

Abstract
Purpose.
The study was done to better understand the biological significance of clusterin co-localization with the exfoliation deposits (XF deposits), and provide insight into a pathogenic mechanism involving activation of the complement system and its pro-inflammatory consequences in patients with exfoliation glaucoma.
Methods.
Exfoliation lens deposits were analyzed by high resolution atomic force microscopy imaging and confocal immunofluorescence. Levels of clusterin and vitronectin, as well as of the complement activation products C3a and soluble C5b-9, were assessed via ELISA.
Results.
Atomic-force microscopy examination of lenses with exfoliation syndrome (XFS) revealed a dense fibrillar network on the anterior, aqueous-bathed surface of the lens, while the epithelial side displayed no discernible structural features at the same resolution. Clusterin colocalized with XF deposits, demonstrating integral association with the fibrils. Levels of activation-derived complement components C3a and soluble C5b-9, as well as the complement inhibitors clusterin and vitronectin, were found significantly elevated (1.7-fold, P < 0.05; 4.1-fold, P < 0.05; 1.8-fold, P < 0.01; and 3.0-fold, P < 0.01, respectively) in aqueous humor from glaucoma patients with XFS compared to non-XFS glaucoma controls.
Conclusions.
The data provide compelling evidence for the activation of the complement system in XFS, highlighting the generation of subproducts with potent proinflammatory activity, which are capable of triggering and chronically maintaining levels of subclinical inflammation, suggesting novel targets for therapeutic intervention. The colocalization of clusterin in exfoliation fibrils suggests a failed attempt to prevent tissue accumulation of protein aggregates, as seen in other protein folding disorders, likely due to the abnormal high levels of misfolded proteins overwhelming its chaperone capacity.
Keywords: exfoliation syndrome, complement system, extracellular chaperones, protein misfolding disorders
This study provides experimental evidence for the involvement of the multifunctional extracellular chaperone clusterin and the generation of proinflammatory complement activation subproducts in exfoliation glaucoma.
Introduction
Exfoliation syndrome (XFS) is an age-related disorder that constitutes the most common identifiable cause of open-angle glaucoma worldwide, accounting for up to 70% of cases in some countries.1,2 Exfoliation material (XFM) also is found in numerous systemic tissues and organs, and its potential association with other diseases is only beginning to be realized. Its importance is expected to expand in view of increasing age and life expectancy of the world population. Abnormal fibrillar deposits, composed of bundles of microfibrils ultrastructurally resembling elastic fibers, are present in aqueous-bathed surfaces of the anterior segment of the eye. Compared to primary open-angle glaucoma, exfoliation glaucoma (XFG) has a more serious clinical course and worse prognosis, being associated typically with higher levels of IOP, greater diurnal pressure fluctuations, marked pressure spikes, higher severity and frequency of optic nerve damage, more rapid visual field loss, poorer response to medications, and the need for more frequent surgical interventions.3 Elevation of the IOP in XFG patients correlates with the presence and severity of glaucomatous optic nerve damage,4 and is caused by an increased aqueous outflow resistance in the trabecular meshwork driven by the gradual buildup of XFM in the juxtacanalicular tissue beneath the inner wall of Schlemm's canal, the site of the greatest resistance to aqueous outflow.
Although the specific pathogenetic mechanism(s) of XFS remain mostly unknown, immunohistochemical and immunoelectron microscopic studies, as well as genomic and proteomic approaches, support the concept of stress-induced elastosis associated with excessive production and/or abnormal cross-linking of elastic tissue components forming fibrillar aggregates of a complex glycoprotein/proteoglycan composition enriched in extracellular matrix, basement membrane, and elastic fiber proteins.1,5 Supporting this notion, genetic studies in multiple populations have identified specific single nucleotide polymorphisms in the lysyl oxidase-like 1 gene (LOXL1), an enzyme that catalyzes the covalent cross-linking of collagen and elastin in connective tissues, as a major risk factor for developing XFS, although additional candidate genes have been reported, including the integral component of exfoliation (XF) fibrils fibrillin-1 (FBN1), latent TGF-β binding protein 2 (LTBP2), microfibril-associated protein 2 (MFAP2), transglutaminase 2 (TGM2), transforming growth factor β1 (TGFB1), and the extracellular chaperone clusterin (CLU).6–9 Proinflammatory cytokines, growth factors, and conditions of oxidative stress and ischemia/hypoxia10,11 have the potential either to stimulate the synthesis of abnormal XF fibrils or induce detrimental cell responses. In fact, oxidative stress and hypoxia have been reported to regulate LOXL1 expression as well as activity and synthesis of elastin and fibrillin, while an increased expression of IL-1α, IL-6, and IL-8 has been linked to glaucoma.12–15
Proteomic analysis of XF fibrils identified the multifunctional glycoprotein clusterin as one of the major contributors to the XF deposits.5,16 This ubiquitous molecule is implicated in multiple physiological and pathologic functions, such as lipid transport, complement regulation, modulation of cell–cell and cell–matrix interactions, and chaperoning activity of misfolded proteins, among others.17,18 In the eye, clusterin normally is expressed in most ocular cells and tissues,16 including corneal and conjunctival epithelium,19,20 corneal endothelium,21 ciliary body, and retina ocular basement membranes and stromal fibers,16 and is present in aqueous and vitreous humors.16,22 Although the proteomic studies clearly demonstrated the increased expression of clusterin in XFM,5 the role of the protein in the disease pathogenesis remains undefined. The present study was done to obtain high definition images of XF fibrils and better understand the biological significance of clusterin colocalization with the deposits. Using a combination of atomic-force microscopy (AFM), classic immunohistochemistry, and fluorescence confocal microscopy, we assessed its topographic location in the fibrils. Quantitative analysis of the levels of activation-derived complement components and fluid-phase physiologic inhibitors in aqueous fluid from patients with XFG pointed to a pathogenic mechanism involving activation of the complement system and its pro-inflammatory consequences.
Materials and Methods
Biological Materials
Anterior lens capsules (ALCs) and aqueous humor (AH) specimens were obtained from patients undergoing glaucoma filtration surgery at the New York Eye and Ear Infirmary (NYEEI). The ALCs were stored in sterile PBS at 4°C until use. The AH specimens were collected, after paracentesis, using a 1-mL syringe with a #30-gauge cannula that was inserted into the anterior chamber, angling the tip slightly anteriorly to avoid touching the iris. Aqueous fluid was aspirated until the anterior chamber began to shallow, yielding typically 30 to 50 μL per patient. After collection, aqueous-containing syringes were capped, snap-frozen in liquid N2, and stored at −80°C until use.
The XFG group consisted of 68 glaucoma patients who had XFM on the lens capsule as determined by slit-lamp examination, while the nonexfoliation control group was composed of 107 patients lacking XFM. Of the cases in the non-XFG group, 80% were diagnosed with open angle glaucoma; the remaining 20% comprised patients diagnosed with angle closure (18%) and other types of glaucoma (2%). The majority of cases (94% of XFG and 89% of non-XFG) received a combination of topical IOP-lowering agents, in accordance with current therapeutic management trends.23 These included α-adrenergic agonists (26.5% of XFG versus 27.0% of non-XFG), β-adrenergic antagonists (61.8% of XFG versus 54.2% of non-XFG), carbonic anhydrase inhibitors (32.3% of XFG versus 46.7% of non-XFG), cholinergics (26.4% of XFG versus 10.3% of non-XFG), and prostaglandin analogs (73.5% of XFG versus 70.0% of non-XFG). Cases with capsular fibrosis, posterior synechiae, or other debris on the anterior lens surface, as well as with a history of uveitis were excluded from the study. All participants signed informed consents approved by NYU and NYEEI Institutional Review Boards, which adhered to the tenets of the Declaration of Helsinki for experiments involving human tissue.
Antibodies and Reagents
Rabbit polyclonal antibodies reacting with vitronectin were purchased from Chemicon International (Temecula, CA, USA). Mouse monoclonal antibodies anti–fibrillin-1 and anti-vitronectin were obtained from Millipore Corporation (Billerica, MA, USA). Mouse monoclonal anti-apolipoprotein B antibodies were procured from Cortex Biochem, Inc. (San Leandro, CA, USA), and Accurate Chemical and Scientific Corporation (Westbury, NY, USA). Goat anti-rabbit IgG conjugated to Alexa Fluor-488 was purchased from Life Technologies/Invitrogen (Carlsbad, CA, USA), whereas goat anti-rabbit IgG conjugated to 30 nm colloidal gold particles was from SPI Supplies (West Chester, PA, USA). The ELISA kits for quantitation of clusterin were purchased from R&D Systems (Minneapolis, MN, USA), for the assessment of C3a and the C5b-9 terminal complex were acquired from Quidel Corp. (San Diego, CA, USA), and for the evaluation of vitronectin from Takara Bio, Inc. (Mountain View, CA, USA). Human clusterin from both recombinant sources and purified from human plasma were purchased from R&D Systems and Creative BioMart (New York, NY, USA), respectively. Rabbit polyclonal antibody recognizing clusterin was obtained from Santa Cruz Biotechnology (H-330; Santa Cruz, CA, USA).
AFM Imaging
The ALCs were placed on clean mica surfaces and allowed to dry in ambient air for 10 minutes to immobilize the tissue, a frequently used procedure in AFM methodologies,24 which does not alter the surface topography.25 The AFM images were acquired using a Nanoscope IIIa multimode scanning probe microscope (Bruker Corp., Santa Barbara, CA, USA). To minimize lateral forces, which can deform the sample and distort the images, all imaging was carried out in Tapping Mode under ambient conditions using single beam silicon cantilevers, 110 to 140 μm in length, with nominal spring constant 20 to 80 N/m, as per manufacturer's specifications (Bruker Corp.). Imaging was performed at scan rates of 1 to 2 Hz collecting 512 samples/line. Oscillation amplitude during scanning was calibrated at 95% of the cantilever's free oscillation amplitude. Scanning and feedback control parameters were optimized continually to ensure good image quality. The surface was examined routinely for damage by increasing the scan size throughout the imaging. The AFM images were processed and analyzed with NanoScope Analysis Version 1.20 software. Width and banding periodicity of XFS fibers, observed in the cross-sections of height mode AFM images, were measured at half maximum of the cross-sectional profiles. The length of the XFS fibers was estimated using linear approximation.
Labeling of Clusterin in XFS Fibrils With Colloidal Gold Particles
The presence of clusterin in XFS ALCs was evaluated by AFM through the use of secondary antibodies conjugated to colloidal gold. After immunoreaction with anti-clusterin antibody (1:400, 4°C, overnight), ALCs were incubated with 30 nm colloidal gold-conjugated anti-rabbit IgG (1:200, 1 hour, room temperature), washed extensively with PBS, air-dried 10 minutes, and imaged by AFM as above.
Immunohistochemical Analysis of Clusterin, Vitronectin, and Fibrillin 1 in XFS Deposits
The 6-μm cryostat sections of XFS ALCs were processed as described previously.5 Briefly, sections were incubated separately for 1 hour at room temperature with either polyclonal antibodies anti-clusterin (1:400), anti-vitronectin (1:100; Chemicon International) or mouse monoclonal anti-fibrillin 1 (1:100) followed by the corresponding biotinylated anti-rabbit or anti-mouse secondary antibodies (Life Technologies/BioSource International, Carlsbad, CA, USA). In all cases, tissue sections subsequently were reacted with ABC complex (Dako, Carpinteria, CA, USA), color developed with diaminobenzidine/H2O2, and sections counterstained with hematoxylin.
Immunofluorescence Microscopy Assessment of Clusterin, Fibrillin 1, and Vitronectin in XFS Deposits
The ALCs were incubated separately with rabbit polyclonal anti-clusterin (1:400), as well as with mouse monoclonal anti-fibrillin 1 (1:1000) and anti-vitronectin (1:1000; Millipore Corporation) antibodies for 1 hour at room temperature. Following PBS washes, lenses were reacted with the respective Alexa Fluor-488 conjugated anti-rabbit IgG and Alexa Fluor-568 conjugated anti-mouse IgG (both at 1:1000 dilution) using standard protocols,26 and mounted on glass slides using Vectashield mounting medium (Vector Laboratories, Burlingame, VA, USA). Images were acquired on a Zeiss LSM 510 laser scanning confocal microscope (NYU School of Medicine Imaging Core Facility) and processed via ImageJ software (National Institutes of Health [NIH], Bethesda, MD, USA; available in the public domain at rsbweb.nih.gov).
Quantitation of Clusterin, Vitronectin, and the Activation-Generated Complement Components C3a and C5b-9 by ELISA
Clusterin, vitronectin, the complement derived anaphylotoxin C3a, as well as the terminal C5b-9 soluble complex were quantitated in AH specimens, thawed immediately before the assays, using capture ELISA in accordance with the respective manufacturers' protocols, as described previously.27 In all cases, assessment of the respective protein concentrations in the XFG and non-XFG specimens was determined by interpolation using standard curves.
Western-Blot and Dot-Blot Analyses
The specificity of H330 anti-clusterin antibody was validated by Western blot analysis using standard methods from our laboratory.26 Briefly, clusterin from recombinant origin and purified from plasma, was electrophoresed on 10% SDS-PAGE and electrotransferred to polyvinylidene difluoride (PVDF) membranes (Immobilon-P; Millipore Corporation) using 10 mM 3-cycloexylamino-1-propanesulfonic acid (CAPS; Sigma-Aldrich Corp., St. Louis, MO, USA) buffer pH 11 containing 10% methanol, as described previously.26 After transfer and blocking with 5% nonfat milk in PBS containing 0.1% Tween 20, the membrane was immunoreacted with the polyclonal anti-clusterin (1:1000 in Tris-buffered saline/0.1% Tween [TBST]) followed by horseradish peroxidase (HRP)–labeled F(ab')2 anti-rabbit IgG (1:5000; GE Healthcare Life Sciences, Pittsburgh, PA, USA). Fluorograms were developed by enhanced chemiluminescence (ECL) with ECL Western blotting detection reagent (GE Healthcare Life Sciences), and exposed to Hyperfilm ECL (GE Healthcare Life Sciences).
The presence of the plasma-associated apolipoprotein B in AH was evaluated by dot-blot analysis. The AH specimens (2 μL each) were added of sterile PBS to a final volume of 50 μL and loaded onto a nitrocellulose membrane assembled into a Bio-Dot Microfiltration Apparatus (Bio-Rad, Hercules, CA, USA). Pooled human plasma (at 1:1000 and 1:10,000 dilutions) was used as a positive control for apoB immunoreactivity. Dot-blot analysis was performed following standard procedures.26 Briefly, after sample application and nitrocellulose blocking for 1 hour with 1% nonfat milk in TBST, membranes subsequently were incubated with mouse monoclonal anti-apoB antibodies (1:1000) followed by HRP-conjugated anti-mouse secondary antibody (GE Healthcare Life Sciences). Immunoreactivity was assessed by ECL, as described above for the Western blot.26
Statistical Analysis
Statistical significance was assessed by t-test using Graph-Pad InStat (GraphPad Software, La Jolla, CA, USA). Values of P ≤ 0.05 were considered significant.
Results
XF Fibers Form Dense but Discontinuous Networks
Detailed evaluation of XF lenses by AFM, a high resolution imaging technique allowing sample analysis in native state,28 revealed a dense fibrillar network on the anterior, aqueous-bathed surface of the lens (Figs. 1A, 1B), while the epithelial side displayed no discernible structural features at the same resolution (Figs. 1D, 1E). Individual fibers exhibited a 20- to 50-nm width with a length extending several micrometers. They appeared to be composed of bead-like subunits with a banding periodicity of 10 to 20 nm (Figs. 1A, 1B, insets), in agreement with previous electron microscopic (EM) assessments.29,30 In contrast to the exfoliation lenses, non-XF cases appeared featureless, and anterior and epithelial sides were virtually indistinguishable from each other (Figs. 1C, 1F). Comparison of the surface morphology of XFM from glaucomatous and nonglaucomatous eyes revealed no obvious differences (Figs. 1A, 1B). Significant variations were found in the density of the fibrillar deposits across the surface of any given XF lens, as illustrated at lower magnification in Figures 1G through 1I, with areas exhibiting high fiber density (Fig. 1G), transition from dense into less covered fields (Fig. 1H), or sparsely covered regions (Fig. 1I).
Figure 1.

Atomic-force microscopy imaging of XF eye lenses. Anterior (A–C, G–I) and epithelial (D–F) side of lens capsules from XF (A, B, D, E, G–I) and non-XF cases (C, F). Insets in (A) and (B) show high magnification images highlighting structural details of XF fibers. Images are representative of seven XFG and four non-XFG cases. The lenses shown in (A, D) and (C, F) are from glaucomatous eyes; (B, E) illustrate a lens capsule from a nonglaucomatous XF eye. The nonhomogeneous distribution of fibril density in XF deposits is illustrated in (G–I). (G) Densely cover area. (H) Transition from densely into more sparsely covered fiber area. (I) Sparsely covered fiber area. In all images z-scale was set at 80 nm to facilitate comparison. Scale bars: 200 nm (A–F), 400 nm (G–I).
Clusterin in XF Material
The topographic distribution of clusterin in XF deposits was investigated by immunofluorescence, standard immunohistochemistry, and confocal microscopy using anti-clusterin polyclonal antibody H330, the specificity of which was validated by Western blot analysis using recombinant and plasma-purified human clusterin (Fig. 2A). Immunofluorescence microscopy using H330 and Alexa-488 conjugated secondary antibodies on free floating ACLs surgical specimens demonstrated an intense clusterin immunoreactivity only in XF deposits which displayed, in difference to non-XF specimens, a patch-like morphology (Figs. 2B, 2C, respectively) consistent with the micro-scale discontinuity of the XF deposits observed in the AFM images. Traditional immunohistochemical light microscopic analysis of 6-μm cryostat sections of XFS ALCs further illustrated the integral distribution of clusterin within XF deposits (Fig. 2D), whereas no reactivity was observed in non-XFG tissues probed with anti-clusterin (Fig. 2E), or in negative controls in which the primary antibody was replaced by saline (Fig. 2F). The XF deposits also were immunoreactive with anti–fibrillin-1, a previously described major component of the XF fibrils (Fig. 2G).1 Confocal microscopy evaluation confirmed the colocalization of clusterin (Fig. 2H) and fibrillin-1 (Fig. 2I) in the deposits, as indicated by the complete overlap of both fluorescence signals (Fig. 2J). Taking into consideration the surface morphology of the deposits and the differences in magnification between the images depicted in Figure 2 with those shown in Figure 1, in which fibrillar structures resolved by AFM represent approximately ×1000 zoom-in image of the patchy deposits seen in Figure 2B, the data strongly suggested that the XF deposits illustrated at lower magnification in immunofluorescence microscopy are, indeed, the areas containing the highest density of XF fibrils.
Figure 2.
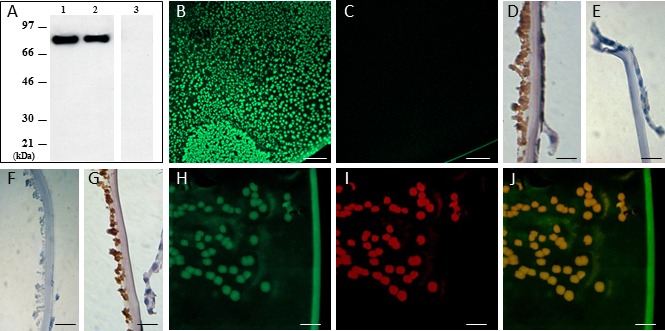
Clusterin in XF anterior lens capsules. Western blot analysis (A) illustrates the specificity of H330 anti-clusterin antibody. Lane 1: human recombinant clusterin. Lanes 2 and 3: human plasma clusterin. Left panel (lanes 1–2): PVDF membrane probed with H330 clusterin antibody. Right panel (lane 3): negative control in which the electrotransferred membrane was incubated with saline in lieu of anti-clusterin. Immunofluorescence microscopy evaluation of clusterin in anterior lens specimens from glaucoma patients with and without XFS is shown in (B) and (C), respectively. Light microscopy analysis illustrates clusterin immunostaining in XFG and non-XFG specimens ([D, E], respectively). Exfoliation glaucoma specimen in which primary antibodies were replaced by saline is shown as a negative control (F). Evaluation of fibrillin-1, a major component of XFM, by light microscopy is illustrated in (G). Confocal microscopy imaging depicts XFM immunolabeled for clusterin (green fluorescence, [H]) and fibrillin-1 (red fluorescence, [I]); both signals colocalize in the XF deposits (J). Scale bars: 200 μm (B, C), 50 μm (D–G), 25 μm in (H–J). Images are representative of nine XFG and six non-XFG lenses for the IF studies, and 13 XFG/four non-XFG controls in the case of the light microscopy analyses.
Clusterin Colocalize With XF Fibers
Combining the high resolution of AFM with the specificity of immunolabeling through the use of secondary antibodies conjugated to 30 nm colloidal gold particles, we were able to demonstrate further the integral co-association of clusterin with XF deposits. The XF fibers (Fig. 3A) appeared, after labeling (Fig. 3B), decorated with the antibody-gold conjugates in an often discontinuous beads-on-a-string pattern (inset) likely reflecting either variation in the density of clusterin epitopes across the fiber length or steric hindrance precluding antibody labeling of close epitopes. Diameter distribution analysis of multiple XF fibrils demonstrated a clear difference between labeled and unlabeled fibers. The data fitted a Gaussian distribution function, showing that clusterin-labeled fiber diameters were approximately three times larger than those of unlabeled structures (33.91 ± 0.11 nm for unlabeled versus 90.64 ± 0.25 nm for labeled fibers; Fig. 3C, red and blue graphs, respectively), consistent with each fiber being surrounded by a single layer of 30 nm antibody-gold conjugates (schematic representation shown in Fig. 3D). Thus, clusterin distribution in the XF lesions was clearly not a random event, but rather a tight colocalization with the actual XF fibrils.
Figure 3.
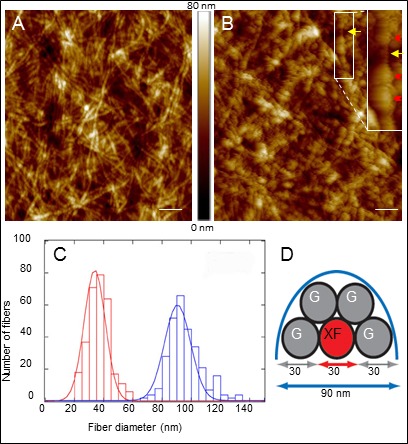
Clusterin colocalize with XF fibers. Atomic-force microscopy image of anterior lens capsules prior (A) and subsequent (B) to clusterin immunolabeling and detection with secondary antibodies conjugated to 30-nm colloidal gold particles. Scale bar: 400 nm; z scale = 80 nm. Inset shows details of unlabeled (yellow arrow) and gold decorated (red arrowheads) fibers at higher resolution. Data are representative of four different XFG lens capsule tissues. (C) Histograms illustrate the diameter distribution of XF fiber prior (red) and after (blue) labeling with colloidal gold. After fitting a Gaussian distribution function to each histogram the diameter of clusterin unlabeled and labeled fibers were estimated at 33.91 ± 0.11 and 90.64 ± 0.25 nm, respectively. (D) Schematic model of a cross-section of XF fibers decorated by gold particles, constructed based on the fiber diameters indicated in (C).
Colocalization of Clusterin and Vitronectin in XF Deposits
Vitronectin also was found to be an integral component of XF deposits, as demonstrated by the complete fluorescence signals overlap of clusterin and vitronectin when analyzed by confocal microscopy (Supplementary Fig. S1). Taking into account the colocalization of clusterin and fibrillin-1, as well as the incorporation of clusterin into the XF fibers illustrated above (Figs. 2, 3, respectively), the data strongly suggested that the three components are associated closely in the XF deposits.
Clusterin and Vitronectin Levels Are Elevated in Aqueous Humor From Glaucoma Patients With XFS
In view of the extensive co-association of clusterin and vitronectin with fibrillar XF deposits, we evaluated the concentration of both proteins in the AH of XFG and non-XFG patients. Despite the expected interindividual variability, both proteins were significantly elevated in XFG patients in comparison with non-XFG cases, as assessed by capture ELISA (Figs. 4A, 4B, respectively). Analysis of aqueous humor revealed a 1.8-fold increase in clusterin levels in XFG patients (n = 68, mean age = 76.8 ± 0.96; 50% men, 50% women) compared to non-XFG cases (n = 107, mean age = 71.6 ± 0.99, 43% men, 57% women, Fig. 4A). In the case of vitronectin, a 3.0-fold increment was observed in XFG patients (n = 34, mean age = 77.4 ± 1.22, 56% men, 44% women) compared to non-XFG cases (n = 37, mean age = 73 ± 1.75, 48.6% men, 51.4% women, Fig. 4B). In both cases, differences were statistically significant (P < 0.01). Since clusterin and vitronectin normally are circulating plasma proteins, their high concentrations might potentially reflect the previously suggested disruption of the physiological blood–aqueous permeability barrier.31 However, the total absence of the blood-associated apolipoprotein-B in AH, as indicated by dot-blot analysis coupled with the sensitive chemiluminescence detection (Supplementary Fig. S2) argue against the magnitude of the barrier leakage, suggesting that local synthesis also is a contributor to the elevated concentrations of clusterin and vitronectin, an issue that warrants further investigation.
Figure 4.
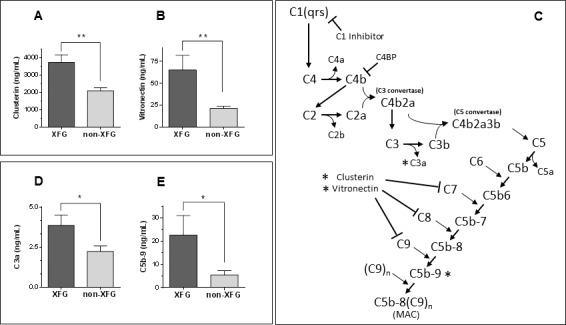
ELISA quantitation of activation-derived complement components and fluid phase complement inhibitors in aqueous humor. (A) Increased clusterin levels (1.8-fold) in aqueous humor from XFG (n = 68) patients compared to non-XFG (n = 107) controls. (B) Elevated vitronectin levels (3.0-fold) in aqueous humor from XFG (n = 34) cases compared to non-XFG (n = 37) controls. (C) Classical complement system activation pathway. The diagram illustrates the activation of the classical complement cascade leading to sequential activation of the different components, primarily by enzymatic cleavage. The sites of action of the known soluble phase complement control proteins C1-inhibitor, C4-binding protein (C4BP), clusterin, and vitronectin43 also are indicated. The activation derived components and regulatory proteins quantitated in aqueous humor in the present studies are highlighted by an asterisk. (D) Quantitative assessment of the activation-derived C3a anaphylotoxin levels in aqueous fluid showed a 1.7-fold increase in XFG (n = 26) patients compared to non-XFG (n = 27) controls. (E) Quantitation of soluble MAC in aqueous fluid revealed a 4.1-fold increase in XFG (n = 35) cases compared to non-XFG (n = 38) controls. In all cases, scale bars represent mean ± SEM. Levels of statistical significance: **P < 0.01, *P < 0.05.
Activation-Derived Complement Components Are Elevated in Aqueous Humor From XFG Patients
Since clusterin and vitronectin are known physiologic modulators of complement activation in fluid phase32,33 (Fig. 4C), their high levels in AH, together with our previous demonstration of the presence of activation-derived components in XF deposits in spite of the absence of terminal complement complex,5 reinforced the notion of the likely involvement of the complement system in XFG. This powerful effector system is composed of more than 20 proteins that, when activated, are sequentially cleaved in a proteolytic cascade (Fig. 4C). The terminal complex typically assembles on biological membranes and is associated with the membranolytic properties of the complement system. Conversely, when activation takes place in extracellular fluid phase, as in aqueous humor, the naturally occurring regulatory proteins vitronectin and clusterin bind to the terminal components resulting in the formation of a soluble nonlytic SC5b-9 complex. We, therefore evaluated, in aqueous humor of XFG and non-XFG patients, the formation of the C3a anaphylotoxin, generated through cleavage of C3, a central component of the system, as well as the formation of the terminal activation product, the membrane attack complex (MAC) C5b-9. As illustrated in Figure 4D, the concentration of C3a, determined by ELISA, was increased significantly (1.7-fold, P < 0.05) in the XFG group (n = 26; mean age, 74.3 ± 1.5; 50% men, 50% women) relative to the non-XFG cases (n = 27; mean age, 69.7 ± 1.8; 44.4% men, 55.6% women), in agreement with our previous demonstration of the presence of activation-derived classical pathway components C1q, C3c, and C4c in the aqueous-bathed XFM.5 Soluble C5b-9 also was significantly increased (4.1-fold, P < 0.05) in XFG samples (n = 35; mean age, 77.6 ± 1.15, 51.4% men, 48.6% women) relative to non-XFG cases (n = 38; mean age, 71.4 ± 1.7; 52.8% men, 47.2% women), as illustrated in Figure 4E. These results, together with the elevated levels of clusterin and vitronectin support the notion of complement activation in fluid phase leading to the assembly of the endpoint soluble MAC in absence of C5b-9 deposition in XFM.5
Discussion
Clusterin is a multifunctional protein that has been implicated in a wide variety of physiological and pathologic processes, including lipid transport, apoptosis, cell–cell and cell–matrix interactions, and stabilization of protein folding following stress-induced denaturation.17,18 As an extracellular molecular chaperone protein, clusterin facilitates folding and conformational maturation of nascent and stress denatured proteins into their active conformations, resembling heat-shock proteins, the most extensively studied molecular chaperones.34 Typically, refolding of a client protein involves interaction between the protein and the chaperone, which has high affinity for hydrophobic, unstructured polypeptide chains, common in denatured proteins. As some of the multiple examples of this chaperone activity, clusterin high affinity interaction with the soluble form of Alzheimer's amyloid-β in vitro was shown to preclude its typical fibrillization and neurotoxicity, a protective effect also seen with other amyloid molecules and prion fragments.17 Paradoxically to its protective chaperone activity, clusterin has been found codeposited in all amyloid lesions tested so far, irrespective of their nature and location,17 including forms restricted to ocular tissues.35 Whether the presence of clusterin in these deposits, as well as in XFS, reflects a failed attempt to prevent the formation of the fibrillar deposits and facilitate normal protein folding remains to be elucidated. Certainly, the intimate association of clusterin with XF fibrils, as proposed previously (Ghiso J, et al. IOVS 2012;53:ARVO E-Abstract 6318) and demonstrated herein is consistent with tight chaperone-client interactions.
One of the multiple functions of clusterin is to act as an inhibitor of the complement system,17 an important mediator of inflammation. Originally thought to be initiated only by binding of immune complexes to C1q, the recognition component of the classical pathway, it became evident that the system could be activated directly, in the absence of antibody, by direct interaction of certain molecules with C3 (alternative pathway), C1q (antibody-independent classical pathway), or by specific lectins on the surface of microorganisms (lectin pathway). Our previous proteomic studies demonstrated the colocalization within XFM of C1q as well as of the activation-derived fragments C3c and C4c, together with absence of alternative pathway generated Bb,5 a clear indication that the deposits only induced activation of the classical pathway. This pathway typically involves the binding of an activator molecule36 to C1q which has the ability to recognize polyionic structures in a broad variety of unrelated targets, including immunoglobulins, DNA, or nonimmunoglobulin proteins, among them serum amyloid P-component and fibronectin,37 which are widely distributed components of XF deposits. Although the precise molecular events leading to classical pathway activation in XFG remain to be elucidated, antibody-independent activation through direct C1q binding has been demonstrated recently in different ocular disorders, including drusen formation in age-related macular degeneration38,39 and experimental glaucoma.40
Because the complement system is a powerful effector mechanism which, through the insertion and polymerization of the C5b-9 complex into cellular membranes, has the potential to severely damage normal host cells, it is under tight regulatory control at various points in the cascade. Formation of terminal MAC is modulated by cell membrane-associated and circulating proteins. In fluid phase, there are only two inhibitors of MAC assembly,41 clusterin and vitronectin. Both molecules exert their inhibitory function primarily by binding to sites within C7, C8, and C9, events that lead to the formation of a fluid phase complex32,42,43 and prevent MAC insertion into lipid membranes. The present demonstration of increased levels of the fluid phase inhibitors and elevated concentrations of soluble terminal complex in XFG aqueous humor together with the absence of C5b-9 complex in the XF deposits5 supports the notion that the increment in clusterin and vitronectin drives the complement cascade toward the production of soluble terminal complex. The existence of both activation derived components together with inhibitors of the system is not surprising and has been shown to coexist at sites of complement activation in other disorders of unrelated origin, including immune complex deposition in autoimmune diseases,44 fibrillar amyloid lesions associated with Alzheimer's disease,45 and other amyloidosis,27,46 as well as abnormal accumulations of extracellular material in macular degeneration.38
It is important to highlight that the detrimental effect of complement activation in vivo not only arises from MAC assembly into lipid bilayers, but from the release of powerful pro-inflammatory anaphylatoxins, like C3a and C5a, byproducts generated at early stages of the system activation. These inflammatory mediators have the potential to exert potent local effects, including smooth muscle contraction, increased vascular permeability and even, when in elevated concentration, leukocyte recruitment and activation, which ultimately may result in cytokine-mediated cellular injury leading to a self-perpetuating cycle of inflammatory events.41 Little is known about the role of complement-related inflammation in the anterior chamber. Normal human aqueous as well as vitreous humors contain all complement components from C1 to C9 with chronic low levels of activation present in normal ocular tissues and fluids, including tears, serving as a primary defense mechanism finely regulated by soluble and membrane-bound intraocular complement regulatory proteins.47,48 In cases of ocular inflammation (uveitis) there is an increase in the levels of activation, as indicated by elevated concentrations of C3a, C4a, and C5a.49,50 Our finding of increased levels of C3a in the aqueous humor of XFG cases, with values almost double to those found in glaucoma patients lacking XFM, reinforce the existence of pro-inflammatory conditions associated with the disease, in agreement with recent reports demonstrating a role of the pro-inflammatory cytokines IL-6 and IL-8 in early stages of XFS.51 Complement activation and its regulation by ocular complement regulatory proteins are known to contribute to the pathology of various ocular diseases, including keratitis, uveitis, and macular degeneration. New evidence also links the complement cascade to glaucoma with recent work indicating altered expression of classical pathway components in the glaucomatous retina, and implicating the system in early synapse loss of retinal ganglion cells in both experimental models and human disease.52–54 Notably, as in the case of pro-inflammatory cytokines, complement pathway involvement appears to take place early in glaucoma pathogenesis,55 and, as indicated by the results presented herein, is exacerbated in XFG. The generation of activation-derived mediators is, without doubt, capable of triggering and chronically maintaining the levels of subclinical inflammation seen in XFS, suggesting that targeting these early molecular events may constitute potential therapeutic avenues to investigate.
Acknowledgments
The authors thank Christopher Teng and Celso Tello for supplying surgical specimens.
Supported in part by NIH Grants AG030539, NS051715, and EY019129, and the Glaucoma Foundation, New York, New York, United States.
Disclosure: I. Doudevski, None; A. Rostagno, None; M. Cowman, None; J. Liebmann, None; R. Ritch, None; J. Ghiso, None
References
- 1. Ritch R, Schlotzer-Schrehardt U. Exfoliation syndrome. Surv Ophthalmol. 2001; 45: 265–315 [DOI] [PubMed] [Google Scholar]
- 2. Ritch R. Exfoliation syndrome: the most common identifiable cause of open-angle glaucoma. J Glaucoma. 1994; 3: 176–178 [PubMed] [Google Scholar]
- 3. Ritch R, Schlötzer-Schrehardt U, Konstas AG. Why is glaucoma associated with exfoliation syndrome? Progr Ret Eye Res. 2003; 22: 253–275 [DOI] [PubMed] [Google Scholar]
- 4. Schlötzer-Schrehardt U, Naumann GO. Ocular and systemic pseudoexfoliation syndrome. Am J Ophthalmol. 2006; 141: 921–937 [DOI] [PubMed] [Google Scholar]
- 5. Ovodenko B, Rostagno A, Neubert TA, et al. Proteomic analysis of exfoliation deposits. Invest Ophthalmol Vis Sci. 2007; 48: 1447–1457 [DOI] [PubMed] [Google Scholar]
- 6. Schlötzer-Schrehardt U. Molecular pathology of pseudoexfoliation syndrome/glaucoma--new insights from LOXL1 gene associations. Exp Eye Res. 2009; 88: 776–785 [DOI] [PubMed] [Google Scholar]
- 7. Burdon KP, Sharma S, Hewitt AW, et al. Genetic analysis of the clusterin gene in pseudoexfoliation syndrome. Mol Vis. 2008; 14: 1727–1736 [PMC free article] [PubMed] [Google Scholar]
- 8. Krumbiegel M, Pasutto F, Mardin CY, et al. Exploring functional candidate genes for genetic association in german patients with pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Invest Ophthalmol Vis Sci. 2009; 50: 2796–2801 [DOI] [PubMed] [Google Scholar]
- 9. Schlotzer-Schrehardt U, Zenkel M, Kuchle M, Sakai LY, Nauman GO. The role of transforming growth factor-b1 and its latent form binding protein in pseudoexfoliation syndrome. Exp Eye Res. 2001; 73: 765–780 [DOI] [PubMed] [Google Scholar]
- 10. Ten VS, Yao J, Ratner V, et al. Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J Neurosci. 2010; 30: 2077–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clarkson AN, Sutherland BA, Appleton I. The biology and pathology of hypoxia-ischemia: an update. Arch Immunol Ther Exp. 2005; 53: 213–225 [PubMed] [Google Scholar]
- 12. Zenkel M, Krysta A, Pasutto F, Juenemann A, Kruse FE, Schlotzer-Schrehardt U. Regulation of lysyl oxidase-like 1 (LOXL1) and elastin-related genes by pathogenic factors associated with peudoexfoliation syndrome. Invest Ophthalmol Vis Sci. 2011; 52: 8488–8495 [DOI] [PubMed] [Google Scholar]
- 13. Gonzalez P, Epstein DL, Borras T. Genes upregulated in the human trabecular meshwork in response to elevated intraocular pressure. Invest Ophthalmol Vis Sci. 2000; 41: 352–361 [PubMed] [Google Scholar]
- 14. Chen KH, Wu CC, Roy S, Lee SM, Liu JH. Increased interleukin-6 in aqueous humor of neovascular glaucoma. Invest Ophthalmol Vis Sci. 1999; 40: 2627–2632 [PubMed] [Google Scholar]
- 15. Wang N, Chintala SK, Fini ME, Schuman JS. Activation of a tissuespecific stress response in the aqueous outflow pathway of the eye defines the glaucoma disease phenotype. Nat Med. 2001; 7: 304–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zenkel M, Kruse FE, Junemann A, Naumann GO, Schlotzer-Schrehardt U. Clusterin deficiency in eyes with pseudoexfoliation syndrome may be implicated in the aggregation and deposition of pseudoexfoliative material. Invest Ophthalmol Vis Sci. 2006; 47: 1982–1990 [DOI] [PubMed] [Google Scholar]
- 17. Calero M, Rostagno A, Frangione B, Ghiso J. Clusterin and Alzheimer's disease. Subcell Biochem. 2005; 38: 273–298 [PubMed] [Google Scholar]
- 18. Calero M, Rostagno A, Matsubara E, Ziokovic B, Frangione B, Ghiso J., Apolipoprotein J. (clusterin) and Alzheimer's disease. Microsc Res Tech. 2000; 50: 305–315 [DOI] [PubMed] [Google Scholar]
- 19. Nishida K, Kawasaki S, Adachi W, Kinoshita S. Apolipoprotein J expression in human ocular surface epithelium. Invest Ophthalmol Vis Sci. 1996; 37: 2285–2292 [PubMed] [Google Scholar]
- 20. Nishida K, Adachi W, Shimizu-Matsumoto A, et al. A gene expression profile of human corneal epithelium and the isolation of human keratin 12 cDNA. Invest Ophthalmol Vis Sci. 1996; 37: 1800–1809 [PubMed] [Google Scholar]
- 21. Dota A, Nishida K, Quantock AJ, Kinoshita S. Clusterin in human corneal endothelium and aqueous humor. Exp Eye Res. 1999; 69: 705–708 [DOI] [PubMed] [Google Scholar]
- 22. Reeder DJ, Stuart WD, Witte DP, Brown TL, Harmony JAK. Local synthesis of apolipoprotein J in the eye. Exp Eye Res. 1995; 60: 495–504 [DOI] [PubMed] [Google Scholar]
- 23. Ritch R. The management of exfoliative glaucoma. In: Nucc C, Ceruli L, IN Osborne, Bagetta G. eds Glaucoma: An Open Window to Neurodegeneration and Neuroprotection. New York, NY: Elsevier, Inc.; 2008: 211–224 [Google Scholar]
- 24. Pountney DL, Lowe R, Quilty M, Vickers JC, Voelcker NH, Gai WP. Annular alpha-synuclein species from purified multiple system atrophy inclusions. J Neurochem. 2004; 90: 502–512 [DOI] [PubMed] [Google Scholar]
- 25. Creasey R, Sharma S, Craig JE, et al. Detecting protein aggregates on untreated human tissue samples by atomic force microscopy recognition imaging. Biophys J. 2010; 99: 1660–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fossati S, Cam J, Meyerson J, et al. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-b variants in cells composing the cerebral vessel walls. Faseb J. 2010; 24: 229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rostagno A, Revesz T, Lashley T, et al. Complement activation in chromosome 13 dementias: similarities with Alzheimer's disease. J Biol Chem. 2003; 277: 49782–49790 [DOI] [PubMed] [Google Scholar]
- 28. Radmacher M, Tillamnn RW, Fritz M, Gaub HE. From molecules to cells: imaging soft samples with the atomic force microscope. Science. 1992; 257: 1900–1905 [DOI] [PubMed] [Google Scholar]
- 29. Cursiefen C, Hammer T, Küchle M, Naumann GO, Schlötzer-Schrehardt U. Pseudoexfoliation syndrome in eyes with ischemic central retinal vein occlusion. A histopathologic and electron microscopic study. Acta Ophthalmol Scand. 2001; 79: 476–478 [DOI] [PubMed] [Google Scholar]
- 30. Hammer T, Schlötzer-Schrehardt U, Naumann GO. Unilateral or asymmetric pseudoexfoliation syndrome? An ultrastructural study. Arch Ophthalmol. 2001; 119: 1023–1031 [DOI] [PubMed] [Google Scholar]
- 31. Conway RM, Schlotzer-Schrehardt U, Kuchle M, Naumann GO. Pseudoexfoliation syndrome: pathological manifestations of relevance to intraocular surgery. Clin Exp Ophthalmol. 2004; 32: 199–210 [DOI] [PubMed] [Google Scholar]
- 32. Milis L, Morris C, Sheehan MC, Charlesworth JA, Pussell BA. Vitronectin-mediated inhibition of complement: evidence for different binding sites for C5b-7 and C9. Clin Exp Immunol. 1993; 92: 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tschopp J, French LE. Clusterin: modulation of complement function. Clin Exp Immunol. 1994; 97: 11–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson MR, Easterbrook-Smith SB. Clusterin is a secreted mammalian chaperone. Trends Biochem Sci. 2000; 25: 95–98 [DOI] [PubMed] [Google Scholar]
- 35. Nishida K, Quantock AJ, Dota A, Choi-Miura NH, Kinoshita S., Apolipoproteins J. and E co-localize with amyloid in gelatinous drop-like and lattice type I corneal dystrophies. Br J Ophthalmol. 1999; 83: 1178–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kishore U, Reid KB. C1q: structure, function, and receptors. Immunopharmacology. 2000; 49: 159–170 [DOI] [PubMed] [Google Scholar]
- 37. Gaboriaud C, Thielens NM, Gregory LA, Rossi V, Fontecilla-Camps JC, Arlaud GJ. Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol. 2004; 25: 368–373 [DOI] [PubMed] [Google Scholar]
- 38. Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001; 73: 887–896 [DOI] [PubMed] [Google Scholar]
- 39. Johnson LV, Forest DL, Banna CD, et al. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc New York Acad Sci U S A. 2011; 108: 18277–18282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ding QJ, Cook AC, Dumitrescu AV, Kuehn MH. Lack of immunoglobulins does not prevent C1q binding to RGC and does not alter the progression of experimental glaucoma. Invest Ophthalmol Vis Sci. 2012; 53: 6370–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morgan BP. The complement system: an overview. In: Morgan BP. ed Complement Methods and Protocols. Clifton, NJ: Humana Press; 2000: 1–13 [DOI] [PubMed] [Google Scholar]
- 42. McDonald JF, Nelsestuen GL. Potent inhibition of terminal complement assembly by clusterin: characterization of its impact on C9 polymerization. Biochemistry. 1997; 36: 7464–7473 [DOI] [PubMed] [Google Scholar]
- 43. Tschopp J, Chonn A, Hertig S, French LE. Clusterin, the human apolipoprotein and complement inhibitor, binds to complement C7, C8β, and the b domain of C9. J Immunol. 1993; 151: 2159–2165 [PubMed] [Google Scholar]
- 44. Morgan BP. Regulation of the complement membrane attack pathway. Crit Rev Immunol. 1999; 19: 173–198 [PubMed] [Google Scholar]
- 45. Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000; 21: 383–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rostagno A, Tomidokoro Y, Lashley T, et al. Chromosome 13 dementias. Cell Mol Life Sci. 2005; 62: 1814–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bora NS, Jha P, Bora PS. The role of complement in ocular pathology. Semin Immunopathol. 2008; 30: 85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Willcox MD, Morris CA, Thakur A, Sack RA, Wickson J, Boey W. Complement and complement regulatory proteins in human tears. Invest Ophthalmol Vis Sci. 1997; 38: 1–8 [PubMed] [Google Scholar]
- 49. Mondino BJ, Sidikaro Y, Sumner H. Anaphylatoxin levels in human vitreous humor. Invest Ophthalmol Vis Sci. 1988; 29: 1195–1198 [PubMed] [Google Scholar]
- 50. Mondino BJ, Sumner H. Anaphylotoxin levels in human aqueous humor. Invest Ophthalmol Vis Sci. 1986; 27: 1288–1292 [PubMed] [Google Scholar]
- 51. Zenkel M, Lewczuk P, Jünemann A, Kruse FE, Naumann GO, Schlötzer-Schrehardt U. Proinflammatory cytokines are involved in the initiation of the abnormal matrix process in pseudoexfoliation syndrome/glaucoma. Am J Pathol. 2010; 176: 2868–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rosen AM, Stevens B. The role of the classical complement cascade in synapse loss during development and glaucoma. Adv Exp Med Biol. 2010; 703: 75–93 [DOI] [PubMed] [Google Scholar]
- 53. Ren L, Danias J. A role for complement in glaucoma. Adv Exp Med Biol. 2010; 703: 95–104 [DOI] [PubMed] [Google Scholar]
- 54. Kuehn MH, Kim CY, Ostojic J, et al. Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp Eye Res. 2006; 83: 620–628 [DOI] [PubMed] [Google Scholar]
- 55. Howel GR, Macalinao DG, Sousa GL, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Inv. 2011; 121: 1429–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]