acid (2 µg/ml) is an inhibitor of certain beta-lactamases.
Methods used to identify antibiotic resistance genes in metagenomic libraries.
Primers used in this study for sequencing and quantitative PCR.