

Abstract
Polypeptides bearing quaternary phosphonium side chains were synthesized via controlled ring-opening polymerization of chlorine-functionalized amino acid N-carboxyanhydride monomers followed by one-step nucleophilic substitution reaction with triethylphosphine. The conformation of the resulting polypeptides can be controlled by modulating the side-chain length and α-carbon stereochemistry. The phosphonium-based poly(l-glutamate) derivatives with 11 σ-bond backbone-to-charge distance adopt stable α-helical conformation against pH and ionic strength changes. These helical, quaternary phosphonium-bearing polypeptides exhibit higher cell-penetrating capability than their racemic and random-coiled analogues. They enter cells mainly via an energy-independent, nonendocytic cell membrane transduction mechanism and exhibit low cytotoxicity, substantiating their potential use as a safe and effective cell-penetrating agent.
Introduction
Cell-penetrating peptides (CPPs) are short, sequence-specific peptides derived from natural resources or artificial constructs and are well-known for their potency to facilitate cellular internalization of a variety of therapeutics, imaging probes, and reporter molecules.1−3 CPPs are often positively charged, and the positive charges are originated exclusively from the primary amino groups of the lysine residue or the guanidino groups of the arginine residue. While the cationic amino and guanidino groups afford electrostatic interactions with negatively charged phospholipid bilayers to potentiate the membrane permeability, they may also induce appreciable damage to biological membranes due to perturbation of the phospholipid structure.1,3 Phosphonium group, another class of cationic functional group, has been recently reported to have lower toxicity toward cell membranes because of its relatively large size.4,5 Substitution of quaternary ammonium with phosphonium in lipid or polymer systems leads to higher cationic charge density, which therefore contributes to enhanced cellular delivery efficiencies toward various cargos.5−11 Considering the potential benefits of phosphonium groups, it would be of great interest to design and develop phosphonium-bearing CPPs. However, since phosphonium is not present in natural amino acids, the synthesis of phosphonium-containing CPPs is challenging and thus has never been reported.
In addition to the functional group type, secondary conformation of CPPs also has dominant roles on their membrane activities and penetration mechanisms. A large number of CPPs adopt helical conformations or form helices upon interacting with cell membranes, presenting a rigid amphiphilic structure to promote translocation across cell membranes.12−14 However, traditional ionic polypeptides synthesized via ring-opening polymerization (ROP) of natural amino acid derived N-carboxyanhydrides (NCAs; such as glutamic acid and lysine) adopt random coil conformation under physiological condition due to the helix-destabilizing side-chain charge repulsion. To overcome this issue, we recently developed a strategy to enhance the side-chain hydrophobicity of polypeptides while reduce the intramolecular electrostatic repulsion by elongating the backbone-to-charge length, and thus, the polypeptides were allowed to adopt stable helical conformation in aqueous solution.15 With amino or guanidino groups on their side-chain terminal, these helical polypeptides showed excellent helix-associated cell-penetrating capabilities and some outperformed classical CPPs such as Arg9 and HIV-TAT by up to 2 orders of magnitude.16 As a result, these polypeptides act as efficient molecular transporters to facilitate the cellular delivery of various cargos, such as DNA and siRNA.17−23
In this contribution, we report the first example of helical polypeptides bearing quaternary phosphonium side chains, aiming to design phosphonium-based CPP derivatives with low cytotoxicity. Recent development of controlled ROP of NCAs24−28 and side-chain functionalization technique29−34 has enabled the facile synthesis of polypeptides with predictable molecular weights (MWs) and demanded side-chain functional groups. Based on this well-developed platform, we attempted and successfully synthesized the phosphonium-based polypeptides via controlled ROP of chlorine-functionalized NCA16,31,34 and subsequent side-chain functionalization with one-step nucleophilic substitution.11 By elongating the charged phosphonium side-chain length, polypeptides with stable α-helical conformation were obtained and were found to outperform their racemic and random-coiled analogues in terms of the cell penetration capabilities. These helical phosphonium-based polypeptides entered cells mainly via the energy-independent nonendocytic pathway, and showed low cytotoxicity as expected.
Experimental Section
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.) and used as received unless otherwise specified. Anhydrous dimethylformamide (DMF) was dried by a column packed with 4 Å molecular sieves and stored in a glovebox. Anhydrous tetrahydrofuran (THF), ethyl acetate (EtOAc), and hexane were dried by a column packed with alumina. Hexamethyldisilazane (HMDS) was dissolved in DMF in a glovebox. SiliaFlash P60 silica gel (particle size 40–63 μm) was purchased from SiliCycle Inc. (Quebec City, Quebec, Canada) and dried at 150 °C under vacuum for 48 h before use. Spectra/Por RC dialysis tubing with a molecular weight cutoff (MWCO) of 1 kDa was purchased from Spectrum Laboratories (Rancho Dominguez, CA, U.S.A.). Pierce BCA assay kit was purchased from ThermoFisher Scientific (Rockford, IL, U.S.A.). 3-(4,5-Dimethylthiahiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Invitrogen (Carlsbad, CA, U.S.A.).
HeLa (human cervix adenocarcinoma) cells were purchased from the American Type Culture Collection (Rockville, MD, U.S.A.) and cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, U.S.A.) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin.
Instrumentation
1H NMR spectra were recorded on a Varian U500 MHz spectrometer. Chemical shifts were reported in ppm and referenced to the solvent proton impurities. Gel permeation chromatography (GPC) experiments were performed on a system equipped with an isocratic pump (Model 1100, Agilent Technology, Santa Clara, CA, U.S.A.), a DAWN HELEOS multiangle laser light scattering detector (MALLS) detector (Wyatt Technology, Santa Barbara, CA, U.S.A.), and an Optilab rEX refractive index detector (Wyatt Technology, Santa Barbara, CA, U.S.A.). The detection wavelength of HELEOS was set at 658 nm. Separations were performed using serially connected size exclusion columns (100, 500, 103, and 104 Å Phenogel columns, 5 μm, 300 × 7.8 mm, Phenomenex, Torrance, CA, U.S.A.) at 60 °C using DMF containing 0.1 mol/L LiBr as the mobile phase. The MALLS detector was calibrated using pure toluene and can be used for the determination of the absolute molecular weights (MWs). The MWs of polymers were determined based on the dn/dc value of each polymer sample calculated offline by using the internal calibration system processed by the ASTRA V software (version 5.1.7.3, Wyatt Technology, Santa Barbara, CA, U.S.A.). Circular dichroism (CD) measurements were carried out on a JASCO J-815 CD spectrometer (JASCO, Easton, MD, U.S.A.). The polypeptide samples were prepared at a concentration of 0.025–0.5 mg/mL in aqueous solution at pH = 7 in general unless otherwise specified. The solution was placed in a quartz cell with a path length of 0.10 cm. The mean residue molar ellipticity of each polypeptide was calculated on the basis of the measured apparent ellipticity by following the literature-reported formulas: Ellipticity ([θ] in deg cm2 dmol–1) = (millidegrees × mean residue weight)/(path length in millimeters × concentration of polypeptide in mg mL–1).35,36 The α-helix contents of the polypeptides were calculated by the following equation: percentage of α-helix = (−[θ]222 nm + 3000)/39000.37 For the ionic strength dependent experiments, polypeptides were dissolved in aqueous solution containing different concentrations of NaCl; for the pH-dependent experiments, the pH of the polypeptide solution was tuned by the addition of concentrated HCl or NaOH solution. Infrared spectra were recorded on a Perkin-Elmer 100 serial FTIR spectrophotometer calibrated with polystyrene film (Perkin-Elmer, Santa Clara, CA, U.S.A.). Lyophilization was performed on a FreeZone lyophilizer (Labconco, Kansas City, MO, U.S.A.).
All chlorine-functionalized polypeptides were synthesized following the literature procedure starting from glutamic acids.16,31
Synthesis of γ-(6-Chlorohexyl)-l-glutamate (CH-l-Glu), γ-(6-Chlorohexyl)-dl-glutamate (CH-dl-Glu), and γ-(3-Chloropropyl)-l-glutamate (CP-l-Glu)
l-Glutamic acid (10.0 g, 68.0 mmol) and 6-chlorohexanol (15 mL, 112.4 mmol) were mixed and stirred at 0 °C followed by dropwise addition of H2SO4 (4.0 mL). The reaction was allowed to warm up to room temperature and kept stirring for 24 h. Saturated Na2CO3 solution was then added to the viscous mixture to adjust the pH to 7. The resulting solid was collected by filtration and purified by recrystallization from isopropanol/H2O (1:1, v/v). Isopropanol was removed under vacuum and water was removed via lyophilization to obtain CH-l-Glu as white solid powder (6.92 g, 38% yield). 1H NMR [DMSO-d6/D2O–DCl (35 wt %), 9:1, v/v]: δ 3.91 (t, 2H, −CH2OOC−), 3.82 (t, 1H, α-H), 3.52 (t, 2H, −CH2Cl), 2.53–2.32 (m, 2H, −CH2CH2COO−), 1.98 (m, 2H, −CH2CH2COO−), 1.64–1.17 (m, 8H, ClCH2(CH2)4CH2O−).
CH-dl-Glu was synthesized using the same method with dl-glutamic acid and 6-chlorohexanol as the staring material (36% yield). 1H NMR [DMSO-d6/D2O–DCl (35 wt %), 9:1, v/v]: δ 3.91 (t, 2H, −CH2OOC−), 3.82 (t, 1H, α-H), 3.52 (t, 2H, −CH2Cl), 2.53–2.32 (m, 2H, −CH2CH2COO−), 1.98 (m, 2H, −CH2CH2COO−), 1.64–1.17 (m, 8H, ClCH2(CH2)4CH2O−).
CP-l-Glu was synthesized using the same method with l-glutamic acid and 3-chloropropanol as the starting material (30% yield). 1H NMR [DMSO-d6/D2O–DCl (35 wt %), 9:1, v/v]: δ 4.05 (t, 2H, −CH2OOC−), 3.83 (t, 1H, α-H), 3.60 (t, 2H, −CH2Cl), 2.54–2.37 (m, 2H, −CH2CH2COO−), 2.00 (m, 2H, −CH2CH2COO−), 2.04–1.95 (m, 2H, ClCH2CH2CH2O−).
Synthesis of γ-(6-Chlorohexyl)-l-glutamic Acid Based N-carboxyanhydride (CH-l-Glu-NCA), γ-(6-Chlorohexyl)-dl-glutamic Acid Based N-Carboxyanhydride (CH-dl-Glu-NCA), and γ-(3-Chloropropyl)-l-glutamic Acid Based N-Carboxyanhydride (CP-l-Glu-NCA),
In a dried 250 mL two-neck round-bottom flask, CH-l-Glu (7.3 g, 27.4 mmol) was added and dried under vacuum for 2 h. Phosgene (15 wt-% in toluene, 39.2 mL, 54.9 mmol) was added along with anhydrous THF (60 mL), and the mixture was stirred at 50 °C for 2 h under the protection of drying tube. The solvent was removed under vacuum to obtain yellow oily liquid. The product was purified by silica column chromatography using EtOAC/hexane (from 100 to 60% hexanes) as the eluent (6.6 g, 83% yield). The resulting CH-l-Glu-NCA was stored at −30 °C in a glovebox. 1H NMR (CDCl3): δ 7.16 (s, 1H, −NH), 4.43 (t, 1H, α-H), 4.05 (t, 2H, −CH2OOC−), 3.51 (t, 2H, −CH2Cl), 2.51 (t, 2H, −CH2CH2COO−), 2.25–2.07 (m, 2H, −CH2CH2COO−), 1.79–1.30 (m, 8H, ClCH2(CH2)4CH2O−). 13C NMR (CDCl3): δ 172.8, 170.0, 152.6, 65.4, 57.0, 45.2, 32.6, 29.7, 28.5, 27.0, 26.6, 25.4.
CH-dl-Glu-NCA was synthesized from CH-dl-Glu using the same method (81% yield). 1H NMR (CDCl3): δ 7.31 (s, 1H, −NH), 4.41 (s, 1H, α-H), 3.99 (s, 2H, −CH2OOC−), 3.45 (s, 2H, −CH2Cl), 2.45 (s, 2H, −CH2CH2COO−), 2.10 (d, 2H, −CH2CH2COO−), 1.75–1.22 (m, 8H, ClCH2(CH2)4CH2O−). 13C NMR (CDCl3): δ 172.8, 170.2, 152.7, 65.3, 57.1, 45.3, 32.5, 29.6, 28.5, 27.0, 26.6, 25.3.
CP-l-Glu-NCA was synthesized from CP-l-Glu using the same method (85% yield). 1H NMR (CDCl3): δ 6.57 (s, 1H, −NH), 4.42 (t, 1H, α-H), 4.27 (t, 2H, −CH2OOC−), 3.62 (t, 2H, −CH2Cl), 2.57 (t, 2H, −CH2CH2COO−), 2.32–2.08 (m, 4H, −CH2CH2COO– and ClCH2CH2CH2O−). 13C NMR (CDCl3): δ 172.7, 170.0, 152.7, 62.2, 57.1, 41.5, 31.5, 29.6, 27.0.
Synthesis of Poly(γ-6-chlorohexyl-l-glutamate) (PCHLG), Poly(γ-6-chlorohexyl-dl-glutamate) (PCHDLG), and Poly(γ-3-chloropropyl-l-glutamate) (PCPLG)
In a glovebox, CH-l-Glu-NCA (100 mg, 0.34 mmol) was dissolved in DMF (1.5 mL) followed by addition of the HMDS-DMF solution (85.8 μL, 0.1 M, M/I = 40). The mixture was stirred at room temperature until the monomer conversion reached above 99% as monitored by FTIR. The polymer was then precipitated with cold methanol and dried under vacuum at 40 °C for 8 h. The polymer PCHLG was obtained as white viscous solid (78–81% yield). 1H NMR (CDCl3/TFA-d, 85:15, v/v): δ 4.60 (m, 1H, α-H), 4.09 (m, 2H, −CH2OOC−), 3.52 (t, 2H, −CH2Cl), 2.50 (m, 2H, −CH2CH2COO−), 2.19–1.90 (m, 2H, −CH2CH2COO−), 1.81–1.30 (m, 8H, ClCH2(CH2)4CH2O−).
PCHDLG was was polymerized from CH-DL-Glu-NCA using the same method (76–80% yield). 1H NMR (CDCl3/TFA-d, 85:15, v/v): δ 4.60 (m, 1H, α-H), 4.10 (m, 2H, −CH2OOC−), 3.52 (t, 2H, −CH2Cl), 2.48 (s, 2H, −CH2CH2COO−), 2.28–1.90 (m, 2H, −CH2CH2COO−), 1.81–1.30 (m, 8H, ClCH2(CH2)4CH2O−).
PCPLG was polymerized from CP-l-Glu-NCA using the same method (72–75% yield). 1H NMR (CDCl3/TFA-d, 85:15, v/v): δ 4.58 (s, 1H, α-H), 4.27 (m, 2H, −CH2OOC−), 3.57 (t, 2H, −CH2Cl), 2.52 (s, 2H, −CH2CH2COO−), 2.23–1.92 (m, 4H, −CH2CH2COO– and ClCH2CH2CH2O−).
Synthesis of Poly(γ-6-(triethylphosphonium)hexyl-l-glutamate) (PHLG-TEP), Poly(γ-6-(triethylphosphonium)hexyl-dl-glutamate) (PHDLG-TEP), and Poly(γ-3-(triethylphosphonium)propyl-l-glutamate) (PPLG-TEP)
PCHLG (86.5 mg, 0.35 mmol of chloro groups) was dissolved in DMF (2.0 mL) and NaI (157 mg, 1.05 mmol) was dissolved in acetonitrile (2.0 mL). Both solutions were transferred to a 25 mL Schlenk tube into which triethylphosphine (TEP, 103 μL, 0.70 mmol) was added. The mixture was stirred at 80 °C for 48 h. After most solvent was removed under vacuum, NaCl aqueous solution (1.0 M, 3.0 mL) was added. The solution was then stirred at room temperature for 3 h to promote ion exchange. The product was purified by dialysis (MWCO = 1 kDa) against distilled (DI) water for 3 days. PHLG-TEP as the final product was obtained as white solid powder after lyophilization (94.7 mg, 74% yield). 1H NMR (TFA-d): δ 4.83 (s, 1H, α-H), 4.24 (m, 2H, −CH2OOC−), 2.68 (m, 2H, −CH2CH2COO−), 2.42–2.14 (m, 10H, −CH2CH2COO– and −P+(CH2CH3)3), 1.82–1.45 (m, 8H, −P+CH2(CH2)4CH2O−), 1.35 (m, 9H, −P+(CH2CH3)3). 31P NMR (TFA-d): δ 39.6.
PHDLG-TEP was synthesized from PCHDLG using the same method (68% yield). 1H NMR (D2O): δ 4.22 (s, 1H, α-H), 3.98 (m, 2H, −CH2OOC−), 2.36 (s, 2H, −CH2CH2COO−), 2.22–1.78 (m, 10H, −CH2CH2COO– and −P+(CH2CH3)3), 1.57–1.23 (m, 8H, −P+CH2(CH2)4CH2O−), 1.09 (s, 9H, −P+(CH2CH3)3). 31P NMR (D2O): δ 39.5.
PPLG-TEP was synthesized from PCPLG using the method (70% yield). 1H NMR (D2O): δ 4.20 (s, 1H, α-H), 4.04 (m, 2H, −CH2OOC−), 2.37 (s, 2H, −CH2CH2COO−), 2.20–1.74 (m, 12H, −CH2CH2COO–, −P+(CH2CH3)3 and −PCH2CH2CH2O−), 1.07 (s, 9H, −P+(CH2CH3)3). 31P NMR (D2O): δ 40.2.
Synthesis of Fluorescein-Dimethylaminopropylamine (FITC-DMAPA)
Fluorescein isothiocyanate (FITC, 30 mg, 0.077 mmol) and 3-(dimethylamino)-1-propylamine (DMAPA, 19 μL, 0.15 mmol) were dissolved in DMF (2.0 mL) and stirred at 50 °C for 24 h with the protection of aluminum foil. After most DMF and unreacted DMAPA were removed under vacuum, the residue was washed three times with methanol. The final product FITC-DMAPA was obtained as orange powder (85% yield). 1H NMR (DMSO-d6): δ 8.40–6.40 (9H, ArH from FITC), 3.52 (m, 2H, −NHC(S)NHCH2−), 2.25 (m, 2H, (CH3)2NCH2−), 2.08 (s, 6H, −N(CH3)2), 1.66 (m, 2H, (CH3)2NCH2CH2−). ESI-MS: m/z Calcd for C26H26N3O5S [M + H]+, 492.16; found, 492.2.
Synthesis of Fluorescein–Polypeptide Conjugates
Fluorescein-labeled polypeptides were synthesized via the nucleophilic reaction of chlorine-functionalized polypeptides with fluorescein-labeled tertiary amine FITC-DMAPA and triethylphosphine. Briefly, PCHLG (30 mg, 0.12 mmol of chloro groups) and FITC-DMAPA (3.0 mg, 5 mol % of chloro groups) were dissolved in DMF (2.0 mL), and NaI (54 mg, 0.36 mmol) was dissolved in acetonitrile (2.0 mL). Both solutions were transferred to a 25 mL Schlenk tube and stirred at 80 °C for 24 h. TEP (36 μL, 0.24 mmol) was then added and the resulting mixture was stirred at 80 °C for another 48 h. After most of the solvent was removed under vacuum, NaCl aqueous solution (1.0 M, 3.0 mL) was added and the mixture was stirred for 3 h to promote ion exchange. The solution was then dialyzed against DI water (MWCO = 1 kDa) for 3 days, and fluorescein-labeled PHLG-TEP was obtained as orange powder after lyophilization. Fluorescein-labeled PHDLG-TEP and PPLG-TEP were synthesized using the same method from PCHDLG and PCPLG, respectively.
Cell Penetration of Polypeptides
Cells were seeded on 96-well plates at 1 × 104 cells/well and cultured for 24 h before they reached confluence. The medium was replaced with serum-free DMEM, into which fluorescein-labeled polypeptides were added at various final concentrations (10, 20, 50, and 100 μg/mL). After incubation for various time periods (0.5, 1, 2, and 4 h), cells were washed with cold phosphate-buffered saline (PBS) containing heparin (20 U/mL) for three times to completely remove surface-bound cationic polymers from the cells. TRAMA-HIV-TAT (20 μg/mL) was used as a control group and incubated for 2 h. Cells were then lysed with 100 μL RIPA lysis buffer at room temperature for 20 min, and the fluorescein content in the lysate was quantified by spectrofluorimetry (λex = 485 nm, λem = 530 nm) while the protein level was measured using the BCA kit. The uptake level was expressed as ng FITC-polypeptide associated with 1 mg of cellular protein.
Fluorescence-Activated Cell Sorting (FACS) Experiment
Cells were seeded on 24-well plates at 5 × 104 cells/well and cultured for 24 h before they reached confluence. The medium was replaced with serum-free DMEM, into which fluorescein-labeled PHLG-TEP was added at the final concentration of 20 μg/mL. After incubation for 4 h at 37 or 4 °C, cells were washed with PBS containing heparin (20 U/mL) for 3 times and trypsin (200 μL/well) was then added to detach the cells from the plate. After the cells were harvested, 4% paraformaldehyde (100 μL) was added to fix the cells. Samples were kept in covered tubes and subjected to flow cytometry analysis (BD FACSCanto, Franklin Lakes, NJ, U.S.A.).
Cell Penetration Mechanisms
To explore the cellular internalization mechanism of polypeptides, cells were preincubated with endocytosis inhibitors including methyl-β-cyclodextrin (mβCD, 5 mM), chlorpromazine (10 μg/mL), or wortmannin (10 μg/mL) for 30 min prior to the addition of fluorescein-polypeptides, and cells were further incubated at 37 °C for 2 h. To block the energy-dependent endocytosis, cells were incubated at 4 °C during a 2 h uptake period. The cell penetration level was determined as described above, and results were expressed as percentage uptake level of the control cells which were incubated with polypeptides at 37 °C for 2 h in the absence of endocytic inhibitors.
CLSM Observation
Cells were seeded on coverslips in a 6-well plate at 1 × 105 cells/well and cultured for 24 h. Fluorescein-labeled PHLG-TEP were added at 20 μg/mL, and the cells were cultured for 4 h at 4 or 37 °C. Following PBS wash for 3 times and fixation by PFA, the endosomes/lysosomes were stained with Lysotracker Red (200 nM, Invitrogen, Grand Island, NY, U.S.A.) and the nuclei were stained with DAPI (2 μg/mL). Cells were then observed by confocal laser scanning microscopy (CLSM, LSM 700, Zeiss, Germany).
Cytotoxicity Measurement
Cells were seeded on 96-well plates at 1 × 104 cells/well and cultured for 24 h at 37 °C before they reached confluence. The medium was replaced with serum-free DMEM, into which polypeptides were added at final concentrations of 200, 150, 100, 50, 20, and 10 μg/mL, respectively. Cells were incubated for 4 h, and the medium was then replaced with fresh serum-containing DMEM. After further incubation at 37 °C for 20 h, cell viability was assessed using the MTT assay.
Results and Discussion
The monomers, γ-chloroalkyl-l-glutamic acid and γ-chloroalkyl-dl-glutamic acid based NCAs, were prepared via monoesterification of the corresponding glutamic acids and subsequent cyclization reaction with phosgene (Scheme 1).16,31,34 During the esterification step, different chloroalkyl alcohols were selected to vary the side-chain lengths and different glutamic acids were used to alternate the α-carbon stereochemistry. The resulting NCAs were light yellow oily liquids and were purified by the silica column chromatography.38 The molecular structures of CH-l-Glu-NCA, CH-dl-Glu-NCA, and CP-l-Glu-NCA were verified by 1H and 13C NMR (Supporting Information, Figures S4–S6).
Scheme 1. Synthesis of Polypeptides with Quaternary Phosphonium Side Chains.

Hexamethyldisilazane (HMDS) was then used to initiate the controlled ROP of the NCAs (Scheme 1).28 All polymerizations were conducted in a glovebox at room temperature with the monomer conversion >99% (monitored by FTIR). The polypeptides containing chlorine side chains were then purified by precipitation with cold methanol and their structures were verified by 1H NMR (Supporting Information, Figures S7–S9). The MWs and molecular weight distributions (MWDs) were determined by gel permeation chromatography (GPC; Table S1 and Supporting Information, Figure S12). The obtained MWs agreed well with the monomer to initiator (M/I) ratios (degree of polymerization ∼ 40), and all three polymers had narrow MWDs (<1.10). Polypeptides containing quaternary phosphonium functional groups were easily obtained through a one-step nucleophilic reaction of the side-chain chlorine groups with tertiary triethylphosphine (TEP; Scheme 1). The structures of three phosphonium-based polypeptides were summarized in Table 1. A quantitative conjugation was revealed by 1H NMR analysis, which showed complete disappearance of the chloromethylene signal (Figure 1, Supporting Information, Figures S10 and S11); 31P NMR also confirmed the completion of the conjugation reaction, as evidenced by the appearance of a new phosphonium signal at ∼40 ppm (Figure 1, Supporting Information, Figures S10 and S11). According to GPC analyses on chlorine-based polypeptides, the MWs of phosphonium-based polypeptides PHLG-TEP, PHDLG-TEP, and PPLG-TEP were determined to be 15, 15, and 13 kDa, respectively.
Table 1. Structures and properties of polypeptides with quaternary phosphonium side chains.
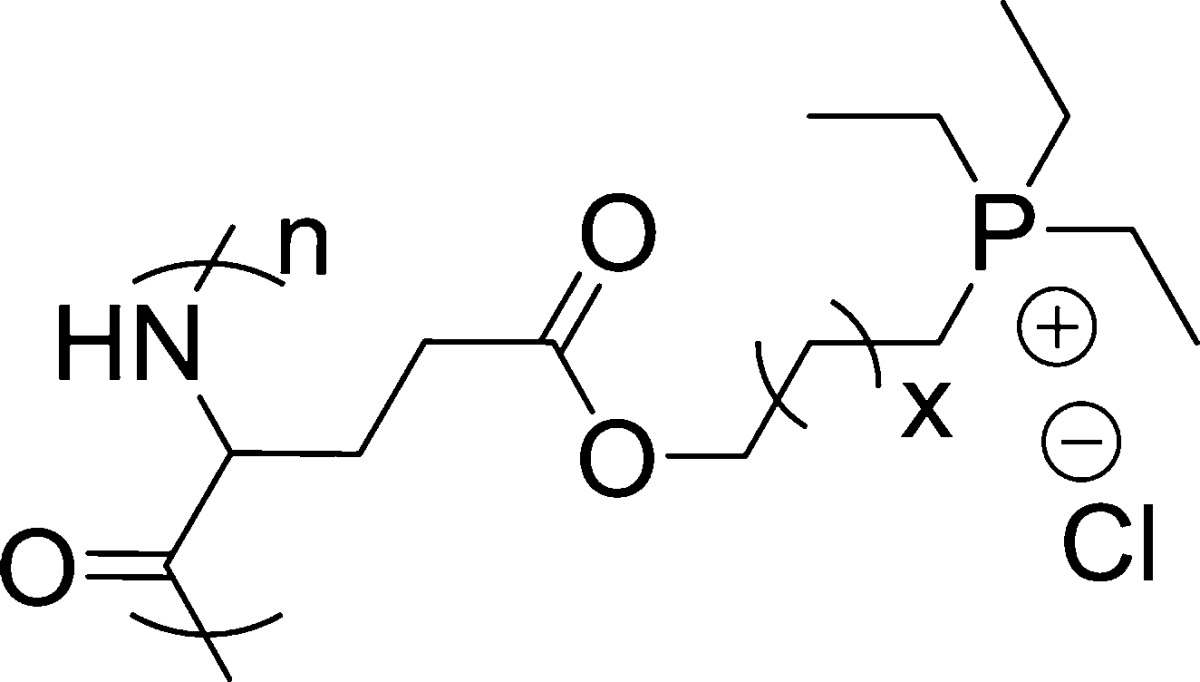
Stereochemistry of the starting glutamic acid.
Determined by CD.
Figure 1.
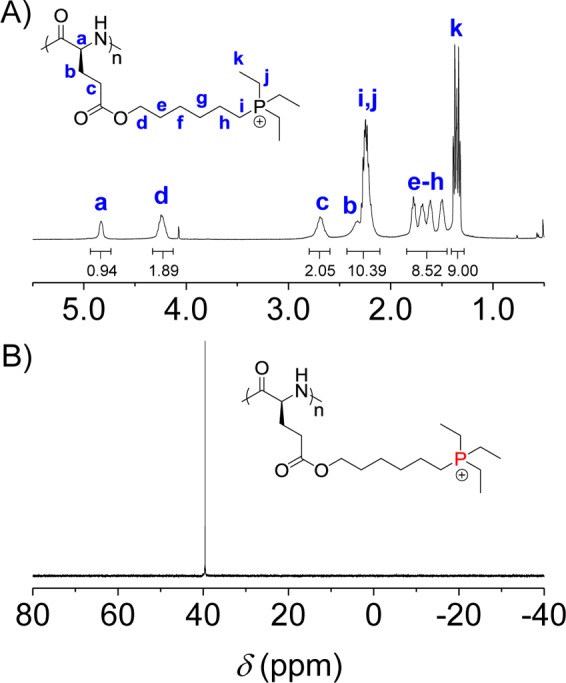
1H (A) and 31P (B) NMR spectra of PHLG-TEP in TFA-d.
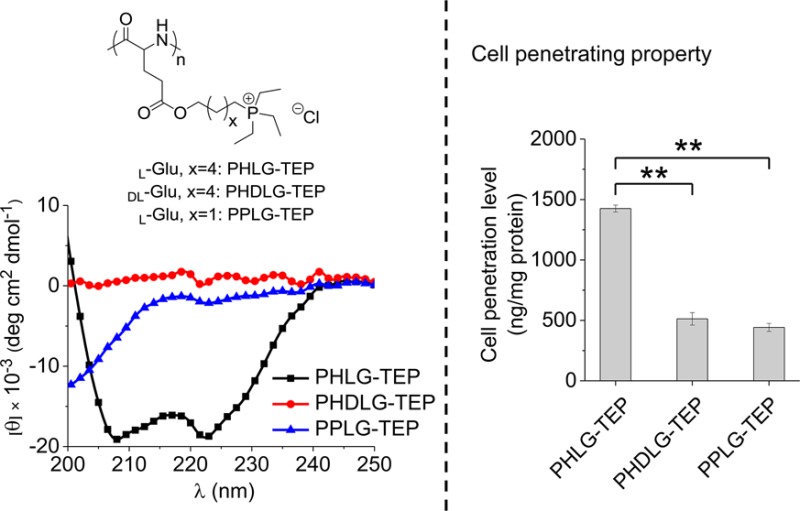
The secondary structure of the resulting polypeptides with different side-chain lengths and α-carbon stereochemistry (l or dl) was studied by circular dichroism (CD) spectroscopy (Figure 2A). PPLG-TEP (Scheme 1), affording 8 σ-bonds between the backbone and the cationic charged phosphonium group, showed a typical random coil conformation due to the side-chain charge repulsion. When the backbone-to-charge distance was increased to 11 σ-bonds, the corresponding polypeptide (PHLG-TEP, Scheme 1) was found to adopt α-helical conformation with 56% helicity, evidenced by the characteristic minima at 208 and 222 nm of its CD curve.15 The formation of the helical conformation of PHLG-TEP with elongated hydrophobic side chains is presumably due to the enhanced hydrophobic interaction and reduced charge repulsion of the side chains. In comparison, PHDLG-TEP (Scheme 1), an analogue of PHLG-TEP prepared with racemic mixture of NCAs, showed no helical conformation.
Figure 2.

(A) CD spectra of polypeptides in aqueous solution at pH 7. The effect of polypeptide concentration (B), pH (C), and ionic strength (D) on the residue molar ellipticity of PHLG-TEP at 222 nm.
The helicity of PHLG-TEP remained unchanged with the change of polypeptide concentration (Figure 2B), demonstrating that PHLG-TEP stayed as monomeric form in aqueous solution.15 PHLG-TEP also exhibited remarkable helical stability over a broad pH range (1–8; Figure 2C), suggesting that the helical polypeptide may well maintain its helix-associated properties at neutral physiological pH as well as acidic pH in some subcellular compartments (e.g., endosome). At pH higher than 9 when the solution turned slightly basic, the precipitation of PHLG-TEP resulted in a decreased CD signal. PHLG-TEP also showed excellent helical stability against ionic strength increment (Figure 2D); its CD pattern and the molar ellipticity at 222 nm remained unchanged at NaCl concentration up to 4 M. The unusual helical stability of phosphonium-containing polypeptide at high ionic strength contrasts the previously reported cationic polypeptides containing primary amino or guanidino groups whose helical structures tend to be distorted upon increased solution ionic strength.15,16
Cationic polypeptides with rigid molecular structures, mostly α-helical conformations, have been previously demonstrated to be cell membrane permeable.16,17,39 Motivated by such finding, we explored the cell penetration properties of the obtained cationic polypeptides bearing quaternary phosphonium side chains. Fluorescein-labeled polypeptides were synthesized via conjugation of fluorescein-modified tertiary amine (Supporting Information, Scheme S1) to the polypeptide side chains. The cellular uptake of the resulting polypeptides was assessed in HeLa cells at 37 °C and was quantified by spectrofluorimetry. For all three phosphonium-bearing polypeptides tested, the helical PHLG-TEP notably outperformed its racemic analogue PHDLG-TEP and random-coiled PPLG-TEP (Figure 3A), which was consistent with our previous findings of helix-dependent membrane activity in amine- and guanidine-based charged, helical polypeptides and further substantiated the importance of helical conformation and structure rigidity on their membrane activity.16,17 In direct comparison with TRAMA-labeled HIV-TAT, a widely used CPP, PHLG-TEP also showed a 2-fold higher cell uptake level. Internalization of large amount of PHLG-TEP was observed for incubation as short as 30 min, demonstrating its potent membrane permeability (Figure 3A). Increase of the incubation time or polypeptide concentration resulted in increased uptake level (Figure 3A and Supporting Information, Figure S13). The penetration mechanism of polypeptides may have appreciable effect on their penetration efficiency and intracellular distribution, which in turn determines their cargo delivery capability when they are used as molecular transporters.40,41 Thus, PHLG-TEP was selected for further elucidation of its cell penetration mechanism. HeLa cells were incubated with PHLG-TEP at 4 °C when the energy-dependent endocytosis was blocked, and the penetration level was compared to that at 37 °C. As shown in Figure 3B, 71 and 61% of the cells had internalized PHLG-TEP at 37 and 4 °C, respectively, and unappreciable difference was noted in terms of the fluorescence shift. The cells were then visualized by CLSM, which showed notable separation of green fluorescence (fluorescein-labeled PHLG-TEP) from red fluorescence (Lysotracker Red-stained endosome) after incubation at both 37 and 4 °C (Figure 3C), indicating that majority of the internalized polypeptides were not entrapped in the endosomes. In support of such observation, a quantitative analysis further revealed that the intracellular levels of the internalized polypeptide made no significant difference at 37 and 4 °C (Figure 3D), which indicated that PHLG-TEP penetrated cells mainly via the energy-independent, nonendocytic, direct transduction. When the cell uptake was performed in the presence of different endocytic inhibitors, including mβCD that inhibits caveolae-mediated endocytosis,42 chlorpromazine that inhibits clathrin-mediated endocytosis,43 and wortmannin that inhibits macropinocytosis,44 none of them showed significant inhibitory effect, which further demonstrated that the penetration mechanism of the helical PHLG-TEP was not associated with endocytosis (Figure 3D). When used as transporters to deliver nucleic acids intracellularly, such nonendocytic mechanism would help to avoid the endosomal/lysosomal entrapment that often leads to cargo degradation and poor transfection efficiency.40 MTT assay was then utilized to monitor the cytotoxicity of the obtained polypeptides. All tested phosphonium-based polypeptides showed desired cell tolerability (>80% viability) at concentration up to 50 μg/mL (Figure 3E), indicating that these phosohonium-based polypeptides are safe yet effective cell-penetrating vehicles. When the concentration was further increased, appreciable toxicity of the helical PHLG-TEP was observed, which may be attributed to the excessive amount of the polypeptides internalized into the cells. With the excellent cell penetrating properties and low cytotoxicity, the phosphonium helical polypeptides would be a promising molecular transporter. As a brief demonstration, PHLG-TEP was evaluated for its capacity in delivering YOYO-1-labeled DNA (YOYO-1-DNA) into HeLa cells. As shown in Supporting Information, Figure S14, YOYO-1-DNA (green fluorescence) was distributed into both cytoplasm and nuclei of HeLa cells, demonstrating effective delivery of the gene cargo into cells.
Figure 3.

Cell-penetrating properties of polypeptides with quaternary phosphonium side chains. (A) Uptake level of fluorescein-labeled polypeptides in HeLa cells at different incubation time (n = 3). The polypeptide concentration was maintained constant at 20 μg/mL. (B) Uptake level of fluorescein-labeled PHLG-TEP in HeLa cells at 37 and 4 °C as monitored by flow cytometry. M1 and M2 phases corresponded to fluorescence-negative and positive cells, respectively. (C) CLSM images of HeLa cells incubated with fluorescein-labeled PHLG-TEP at 37 and 4 °C for 4 h (bar = 20 μm). (D) Uptake level of fluorescein-labeled PHLG-TEP in HeLa cells at 4 °C or in the presence of various endocytic inhibitors (n = 3). NS denotes no significant differences. (E) Cytotoxicity of polypeptides toward HeLa cells following 20 h treatment as determined by the MTT assay (n = 3).
Conclusions
In summary, we designed and synthesized polypeptides that have long hydrophobic side chains and terminal quaternary phosphonium positive charged groups for the first time via controlled ring-opening polymerization of chlorine-functionalized glutamic acid based N-carboxyanhydrides followed by nucleophilic substitution of the pendant chlorine groups with triethylphosphine. The side-chain length as well as the α-carbon stereochemistry of the polypeptides could be easily modulated, leading to polypeptides with different secondary structures. PHLG-TEP, a phosphonium-containing polypeptide having a sufficient long backbone-to-charge distance (11 σ-bonds), adopted helical conformation with great stability against the change of pH and ionic strength of the polypeptide aqueous solution. The helical PHLG-TEP showed desired cell penetration capability that was mediated mainly via the nonendocytic transduction, and notably outperformed its racemic and random-coiled analogues, which demonstrated the importance of α-helix in mediating membrane penetration. Along with its low cytotoxicity, this novel class of cationic, phosphonium-based, helical polypeptides would provide an important addition to synthetic cell penetrating peptides and exhibit promising potentials for biomedical application.
Acknowledgments
This work is supported by NSF (CHE 11-53122) and NIH (Director’s New Innovator Award 1DP2OD007246).
Supporting Information Available
NMR spectra of amino acids and NCA monomers (Figures S1–S6), NMR spectra and GPC traces of polypeptides with chloroalkyl side chains (Figures S7–S9 and S12), NMR spectra of polypeptides with quaternary phosphonium side chains (Figures S10 and S11), the dosage-dependent uptake level of fluorescein-labeled PHLG-TEP (Figure S13) and DNA delivery using PHLG-TEP as molecular transporter (Figure S14). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zorko M.; Langel U. Adv. Drug Delivery Rev. 2005, 57, 529–545. [DOI] [PubMed] [Google Scholar]
- Stewart K. M.; Horton K. L.; Kelley S. O. Org. Biomol. Chem. 2008, 6, 2242–2255. [DOI] [PubMed] [Google Scholar]
- Fonseca S. B.; Pereira M. P.; Kelley S. O. Adv. Drug Delivery Rev. 2009, 61, 953–964. [DOI] [PubMed] [Google Scholar]
- Floch V.; Loisel S.; Guénin E.; Hervé A. C.; Clément J. C.; Yaouanc J. J.; des Abbayes H.; Férec C. J. Med. Chem. 2000, 43, 4617–4628. [DOI] [PubMed] [Google Scholar]
- Guénin E.; Hervé A. C.; Floch V.; Loisel S.; Yaouanc J. J.; Clément J. C.; Férec C.; des Abbayes H. Angew. Chem., Int. Ed. 2000, 39, 629–631. [PubMed] [Google Scholar]
- Stekar J.; Nössner G.; Kutscher B.; Engel J.; Hilgard P. Angew. Chem., Int. Ed. 1995, 34, 238–240. [Google Scholar]
- Picquet E.; Le Ny K.; Delépine P.; Montier T.; Yaouanc J. J.; Cartier D.; des Abbayes H.; Férec C.; Clément J. C. Bioconjugate Chem. 2005, 16, 1051–1053. [DOI] [PubMed] [Google Scholar]
- Wang L.; Xu X.; Guo S.; Peng Z.; Tang T. Int. J. Biol. Macromol. 2011, 48, 375–380. [DOI] [PubMed] [Google Scholar]
- Hemp S. T.; Allen M. H.; Green M. D.; Long T. E. Biomacromolecules 2011, 13, 231–238. [DOI] [PubMed] [Google Scholar]
- Hemp S. T.; Smith A. E.; Bryson J. M.; Allen M. H.; Long T. E. Biomacromolecules 2012, 13, 2439–2445. [DOI] [PubMed] [Google Scholar]
- Ornelas-Megiatto C.; Wich P. R.; Fréchet J. M. J. J. Am. Chem. Soc. 2012, 134, 1902–1905. [DOI] [PubMed] [Google Scholar]
- Deshayes S.; Heitz A.; Morris M. C.; Charnet P.; Divita G.; Heitz F. Biochemistry 2004, 43, 1449–1457. [DOI] [PubMed] [Google Scholar]
- Veldhoen S.; Laufer S. D.; Trampe A.; Restle T. Nucleic Acids Res. 2006, 34, 6561–6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M. C.; Deshayes S.; Heitz F.; Divita G. Biol. Cell 2008, 100, 201–217. [DOI] [PubMed] [Google Scholar]
- Lu H.; Wang J.; Bai Y.; Lang J. W.; Liu S.; Lin Y.; Cheng J. Nat. Commun. 2011, 2, 206. [DOI] [PubMed] [Google Scholar]
- Tang H.; Yin L.; Kim K. H.; Cheng J. Chem. Sci. 2013, 4, 3839–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielson N. P.; Lu H.; Yin L.; Li D.; Wang F.; Cheng J. Angew. Chem., Int. Ed. 2012, 51, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielson N. P.; Lu H.; Yin L.; Kim K. H.; Cheng J. Mol. Ther. 2012, 20, 1599–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L.; Song Z.; Kim K. H.; Zheng N.; Tang H.; Lu H.; Gabrielson N.; Cheng J. Biomaterials 2013, 34, 2340–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L.; Song Z.; Qu Q.; Kim K. H.; Zheng N.; Yao C.; Chaudhury I.; Tang H.; Gabrielson N. P.; Uckun F. M.; Cheng J. Angew. Chem., Int. Ed. 2013, 52, 5757–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L.; Song Z.; Kim K. H.; Zheng N.; Gabrielson N. P.; Cheng J. Adv. Mater. 2013, 25, 3063–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L.; Tang H.; Kim K. H.; Zheng N.; Song Z.; Gabrielson N. P.; Lu H.; Cheng J. Angew. Chem., Int. Ed. 2013, 52, 9182–9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen J.; Zhang Y.; Gabrielson N. P.; Yin L.; Guan L.; Chaudhury I.; Lu H.; Wang F.; Cheng J. Biomater. Sci. 2013, 1, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deming T. J. Nature 1997, 390, 386–389. [DOI] [PubMed] [Google Scholar]
- Dimitrov I.; Schlaad H. Chem. Commun. 2003, 2944–2945. [DOI] [PubMed] [Google Scholar]
- Aliferis T.; Iatrou H.; Hadjichristidis N. Biomacromolecules 2004, 5, 1653–1656. [DOI] [PubMed] [Google Scholar]
- Vayaboury W.; Giani O.; Cottet H.; Deratani A.; Schue F. Macromol. Rapid Commun. 2004, 25, 1221–1224. [Google Scholar]
- Lu H.; Cheng J. J. Am. Chem. Soc. 2007, 129, 14114–14115. [DOI] [PubMed] [Google Scholar]
- Engler A. C.; Lee H.-i.; Hammond P. T. Angew. Chem., Int. Ed. 2009, 48, 9334–9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habraken G. J. M.; Koning C. E.; Heuts J. P. A.; Heise A. Chem. Commun. 2009, 3612–3614. [DOI] [PubMed] [Google Scholar]
- Tang H.; Zhang D. Biomacromolecules 2010, 11, 1585–1592. [DOI] [PubMed] [Google Scholar]
- Sun J.; Schlaad H. Macromolecules 2010, 43, 4445–4448. [Google Scholar]
- Lu H.; Bai Y.; Wang J.; Gabrielson N. P.; Wang F.; Lin Y.; Cheng J. Macromolecules 2011, 44, 6237–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Zhang D. Polym. Chem. 2011, 2, 1542–1551. [Google Scholar]
- Adler A. J.; Greenfield N. J.; Fasman G. D. Methods Enzymol. 1973, 27, 675–735. [DOI] [PubMed] [Google Scholar]
- Greenfield N. J. Nat. Protoc. 2006, 1, 2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow J. A.; Segall M. L.; Lund-Katz S.; Phillips M. C.; Knapp M.; Rupp B.; Weisgraber K. H. Biochemistry 2000, 39, 11657–11666. [DOI] [PubMed] [Google Scholar]
- Kramer J. R.; Deming T. J. Biomacromolecules 2010, 11, 3668–3672. [DOI] [PubMed] [Google Scholar]
- Tang H.; Yin L.; Lu H.; Cheng J. Biomacromolecules 2012, 13, 2609–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil I. A.; Kogure K.; Akita H.; Harashima H. Pharmacol. Rev. 2006, 58, 32–45. [DOI] [PubMed] [Google Scholar]
- Gratton S. E. A.; Ropp P. A.; Pohlhaus P. D.; Luft J. C.; Madden V. J.; Napier M. E.; DeSimone J. M. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 11613–11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano M.; Teodósio C.; Paiva A.; Simões S.; Pedroso de Lima M. C. Biochem. J. 2005, 390, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rejman J.; Bragonzi A.; Conese M. Mol. Ther. 2005, 12, 468–474. [DOI] [PubMed] [Google Scholar]
- Muscella A.; Elia M. G.; Greco S.; Storelli C.; Marsigliante S. J. Cell. Physiol. 2003, 195, 234–240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.