

Abstract

Difluorinated cyclooctynes are important reagents for labeling azido-biomolecules through copper-free click chemistry. Here, a safe, scalable synthesis of a difluorinated cyclooctyne is reported, which involves a key homologation/ring-expansion reaction. Sequential ring expansions were also employed to synthesize and study a novel difluorinated cyclononyne.
Copper-free click chemistry, the reaction between an azide and a strained cycloalkyne (Figure 1A), has become a popular ligation strategy for applications that are sensitive to metals, precluding the use of the copper-catalyzed azide–alkyne cycloaddition (CuAAC, click chemistry, Figure 1B).1 To date, the study of live cells and organisms has been the premier application of cyclooctyne reagents;2 however, the simple reaction conditions and purification procedures of copper-free click chemistry have resulted in its expansion to a variety of disciplines including surface immobilization,3 gel synthesis,4 oligonucleotide functionalization,5 dendrimer modification,6 and the decoration of nanostructures.7
Figure 1.

(A) Copper-free click chemistry. (B) Copper-catalyzed azide–alkyne cycloaddition (CuAAC). (C) Selected cyclooctyne reagents for copper-free click chemistry.
The initial report of copper-free click chemistry involved the use of cyclooctyne 1 (OCT, Figure 1C) for the labeling of glycan-associated azides.8 This reaction was selective and mild, but its relatively slow rate remained a liability for many applications. Since that time, our group and others have focused on enhancing the rate of the azide-cyclooctyne cycloaddition by applying classical physical organic chemistry principles to further activate the strained alkynes for reaction with azides. Through these experimental efforts, two rate-enhancement strategies have emerged: the addition of electron-withdrawing groups at the propargylic position (MOFO 2, DIFO 3)2a,9 and the augmentation of strain energy through fused aryl (DIBO 4, DIBAC 5, BARAC 6)10 or cyclopropyl (BCN 7) rings.11 The second-order rate constant for the [3 + 2] cycloaddition is an important consideration in the choice of a cycloalkyne reagent, as rapid reaction kinetics allow for low concentrations of reagents and offer opportunities for real-time labeling. However, superior reaction kinetics do not come without liabilities. For example, BARAC has a limited half-life (24 h) in phosphate-buffered saline due to hydrolysis of the strained central amide10c and undergoes rearrangement to indole products under acidic conditions.12 The balance between reactivity and stability is a significant challenge, as efforts to synthesize new cyclooctyne reagents are pursued.13
Despite slower reaction kinetics than BARAC, difluorinated cyclooctynes have remained useful reagents for copper-free click chemistry. DIFO is an exceptional cyclooctyne for labeling glycan-associated azides on the enveloping layer of zebrafish.2b,14 Also, propargylic difluorination is a much less sterically encumbering rate-enhancement strategy than aryl-ring fusion. Smaller reagents are advantageous for reaching hindered azides as well as for the modification of biomolecules with cycloalkynes. Recent work exemplifies this as Lemke and co-workers have discovered that cyclooctyne, but not the larger dibenzocyclooctyne, can be site-specifically incorporated into proteins through amber stop codon techniques.15
The major limitation of DIFO surrounds its synthetic intractability, particularly when compared to other biaryl-based cyclooctynes with similar reaction kinetics (DIBO, DIBAC). Previous work from our group addressed this problem through the synthesis of difluorinated cyclooctyne 8 (DIFO2, Scheme 1), where difluorinated cyclooctanone 9 was a key intermediate.16 The synthesis of DIFO2 was much improved; however, its reported starting material 1,3-cyclooctadione (10) is not readily available in large quantities. Efforts to improve the safety and scalability of 1,3-cyclooctadione have been pursued but remain low yielding.17
Scheme 1. Retrosynthetic Analysis of Difluorinated Cycloalkynes from Cyclic 1,3-Diketones.

Here, an alternative approach to DIFO2 that begins from the much more synthetically accessible 1,3-cycloheptadione (12) and relies upon a key homologation with trimethylsilyl diazomethane is described. Additionally, two sequential ring expansions were employed to synthesize cyclononanone 13, which could be readily transformed into difluorinated cyclononyne 14 (DIFN). Cycloheptadione 12 was prepared in three steps from cyclopentanone and dichloroacetyl chloride as previously reported.18 This procedure has been used to safely produce up to 50 g of 1,3-cycloheptadione.18b The gem-difluoro group was installed by treatment of 12 with cesium carbonate and Selectfluor to give 2,2-difluoro-1,3-cycloheptadione (15) (Scheme 2). Desymmetrization of 15 was achieved through a mono-Wittig reaction with phosphonium salt 16 to yield compound 17. Hydrogenation of the resulting olefin quantitatively gave ketone 11, which was treated with trimethylsilyl diazomethane, a safe diazo species,19 in the presence of trimethylaluminum to yield the eight-membered ring scaffold. The trimethylsilyl group was hydrolyzed by treatment with acid to produce difluorinated ketone 9, which can be taken on to difluorinated cyclooctyne 8 as previously reported.16 Comparison of the two DIFO2 syntheses reveals that the homologation approach has a lower overall yield. This is primarily due to the desymmetrization reaction between 15 and 16 where it is difficult to suppress the competing double Wittig olefination.20 Despite this limitation, the ring expansion approach is the preferred method of DIFO2 synthesis within our group, as it precludes the need for potentially explosive or highly pyrophoric reagents such as periodic acid and diethyl zinc.
Scheme 2. Homologation Approach to DIFO2.

The homologation strategy also provided a facile opportunity to access a difluorinated cyclononyne analog through consecutive ring expansions. Cyclononynes have significantly less strain energy than their eight-membered ring congeners; however, they offer valuable insight into the fundamental physical organic chemistry of copper-free click chemistry, particularly when all other aspects of the cycloalkynes are identical. Additionally, slower azide-reactive reagents may prove useful for applications where proximity-induced reactivity is desired, such as the study of protein–protein interactions or binding-promoted ligations.21
To synthesize the target cyclononyne, compound 9 was treated with trimethylsilyl diazomethane in the presence of trimethylaluminum followed by acidic conditions to remove the resulting trimethylsilyl group (Scheme 3). Ketone 13 was readily isolated and converted to the desired alkyne through in situ vinyl triflate formation followed by syn-elimination of triflic acid to yield the target cyclononyne (DIFN, 14).
Scheme 3. Synthesis of Difluorinated Cyclononyne 14.

With DIFN in hand, the second-order rate constant for the reaction of 14 with benzyl azide was measured using an NMR assay (Scheme S1). The increased ring size led to a reaction rate that is 3 orders of magnitude less than that of DIFO2 (5.92 × 10–5 ± 2 × 10–7 M–1 s–1 vs 4.2 × 10–2 M–1 s–1 for 14 and 8, respectively).16 This second-order rate constant is similar to previously reported monobenzocyclononynes (10–4 to 10–5 M–1 s–1).22 Interestingly, our previous work has shown that difluorinated cyclooctynes are over 10 times more reactive with benzyl azide than monobenzocyclooctynes13 suggesting that electronic modulation, bond polarization, and/or transition state stabilization23 due to propargylic difluorination have a large effect only when an alkyne is highly strained. Recent computational work by Alabugin and co-workers23c has indicated that minimal reactant destabilization (i.e., low strain energies) can be completely compensated for by transition-state stabilization in copper-free click chemistry. DIFN and other published cyclononynes with experimentally characterized azide reactivity22 currently do not support Alabugin’s conclusion as DIFN’s transition-state stabilizing gem-difluoro group does not result in increased reactivity with azides. However, these cyclononynes have not been explicitly designed to take advantage of transition-state stabilization. Alabugin’s work exemplifies that although the array of copper-free click chemistry reagents is rapidly growing, a larger set is still necessary to understand the importance of each chemical interaction during the cycloaddition reaction.
Toward this end, and in hopes of achieving highly azide-reactive reagents, the homologation approach was chosen as a key retrosynthetic step in the synthesis of tri- and tetrafluorinated cyclooctynes. Properly substituted ketone intermediates 18 and 19 became our target ring-expansion products (Scheme 4). These derivatized cyclooctanones could be converted to tetra- and trifluorinated cyclooctynes, respectively, by established methods.1a,2a,9 Tetrafluorinated ketone 20 and trifluorinated ketone 22 were prepared through established α-fluorination methods of imine (for 20)24 or silyl enol ether (for 22 via 21)2a formation followed by treatment with Selectfluor. However, upon treatment of 20 and 22 with an array of diazo species in the presence of a variety of Lewis acids, the desired ring-expansion products were not obtained.25 Instead, primarily oxirane formation (23, 24) was observed, indicating that 1,2 migration of an electron-deficient C–CFn bond is disfavored. The high regioselectivity observed in the homologation of 11 also supports this hypothesis. Thus, it appears that the homologation strategy is not viable for the preparation of highly fluorine-substituted (F atoms >2) cycloalkynes.
Scheme 4. Synthesis and Reactivity of Tri- and Tetrafluorinated Cycloheptanone Homologation Substrates.

A safely scalable synthesis of the second-generation difluorinated cyclooctyne 8, a reagent that has seen success in the labeling of azides in living organisms as well as use in materials science, was devised. The central step in this new synthesis is a one-carbon ring expansion employing trimethylsilyl diazomethane, which proceeds readily on substituted difluorocycloheptanone and the larger difluorocyclooctanone substrates; however, the homologation reaction was suppressed when fluorine atoms were added to both α positions of the ketone. Thus, the homologation methodology is restricted to the synthesis of gem-difluorocycloalkynes. Using this strategy, novel difluorinated cyclononyne 14 was synthesized and found to react with benzyl azide 3 orders of magnitude more slowly than difluorinated cyclooctyne 8. These data indicate that the primary importance in the design and synthesis of reactive cycloalkynes for copper-free click chemistry is ring strain, followed by electronic activation and steric effects. The synthesis and analysis of difluorinated cyclononyne 14 represents another important addition to the collection of copper-free click chemistry reagents, as it will further guide the design of new more reactive reagents, such as transition-state stabilized or additionally fluorinated cycloalkynes. New synthetic strategies toward the putatively highly reactive tri- and tetrafluorinated cyclooctynes are underway.
Acknowledgments
This work was supported by a grant to C.R.B. from the NIH (GM058867), an ACS Organic Division Fellowship to E.M.S., and a predoctoral Ruth L. Kirschstein National Research Service Award to G.d.A. We thank Jason Kingsbury and Victor Rendina for helpful discussions regarding the homologation of 20 and 22.
Supporting Information Available
Procedures and characterization for all new compounds, kinetic analysis of DIFN, and further discussion of the ring expansion of tri- and tetrafluorinated ketones. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Massachusetts Institute of Technology, Cambridge, MA 02139.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Jewett J. C.; Bertozzi C. R. Chem. Soc. Rev. 2010, 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Debets M. F.; van der Doelen C. W. J.; Rutjes F. P. J. T.; van Delft F. L. ChemBioChem 2010, 11, 1168–1184. [DOI] [PubMed] [Google Scholar]; c Sletten E. M.; Bertozzi C. R. Angew. Chem., Int. Ed. 2009, 48, 6974–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Baskin J. M.; Prescher J. A.; Laughlin S. T.; Agard N. J.; Chang P. V.; Miller I. A.; Lo A.; Codelli J. A.; Bertozzi C. R. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Laughlin S. T.; Baskin J. M.; Amacher S. L.; Bertozzi C. R. Science 2008, 320, 664–667. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yao J. Z.; Uttamapinant C.; Poloukhtine A.; Baskin J. M.; Codelli J. A.; Sletten E. M.; Bertozzi C. R.; Popik V. V.; Ting A. Y. J. Am. Chem. Soc. 2012, 134, 3720–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Dieterich D. C.; Hodas J. J.; Gouzer G.; Shadrin I. Y.; Ngo J. T.; Triller A.; Tirrell D. A.; Schuman E. M. Nat. Neurosci. 2010, 13, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Beatty K. E.; Fisk J. D.; Smart B. P.; Lu Y. Y.; Szychowski J.; Hangauer M. J.; Baskin J. M.; Bertozzi C. R.; Tirrell D. A. ChemBioChem 2011, 12, 2137–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kuzmin A.; Poloukhtine A.; Wolfert M. A.; Popik V. V. Bioconjugate Chem. 2011, 21, 2076–2085. [DOI] [PubMed] [Google Scholar]; b Wendein C.; Singh I.; Rinnen S.; Schulz C.; Arlinghaus H. F.; Burley G. A.; Ravoo B. J. Chem. Sci. 2012, 3, 2479–2484. [Google Scholar]; c Wammes A. E. M.; Fischer M. J. E.; de Mol N. J.; van Eldijk M. B.; Rutjes F. P. J. T.; van Hest J. C. M.; van Delft F. L. Lab Chip 2012, 13, 1863–1867. [DOI] [PubMed] [Google Scholar]
- a Johnson J. A.; Baskin J. M.; Bertozzi C. R.; Koberstein J. T.; Turro N. J. Chem. Commun. 2008, 3064–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]; b DeForest C. A.; Polizzotti B. D.; Anseth K. S. Nat. Mater. 2009, 8, 659–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a van Delft P.; Meeuwenoord N. J.; Hoogendoorn S.; Dinkelaar J.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V. Org. Lett. 2010, 12, 5486–5489. [DOI] [PubMed] [Google Scholar]; b Jayaprakash K. N.; Peng C. G.; Butler D.; Varghese J. P.; Maier M. A.; Rajeev K. G.; Manoharan M. Org. Lett. 2010, 12, 5410–5413. [DOI] [PubMed] [Google Scholar]; c Singh I.; Freeman C.; Heaney F. Eur. J. Org. Chem. 2011, 33, 6739–6746. [Google Scholar]
- a Ornelas C.; Broichhagen J.; Weck M. J. Am. Chem. Soc. 2010, 132, 3923–3931. [DOI] [PubMed] [Google Scholar]; b Huang B.; Desai A.; Zong H.; Tang S.; Leroueil P.; Baker J. R. Tetrahedron Lett. 2011, 52, 1411–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ledin P. A.; Friscourt F.; Guo J.; Boons G. J. Chem.—Eur. J. 2011, 17, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lallana E.; Fernandez-Megia E.; Riguera R. J. Am. Chem. Soc. 2009, 131, 5748. [DOI] [PubMed] [Google Scholar]; b Chakrabarty R.; Stang P. J. Am. Chem. Soc. 2012, 134, 14738–14741. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gobbo P.; Novoa S.; Biesinger M. C.; Workentin M. S. Chem. Commun. 2013, 49, 3982–3984. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. J. Am. Chem. Soc. 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Baskin J. M.; Prescher J. A.; Lo A.; Bertozzi C. R. ACS Chem. Biol. 2006, 1, 644–648. [DOI] [PubMed] [Google Scholar]
- a Ning X.; Guo J.; Wolfert M. A.; Boons G. J. Angew. Chem., Int. Ed. 2008, 47, 2253–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Debets M. F.; Van Berkel S. S.; Schoffelen S.; Rutjes F. P. J. T.; Van Hest J. C. M.; Van Delft F. L. Chem. Commun. 2010, 46, 97–99. [DOI] [PubMed] [Google Scholar]; c Jewett J. C.; Sletten E. M.; Bertozzi C. R. J. Am. Chem. Soc. 2010, 132, 3688–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dommerholt J.; Schmidt S.; Temming R.; Hendriks L. J. A.; Rutjes F. P. J. T.; van Hest J. C. M.; Lefeber D. J.; Friedl P.; van Delft F. L. Angew. Chem., Int. Ed. 2010, 49, 9422–9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chigrinova M.; McKay C. S.; Beaulieu L. P. B.; Udachin K. A.; Beauchemin A. M.; Pezacki J. P. Org. Biomol. Chem. 2013, 11, 3436–3441. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Nakamura H.; Jewett J. C.; Bertozzi C. R. J. Am. Chem. Soc. 2010, 132, 11799–11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dehnert K. W.; Beahm B. J.; Huynh T. T.; Baskin J. M.; Laughlin S. T.; Wang W.; Wu P.; Amacher S. L.; Bertozzi C. R. ACS Chem. Biol. 2011, 6, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Baskin J. M.; Dehnert K. W.; Laughlin S. T.; Amacher S. L.; Bertozzi C. R. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10360–10365. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dehnert K. W.; Baskin J. M.; Laughlin S. T.; Beahm B. J.; Naidu N. N.; Amacher S. L.; Bertozzi C. R. ChemBioChem 2012, 13, 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plass T.; Milles S.; Koehler C.; Schultz C.; Lemke E. A. Angew. Chem., Int. Ed. 2011, 50, 3878–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codelli J. A.; Baskin J. M.; Agard N. J.; Bertozzi C. R. J. Am. Chem. Soc. 2008, 130, 11486–11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims E. A.; DeForest C. A.; Anseth K. S. Tetrahedron Lett. 2011, 52, 1871–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Do N.; McDermott R. E.; Ragan J. A. Org. Synth. 2008, 85, 138–146. [Google Scholar]; b Ragan J. A.; Makowski T. W.; am Ende D. J.; Clifford P. J.; Young G. R.; Conrad A. K.; Eisenbeis S. A. Org. Process Res. Dev. 1998, 2, 379–381. [Google Scholar]
- Hahimoto N.; Aoyama T.; Shioiri T. Tetrahedron Lett. 1980, 21, 4619–4622. [Google Scholar]
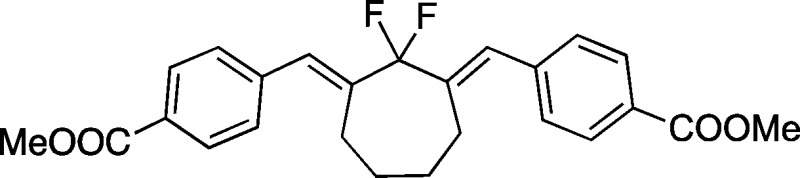
- The double Wittig olefination
product observed
in the reaction of 15 and 16.
- a Sharma A. K.; Heemstra J. M. J. Am. Chem. Soc. 2011, 133, 12426–12429. [DOI] [PubMed] [Google Scholar]; b Sharma A. K.; Kent A. D.; Heemstra J. A. Anal. Chem. 2012, 84, 6104–6109. [DOI] [PubMed] [Google Scholar]
- a Tummatorn J.; Batsomboon P.; Clark R. J.; Alabugin I. V.; Dudley G. B. J. Org. Chem. 2012, 77, 2093–2097. [DOI] [PubMed] [Google Scholar]; b de Almeida G.; Sletten E. M.; Nakamura H.; Palaniappan K. K.; Bertozzi C. R. Angew. Chem., Int. Ed. 2012, 51, 2443–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cheong P. H. Y.; Paton R. S.; Bronner S. M.; Im G. Y. J.; Garg N. K.; Houk K. N. J. Am. Chem. Soc. 2010, 132, 1267–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Im G. Y. J.; Bronner S. M.; Goetz A. E.; Paton R. S.; Cheong P. H. Y.; Houk K. N.; Garg N. K. J. Am. Chem. Soc. 2010, 132, 17933–17944. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gold B.; Dudley G. B.; Alabugin I. V. J. Am. Chem. Soc. 2013, 135, 1558–1569. [DOI] [PubMed] [Google Scholar]
- Pravst I.; Zupan M.; Stavber S. Synthesis 2005, 3140–3146. [Google Scholar]
- See Supporting Information for further details on the attempted ring expansion of 20 and 22.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.