

Abstract
The dry antibiotic development pipeline coupled with the emergence of multidrug resistant Gram-negative ‘superbugs’ has driven the revival of the polymyxin lipopeptide antibiotics. Polymyxin resistance implies a total lack of antibiotics for the treatment of life-threatening infections. The lack of molecular imaging probes that possess native polymyxin-like antibacterial activity is a barrier to understanding the resistance mechanisms and the development of a new generation of polymyxin lipopeptides. Here we report the regioselective modification of the polymyxin B core scaffold at the N-terminus with the dansyl fluorophore to generate an active probe that mimics polymyxin B pharmacologically. Time-lapse laser scanning confocal microscopy imaging of the penetration of probe (1) into Gram-negative bacterial cells revealed that the probe initially accumulates in the outer membrane and subsequently penetrates into the inner membrane and finally the cytoplasm. The implementation of this polymyxin-mimetic probe will advance the development of platforms for the discovery of novel polymyxin lipopeptides with efficacy against polymyxin-resistant strains.
Introduction
The dry antibacterial drug development pipeline has driven the revival of polymyxin B and E (also known as colistin) as a last line of defense against infections caused by multidrug-resistant (MDR) Gram-negative ‘superbugs’, Acinetobacter baumannii, Pseudomonas aeruginosa, and Klebsiella pneumoniae.1 These microorganisms are among the 6 top-priority dangerous ‘superbugs’ identified by Infectious Diseases Society of America as requiring the most urgent attention for discovery of novel antibiotics.2 The emergence of pandrug-resistant (PDR) strains which are even resistant to polymyxins has prompted fears that life-threatening infections will soon become untreatable.3 Investigations into the mechanisms of polymyxin action and resistance are warranted in order to provide a platform for the development of next-generation superior polymyxin lipopeptides with efficacy against these PDR strains. Unfortunately, the unavailability of polymyxin imaging probes with native activities is a significant barrier.4
The initial cellular target of polymyxins is the lipopolysaccharide (LPS) component of the bacterial outer membrane. The binding of polymyxin to LPS involves an initial electrostatic interaction of the cationic Dab side-chains with the phosphate groups of the lipid A component of LPS, displacing divalent cations (Ca2+ and Mg2+) that bridge adjacent LPS molecules.3−5 Hydrophobic interactions take place between the N-terminal fatty acyl tail of the polymyxin molecule and the lipid A fatty acyl chains. The fatty acyl tail of polymyxins is crucial for their antibacterial activity, as polymyxin nonapeptide (derived by proteolytic cleavage of the fatty acyl-Dab1 from the N-terminus of the polymyxin) is devoid of antibacterial activity.6 Our structure–activity relationship (SAR) model of the interaction of polymyxin B with lipid A indicated a key role for the diaminobutyric acid (Dab) side chains for binding to the lipid A phosphate groups.4 We have previously highlighted the deficiencies of directly amine-coupling dansyl groups onto the Dab side chains in semisynthetic preparations of dansyl-polymyxin B.7 Analysis of these semisynthetic dansyl-polymyxin B preparations revealed the existence of mono-, di-, tri-, and tetra-dansyl substituted species.7 Furthermore, as polymyxin B is composed of two major components (B1 and B2), the potential for either of these components to be substituted at any of the five Dab side chains with up to four dansyl molecules results in a highly variable mixture of dansylated derivatives.7 There is little value in using these semisynthetic preparations as imaging probes since they lack native antibacterial activity.
Here we have designed and synthesized a fluorescent dansyl-polymyxin lipopeptide probe which retains the native antibacterial activity of the polymyxin B parent structure; and demonstrated its considerable utility for imaging polymyxin penetration and localization in Gram-negative bacterial cells. The availability of this probe will greatly facilitate molecular imaging studies on both the mode of action and the pharmacokinetics; and contribute toward the development of a new generation of polymyxin lipopeptides with superior activity against polymyxin-resistant Gram-negative ‘superbugs’.
Results
Probe Design and Synthesis
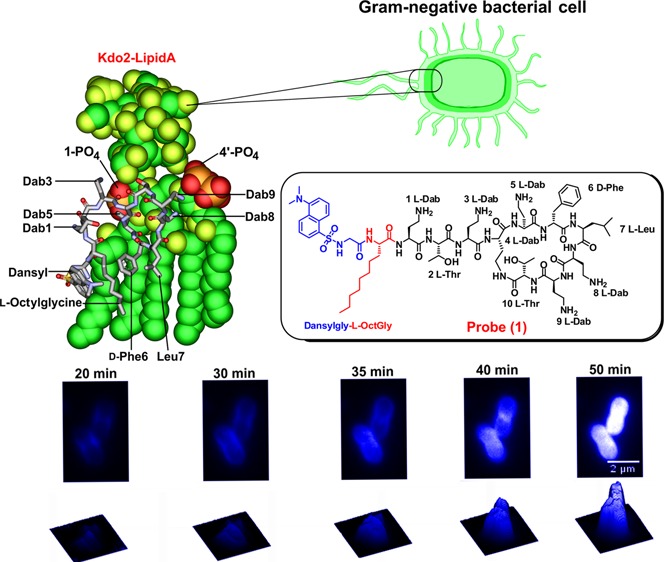
The design and synthesis of a dansyl molecular probe through regioselective modification of the polymyxin B core structure was undertaken with the aforementioned lipid A interaction principles in mind, in order to mimic the polymyxin B structure as closely as possible and to maintain its native antibacterial activity. The dansyl group was utilized for the fluorescent probe as it has suitable spectral properties and its relatively small size would reduce the chance of steric effects. The dansyl moiety incorporates a natpthyl structure, together with a sulfonamide group which conveys electron withdrawing properties. We reasoned that the best point for the attachment of the dansyl group to the polymyxin core structure would be at the N-terminal fatty acyl region (Figure 1). Therefore, the strategy we employed was to replace the N-terminal fatty acyl group of polymyxin B with the amino acid l-octylglycine, where the eight carbon fatty acyl chain emulated the N-terminal fatty acyl chain of polymyxin B, while the Nα-amino group would provide a convenient point of attachment for the dansyl group (eliminating the need for additional orthogonal protection during the synthesis). Antimicrobial activity was screened against a panel of ATCC and recent clinical isolates of polymyxin-susceptible and -resistant strains of P. aeruginosa, A. baumannii, and K. pneumoniae (Table 1). The probe showed very promising activity against the polymyxin-susceptible strains. The static time-kill kinetic experiments showed that at 0.5× and 5× MIC probe (1) achieved a ∼3-log reduction of the viable cell count at 1 h which is comparable to colistin at an initial inoculum of ∼106 cfu/mL (Figure S2A), whereas with an initial inoculum of ∼108cfu/mL, both probe (1) and colistin were inactive at 0.5× MIC and only showed killing effect at 5× MIC (Figure S2B). We have previously observed this inoculum effect with colistin treatment of this strain.22 Regrowth of bacteria in both probe (1) and colistin treated cells was observed possibly due to the emergence of the polymyxin-resistant subpopulation.22
Figure 1.
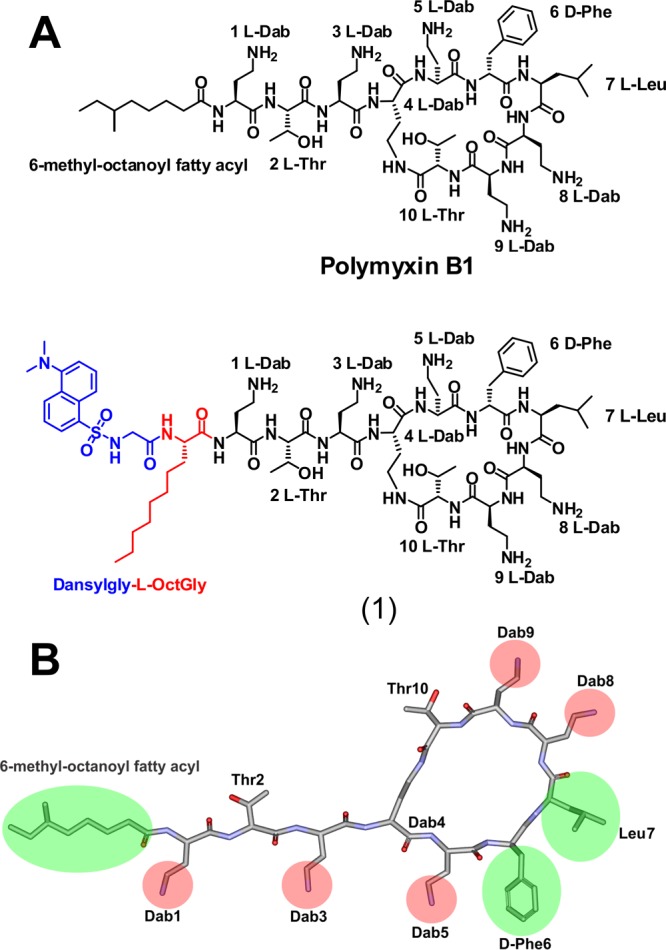
(A) Chemical structures of polymyxin B and dansyl polymyxin B probe (1). (B) SAR model of the polymyxin B core scaffold showing regions that can be modified with the dansyl fluorophore. The green shaded regions can tolerate modification without much activity loss, whereas substitution of the red shaded regions results in the loss of activity.
Table 1. Comparison of the Antimicrobial Activity of the Dansyl-Polymyxin Probe (1) with Polymyxin B and Colistin.
| minimum inhibitory concentration (mg/L) |
||||
|---|---|---|---|---|
| bacteria | strain | polymyxin B | colistin | probe (1) |
| K. pneumoniae | ATCC 13883 | 1 | 1 | 8 |
| FADDI-03 | <0.125 | 1 | 8 | |
| FADDI-04 Col Ra | 128 | >128 | >32 | |
| FADDI-05 Col Ra | >32 | 128 | >32 | |
| A. baumannii | ATCC 19606 | 1 | 1 | 4 |
| FADDI-06 | 0.5 | 0.5 | 8 | |
| ATCC 19606 Col Ra | 128 | 128 | 8 | |
| FADDI-07 | 8 | 16 | 16 | |
| P. aeruginosa | ATCC 27853 | 1 | 1 | 4 |
| FADDI-08 | 1 | 1 | 4 | |
| FADDI-09 | 1 | 2 | 4 | |
| FADDI-10 Col Ra | >32 | >128 | 4 | |
| FADDI-11 Col Ra | >32 | >128 | 4 | |
Col R refers to polymyxin resistant strains.
Transmission Electron Microscopy (TEM)
TEM imaging of K. pneumoniae ATCC 13883 cells treated with probe (1) at 0.5× MIC revealed the formation of numerous protrusions or blebs extending from the outer membrane of the cells that possibly represent outer membrane fragments (Figure 2A). Notably, a similar blebbing effect was observed with Gram-negative bacterial cells treated with polymyxin B and colistin.23 The ability of probe (1) to disrupt the other membrane of K. pneumoniae ATCC 13883 appeared comparable to that of polymyxin B and colistin.23
Figure 2.
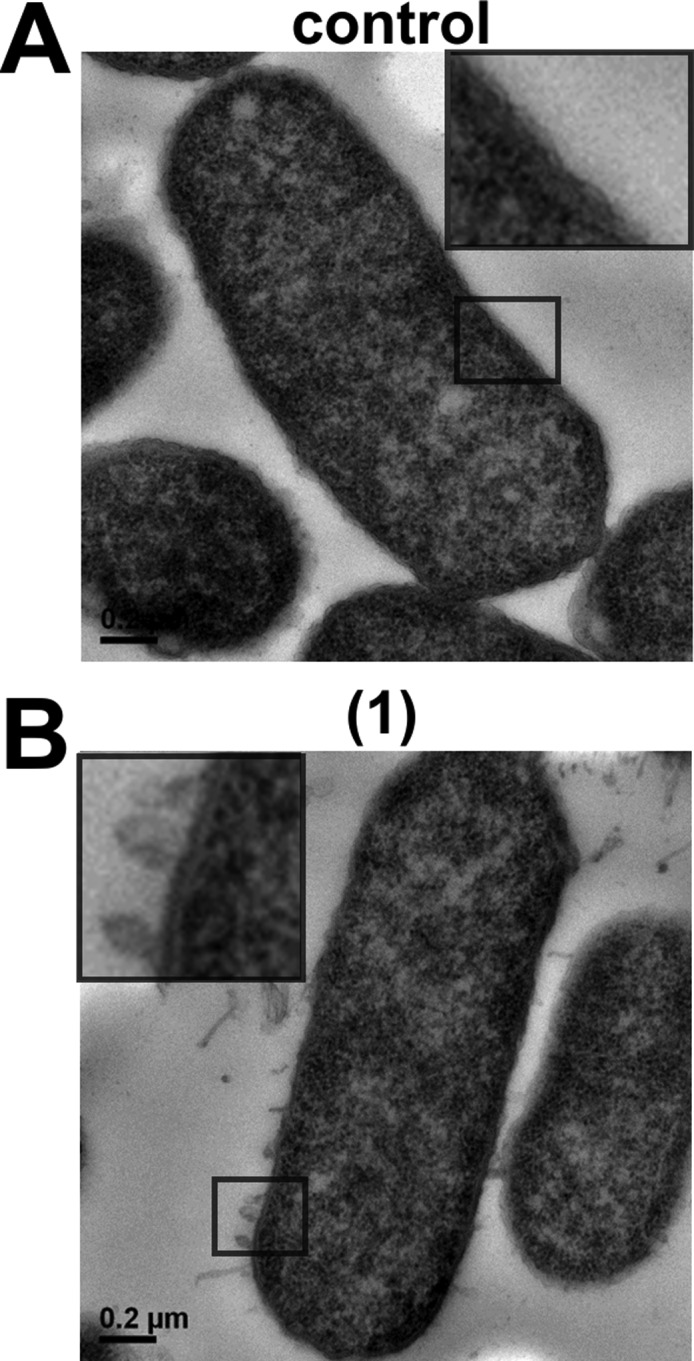
Transmission electron microcopy of (A) untreated K. pneumoniae ATCC 13883 cells. (B) K. pneumoniae ATCC 13883 cells treated with probe (1) (0.5× MIC). The box shows a magnified region of the outer membrane.
NMR Structure Determination in Dodecylphosphocholine (DPC) Micelles
Probe (1) was first examined by recording a series of two-dimensional NMR experiments in an acetate buffer at pH 4.5. The signals in the spectra were assigned using the standard sequential assignment procedure from analysis of 2D NOESY and TOCSY NMR spectra combined with the 2D 13C HSQC and 13C H2BC spectra to facilitate the assignment the side chains, particularly the overlapped protons of the lipid tail. The 2D NOESY spectra (Figure S3A) yielded few small NOEs with the same sign as the diagonal (spin diffusion regime) that were insufficient to determine a structure consistent with that reported previously for polymyxin B in water.5 Moreover, the NOEs involving the aromatic resonances of the dansyl were of opposite sign (extreme narrowing condition) which shows that the dansyl group tumbles faster and independently of the rest of the peptide. The assignments are similar to that reported for the parent peptide at pH 4.0;5 however, Dab9 was not observed at 25 °C but the broadened amide and side chain signals were assigned from spectra recorded at 10 °C. The 1H and 13C assignments are reported in Table S1. Addition of substoichiometric quantities of LPS to a sample of probe (1) resulted in immediate precipitation and a concomitant signal decrease in the 1D 1H NMR spectra without chemical shift change (data not shown) indicating the formation of an insoluble probe (1)-LPS aggregate. These observations are in contrast to the addition of LPS to the polymyxin B peptide where moderate signal broadening and efficient transferred NOE effects to the LPS aggregate were observed that were sufficient to determine the LPS bound structure.5 This is potentially due to an increased avidity of probe (1) for the LPS aggregates due to the dansyl-substituent. LPS is found in Gram-negative bacteria embedded in a phospholipid bilayer. We thus recorded NMR data on probe (1) in the presence of an excess of deuterated n-dodecylphosphocholine (DPC) micelles as a model membrane mimetic to better reflect the functional environment of probe (1). Broader signals were observed in the 1D spectrum and an increase in the number and size of the cross peaks in the 2D NOESY spectra indicated binding to the micelle (Figure S3B). Assignments of the broader signals (Table S2), were carried out as before (vide supra) and partly facilitated by comparing the 2D 13C HSQC spectrum to that from the peptide dissolved in water. Given the high quality of the NMR spectra (Figure S3B,C), we then determined the structure of probe (1) in DPC micelles using a total of 253 distance restraints derived from the 2D NOESY spectra in conjunction with restrained simulated annealing using the program CYANA 2.1. Superposition of the best CYANA 20 structures, ranked according to the lowest overall target function, reveals a well converged ensemble with an overall backbone root-mean-square distance (r.m.s.d.) of 0.34 Å (Figure 3A). Statistics are shown in Table S3. The three-dimensional structure of probe (1) in DPC micelles essentially resembles that reported for polymyxin B, indicating that introduction of the dansyl group does not markedly perturb the native structure.5 Similar to the polymyxin B NMR structure, the side chains of Dab1, Dab5, and Dab8 project to one side of the structure forming a line of positive charge. Interestingly, 18 NOEs were observed between the lipid OctylGly side chain and the side chain of d-Phe6 that directs the OctylGly side chain and the hydrophobic side chains of d-Phe6 and Leu7 downward, presumably into the membraneous environment of the micelle. The protons of the napthalene side chain showed numerous NOEs to the octyl lipid side chain and two long-range NOEs to the aromatic ring of d-Phe6. These position the dansyl group also downward, increasing the hydrophobic face that projects into the micelle. To investigate the interaction with LPS, we titrated in aliquots of LPS to a final probe (1)/LPS w/w ratio of 0.75:1.0 without precipitation (vide supra) and noted several small amide shifts in fast exchange for Dab1, Thr2, Dab9, Thr12, and Leu7. While more extensive data is required to investigate the probe (1)/LPS interface, no additional NOE cross-peaks were observed in the 2D NOESY spectra suggesting that the structure of probe (1) does not change substantially upon transient binding to the LPS aggregate or LPS bound micelle. DPC and 1,2-dicaproyl-sn-glycero-3-phosphocholine (DHPC) micelles have been previously used as membrane mimetics to investigate the NMR structure of monomeric LPS24 and the interaction of LPS with polymyxin peptides by NMR.25 To access whether the structure of probe (1) in DPC is a plausible model for a LPS bound complex we took our NMR structure as a starting model and docked it into the LPS structure from E. coli LPS. The docking model revealed that most of the interactions seen with the polymyxin B-LPS complex are also evident with LPS bound to the DPC structure of probe (1) (Figure 3B). The molecular model indicates that electrostatic interactions with the 1-phosphorester group on lipid A are not hampered by the dansyl group. The model further suggests that the hydrophobic dansyl group interacts with the apolar environment formed by the fatty acyl chains of lipid A. This would suggest that probe (1) potentially binds to LPS in a similar configuration as polymyxin B, which is coincident with its comparable antibacterial activity.
Figure 3.

(A) Stereo view of a superposition of the top 20 lowest energy CYANA conformers of of probe (1). The backbone is shown in red and the side chains are displayed as heavy atom representation. The bridging side chain of Dab4 is shown in yellow. (B) Molecular model of the probe (1)-LPS complex.
Imaging of Lipopeptide Probe Penetration into Gram-Negative Bacteria
The penetration of probe (1) into K. pneumoniae ATCC 13883 cells was monitored by CLSM. The membrane selective dye FM4–64 and the nucleic acid selective dye SYTOX green were employed to visualize the outer membrane and intracellular nucleic acid of the bacterial cells, respectively. The time-lapse imaging studies revealed that probe (1) initially accumulated in the outer membrane, penetrated the cell, and then became homogenously distributed into the cytoplasm (Figures 4 and 5; Movie S1). Notably, FM4–64 emission decreased after 14 min, suggesting probe (1) disrupted the outer membrane integrity over this time period (Figure 5). Intriguingly, confocal imaging and spectrophotometric lysis assay experiments with spheroplasts isolated from K. pneumoniae ATCC 13883 revealed that probe (1) also accumulated within and disrupted the inner membrane structure (Figure S4). In order to observe any concentration-dependent effects, the probe treated cells were visualized after treatment with a low 0.5× MIC or high 5× MIC concentration (Figure S5). The imaging results demonstrated that at the lower sub-MIC concentration probe (1) tended to accumulate on the surface of the bacterial cell and partly penetrated into the outer membrane (Figure S5 iii and iv), whereas at the high 5× MIC concentration, probe (1) accumulated on the surface of the bacterial cell and entered into the cytoplasm (Figure S5 vii and viii).
Figure 4.
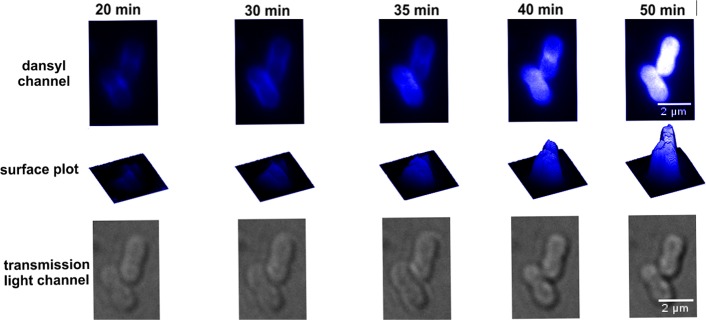
Confocal laser scanning microscopy time-lapse imaging of K. pneumoniae ATCC 13883 cells treated with probe (1) at 5× MIC.
Figure 5.

Laser scanning confocal microscopy time-lapse imaging of K. pneumoniae ATCC 13883 cells treated with probe (1) at 5× MIC, the membrane probe FM4–64 and the nucleic acid selective probe SYTOX green.
Discussion
In recent times the world has seen the emergence of bacterial pathogens that are resistant to almost all available antibiotics.26−29 Over the last 30 years only 2 novel antibiotic classes have been introduced into the clinic (linezolid and daptomycin), illustrating that the post-antibiotic era is fast approaching.30 Many pharmaceutical companies have lost interest in producing new antibiotics, preferring to focus on the development of products for the treatment of more profitable areas such as chronic conditions and lifestyle issues.31 Consequently, there has been a decline in the discovery and development of novel antibiotics, and as a result we have seen the increasing emergence of MDR ‘superbugs’: this dire situation has been dubbed the ‘perfect storm’.32 Currently, polymyxin B and colistin are being used as the last-line therapy against life-threatening infections caused by MDR Gram-negative bacterial infections.33−36 Resistance to polymyxins can rapidly emerge in vitro to P. aeruginosa, A. baumannii, and K. pneumoniae.37−40,10 Moreover, polymyxin resistance in community and hospital acquired infections is being increasingly reported.38,41−45 Unfortunately, polymyxin resistance indicates a total lack of treatment options for these problematic Gram-negative infections. Therefore, there is an urgent unmet medical need to develop new and safer polymyxin lipopeptide antibiotics to combat the increasing incidence of MDR pathogens, especially in the hospital setting. The time has come for a global commitment to develop new antibiotics. This study focuses on the development of fluorescent polymyxin probe with native antibacterial activities, which may prove to be of considerable utility for elucidating the penetration of polymyxins into the outer and inner membranes of the Gram-negative bacterial cell.
This is the first report of a polymyxin probe which maintains the pharmacological properties of the parent compound polymyxin B. Previous reports of polymyxin probes employed either inactive nonapeptide derivatives or dansylated polymyxin B, which was derived by nonspecifically reacting polymyxin B with dansyl-chloride.46−49 Commercial preparations of 4-bora-3a,4a-diaza-s-indacene (BODIPY) labeled polymyxin B displayed markedly reduced antibacterial activity compared to polymyxin B.50 Moreover, our group previously reported a fully synthetic [dansyl-Lys]1polymyxin B3 probe that was devoid of antibacterial activity.7 These findings are consistent with our understanding of polymyxin SAR wherein the dansyl modification of the Dab side chains inactivates antibacterial activity (Figure 1B).7 Here we present data that supports our design strategy to employ total synthesis for the regioselective incorporation of the dansyl group into the hydrophobic N-terminal center of the polymyxin B core scaffold, which has a minimal impact on the native antibacterial activity of polymyxin B. The probe we designed and synthesized displayed antibacterial and pharmacological activity comparable to that of polymyxin B and colistin. Notably, probe (1) displayed activity against polymyxin-resistant P. aeruginosa and A. baumannii strains (Table 1). It is understood that Gram-negative pathogens resist the action of polymyxins by introducing cationic modifications onto the phosphate groups on the lipid A component of LPS.51−56 The most common mechanism involves esterification of lipid A phosphates with aminoarabinose or ethanolamine.51−56 This molecular tailoring serves to reduce the net negative charge of the outer membrane surface, thereby repelling the electrostatic attraction with positively charged polymyxin molecules.57 The molecular model of the probe (1)-LPS complex implies that the combination of the l-octylglycine and the dansyl substituent at the N-terminus provides additional hydrophobic interactive forces that compensate for the electrostatic repulsion of the aminoarabinose phosphate modifications (Figure 3B).
The key role of the outer membrane of Gram-negative bacteria is to act as a permeability barrier (Figure 6, step 1).58 Polymyxins are believed to exert their primary antimicrobial mode of action by permeabilizing the Gram-negative bacterial outer membrane via a direct interaction with the lipid A component of the LPS. The disruptive effect of polymyxins on the outer membrane is thought to involve a two-stage interaction mechanism. In stage one, it is purported that the cationic side-chains of the five Dab residues on the polymyxin molecule electrostatically bind to the anionic phosphate groups on the lipid A core, thereby displacing the divalent cations that normally bridge adjacent lipid A molecules that serve to maintain membrane integrity (Figure 6, step 2).4,58−62 This initial electrostatic interaction allows the N-terminal fatty acyl chain of the polymyxin molecule to insert into the fatty acyl chain layer of the lipid A molecules. This ‘self-promoted’ uptake mechanism is believed to produce disruption of the outer membrane structures, which leads to bacterial cell death (Figure 6, step 3).4,59−62 Subsequent events remain unclear; however, our group has recently shown that polymyxin B and colistin inhibit the NDH-2 oxidoreductase inner membrane respiratory enzyme, which also may contribute toward their bactericidal effect (Figure 6, step 4).20 The CLSM time-lapse imaging results with probe (1) help validate this mechanistic model. Based on the imaging data, it is evident that polymyxins initially accumulate in the outer membrane, followed by a gradual penetration into the inner membrane and finally entering the cytoplasm in K. pneumoniae. These findings are consistent with the secondary mode of action of polymyxin B which involves inhibitory activity against the inner membrane NDH-2 enzyme.20 It will be most interesting to elucidate the intracellular target(s) for the polymyxins, which to date remain uncharacterized.
Figure 6.

Schematic diagram depicting the putative mode of action of polymyxins at the levels of the outer and inner membranes of the Gram-negative bacterial cell. Step 1: Polymyxins target the outer membrane of Gram-negative bacteria. Step 2: The positively charged polymyxins displace divalent cations that bridge adjacent LPS molecules. Step 3: The electrostatic interactions weaken the stability of the outer membrane and the hydrophobic insertion destabilizes the outer membrane through hydrophobic expansion producing damage to the outer membrane. Step 4: Polymyxins penetrate into the inner membrane and inhibit the respiratory enzyme NDH-2.20
In summary, for the first time we have designed and synthesized a regioselectively modified dansyl polymyxin probe, which maintains the pharmacological properties of the parent compound polymyxin B. Our CLSM imaging data garnered with the novel polymyxin probe helps confirm our mechanistic insights into the penetration of polymyxins into the outer and inner membrane barriers of the Gram-negative bacterial cell. These fundamental insights will help seed the foundations that enable rational drug development around this important class of lipopeptide antibiotics, which may be the answer to MDR ‘superbugs’.
Experimental Procedures
Chemicals
Polymyxin B (sulfate), colistin (sulfate), lipopolysaccharides (LPS), dansylglycine, triisopropylsilane (TIPS), diphenylphosphorylazide (DPPA), and diisopropylethylamine (DIPEA) were obtained from Sigma-Aldrich (Sydney, NSW, Australia). All other reagents were of the highest purity and commercially available. Stock solutions of polymyxin B and colistin were prepared in Milli-Q water (Millipore, North Ryde, NSW, Australia) and filtered through 0.22-μm syringe filters (Sartorius, Melbourne, Vic, Australia). Solutions were stored at 4 °C for up to one month.8 Piperidine, diisopropylethylamine (DIPEA), trifluoroacetic acid (TFA), and 1H-benzotriazolium-1-[bis(dimethylamino)methylene]-5-chloro-hexafluorophosphate-(1)-3-oxide (HCTU) were purchased from Auspep (Melbourne, Australia). Fmoc-Dab(Boc)-OH and Fmoc-Dab(ivDde)-OH were obtained from Chem-Impex International (USA). Fmoc-Thr(tBu)-OH was from Mimotopes (Melbourne, Australia). Fmoc-OctGly-OH was obtained from Try-lead Chem (China). Dimethylformamide (DMF), methanol (MeOH), diethyl ether, dichloromethane (DCM), hydrochloric acid, and acetonitrile were obtained from Merck (Melbourne, VIC, Australia), and Fmoc-Thr(tBu)-TCP-Resin from Intavis Bioanyltical Instruments, Germany. Dodecylphosphocholine (purity >99%) was purchased from Avanti Polar Lipids (USA, Alabama).
Synthesis of Polymyxin Probe (1)
Synthesis of the protected linear peptide (residues 1–10 and N-terminal octylglycine) was carried out on a CEM Liberty Microwave automated peptide synthesizer using standard Fmoc solid phase peptide chemistry (Figure S1). Specifically, synthesis was undertaken using TCP-Resin, preloaded with Fmoc-Thr(tBu)-OH (loading 0.78 mmol/g), 0.1 mmol scale (128 mg of resin). Coupling of the Fmoc-amino acids was performed using the default instrument protocol: 5 equiv (relative to resin loading) of Fmoc amino acid and HCTU in DMF with activation in situ, using 10 equiv of DIPEA. This was carried out for 2 min at room temperature, then for 4 min at 50 °C (25W microwave power). Fmoc deprotection was performed using the default instrument protocol: 20% piperidine in dimethylformamide (1 × 30 s, 1 × 3 min) at 75 °C (35W microwave power). The resin was then removed from the instrument and transferred to a synthesis syringe for manual coupling of the dansylglycine. This was carried out using 2 equiv (relative to the loading of the resin) of dansylglycine, DIC, and HOBt, preactivated in 50% DCM/DMF for 10 min followed by reaction for 1 h. The resin was washed with DMF (4 × 2 min) then treated with 2% hydrazine in DMF (4 × 15 min) to remove the ivDde group. The resin was washed with MeOH (2 × 2 min) and diethyl ether (1 × 2 min), then air-dried under vacuum suction. The protected linear peptide was then cleaved from the resin by washing the resin with 1% TFA in DCM (1 × 5 min, 3 × 10 min). The resulting residue was dissolved in 50% acetonitrile/water and freeze-dried overnight. The crude protected linear peptide obtained was used in the next step without further workup. The protected linear peptide was dissolved in DMF (10 mL) to which DPPA, 0.3 mmol, 0.65 μL (3 equiv relative to the loading of the resin), and DIPEA, 0.6 mmol, 104 μL (6 mol equiv relative to the loading of the resin), were added. This solution was stirred at room temperature overnight. The reaction solution was then concentrated under vacuum overnight. The resulting residue was taken up in a solution of 5% TIPS in TFA and stirred at room temperature for 1.5 h. The TFA was removed under a stream of nitrogen and the crude cyclic peptide precipitated with cold diethyl ether. The resulting precipitate was collected by centrifugation and air-dried in a fume hood to give the crude cyclic peptide as a residue. The resulting residue was taken up in Milli-Q water and desalted using a Vari-Pure IPE SAX column. The crude cyclic peptide was subjected to reversed-phase (RP) HPLC purification. Fractions collected were analyzed by LC-MS performed on a Shimadzu 2020 LCMS system, incorporating a photodiode array detector (214 nm) coupled directly to an electrospray ionization source and a single quadrupole mass analyzer. RP-HPLC was carried out employing a Phenomenex column (Luna C8(2), 100 × 2.0 mm ID) eluting with a gradient of 80% acetonitrile in 0.05% aqueous TFA, over 10 min at a flow rate of 0.2 mL/min. Mass spectra were acquired in positive ion mode with a scan range of 200–2000 m/z. Crude cyclic peptides were purified by RP-HPLC on a Agilent 1200 quaternary pump system, photodiode array detector (214 nm), employing a Phenomenex Axia column (Luna C8(2), 50 × 21.3 mm ID) eluting with a gradient of 60% acetonitrile in 0.1% aqueous TFA, over 60 min at a flow rate of 5 mL/min. The combined fractions were freeze-dried for two days to give the dansyl-polymyxin TFA salt as a white solid in a yield of 19.0 mg. The purity was >95% as estimated by RP-HPLC at 214 nm. The identity of probe (1) was confirmed as having the correct molecular weight by ESI-MS analysis: m/z (monoisotopic) calculated; C71H116N19O16S [M+H]+ 1523.86, [M+2H]2+ 762.43, [M+3H]3+ 508.62. Observed: [M+H]+ 1523.20, [M+2H]2+ 762.45, [M+3H]3+ 508.70.
Bacterial Strains and Growth Conditions
Bacterial strains were purchased from the American Type Culture Collection (Manassas, VA, USA). K. pneumonia ATCC 13883 was used as a reference strain (colistin minimum inhibitory concentration (MIC) 0.5 μg/mL). A paired colistin-resistant strain (colistin MIC > 128 μg/mL) was selected from this reference strain in the presence of 10 mg/L colistin. All bacteria were stored at −80 °C in tryptone soya broth (TSB, Oxoid Australia). Prior to experiments, parent strains were subcultured onto nutrient agar plates (Medium Preparation Unit, University of Melbourne, VIC, Australia). Overnight broth cultures were subsequently grown in 5 mL of cation-adjusted Mueller-Hinton broth (CaMHB, Oxoid, Adelaide, SA, Australia), from which a 1 in 100 dilution was performed in fresh broth to prepare mid-logarithmic cultures according to the optical density at 500 nm (OD500 nm = 0.4 to 0.6). All broth cultures were incubated at 37 °C in a shaking incubator (180 rpm).
Determination of MICs and Static Killing Kinetics
The MICs against K. pneumoniae, A. baumannii, and P. aeruginosa strains were determined by the broth microdilution method.9 Experiments were performed with CaMHB in 96-well polypropylene microtiter plates. Wells were inoculated with 100 μL of bacterial suspension prepared in CaMHB (containing ∼106 colony forming units (cfu) per mL) and 100 μL of CaMHB containing increasing concentrations of polymyxins (0 to 128 mg/L). The MICs were defined as the lowest concentration at which visible growth was inhibited following 18 h incubation at 37 °C. To determine the killing kinetics by probe (1), time-kill experiments of K. pneumoniae ATCC 13883 at initial inoculum ∼106 and ∼108 cfu/mL were performed as previously described.10 Briefly, samples were treated with 0.5 × and 5 × MIC as indicated and incubated in a shaking water bath at 37 °C. Viable counting was performed on samples (50 μL) collected at 1, 2, 4, 8, and 24 h. Colistin treatment was used as control. At the indicated time points, aliquots of treated cells were harvested, suitable dilutions were performed, and then the cells were plated onto nutrient agar plates by a WASP2 spiral plater (Don Whitley Scientific Ltd., United Kingdom). After overnight incubation of the plates at 35 °C, cfu values were enumerated by a ProtoCOL colony counter (Don Whitley Scientific Ltd., United Kingdom). The limit of detection was 20 cfu/mL (equivalent to 1 colony per plate), whereas the limit of quantification was 400 cfu/mL (equivalent to 20 colonies per plate).
NMR Solution-State Structure Determination in Dodecylphosphocholine (DPC) Micelles and Modeling of the Probe-LPS Complex
NMR Spectroscopy
All NMR experiments were conducted on a Bruker AVANCE NMR spectrometer operating at a field strength of 14.1 T equipped with a cryoprobe and a Z axis gradient. For experiments recorded in water, 1–3 mM probe (1) was dissolved into 330 μL of 50 mM acetate buffer, pH 4.5, containing 90% H2O/10% D2O and placed in a Shigemi microcell. 2D TOCSY (mixing time 30 and 80 ms), 2D 13C HSQC, 2D multiplicity edited 13C HSQC, 2D 13C H2BC, and 2D NOESY (mixing times 200 - 400 ms) were recorded at 25 °C.11 A 3–9–19 WATERGATE sequence was used for solvent suppression in the homonuclear experiments and gradients were used to select coherences in the heteronuclear experiments.12 For experiments recorded in n-dodecylphosphocholine (DPC) micelles, 1–2 mM probe (1) was dissolved in the same buffer as previously and included 150 mM deuterated DPC (d38-DPC) (Novachem, VIC, Australia). Chemical shifts were referenced to 2,2-dimethyl-2-silapentane-5-sulfonic acid. Samples were vortexed prior to acquisition and the following experiments recorded at 20 °C; 2D TOCSY (mixing time 50 ms), 2D 13C HSQC, 2D multiplicity edited 13C HSQC, and 2D NOESY (mixing times 50 - 200 ms). NMR data was processed using NMRPipe and anaylsed using SPARKY (http://www.cgl.ucsf.edu/home/sparky/) or XEASY.13,14 LPS from E. coli 0111:B4 Sigma-Aldrich (Sydney, NSW, Australia) was titrated into NMR samples by the addition of small volumes from a freshly vortexed stock solution containing 4.4 mg LPS dissolved in 50 μL acetate buffer, pH 4.5. Samples were vortexed prior to NMR acquisition and signal perturbations followed by recording 1D 1H spectra with WATERGATE for solvent suppression.12
Structure Calculations
Structure calculations were performed using restrained simulated annealing in torsion angle space using the program CYANA 2.1.15 NOEs in the 2D NOESY data (150 ms mixing time) were assigned using the automated assignment protocol NOEASSIGN. Nonstandard residues were built and minimized using MAESTRO (http://www.schröinger.com) and new library entries written for the dansyl, Dab, and octylglycine residues. The sign of the y coordinate was swapped for residues with inverted (D) chirality at the CA position. 200 structures were calculated in each of the seven cycles and the top 20 selected based on lowest overall target function. No ’rama’ potential was included in the structure calculations. Structures were visualized using MOLMOL.16
Modeling of the Probe (1)-LPS Complex
The model of probe (1) in complex with LPS was constructed using Accelrys Discovery Studio v 2.1 (Accelrys, San Diego, CA). The coordinates of the NMR solution structure of PmB when bound to E. coli LPS were obtained from Pristovsek and Kidric5 and the dansyl-group was modeled onto the structure. The coordinates of E. coli LPS were derived from the crystal structure of FhuA, the receptor for ferrichrome-iron in E. coli with bound LPS (PDB code:1QFF).4,17 The modeled probe structure was manually docked onto the LPS molecule and the complex was energy minimized in vacuum. The modeling process took into account the electrostatic interactions between the positively charged Dab amine groups on polymyxin scaffold and the negatively charged phosphoester groups on the lipid A, and in addition maximized the reduction of solvent-exposed hydrophobic area on all molecules. Molecular representations were rendered using the POVRAY software package Persistence of Vision Raytracer (Version 3.6; http://www.povray.org).
Confocal Laser Scanning Microscopy (CLSM)
CLSM was performed using a LSM 780, Carl Zeiss Microscope (Jena, Germany). The bacterial cells were washed and reconstituted with 10 mM Tris-HCl (pH 7.5) to make suspension of ∼108cfu/mL. The cells were stained with FM4–64 (1.26 μM) and SYTOX green (2.5 μM) (Invitrogen, Life Technologies, Mulgrave, VIC, Australia) for 5 min as suggested by the manufacturer,18 and probe (1) for 1 h. There was no deleterious effect on growth of the organisms at these concentrations of FM4–64 or SYTOX green (data not shown). The cells were fixed with 3.5% paraformaldehyde and kept at 4 °C until microscopic examination. Fluorophore excitations were induced with 405, 488, and 561 nm wavelength lasers for probe (1), SYTOX green, and FM4–64, respectively; and images were collected at 455–490 nm, 516–551 nm, and 647–690 nm correspondingly. There was no autofluorescence detected, nor cross detection of the probes with these collection parameters. The uptake of probe (1) was studied in a time-lapse experiment using a DeltaVision Elite System (GE Healthcare Company, Washington, US) in a heated chamber at 35 °C. The cells were pretreated with FM4–64 and SYTOX green, and then embedded on a 1% agarose pad containing 10 mM Tris-HCl pH 7.5. The probe (1) was delivered at the edge of gel and the images were taken within ∼5 mm of the edge. Images were collected every 1 min. Three standard filter sets were employed: the 4,6-diamidino-2-phenylindole filter (for detection of probe (1), 360–400 nm excitation and 457–507 nm emission), the A594 filter (for detection of FM4–64, 575–600 nm excitation and 632–692 nm emission), and the fluorescein isothiocyanate filter (for detection of SYTOX green, 461–489 nm excitation and 501–549 nm emission).18 These filters were chosen to avoid cross detection between the probes.
Cell Lysis Assay
To study the activity of probe (1) on the inner membrane a spectrophotometric spheroplast lysis assay was employed as previously described with minor modifications.19 Briefly, spheroplasts were prepared from midlogarithmic phase K. pneumoniae ATCC 13883 cells using the lysozyme-EDTA method,20 and resuspended in 100 mM phosphate buffer (pH 7.5) containing 20% sucrose to an OD500 nm of ∼1. Following lipopeptide treatment, spheroplast lysis was followed by monitoring the decrease in absorbance at 500 nm every 15 s for 1 h. Taurocholic acid treatment was employed as a control.
Electron Microscopy
Approximately 108 cfu/mL of K. pneumoniae ATCC 13883 midlog phase culture were treated with probe (1) at 0.5× and 5× MIC for 1 h. For transmission electron microscopy, the cells were fixed in 1% osmium tetroxide in 100 mM phosphate buffer (pH 7.5) for 1 h. The cells were rinsed three times in 100 mM phosphate buffer (pH 7.5) for 10 min, and then dehydrated in increasing concentrations of ethanol. The cells were infiltrated with increasing concentrations of LR white resin in ethanol consisting of 25, 50, 75, and 100% (w/v) resin for 6 h per step. After a second change of 100% (w/v) resin, cells were embedded in fresh resin in gelatin capsules and allowed to gently sink to the bottom to form a loose pellet. The gelatin capsules were capped to exclude air and the resin polymerized in an oven at 60 °C for 24 h. The embedded cells in blocks were sectioned using a diamond knife on a Leica Ultracut R microtome and ultrathin sections of 90 nm were collected onto pioloform-coated 100 mesh hexagonal copper grids. Grid sections were sequentially stained with saturated uranyl acetate for 10 min and Triple Lead Stain for 5 min.21 Cells were viewed in a Phillips CM120 Biotwin transmission electron microscope at 120 kV. Images were captured with a GatanMultiskan 600 CW digital camera at a resolution of 1024 × 1024 pixels.
Acknowledgments
J.L., T.V., R.L.N., P.E.T., and K.D.R. are supported by a research grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01AI098771). J.L., T.V., R.L.N., and P.E.T. are also supported by the Australian National Health and Medical Research Council (NHMRC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. J.L. is an Australian NHMRC Senior Research Fellow. T.V. is an Australian NHMRC Industry Career Development Level 1 Research Fellow.
Glossary
Abbreviations
- BODIPY
4-bora-3a,4a-diaza-s-indacene
- cfu
colony forming units
- CaMHB
cation-adjusted Mueller-Hinton broth
- DMF
dimethylformamide
- DPPA
diphenylphosphorylazide
- DIPEA
diisopropylethylamine
- DCM
dichloromethane
- Dab
diaminobutyric acid
- HSQC
heteronuclear single quantum coherence
- LPS
lipopolysaccharide
- MeOH
methanol
- MIC
minimum inhibitory concentration
- MDR
multidrug resistant
- NOESY
nuclear Overhauser effect spectroscopy
- TFA
trifluoroacetic acid
- TIPS
triisopropylsilane
- TOCSY
total correlation spectroscopy
- WATERGATE
water suppression by gradient tailored excitation
Supporting Information Available
Figure S1 shows the general scheme for the solid-phase synthesis of the dansyl-polymyxin probe (1). Figure S2 shows the static time-kill kinetics of probe (1) and colistin against K. pneumoniae ATCC 13883. Figure S3 shows the 2D NOESY spectrum of proble (1) in water and DPC micelles. Figure S4 shows laser scanning confocal microscopy imaging of K. pneumoniae ATCC 13883 cells following treatment with probe (1), SYTOX green, and FM4–64. Figure S5 shows spectrophotmetric and CLSM imaging of spheroplastlysis of K. pneumoniae ATCC 13883 following treatment with probe (1). Movie S1 shows the time-lapse confocal imaging of the penetration of probe (1) into K. pneumoniae ATCC 13883 cells. Tables S1 and S2 document 1H and 13C observed chemical shifts for probe (1) in buffer and when bound to DPC micelles. Table S3 documents NMR statistics. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Boucher H. W.; Talbot G. H.; Benjamin D. K. Jr.; Bradley J.; Guidos R. J.; Jones R. N.; Murray B. E.; Bonomo R. A.; Gilbert D. (2013) 10 x ’20 Progress--development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin. Infect. Dis. 56, 1685–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot G. H.; Bradley J.; Edwards J. E. Jr.; Gilbert D.; Scheld M.; Bartlett J. G. (2006) Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 42, 657–68. [DOI] [PubMed] [Google Scholar]
- Velkov T.; Roberts K. D.; Nation R. L.; Thompson P. E.; Li J. (2013) Pharmacology of polymyxins: new insights into an ’old’ class of antibiotics. Future Microbiol. 8, 711–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkov T.; Thompson P. E.; Nation R. L.; Li J. (2010) Structure-activity relationships of polymyxin antibiotics. J. Med. Chem. 53, 1898–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pristovsek P.; Kidric J. (2004) The search for molecular determinants of LPS inhibition by proteins and peptides. Curr. Top. Med. Chem. 4, 1185–201. [DOI] [PubMed] [Google Scholar]
- Ofek I.; Cohen S.; Rahmani R.; Kabha K.; Tamarkin D.; Herzig Y.; Rubinstein E. (1994) Antibacterial synergism of polymyxin B nonapeptide and hydrophobic antibiotics in experimental gram-negative infections in mice. Antimicrob. Agents Chemother. 38, 374–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soon R. L.; Velkov T.; Chiu F.; Thompson P. E.; Kancharla R.; Roberts K.; Larson I.; Nation R. L.; Li J. (2011) Design, synthesis, and evaluation of a new fluorescent probe for measuring polymyxin-lipopolysaccharide binding interactions. Anal. Biochem. 409, 273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deris Z. Z.; Yu H. H.; Davis K.; Soon R. L.; Jacob J.; Ku C. K.; Poudyal A.; Bergen P. J.; Tsuji B. T.; Bulitta J. B.; Forrest A.; Paterson D. L.; Velkov T.; Li J.; Nation R. L. (2012) The combination of colistin and doripenem is synergistic against Klebsiella pneumoniae at multiple inocula and suppresses colistin resistance in an in vitro pharmacokinetic/pharmacodynamic model. J. Antimicro. Agents. Chemother. 56, 5103–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M.; Iida K.; Matsuo T. (1969) Electron microscopic studies on mode of action of polymyxin. J. Bacteriol. 97, 448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Sass H. J.; Zahringer U.; Grzesiek S. (2008) Structure and dynamics of 13C,15N-labeled lipopolysaccharides in a membrane mimetic. Angew. Chem., Int. Ed. 47, 9870–4. [DOI] [PubMed] [Google Scholar]
- Mares J.; Kumaran S.; Gobbo M.; Zerbe O. (2009) Interactions of lipopolysaccharide and polymyxin studied by NMR spectroscopy. J. Biol. Chem. 284, 11498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapor M. J.; Erwin D.; Erowele G.; Wortmann G. (2008) Emergence of multidrug resistance in bacteria and impact on antibiotic expenditure at a major army medical center caring for soldiers wounded in Iraq and Afghanistan. Infect. Control Hosp. Epidemiol. 29, 661–3. [DOI] [PubMed] [Google Scholar]
- van Veen H. W.; Margolles A.; Putman M.; Sakamoto K.; Konings W. N. (1999) Multidrug resistance in lactic acid bacteria: molecular mechanisms and clinical relevance. Antonie Van Leeuwenhoek 76, 347–52. [PubMed] [Google Scholar]
- Poole K. (2001) Multidrug resistance in Gram-negative bacteria. Curr. Opin. Microbiol. 4, 500–8. [DOI] [PubMed] [Google Scholar]
- Nikaido H. (2009) Multidrug resistance in bacteria. Annu. Rev. Biochem. 78, 119–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunkhorn S. (2013) Antibacterial drugs: New antibiotics on the horizon?. Nat. Rev. Drug Discovery 12, 99. [DOI] [PubMed] [Google Scholar]
- Arrowsmith J. (2012) A decade of change. Nat. Rev. Drug Discovery 11, 17–8. [DOI] [PubMed] [Google Scholar]
- Gould I. M. (2009) Antibiotic resistance: the perfect storm. Int. J. Antimicrob. Agents 34(Suppl 3), S2–5. [DOI] [PubMed] [Google Scholar]
- Falagas M. E.; Rafailidis P. I.; Matthaiou D. K.; Virtzili S.; Nikita D.; Michalopoulos A. (2008) Pandrug-resistant Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii infections: characteristics and outcome in a series of 28 patients. Int. J. Antimicrob. Agents 32, 450–4. [DOI] [PubMed] [Google Scholar]
- Antoniadou A.; Kontopidou F.; Poulakou G.; Koratzanis E.; Galani I.; Papadomichelakis E.; Kopterides P.; Souli M.; Armaganidis A.; Giamarellou H. (2007) Colistin-resistant isolates of Klebsiella pneumoniae emerging in intensive care unit patients: first report of a multiclonal cluster. J. Antimicrob. Chemother. 59, 786–90. [DOI] [PubMed] [Google Scholar]
- Falagas M. E.; Bliziotis I. A. (2007) Pandrug-resistant Gram-negative bacteria: the dawn of the post-antibiotic era?. Int. J. Antimicrob. Agents 29, 630–636. [DOI] [PubMed] [Google Scholar]
- Valencia R.; Arroyo L. A.; Conde M.; Aldana J. M.; Torres M. J.; Fernandez-Cuenca F.; Garnacho-Montero J.; Cisneros J. M.; Ortiz C.; Pachon J.; Aznar J. (2009) Nosocomial outbreak of infection with pan-drug-resistant Acinetobacter baumannii in a tertiary care university hospital. Infect. Control Hosp. Epidemiol. 30, 257–63. [DOI] [PubMed] [Google Scholar]
- Bergen P. J.; Bulitta J. B.; Forrest A.; Tsuji B. T.; Li J.; Nation R. L. (2010) Pharmacokinetic/pharmacodynamic investigation of colistin against Pseudomonas aeruginosa using an in vitro model. Antimicrob. Agents Chemother. 54, 3783–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Rayner C. R.; Nation R. L.; Owen R. J.; Tan K. E.; Spelman D. (2006) Hetero-resistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 50, 2946–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen R. J.; Li J.; Nation R. L.; Spelman D. (2007) In vitro pharmacodynamics of colistin against Acinetobacter baumannii clinical isolates. J. Antimicrob. Chemother. 59, 473–7. [DOI] [PubMed] [Google Scholar]
- Tam V. H.; Schilling A. N.; Vo G.; Kabbara S.; Kwa A. L.; Wiederhold N. P.; Lewis R. E. (2005) Pharmacodynamics of polymyxin B against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49, 3624–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko K. S.; Suh J. Y.; Kwon K. T.; Jung S. I.; Park K. H.; Kang C. I.; Chung D. R.; Peck K. R.; Song J. H. (2007) High rates of resistance to colistin and polymyxin B in subgroups of Acinetobacter baumannii isolates from Korea. J. Antimicrob. Chemother. 60, 1163–7. [DOI] [PubMed] [Google Scholar]
- Li J.; Nation R. L.; Turnidge J. D.; Milne R. W.; Coulthard K.; Rayner C. R.; Paterson D. L. (2006) Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 6, 589–601. [DOI] [PubMed] [Google Scholar]
- Poirel L.; Labarca J.; Bello H.; Rioseco M. L.; Bernabeu S.; Nordmann P. (2014) Emergence of the 16S rRNA methylase RmtG in an extended-spectrum-beta-lactamase-producing and colistin-resistant Klebsiella pneumoniae isolate in Chile. Antimicrob. Agents Chemother. 58, 618–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahador A.; Taheri M.; Pourakbari B.; Hashemizadeh Z.; Rostami H.; Mansoori N.; Raoofian R. (2013) Emergence of rifampicin, tigecycline, and colistin-resistant Acinetobacter baumannii in Iran; spreading of MDR strains of novel International Clone variants. Microb. Drug Resist. 19, 397–406. [DOI] [PubMed] [Google Scholar]
- Kontopidou F.; Plachouras D.; Papadomichelakis E.; Koukos G.; Galani I.; Poulakou G.; Dimopoulos G.; Antoniadou A.; Armaganidis A.; Giamarellou H. (2011) Colonization and infection by colistin-resistant Gram-negative bacteria in a cohort of critically ill patients. Clin. Microbiol. Infect. 17, E9–E11. [DOI] [PubMed] [Google Scholar]
- Moore R. A.; Bates N. C.; Hancock R. E. (1986) Interaction of polycationic antibiotics with Pseudomonas aeruginosa lipopolysaccharide and lipid A studied by using dansyl-polymyxin. Antimicrob. Agents Chemother. 29, 496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubery H.; Ofek I.; Cohen S.; Eisenstein M.; Fridkin M. (2002) Modulation of the hydrophobic domain of polymyxin B nonapeptide: effect on outer-membrane permeabilization and lipopolysaccharide neutralization. Mol. Pharmacol. 62, 1036–42. [DOI] [PubMed] [Google Scholar]
- Tsubery H.; Ofek I.; Cohen S.; Fridkin M. (2000) Structure-function studies of polymyxin B nonapeptide: implications to sensitization of gram-negative bacteria. J. Med. Chem. 43, 3085–92. [DOI] [PubMed] [Google Scholar]
- Tsubery H.; Ofek I.; Cohen S.; Fridkin M. (2001) N-terminal modifications of Polymyxin B nonapeptide and their effect on antibacterial activity. Peptides 22, 1675–81. [DOI] [PubMed] [Google Scholar]
- Benincasa M.; Pacor S.; Gennaro R.; Scocchi M. (2009) Rapid and reliable detection of antimicrobial peptide penetration into gram-negative bacteria based on fluorescence quenching. Antimicrob. Agents Chemother. 53, 3501–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn J. S. (2008) The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 16, 284–90. [DOI] [PubMed] [Google Scholar]
- Gunn J. S.; Lim K. B.; Krueger J.; Kim K.; Guo L.; Hackett M.; Miller S. I. (1998) PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol. Microbiol. 27, 1171–82. [DOI] [PubMed] [Google Scholar]
- Gunn J. S.; Ryan S. S.; Van Velkinburgh J. C.; Ernst R. K.; Miller S. I. (2000) Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect. Immun. 68, 6139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurek K. N.; Sampaio J. L.; Kiffer C. R.; Sinto S.; Mendes C. M.; Hancock R. E. (2009) Involvement of pmrAB and phoPQ in polymyxin B adaptation and inducible resistance in non-cystic fibrosis clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 4345–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Cromie M. J.; Hsu F. F.; Turk J.; Groisman E. A. (2004) PhoP-regulated Salmonella resistance to the antimicrobial peptides magainin 2 and polymyxin B. Mol. Microbiol. 53, 229–41. [DOI] [PubMed] [Google Scholar]
- Trent M. S.; Ribeiro A. A.; Lin S.; Cotter R. J.; Raetz C. R. (2001) An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J. Biol. Chem. 276, 43122–31. [DOI] [PubMed] [Google Scholar]
- Soon R. L.; Nation R. L.; Cockram S.; Moffatt J. H.; Harper M.; Adler B.; Boyce J. D.; Larson I.; Li J. (2011) Different surface charge of colistin-susceptible and -resistant Acinetobacter baumannii cells measured with zeta potential as a function of growth phase and colistin treatment. J. Antimicrob. Chemother. 66, 126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R. E. (1997) Antibacterial peptides and the outer membranes of gram-negative bacilli. J. Med. Microbiol. 46, 1–3. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. (1997) Peptide antibiotics. Lancet 349, 418–22. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. (1997) The bacterial outer membrane as a drug barrier. Trends Microbiol. 5, 37–42. [DOI] [PubMed] [Google Scholar]
- Hancock R. E.; Lehrer R. (1998) Cationic peptides: a new source of antibiotics. Trends Biotechnol. 16, 82–8. [DOI] [PubMed] [Google Scholar]
- Li J.; Milne R. W.; Nation R. L.; Turnidge J. D.; Coulthard K. (2003) Stability of colistin and colistin methanesulfonate in aqueous media and plasma as determined by high-performance liquid chromatography. Antimicrob. Agents Chemother. 47, 1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne P. A. (2013) Clinical and Laboratory Standards Institute. 23rd Informational Supplement M100-S23.
- Poudyal A.; Howden B. P.; Bell J. M.; Gao W.; Owen R. J.; Turnidge J. D.; Nation R. L.; Li J. (2008) In vitro pharmacodynamics of colistin against multidrug-resistant Klebsiella pneumoniae. J. Antimicrob. Chemother. 62, 1311–1318. [DOI] [PubMed] [Google Scholar]
- Nyberg N. T.; Duus J. Ã.; Sãrensen O. W. (2005) Heteronuclear two-bond correlation: suppressing heteronuclear three-bond or higher NMR correlations while enhancing two-bond correlations even for vanishing 2J(CH). J. Am. Chem. Soc. 127, 6154–6155. [DOI] [PubMed] [Google Scholar]
- Piotto M.; Saudek V.; Sklenar V. (1992) Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661–5. [DOI] [PubMed] [Google Scholar]
- Bartels C.; Xia T. H.; Billeter M.; Guntert P.; Wuthrich K. (1995) The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 6, 1–10. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–93. [DOI] [PubMed] [Google Scholar]
- Guntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–78. [DOI] [PubMed] [Google Scholar]
- Koradi R.; Billeter M.; Wuthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14(51–5), 29–32. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Welte W.; Hofmann E.; Lindner B.; Holst O.; Coulton J. W.; Diederichs K. (2000) A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure 8, 585–92. [DOI] [PubMed] [Google Scholar]
- Pogliano J.; Pogliano N.; Silverman J. A. (2012) Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 194, 4494–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaara M.; Vaara T. (1981) Outer membrane permeability barrier disruption by polymyxin in polymyxin-susceptible and -resistant Salmonella typhimurium. J. Antimicrob. Chemother. 19, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deris Z. Z.; Akter J.; Sivanesan S.; Roberts K. D.; Thompson P. E.; Nation R. L.; Li J.; Velkov T. (2014) A secondary mode of action of polymyxins against Gram-negative bacteria involves the inhibition of NADH-quinone oxidoreductase activity. J. Antibiot. 67, 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATO T. (1968) A modified method for lead staining of thin sections. J. Electron Microsc. 17, 158–159. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.