

Abstract
The rational design, synthesis, and validation of a significantly improved insoluble polymer-supported siloxane-transfer agent has been achieved that permits efficient palladium-catalyzed cross-coupling reactions. The cross-linked polystyrene support facilitates product purification with excellent siloxane recycling. Drawbacks of a previous polymer-supported siloxane-transfer agent, relating to reaction efficiency and polymer stability after repeated cycles, have been addressed.
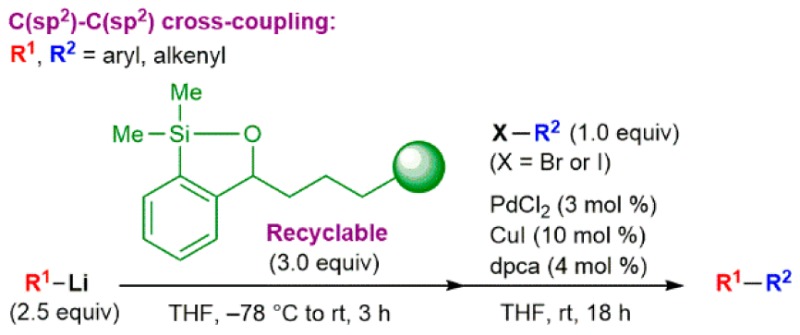
Palladium-catalyzed cross-coupling reactions (CCRs) of organometallic reagents with electrophiles are among the most important transformations in organic synthesis for the construction of natural products, biologically active compounds, and diverse functional materials.1 Recently, we reported a highly atom-efficient process for intermolecular cross-coupling of aryl- and alkenylorganolithiums with aryl and alkenyl iodides exploiting a new class of silicon reagents, termed siloxane-transfer agents (cf. compound 1, Scheme 1).2 This tactic permits the direct use of readily available organolithium reagents in cross-coupling reactions and, in turn, eliminates the need for additional synthetic manipulations and/or the isolation of suitable nucleophilic coupling partners (i.e., Suzuki organoborons,3 Negishi organozincs,4 Stille organotins,5 and Hiyama/Denmark organosilicons6). Importantly, the use of siloxane-transfer agents offers a solution to the intrinsic limitation of Murahashi CCR7 and the recent Feringa protocol8 where slow addition of the organolithium reagent is required to avoid the competing formation of homocoupled products resulting from fast lithium–halogen exchange. Although the reported transfer agents 1 proved highly effective in CCRs, recovery of these congeners following the desired transformation via either chromatographic separation or acid–base extraction via inclusion of a Brønsted base in the transfer agent in some cases proved less than optimal. The development of an effective polymer-supported siloxane transfer agent (PSTA) would simplify both product purification and siloxane recycling, facilitating use of the siloxane tactic by the scientific community and further advancing the siloxane based bond forming process9 for a variety of applications (e.g., coating of siloxane polymer in flow microreactors).
Scheme 1. CCRs Employing Siloxane-Transfer Agent.
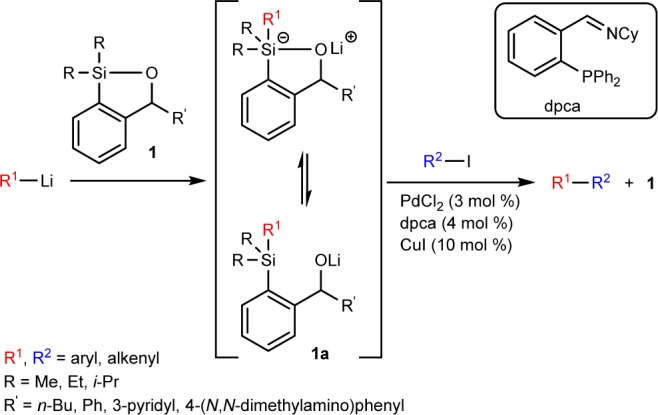
Recently, we reported a soluble polynorbornene-supported siloxane-transfer agent10 (PSTA-I, Scheme 2) employing the ring-opening metathesis polymerization (ROMP) technique. While this polymer proved effective in mediating palladium-catalyzed cross-coupling reactions, there was a decrease in reaction efficiency after repeated cycles in conjunction with reagent-induced increase in the polymer dispersity. In recent years, the trend emphasized in synthetic organic chemistry has been harmonization with the environment and reaction efficiency; thus, recent work has been aimed at developing new solid supports that could be recycled indefinitely with no loss in reaction efficiency. Herein, we report the rational design, synthesis, and validation of a second-generation polymer-supported siloxane-transfer agent for efficient Pd-catalyzed cross-coupling reactions with excellent recyclability.
Scheme 2. Design of Second-Generation PSTA.

Attention was first directed toward the design of a more effective solid support for the transfer agent (Scheme 2). During our study employing PSTA-I we observed, in repeated Pd-catalyzed cross-coupling cycles, an increase in the number average molecular weight and higher polydispersity index of the recovered polymer as determined by gel permeation chromatography. This result is likely due to polymer cross-linking, in which the oxyanion (alkoxide 1a, Scheme 1) formed in solution after the addition of the organolithium attacks a nearby silicon atom of another siloxane unit on a different polymer chain. We thus reasoned that we could suppress this process by attaching the transfer agent onto an insoluble support, thus eliminating the interaction between the freely moving polymer chains in solution. Furthermore, the use of a co-monomer would place the siloxane units further away from each other, minimizing the undesired behavior. Removal of the unsaturation in the polymer chain, given the possible vulnerability of the olefinic backbone to chemical degradation, was also viewed as desirable. Finally, a longer linker connecting the siloxane unit to the polymer backbone would likely reduce any steric effect caused by the bulky polymer backbone. On the basic of these considerations, we decided to employ a cross-linked polystyrene polymer (Scheme 2) as an insoluble support for the design of a second-generation PSTA.
We began the synthesis of the designed cross-linked polystyrene-supported transfer agent via treatment of commercially available 2-bromobenzaldehyde with allylmagnesium chloride to furnish benzylic alcohol 3 (95%), which upon lithiation with n-BuLi, followed by anion capture with Me2SiHCl and treatment with H2O, led to siloxane 4 in 45% yield (Scheme 3). Siloxane 4 was then converted to aryl bromide 5 in 85% yield by hydroboration with 9-BBN, followed by reaction with p-dibromobenzene in the presence of the palladium catalyst Pd(dppf)Cl2.11 In turn, 5 was converted smoothly to styrene 6 via Suzuki reaction12 employing potassium vinyltrifluoroborate under Pd-catalyzed cross-coupling condition. Suspension copolymerization13 of 6, styrene, and the flexible tetrahydrofuran-based cross-linker 7 (in a molar ratio of 12:87:1, respectively) was then achieved with benzoyl peroxide to provide polystyrene-supported siloxane transfer agent PSTA-II as well-defined beads.14 From the design perspective, the employed cross-linker 7 contains a highly stable phenyl ether linkage, leading to a gel-type resin that exhibits excellent swelling properties.15 The siloxane loading of PSTA-II was reasoned to be nearly identical to the silicon loading (1.5 mmol/g), the latter indicated by elemental analysis.
Scheme 3. Synthesis of Second-Generation PSTA.

To evaluate PSTA-II as a viable cross-coupling transfer agent, we employed conditions similar to those previously reported for PSTA-I.10 Pleasingly, PSTA-II efficiently mediated the palladium-catalyzed cross-coupling reaction between phenyllithium and 4-iodoanisole, furnishing the cross-coupling product 10 in 96% isolated yield, importantly with no evidence of homocoupling.16 Following the coupling reaction, the polymer was removed from the product by simple filtration. After several washings with organic solvents, the recovered polymer was then reused five additional times, providing excellent yield of the desired cross-coupling product 10, with no loss in reaction efficiency after repeated cycles (Scheme 4).
Scheme 4. Recyclability of PSTA-II Using the Same Nucleophile and Electrophile.
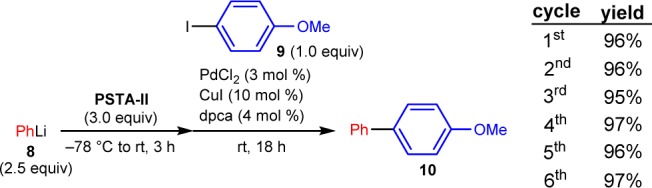
Siloxane loading was 1.5 mmol/g
We next explored the possibility of repeated recycling of PSTA-II in cross-coupling reactions with different nucleophiles and electrophiles (Scheme 5). 4-tert-Butylphenyllithium was cross-coupled with 4-iodoanisole to provide the desired product 10b in 95% isolated yield. The recovered polymer was then employed in the second cross-coupling reaction between phenyllithium and 4-iodoanisole, furnishing the desired product 10 in 97% isolated yield. Pleasingly, the first cross-coupling product 10b was not detected in the second cycle, demonstrating the ability to reuse PSTA-II with a different nucleophilic coupling partner without cross-contamination. The recovered polymer above was then recycled successfully in a sequence of eight cross-coupling reactions employing different nucleophilic and electrophilic coupling partnerts for a total of nine iterations (Scheme 5).17 Cross-coupling between arylorganolithium reagents and aryl or alkenyl iodides proceeded smoothly to provide the CCR products in excellent yields. Electron-rich and electron-deficient substrates were well tolerated; even an azaheterocycle proceeded well (entry 7). Cross-coupling reactions between phenyllithium and electron-deficient aryl bromides also readily provided the desired products in good yield (entries 8 and 10). Equally successful, CCRs employing alkenylorganolithiums proceeded in good yield with retention of the alkene geometry (entries 4 and 5). In all cases, no evidence of homocoupled products was observed. Pleasingly, cross-coupling reactions employing recovered PSTA-II are as high yielding as those employing the freshly made PSTA-I. Furthermore, PSTA-II offers an operationally convenient protocol in which the polymer could be removed directly from the CCR products via simple filtration without the need to introduce additional solvent to induce polymer precipitation. Importantly, PSTA-II permits the cross-coupling of substrates containing highly sensitive functional groups such as a nitrile and ketone (entries 6, 8, 9, and 10), demonstrating the unique advantage offered by this cross-coupling protocol over other reported CCR methods7,8 employing organolithium reagents.
Scheme 5. Recyclability of PSTA-II Using Multiple Nucleophiles and Electrophiles.
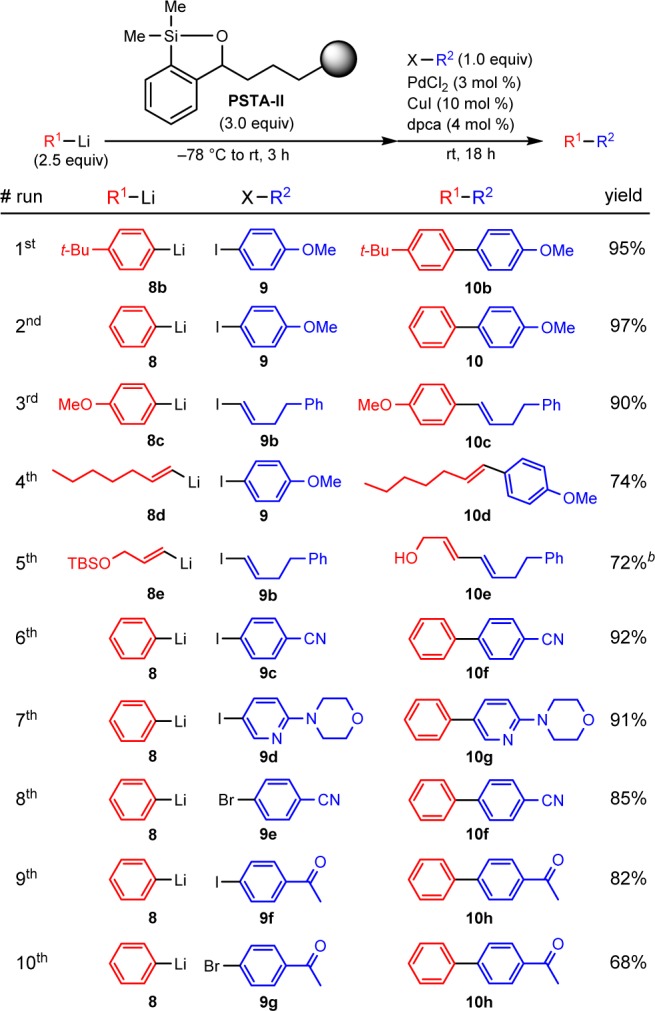
Siloxane loading was 0.74 mmol/g. All reactions were performed on 0.2 mmol scale of the aryl or alkenyl halides.
After polymer removal, product mixture was treated with TBAF to remove the silyl group prior to purification.
In summary, the rational design, synthesis, and validation of a greatly improved second-generation insoluble polymer-supported siloxane-transfer agent for use in palladium-catalyzed cross-coupling reactions has been achieved. Importantly, the cross-linked polystyrene support significantly simplifies product purification and permits excellent siloxane polymer reuse with diverse nucleophiles and electrophiles, rendering this tactic a powerful new tool in organic synthesis that could provide “greener” and more sustainable chemical processes. Studies to expand the application of polymer-supported siloxane-transfer agent in other bond-forming processes continue in our laboratory.
Acknowledgments
Financial support was provided by the NIH through Grant No. GM-29028. We also thank Bruno Melillo, Stephen P. Brown, and Dr. Rakesh Kohli at Department of Chemistry, University of Pennsylvania, for helpful suggestions on polymer selection, polymer handling, and assistance with HRMS, respectively.
Supporting Information Available
Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere A., Diederich F., Eds.; Wiley-VCH: Weinheim, 2004. [Google Scholar]; b Seechurn C. C. J.; Kitching M. O.; Colacot T. J.; Snieckus V. Angew. Chem., Int. Ed. 2012, 51, 5062. [DOI] [PubMed] [Google Scholar]
- a Smith A. B. III; Hoye A. T.; Martinez-Solorio D.; Kim W.-S.; Tong R. J. Am. Chem. Soc. 2012, 134, 4533. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Martinez-Solorio D.; Hoye A. T.; Nguyen M. H.; Smith A. B. III. Org. Lett. 2013, 15, 2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Miyaura N.; Yamada K.; Suzuki A. Tetrahedron Lett. 1979, 20, 3437. [Google Scholar]; b Miyaura N.; Suzuki A. J. Chem. Soc., Chem. Commun. 1979, 866. [Google Scholar]; Reviews:; c Miyaura N.; Suzuki A. Chem. Rev. 1995, 95, 2457. [Google Scholar]; d Suzuki A.; Yamamoto Y. Chem. Lett. 2011, 40, 894. [Google Scholar]
- a Negishi E.; Baba S. Chem. Commun. 1976, 596. [Google Scholar]; b Baba S.; Negishi E. J. Am. Chem. Soc. 1976, 98, 6729. [Google Scholar]; Reviews:; c Negishi E. Acc. Chem. Res. 1982, 15, 340. [Google Scholar]; d Negishi E.; Hu Q.; Huang Z.; Qian M.; Wang G. Aldrichimica Acta 2005, 38, 71. [Google Scholar]
- a Milstein D.; Stille J. K. J. Am. Chem. Soc. 1978, 100, 3636. [Google Scholar]; b Milstein D.; Stille J. K. J. Am. Chem. Soc. 1979, 101, 4992. [Google Scholar]; Reviews:; c Stille J. K. Angew. Chem., Int. Ed. Engl. 1986, 25, 508. [Google Scholar]; d Mitchell T. N. Synthesis 1992, 803. [Google Scholar]
- a Hatanaka Y.; Hiyama T. J. Org. Chem. 1988, 53, 918. [Google Scholar]; b Hatanaka Y.; Hiyama T. Synlett 1991, 845. [Google Scholar]; c Hiyama T.; Hatanaka T. Pure Appl. Chem. 1994, 66, 1471. [Google Scholar]; d Hiyama T. J. Organomet. Chem 2002, 653, 58. [Google Scholar]; e Nakao Y.; Imanaka H.; Sahoo A. K.; Yada A.; Hiyama T. J. Am. Chem. Soc. 2005, 127, 6952. [DOI] [PubMed] [Google Scholar]; f Nakao Y.; Takeda M.; Matsumoto T.; Hiyama T. Angew. Chem., Int. Ed. 2010, 49, 4447. [DOI] [PubMed] [Google Scholar]; g Chen J.; Tanaka M.; Sahoo A. K.; Takeda M.; Yada A.; Nakao Y.; Hiyama T. Bull. Chem. Soc. Jpn. 2010, 83, 554. [Google Scholar]; h Nakao Y.; Hiyama T. Chem. Soc. Rev. 2011, 40, 4893. [DOI] [PubMed] [Google Scholar]; i Tang S.; Takeda M.; Nakao Y.; Hiyama T. Chem. Commun. 2011, 47, 307. [DOI] [PubMed] [Google Scholar]; j Denmark S. E.; Choi J. Y. J. Am. Chem. Soc. 1999, 121, 5821. [Google Scholar]; k Denmark S. E.; Sweis R. F. J. Am. Chem. Soc. 2004, 126, 4876. [DOI] [PubMed] [Google Scholar]; Reviews:; l Denmark S. E.; Sweis R. F. Acc. Chem. Res. 2002, 35, 835. [DOI] [PubMed] [Google Scholar]; m Denmark S. E.; Regens C. S. Acc. Chem. Res. 2008, 41, 1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Murahashi S.-I.; Tanba Y.; Yamamura M.; Moritani I. Tetrahedron Lett. 1974, 15, 3749. [Google Scholar]; b Murahashi S.-I.; Tanba Y.; Yamamura M.; Yoshimura N. J. Org. Chem. 1978, 43, 4099. [Google Scholar]; c Murahashi S.-I. J. Organomet. Chem. 2002, 653, 27. [Google Scholar]
- a Giannerini M.; Fananas-Mastral M.; Feringa B. L. Nature Chem. 2013, 5, 667. [DOI] [PubMed] [Google Scholar]; b Hornillos V.; Giannerini M.; Vila C.; Fananas-Mastral M.; Feringa B. L. Org. Lett. 2013, 15, 5114. [DOI] [PubMed] [Google Scholar]; c Giannerini M.; Hornillos V.; Vila C.; Fananas-Mastral M.; Feringa B. L. Angew. Chem., Int. Ed. 2013, 52, 13329. [DOI] [PubMed] [Google Scholar]
- For C–N bond formation mediated by siloxane transfer agent, see:Nguyen M. H.; Smith A. B. III. Org. Lett. 2013, 15, 4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M. H.; Smith A. B. III. Org. Lett. 2013, 15, 4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaura N.; Ishiyama T.; Sasaki H.; Ishikawa M.; Satoh M.; Suzuki A. J. Am. Chem. Soc. 1989, 111, 314. [Google Scholar]
- Molander G. A.; Brown A. R. J. Org. Chem. 2006, 71, 9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. E.; Paech K.; Zhou W. J.; Kurth M. J. J. Org. Chem. 1998, 63, 5094. [Google Scholar]
- The polymer is highly stable. Samples were stored in capped vial on benchtop at room temperature during the study and showed no sign of decomposition.
- a Toy P. H.; Janda K. D. Tetrahedron Lett. 1999, 40, 6329. [Google Scholar]; b Toy P. H.; Reger T. S.; Garibay P.; Garno J. C.; Malikayil J. A.; Liu G.; Janda K. D. J. Comb. Chem. 2001, 3, 117. [DOI] [PubMed] [Google Scholar]
- Three equivalents of siloxane transfer agent were employed to drive the reaction to completion with full consumption of the aryl halide.
- The Si loading of the recovered polymer sample after nine iterations was determined to be 0.78 mmol/g.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.