

Abstract

An intramolecular Huisgen cycloaddition of an interconverting set of isomeric allylic azides with alkynes affords substituted triazoles in high yield. The stereoisomeric vinyl-substituted triazoloxazines formed depend on the rate of cycloaddition of the different allylic azide precursors when the reaction is carried out under thermal conditions. In contrast, dimerized macrocyclic products were obtained when the reaction was done using copper(I)-catalyzed conditions, demonstrating the ability to control the reaction products through changing conditions.
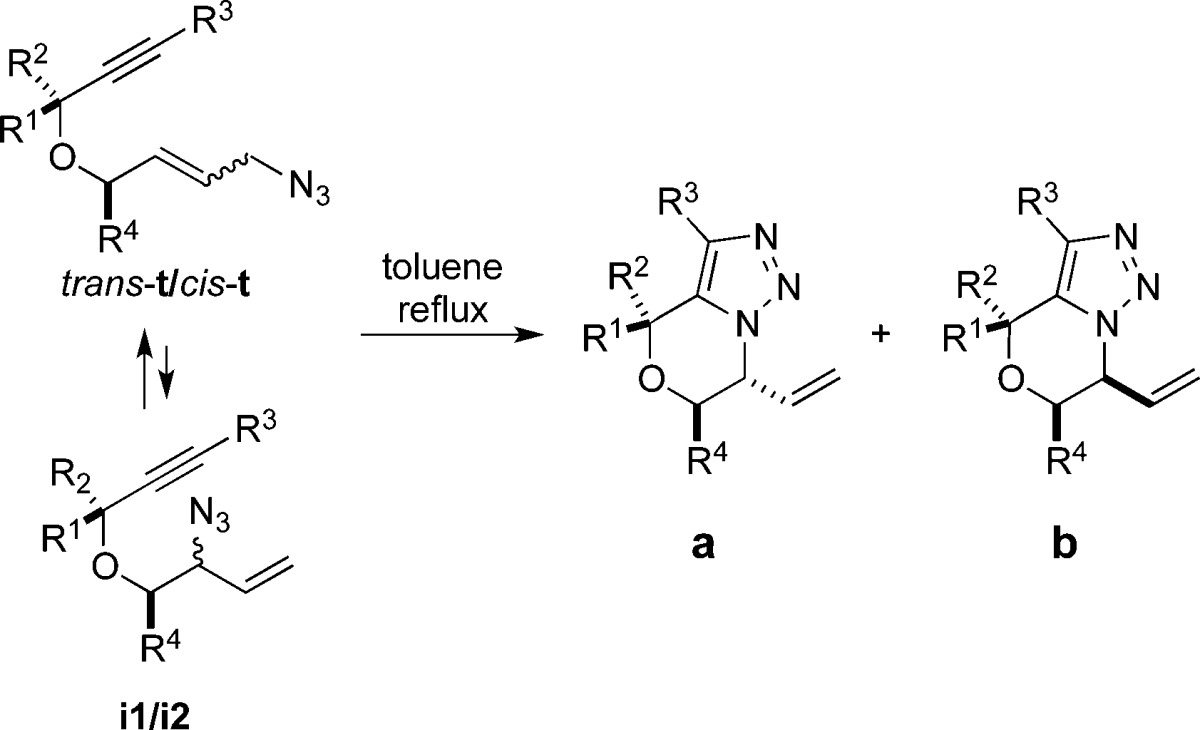
The 1,3-dipolar Huisgen azide–alkyne cycloaddition (AAC) is one of the most commonly used modern organic reactions.1−5 Allylic azides represent a particularly interesting partner in AAC chemistry because such compounds undergo facile 1,3-allylic azide rearrangements at room temperature (known as Winstein rearrangements6), and the resulting allyl triazoles can be converted into diverse products.7 The Winstein rearrangement typically provides access to constitutionally isomeric (regiosiomeric) allylic azides, which can be differentially reacted in a subsequent transformation; an early example of this was the rearrangement/AAC sequence reported by Sharpless and later investigated by Batra (Figure 1).7,8 Recently, we reported that equilibrating allylic azide stereoisomers can selectively participate in a downstream azido-Schmidt reaction sequence, affording diastereomerically enriched lactam products a and b (Figure 1); this strategy was used in the preparation of an intermediate useful in the total synthesis of pinnaic acid.9 In this paper, we describe a sequence in which a Winstein rearrangement is combined with an intramolecular Huisgen cyclization, paying particular attention to the effects of substrate structure and reaction conditions on the product structure (Figure 1). A particularly interesting outcome of these experiments is that the nature of the final product is in some cases sensitive to reaction conditions.
Figure 1.

Examples of selective reactions involving Winstein rearrangements.
AAC reactions may be carried out thermally or using transition-metal catalysts, notably Cu(I) and Ru(II).2,3 A high measure of product control (regiocontrol) is possible through transition-metal catalysis of the reaction, with 1,4-dialkyltriazoles predominantly formed using copper(I) promotion2 and 1,5-isomers using ruthenium(II) catalysts.3 In an intramolecular context, the 1,5-isomer is formed under either thermal or catalyzed versions.1c,5,10
An equilibrating mixture of terminal azides cis-t/trans-t with internal i1/i2 isomers, containing an existing stereogenic center elsewhere in the molecule, could in principle afford three triazole products (Scheme 1). Thus, diastereomers x and y would arise from i1 or i2, respectively. If one assumed rapid equilibrium of azides relative to cyclization (i.e., Curtin–Hammet conditions; but see discussion below) and that the R group in the tether would prefer an equatorial orientation in the transition structure, the ratio of products x/y would reflect the relative energies of transition states containing an equatorial or axial vinyl group. In addition, terminal azide cis-t might form a constitutional isomer z if the cis double bond could be accommodated in the fused ring, although low yields of this material would be expected given the low amounts of cis olefin present in the equilibrium mixure of azides (usually <10%9). Finally, for most ring sizes (and all of those investigated here), azide trans-t containing a trans double bond would not be expected to cyclize to any appreciable extent.
Scheme 1.

We prepared a series of alkynyl azides 1–10 linked by a three-atom oxygen-containing tether (Scheme 2). In general, alkylation of a propargylic alcohol with 1,4-dibromobutene gives an allylic bromide that is then subjected to simple substitution with NaN3. Each compound as drawn represents a mixture of equilibrating azides with the thermodynamic ratio of terminal and internal azides (pairs of enantiomers or diastereomers) indicated in Table 1. The azides were initially isolated as mixtures that were purified by chromatography to yield the trans-t isomers. 1H NMR indicated that such samples generally reattained equilibrium in about a week at room temperature (see the Supporting Information for the specific case of compound 7; reported half-lives range from hours to days6,7a).
Scheme 2. General Route to Allylic Azides.

Table 1. Intramolecular AAC of Allylic Azidesa.
| entry | azide (R1, R2, R3, R4) | ter/int ratiob | triazole (yield, %) | dr (a/b)c |
|---|---|---|---|---|
| 1 | 1 (H, H, H, H) | 83:17 | 11 (72) | |
| 2 | 2 (Me, H, H, H) | 67:33 | 12 (85) | 1.7:1 |
| 3 | 3 (Ph, H, H, H) | 84:16 | 13 (83) | 1.4:1 |
| 4 | 4 (Ph, Me, H, H)12 | 64:36 | 14 (76) | 1.3:1e |
| 5 | 5 (Me, H, Et, H) | 69:31 | 15 (93) | 1.9:1 |
| 6 | 6 (Et, H, Me, H) | 86:14 | 16 (88) | 1.5:1 |
| 7 | 7 (Me, H, Ph, H) | 81:19 | 17 (84) | 2:1 |
| 8 | 8 (iPr, H, Ph, H) | 74:26 | 18 (84) | 1.5:1d |
| 9 | 9 (H, H, H, Me) | 88:12 | 19 (79) | 1:1 |
| 10 | 10 (H, H, H, Ph) | 74:26 | 20 (82) | 1:1e |
Conditions: toluene, reflux, 1–2 h (except for entry 1: CHCl3, reflux, 4 h).
Equilibrium ratio as determined by NMR analysis of purified allylic azides; compounds attained equilibrium over 1 week at room temperature.11
Ratio determined by NMR analysis of crude reaction mixtures.
The relative stereochemistry of triazoles 18a and 18b was confirmed by X-ray crystallography (Supporting Information).
Inseparable mixture.
Upon heating the unsubstituted azide 1, a 72% yield of 11 was obtained (Table 1, entry 1). The product exclusively resulted from cycloaddition of the internal azide, which was present in <20% of equilibrated 1. Thus, the rearrangement occurred at a sufficient rate to allow for successful cyclization from the starting azide mixture.
We then subjected azides 2–10 to similar conditions. Although good yields and separable compounds were generally obtained, stereoselectivities were poor, topping out at only 2:1 for two cases (Table 1, entries 5 and 7). Compound 2 also reacted slowly (ca. 20 days for similar conversion) at room temperature to afford 12 with similar diastereoselectivity.
The poor stereoselectivities observed in these reactions were not surprising. In both transition states A and B, the emerging vinyl group has a 1,3-relationship with an oxygen atom, leading to small energy differences between axial and equatorial placement of the vinyl group (Figure 2; here, R1 and R4 are presumed to both be equatorial). In examples where R3 is an alkyl group, A1,3 strain would lead to alternatives where R3 is in an axial position, but those cases were similarly nonselective (Table 1, entries 5 and 6).
Figure 2.
Equatorial vs axial orientation of vinyl group in transition states arising from compounds 1–10, leading to isomers a and b, respectively.
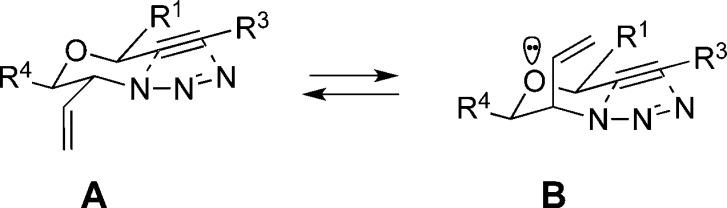
We then prepared azido alkynes bearing all-carbon tethers in which the stereocenter in the product would emerge in a 1,2 or 1,3 relationship to an existing atom (Table 2; see the Supporting Information for routes to compounds 25–29). Although some of these examples gave better ratios favoring all-equatorial transition states, the overall results were still modest. This may reflect the moderate penalty for placing a vinyl group in an axial orientation (A values between 1.49 and 1.68,12,13 Figures 3a,b) or the intervention of flattened reactive conformations (e.g., entry 2 in Table 2). We examined the effect of additional ring systems with mixed results. In most cases, modest stereoselectivities were still observed, although a 6.2:1 ratio was obtained for the formation of 28a (Table 2, entry 4). This can be understood by considering a transition state that contains a 1,3-diaxial interaction both involving axial alkyl groups across the resident cyclohexane ring and leads to the minor product (Figure 3c). In contrast, the trans isomer 27a, which is formed with poorer selectivity, is hampered by an analogous 1,3-transdiaxial interaction arising from the vinyl group and a hydrogen atom (Figure 3b).
Table 2. Intramolecular AAC of Allylic Azidesa.
Conditions: toluene, reflux, 2–8 h.
Starting azides exist as mixtures of terminal and internal azide stereoisomers; ratios noted indicate specific examples used in the experiment noted (1H NMR).11
Ratio determined by NMR analysis of crude reaction mixtures.
The relative stereochemistry of triazoles 27a, 28a, and 29a was determined by X-ray crystallography (Supporting Information).
Inseparable mixtures.
Figure 3.

Steric interactions encountered en route to disfavored isomers, specifically compounds (a) 25b, (b) 27b, and (c) 28b.
As noted above, 1H NMR studies indicated that the allylic rearrangement reaction occurred at room temperature to reach equilibrium in about a week. In addition, we note that no 1,3-dipole cycloaddition was apparent in these samples when examples bearing terminal alkyne substitution, like 7, were examined. On the other hand, alkynes without terminal substitution (i.e., 1–10 where R3 = H) were observed to undergo cyclization at a somewhat slower, but comparable, rate (e.g., about 40% of product being observed at 6.5 days for compound 1, when the terminal–internal azide equilibrium was also reached9b). Assuming that the relative rates are similar at higher temperatures, these data suggest that while terminally substituted alkyne substrates are reacting under Curtin–Hammett conditions, those with a terminal H atom are in a mixed kinetic regime.14
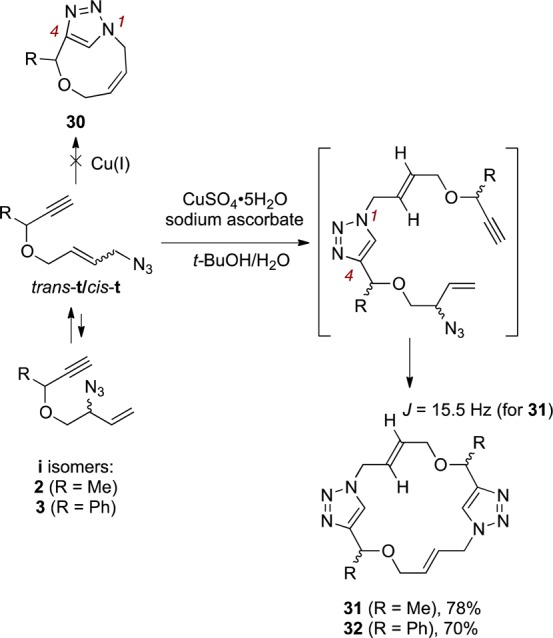
An interesting result was obtained when we treated 2 with CuSO4·5H2O (Scheme 3). Thus, a single new compound was obtained in 78% yield (R = Me), which was assigned as 31. The trans geometry of the double bonds in 31 was assigned based on the vicinal J coupling constant (15.5 Hz); both vinyl protons coincide in the 1H NMR spectrum of 32 so in this case trans geometry is presumed by analogy. In each case, only a single set of resonances was present in both 1H and 13C spectra, but it was not possible to unambiguously assign relative stereochemistry to either product (or, indeed, completely rule out the possibility that mixtures of stereoisomers were obtained). The formation of 1,4-disubstituted triazoles is in accord with literature expectations for the Cu-catalyzed reaction leading preferentially to 1,4-disubstitued triazoles.15 In this case, the cyclophane 30 could potentially arise from intramolecular formation of the 1,4-disubstituted triazole, but is presumably disfavored due to ring strain as well as the low population of cis-t allylic azide. Accordingly, it is likely that intermolecular reaction of the terminal azide occurs affording the intermediate shown, which is then followed by equilibration to terminal azide and subsequent cycloaddition/macrocyclization.
Scheme 3.
The oxazepine 34 and macrocylic triazole 35 were obtained from the equilibrating mixture of homologous azides 33 in 81% and 78% yields, respectively (Scheme 4). However, attempts to obtain the ring systems containing an additional carbon intervening between the reacting groups were not successful.
Scheme 4.

The triazoles obtained in this study represent interesting classes of heterocyclic compounds that contain an alkene moiety for further manipulation. Although the stereoselectivities obtained in this work were slight, the ability to separate products and select between several equilibrating azides by modification of reaction conditions is noteworthy and suggests future applications for such azides in chemical synthesis.
Acknowledgments
We are grateful to the National Institute of General Medical Sciences P41 GM089164 for financial support. We thank Patrick Porubsky for HRMS, Victor Day for X-ray crystallography (the diffractometer and software used in this study were purchased through NSF–MRI Grant No. CHE-0923449), and Gurpreet Singh and Digamber Rane for helpful suggestions (all at the University of Kansas).
Supporting Information Available
Experimental procedures, full spectroscopic data, and NMR spectra for all new compounds; X-ray data for structures of compounds 18a, 18b, 27a, 28a, and 29a. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For recent reviews, see:; a Meldal M.; Tornøe C. W. Chem. Rev. 2008, 108, 2952. [DOI] [PubMed] [Google Scholar]; b Hein J. E.; Fokin V. V. Chem. Soc. Rev. 2010, 39, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Majumdar K. C.; Ray K. Synthesis 2011, 3767. [Google Scholar]; d Sletten E. M.; Bertozzi C. R. Acc. Chem. Res. 2011, 44, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Mohan A.; Sankararaman S. Isr. J. Chem. 2012, 52, 92. [Google Scholar]
- For Cu(I)-catalyzed AAC, see:; a Tornøe C. W.; Christensen C.; Meldal M. J. Org. Chem. 2002, 67, 3057. [DOI] [PubMed] [Google Scholar]; b Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596. [DOI] [PubMed] [Google Scholar]; c Himo F.; Lovell T.; Hilgraf R.; Rostovtsev V. V.; Noodleman L.; Sharpless K. B.; Fokin V. V. J. Am. Chem. Soc. 2005, 127, 210. [DOI] [PubMed] [Google Scholar]; d Angell Y.; Burgess K. J. Org. Chem. 2005, 70, 9595. [DOI] [PubMed] [Google Scholar]; e Chandrasekhar S.; Rao C. L.; Nagesh C.; Reddy C. R.; Sridhar B. Tetrahedron Lett. 2007, 48, 5869. [Google Scholar]
- For Ru(II)-catalyzed AAC, see:; a Zhang L.; Chen X.; Xue P.; Sun H. H. Y.; Williams I. D.; Sharpless K. B.; Fokin V. V.; Jia G. J. Am. Chem. Soc. 2005, 127, 15998. [DOI] [PubMed] [Google Scholar]; b Boren B. C.; Narayan S.; Rasmussen L. K.; Zhang L.; Zhao H.; Lin Z.; Jia G.; Fokin V. V. J. Am. Chem. Soc. 2008, 130, 8923. [DOI] [PubMed] [Google Scholar]; c Zhang J.; Kemmink J.; Rijkers D. T.; Liskamp R. M. Org. Lett. 2011, 13, 3438. [DOI] [PubMed] [Google Scholar]
- For other ACC catalysts, see:; a McNulty J.; Keskar K.; Vemula R. Chem.—Eur. J. 2011, 17, 14727. [DOI] [PubMed] [Google Scholar]; b McNulty J.; Keskar K. Eur. J. Org. Chem. 2012, 2012, 5462. [Google Scholar]; c Kwok S. W.; Fotsing J. R.; Fraser R. J.; Rodionov V. O.; Fokin V. V. Org. Lett. 2010, 12, 4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For related examples using thermal conditions, see:; a Li R.; Jansen D. J.; Datta A. Org. Biomol. Chem. 2009, 7, 1921. [DOI] [PubMed] [Google Scholar]; b Declerck V.; Toupet L.; Martinez J.; Lamaty F. J. Org. Chem. 2009, 74, 2004. [DOI] [PubMed] [Google Scholar]
- a Gagneux A.; Winstein S.; Young W. G. J. Am. Chem. Soc. 1960, 82, 5956. [Google Scholar]; b VanderWerf C. A.; Heasley V. L. J. Org. Chem. 1966, 31, 3534. [Google Scholar]
- a Feldman A. K.; Colasson B.; Sharpless K. B.; Fokin V. V. J. Am. Chem. Soc. 2005, 127, 13444. [DOI] [PubMed] [Google Scholar]; b Mishra A.; Hutait S.; Bhowmik S.; Rastogi N.; Roy R.; Batra S. Synthesis 2010, 2731. [Google Scholar]
- For recent examples of allylic azides in synthesis, see:; a Cardillo G.; Fabbroni S.; Gentilucci L.; Perciaccante R.; Piccinelli F.; Tolomelli A. Org. Lett. 2005, 7, 533. [DOI] [PubMed] [Google Scholar]; b Gagnon D.; Lauzon S.; Godbout C.; Spino C. Org. Lett. 2005, 7, 4769. [DOI] [PubMed] [Google Scholar]; c Takasu H.; Tsuji Y.; Sajiki H.; Hitota K. Tetrahedron 2005, 61, 11027. [Google Scholar]; d Sá M. M.; Ramos M. D.; Fernandes L. Tetrahedron 2006, 62, 11652. [Google Scholar]; e Klepper F.; Jahn E.-M.; Hickmann V.; Craell T. Angew. Chem., Int. Ed. 2007, 46, 2325. [DOI] [PubMed] [Google Scholar]; f Lauson S.; Tremblay F.; Gagnnon D.; Godbout C.; Chabot C.; Mercier-Shanks C.; Perreault S.; DeSevè H.; Spino C. J. Org. Chem. 2008, 73, 6239. [DOI] [PubMed] [Google Scholar]; g Chang Y.-K.; Lo H.-J.; Yan T.-H. Org. Lett. 2009, 11, 4278. [DOI] [PubMed] [Google Scholar]; h Cakmak M.; Mayer P.; Trauner D. Nat. Chem 2011, 3, 543. [DOI] [PubMed] [Google Scholar]; i Craig D.; Harvey J. W.; O’Brien A. G.; White A. J. P. Org. Biomol. Chem. 2011, 9, 7057. [DOI] [PubMed] [Google Scholar]
- a Liu R.; Gutierrez O.; Tantillo D. J.; Aubé J. J. Am. Chem. Soc. 2012, 134, 6528. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu R. Ph.D. Dissertation, University of Kansas, Lawrence, KS, 2012. [Google Scholar]
- Ellison A.; Boyer R.; Hoogestraat P.; Bell M. Tetrahedron lett. 2013, 54, 6005. [Google Scholar]
- We estimate the cis-t isomer to be formed in 0–5% yield, although it is generally hard to quantify due to overlapping signals in the 1H NMR.
- For 1-methyl-1-phenylcyclohexane, the chair conformer with an axial phenyl group is favored by 0.32 kcal/mol over the alternative chair:Eliel E. L.; Manoharan M. J. Org. Chem. 1981, 46, 1959. [Google Scholar]
- Buchanan G. W. Can. J. Chem. 1982, 60, 2908. [Google Scholar]
- Seeman J. I. Chem. Rev. 1983, 83, 83–134. [Google Scholar]
- a Angell Y.; Burgess K. J. Org. Chem. 2005, 70, 9595. [DOI] [PubMed] [Google Scholar]; b Chandrasekhar S.; Rao C. L.; Nagesh C.; Reddy C. R.; Sridhar B. Tetrahedron Lett. 2007, 48, 5869. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.