

Abstract
Very few hydride complexes are known in which the metals have a high-spin electronic configuration. We describe the characterization of several high-spin iron(II) hydride/deuteride isotopologues and their exchange reactions with one another and with H2/D2. Though the hydride/deuteride signal is not observable in NMR spectra, the choice of isotope has an influence on the chemical shifts of distant protons in the dimers through the paramagnetic isotope effect on chemical shift. This provides the first way to monitor the exchange of H and D in the bridging positions of these hydride complexes. The rate of exchange depends on the size of the supporting ligand, and this is consistent with the idea that H2/D2 exchange into the hydrides occurs through the dimeric complexes rather than through a transient monomer. The understanding of H/D exchange mechanisms in these high-spin iron hydride complexes may be relevant to postulated nitrogenase mechanisms.
Short abstract
The paramagnetic isotope effect on chemical shift (PIECS) in dimeric high-spin iron hydride complexes enabled detailed characterization of their structure, flexibility, and H/D exchange reactions.
Introduction
Though thousands of transition-metal hydride complexes are known, relatively few of them have unpaired electrons.1 Even fewer have metals with a high-spin electronic configuration, since hydride is a strong-field ligand and since hydride complexes are often supported by strong-field ancillary ligands. In a series of publications, we described exceptional di(μ-hydrido)diiron(II) complexes supported by bulky β-diketiminate ligands (Chart 1).2−6 In these complexes, the iron(II) ions had high-spin electronic configurations because of the low metal coordination number and the π-donor character of the anionic β-diketiminate.7−9 All of the iron(II) examples were dimers in the solid state, and the two pseudotetrahedral metal centers were bridged by hydride ligands (Chart 1, upper left). With the bulkiest β-diketiminate (LtBu), the dimer dissociated in solution to give a three-coordinate monomer, as shown by a combination of magnetic, spectroscopic, and kinetics studies.2,10
Chart 1. Diagram of the Fe2(μ-H)2 Core And Three β-Diketiminate Ligands That Form Crystallographically Characterized Complexes with This Core.

The high-spin electronic configuration of the complexes presents characterization challenges that are distinctive to paramagnetic species. Namely, the resonances in the 1H NMR spectra are broadened and highly shifted, and these chemical shifts do not correlate with structure in the manner that is familiar from diamagnetic complexes.11 The relaxation of 1H nuclei directly bonded to the paramagnetic metal is particularly rapid, and to our knowledge no metal-bound 1H nuclei have been detected in NMR spectra of hydride complexes with a paramagnetic ground state.12,13 Another challenge is that the paramagnetic hydride complexes in Chart 1 are highly reactive: for example, they cleave B–C bonds14 and reductively eliminate H2 with light or with added ligands3 including N2.8,15 Though the high reactivity of the hydrides makes them difficult to handle, their reactivity is also an opportunity to form new C–H bonds, because the M–H bonds undergo rapid [1,2]-addition to practically all multiple bonds in organic molecules.3 This reactivity can be attributed to Fe–H bond weakening as a result of the partial population of Fe–H σ* orbitals in the high-spin d6 electronic configuration.
One of the important reactions of coordinatively unsaturated metal-hydride complexes is the exchange of the hydride hydrogens with free H2.16 This reaction has biological relevance because of the H/D exchange of H2 protons with solvent protons in nitrogenase enzymes, which occurs only in the presence of N2.17 This specificity has been used to suggest that hydride species are key intermediates during N2 reduction.18 Very recently, deuterium atoms from D2 were incorporated into ethylene produced from acetylene reduction by nitrogenase.19 However, the mechanisms are difficult to evaluate without “model” studies on well-characterized synthetic hydride complexes, particularly with iron.20 Unfortunately, the aforementioned inability to observe resonances for H and D bound to a metal has so far prevented the monitoring of H/D exchange in paramagnetic hydride complexes.
In this article, we describe synthetic and characterization advances for two high-spin iron(II)-hydride complexes, supported by the β-diketiminate ligands LtBu and LMe (Chart 1). In an interesting twist, these studies benefit from unusually large (up to 5.7 ppm) isotope effects on the 1H NMR chemical shifts of the distant protons upon hydride deuteration, a phenomenon that occurs only in the dimeric hydride complexes in our system. This tool for differentiating isotopologues enables the revision of the 1H NMR assignments in two previously reported hydride complexes. In addition, this is the only way to distinguish hydride from deuteride isotopologues, and this discovery enables us to monitor H/D exchange in this system for the first time. The results show that the rates of H/D exchange between hydride complexes, and between these hydride complexes and H2, are greatly influenced by the size of the supporting ligand. Mechanistic considerations lead to new insights into the distinctive reactivity of high-spin hydride complexes.
Results
Spectroscopic Comparison of Protiated and Deuterated LtBuFeH in Monomeric and Dimeric Forms
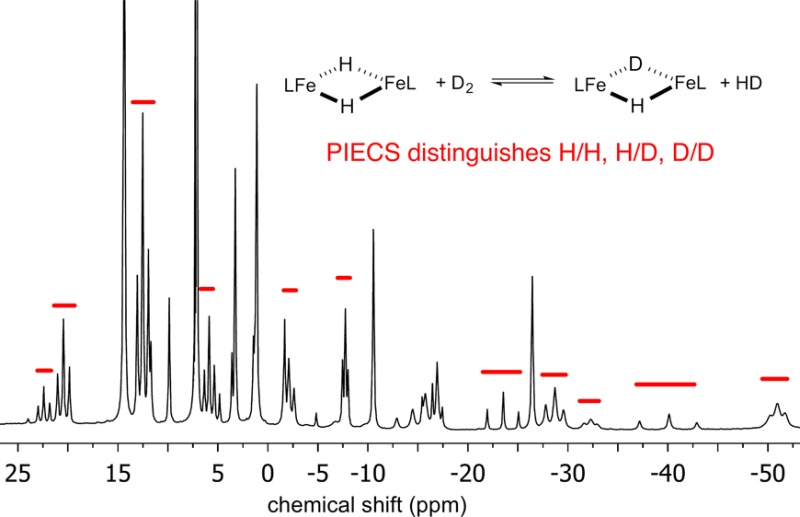
The synthesis of [LtBuFe(μ-D)]2 (1-D2) was reported previously, and initial characterization of 1-D2 by 1H NMR spectroscopy in C6D6 suggested that the deuteride and the hydride complexes had identical 1H NMR spectra.5 However, closer investigation has shown that the resonances have different chemical shifts in the different isotopologues. A 1H NMR spectrum of an equimolar mixture of 1 and 1-D2 in C6D6 showed that the differences were not from temperature or medium effects (Figure 1). Close examination of the 1H NMR spectrum revealed several envelopes of nearby resonances, and the components of each envelope had a 1:2:1 ratio of integrations. For example, resonances at δ −37.2 (resonance assigned to 1), −40.1, and −43.0 (resonance assigned to 1-D2) ppm were observed in a 1:2:1 ratio, rather than the single resonance at δ −37.2 ppm in 1. When this experiment was repeated, starting from a different mixture of isotopologues that contained more 1 than 1-D2, the 1H NMR spectrum showed the same number of resonances, but the integrations were no longer 1:2:1 and favored the hydride resonance at δ −37.2 ppm. The resonances located between 1 and 1-D2 are most reasonably assigned to {LtBuFe}2(μ-H)(μ-D) (1-D). These experiments also indicate that mixing of 1 and 1-D2 rapidly yields an equilibrium mixture of 1, 1-D, and 1-D2. The difference between the chemical shifts of distant protons in different isotopologues has been described previously in a number of paramagnetic complexes,21 and Theopold has termed this paramagnetic isotope effect on chemical shift (PIECS).22
Figure 1.

(a) 1H NMR spectrum of [LtBuFe(μ-H)]2 (1) in C6D6. This particular sample has a 7% impurity of the oxo complex {LtBuFe}2(μ-O). Peaks are marked as follows: dimer D, monomers M, oxo impurity I, and solvent and solvent impurities S. (b) 1H NMR spectrum from mixing equimolar amounts of [LtBuFe(μ-H)]2 (1) and [LtBuFe(μ-D)]2 (1-D2) in C6D6 for 45 min. All three isotopologues of the dimer (H/H, H/D, and D/D) are visible in (b), as several groups of three nearby peaks in a statistical 1:2:1 ratio. Only the parts of the spectra from δ 45 to −70 ppm are shown for clarity.
The discovery that 1, 1-D, and 1-D2 exhibited PIECS enabled the use of 1H NMR spectroscopy to accurately determine the amount of deuterium incorporation into 1-D2. This was done by comparing the integrations of the three isotopologues in the 1H NMR spectrum. Compound 1-D2 typically had greater than 90% deuterium incorporation into the hydride positions, which is consistent with the level of deuteration previously reported. (In earlier studies this determination was done indirectly, using mass spectrometric analysis of 3-hexene-d1 generated from treating 1-D2 with 3-hexyne and then acid.)5
Note that PIECS is not observed for some of the peaks in the spectrum. Seven of these peaks are assigned to monomeric LtBuFeH, which is in equilibrium with 1, as previously shown.2 This is the number of resonances expected for LtBu in an environment having C2v symmetry, when the N-aryl bonds have hindered rotation that makes the two methyl groups of the isopropyl substituents inequivalent. By process of elimination, the PIECS of the peaks of 1 enabled the assignment of 18 resonances each to the 1, 1-D, and 1-D2 isotopologues of the dimers, giving 54 resonances in total. Therefore, the 1H NMR spectrum of isotopically pure 1 contains 25 resonances, where 7 peaks may be assigned to LtBuFeH and 18 peaks may be assigned to the dimer.
It is notable that 1H NMR spectra of a mixture of the three isotopologues always showed some additional resonances that neither exhibit PIECS nor can be assigned to the monomer LtBuFeH. These resonances had previously been assigned as resonances of 1.2 Comparison with literature 1H NMR spectra indicated that these additional resonances were associated with a persistent impurity, {LtBuFe}2(μ-O), which can come from the reaction of 1 with trace H2O to give {LtBuFe}2(μ-O).23 We were not able to completely avoid or eliminate the oxo impurity, but careful handling gave samples of 1 that were below 10% oxo impurity, with typical samples between 4 and 8%. The oxo impurity in 1-D2 was typically higher (around 20%), due to the multiple D2 additions necessary for full deuterium incorporation.
Solid 1 and 1-D2 were also evaluated using Mössbauer spectroscopy. The zero-field Mössbauer spectrum of solid 1 at 80 K was previously reported to have δ = 0.59 mm/s and |ΔEQ| = 1.58 mm/s.5 However, the published data were reexamined after the discovery of the persistent oxo impurity. The oxo impurity was modeled using parameters that were fixed at literature values of δ = 0.64 mm/s and |ΔEQ| = 1.42 mm/s,23 while the amount of the oxo impurity and the parameters of the major component were refined to give the best fit to the data. Complex 1 was determined to be 71% of the earlier sample, and the major component had δ = 0.57 mm/s and |ΔEQ| = 1.63 mm/s. A new sample of 1 with <5% oxo impurity by NMR spectroscopy yielded a zero-field Mössbauer spectrum at 80 K with parameters δ = 0.58 mm/s and |ΔEQ| = 1.62 mm/s, which are the same within the experimental uncertainty of ±0.02 mm/s.
The zero-field Mössbauer spectrum of solid 1-D2 at 80 K is shown in Figure 2. The slightly asymmetric two-line pattern was modeled with a two-component fit, of which the major component was found to have δ = 0.58 mm/s and |ΔEQ| = 1.74 mm/s, accounting for 79% of the sample. We assign this subspectrum to compound 1-D2, whereas the second doublet was fixed to the properties of the oxo impurity (δ = 0.64 mm/s and |ΔEQ| = 1.42 mm/s). The isomer shifts of 1 and 1-D2 are identical, but there was a variation of 0.12 mm/s in the quadrupole splitting values. The reason for the difference in the quadrupole splitting is unknown at this time. Unfortunately, attempts to fit the variable-field Mössbauer spectra of 1 and of 1-D2 did not yield a comprehensible model that gave additional insight. The isomer shifts of 1 and 1-D2 lie in the range of δ 0.47–0.90 mm/s observed in other high-spin Fe(II) diketiminate complexes.23,24
Figure 2.
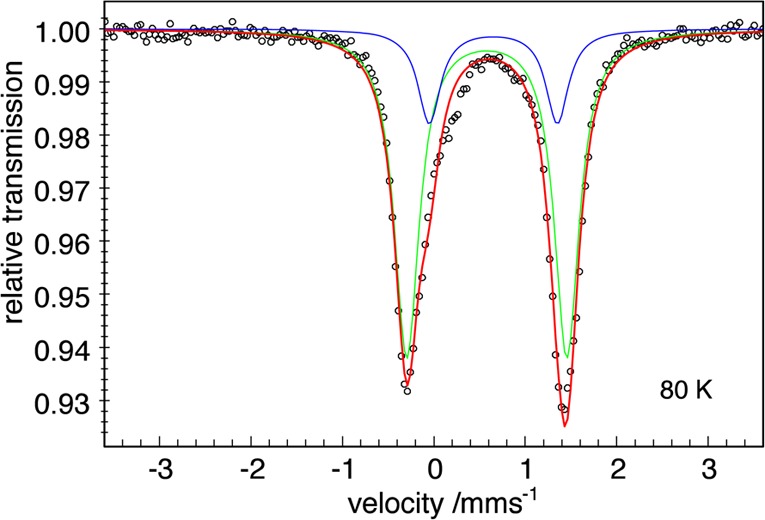
Zero-field Mössbauer spectrum of [LtBuFe(μ-D)]2 (1-D2) recorded at 80 K. The signal with δ = 0.58 mm/s and |ΔEQ| = 1.74 mm/s accounted for 79% of the sample. The blue line represents the contribution of the oxo impurity, the green line represents the contribution of 1-D2, the red line represents the sum, and the black circles are the data.
Synthesis and Characterization of LMeFeBr(THF) (2)
We now shift to iron(II) complexes of the smaller diketiminate ligand LMe, which we have previously derived from the iron chloride precursors LMeFe(μ-Cl)2Li(THF) (THF = tetrahydrofuran) and [LMeFe(μ-Cl)]2.25 However, the relative ease of removing Br salts encouraged us to prepare LMeFeBr(THF) (2) in 82% yield from LMeK and FeBr2. During the preparation of our work, compound 2 was reported by Tonzetich and Lippard,26 using a very similar prepatory method with LMeNa. Our characterization of 2 by NMR and X-ray diffraction (Supporting Information) is indistinguishable from the literature.26 However, Mössbauer data have not been reported previously for this compound. The zero-field Mössbauer spectrum of 2 at 80 K (Figure S-1, Supporting Information) has a doublet with δ = 0.89 mm/s and |ΔEQ| = 2.36 mm/s, which is nearly identical to that of LMeFe(μ-Cl)2Li(THF).24e
THF was removed from 2 by dissolving it in noncoordinating solvents; this gave [LMeFe(μ-Br)]2, which precipitated from solution as an orange powder. This mirrors the behavior of [LMeFe(μ-Cl)]2, which also has low solubility.25b,25c Tonzetich and Lippard also reported this behavior, and they reported that LMeFeBr(THF) had different electronic absorption spectra in THF versus toluene, which resulted from the formation of [LMeFe(μ-Br)]2.26 Here, the Mössbauer spectrum of orange [LMeFe(μ-Br)]2 derived from 2 was recorded at 80 K (Figure S-2, Supporting Information). One quadrupole doublet with δ = 0.91 mm/s and |ΔEQ| = 2.64 mm/s was observed. The isomer shift of [LMeFe(μ-Br)]2 is the same as 2, but the quadrupole splitting is larger, consistent with a slightly different geometry at iron.
Synthesis of [LMeFe(μ-H)]2 (3) Using H2
The successful synthesis of [LtBuFe(μ-H)]2 from addition of H2 to an iron(I) source5 prompted us to use this synthetic method for an improved synthesis of [LMeFe(μ-H)]2 and [LMeFe(μ-D)]2. However, the order of addition of reagents was important, as reduction of 2 with KC8 in Et2O followed by H2 addition yielded many unidentified resonances in the crude 1H NMR spectrum. This suggests that the transient iron(I) species formed by KC8 reduction rapidly decomposes in the absence of a trap.27 Therefore, a degassed solution of 2 in THF was exposed to 14 equiv of H2 gas prior to addition of KC8. After 3 h, volatile materials were removed from the brown reaction mixture, and 3 was isolated in 56% yield following workup. The identity of 3 was established by comparing its 1H NMR spectrum to the spectrum reported in the literature.4 Again, the iron-bound hydrogen atoms are not visible by 1H NMR spectroscopy due to close proximity to the paramagnetic iron atoms.
The deuterated isotopologue 3-D2 was synthesized using the above method with D2 gas in 58% yield. The 1H NMR spectrum of 3-D2 (Figure 3) revealed that 3 and 3-D2 exhibited PIECS as with the hydride dimers described above. Thus, 1H NMR spectroscopy could similarly be used to determine the amount of deuterium incorporated into the hydride ligands. (Figure 6 below shows mixtures of isotopologues.) The bridging ligands in samples of 3-D2 were typically greater than 90% deuterated, as judged by 1H NMR spectroscopy.
Figure 3.

1H NMR spectra of (bottom) [LMeFe(μ-H)]2 (3) and (top) [LMeFe(μ-D)]2 (3-D2) in C6D6.
Figure 6.

1H NMR spectra of [LMeFe(μ-H)]2 (3), {LMeFe}2(μ-H)(μ-D) (3-D), and [LMeFe(μ-D)]2 (3-D2) isotopologues in C6D6 during gas exchange. The columns on the right indicate the order and type of gas that was added to give the observed spectrum.
In the 1H NMR spectrum of 3 at room temperature, only four paramagnetically shifted peaks were observed, a number that is well short of the seven resonances expected for 3 in a dimeric structure with D2d or D2h symmetry. Therefore, we hypothesized that additional resonances were hidden at room temperature. In addition, the integrations of the peaks are inconsistent with the original assignments for the 1H NMR spectrum of 3,4 so further investigations were pursued. 1H NMR spectra of 3 between 26 and 85 °C in C6D6 are shown in Figure 4a. The peak at δ 13 ppm at 26 °C corresponds to the backbone methyl and meta-aryl protons, two resonances that are only distinct above 60 °C (a close-up is shown in Figure S-3, Supporting Information). A previously undetected resonance for isopropyl methyl groups integrating to 24 protons (which had been hidden under the residual benzene at room temperature) became visible above 40 °C. The other resonance for isopropyl methyl protons, a broad peak at δ −25 ppm in the 26 °C spectrum, sharpened at elevated temperatures. Finally, a new broad resonance for the isopropyl methine protons at δ −1.5 ppm was observed above 70 °C. This resonance is broadened into the baseline at room temperature, explaining why it had not been identified in previous studies. The remaining resonances at δ −24 and −56 ppm corresponded to the para-aryl protons and the backbone protons, respectively, completing the catalogue of resonances with the expected integrations (see Experimental Section).
Figure 4.

(a) Variable-temperature 1H NMR spectra of [LMeFe(μ-H)]2 (3) between 26 and 85 °C in C6D6. (b) Variable-temperature 1H NMR spectra of 3 between −90 and 20 °C in toluene-d8. The asterisks indicate the resonance that splits with a coalescence temperature of 0 °C.
The appearance and sharpening of resonances in the high-temperature 1H NMR spectra of 3 suggested the possibility of a fluxional process in solution. Therefore, low-temperature 1H NMR spectra were measured between −90 and 20 °C in toluene-d8 (Figure 4b). The broad isopropyl methyl resonance that appeared at δ −25 ppm in the 1H NMR spectrum of 3 at 20 °C split into two broad resonances at δ −27.6 and −48.3 ppm in the 1H NMR spectrum at −75 °C. These resonances moved together and became broader as the sample was warmed, with a coalescence temperature of 0 °C. The barrier for this fluxional process, assuming that the resonances at −75 °C are in the slow-exchange limit, is ΔG‡ = 10.5 kcal/mol.28 Decoalescence of other peaks was not observed within this temperature range, likely because there was a smaller difference between the frequencies in the slow-exchange limit. The possible nature of this fluxional process is discussed below.
Mössbauer Spectroscopy of [LMeFe(μ-H)]2 and [LMeFe(μ-D)]2
The purity and electronic structure of solid 3 and 3-D2 were evaluated using Mössbauer spectroscopy. The zero-field Mössbauer spectra of solid 3 and 3-D2 at 80 K are shown in Figure 5. Compound 3 had δ = 0.51 mm/s and |ΔEQ| = 2.05 mm/s. The deuterated isotopologue 3-D2 had an indistinguishable Mössbauer spectrum, with δ = 0.51 mm/s and |ΔEQ| = 2.10 mm/s. There were impurities of 13 and 6%, respectively, which are discussed in detail in the Supporting Information. One impurity doublet in each case has parameters similar to those in the literature iron(I) benzene compound LMeFe(η6-C6H6),27 which has δ = 0.70 mm/s and |ΔEQ| = 0.74 mm/s (Figure S-4, Supporting Information). We also note that this nearly NMR-silent impurity was the major species in the Mössbauer spectrum previously attributed to 3.3
Figure 5.
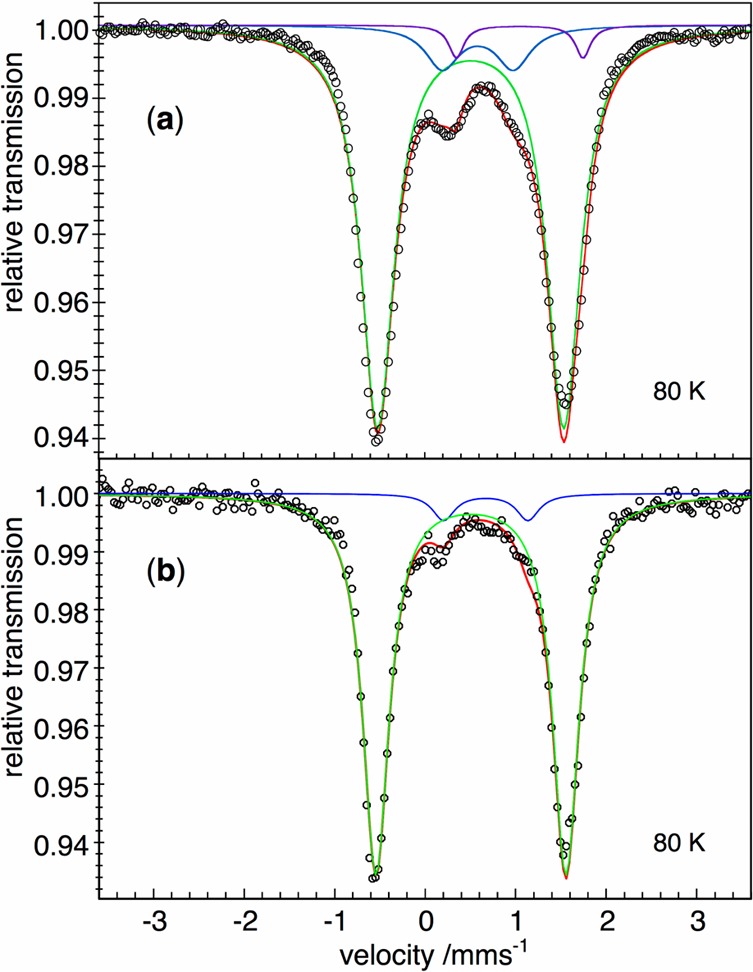
(a) Mössbauer spectrum of [LMeFe(μ-H)]2 (3). (b) Mössbauer spectrum of [LMeFe(μ-D)]2 (3-D2). Both spectra were recorded at 80 K, with zero field. The black circles are the data, and the red lines represent the sums of a major doublet for 3 (green) and impurities (blue, purple) that are discussed in the Supporting Information.
Intermolecular Hydride Exchange in Isotopologues, and Exchange of Hydrides with H2
As reported above, a 1:2:1 mixture of 1, 1-D, and 1-D2 was obtained upon mixing an equimolar mixture of 1 and 1-D2 in C6D6, and PIECS enabled all three isotopologues to be distinguished in the paramagnetic 1H NMR spectrum. Equilibrium was established within 45 min at room temperature. An analogous experiment was performed using an equimolar solution of 3 and 3-D2 maintained at 30 °C in C6D6. A 1:2:1 mixture of 3, {LMeFe}2(μ-H)(μ-D) (3-D), and 3-D2 was observed after 2 h. This shows that the inability of 3 to form monomeric LMeFeH in solution14 does not hinder hydride exchange between isotopologues.
The exchange of hydride and deuteride ligands with D2 and H2 was also examined. Compound 3 was treated with an excess of D2 (1 atm) in C6D6 to give a mixture of 3-D and 3-D2. Equilibrium was established immediately upon mixing (Figure 6A). When this mixture was treated with fresh D2, the equilibrium was pushed all the way to the fully deuterated isotopologue, 3-D2 (Figure 6B). Two treatments with H2 caused the sample to revert to 3 in quantitative yield (Figure 6C,D), showing that the exchange is reversible. In contrast to the immediate exchange in 3, treatment of 1 in C6D6 with D2 (1 atm) produced the deuterated isotopologue only after much longer amounts of time. Compound 1-D2 was finally observed in quantitative yield after 50 h at room temperature. Hydride exchange between H2 and 1-D2 is reversible, as treatment of 1-D2 with H2 (1 atm) produced 1 in quantitative yield under the same conditions and time. Treatment of 1 with 8 atm of D2 was faster but required 24 h to yield 1-D2 in quantitative yield. A qualitative summary of hydride ligand exchange rates is given in Table 1.
Table 1. Times for Exchange of Hydride Ligands between Isotopologues, And for Exchange of Hydride with 1 atm of H2/D2 Gas, Giving Qualitative Times to Reach Equilibriuma.
| compound | isotopologue exchange | gas exchange |
|---|---|---|
| [LtBuFe(μ-H)]2 (1) | <45 min | 2 d |
| [LMeFe(μ-H)]2 (3) | 2 h | <1 min |
| [LtBuCo(μ-H)]2 | none |
Each solution was shaken for the duration of the experiment.
Lack of H/D Exchange in Analogous Cobalt Hydrides
We have also reported the dimeric cobalt(II) hydride complex [LtBuCo(μ-H)]2.8 This compound is much less reactive than the iron analogues described above; for example, it does not react with alkenes or Lewis bases.8b The low reactivity was attributed to the greater stability of the dimer and/or to the decreased lability of the Co–H bonds. Therefore, it was interesting to evaluate the analogous cobalt compounds for intermolecular H/D exchange.
A sample of [LtBuCo(μ-D)]2 was prepared from [LtBuCo(μ-F)]2 and Et3SiD, using a method analogous to that used to synthesize the protiated analogue.8 Several peaks were shifted by 0.2–0.5 ppm from those in [LtBuCo(μ-H)]2, as verified by spiking the sample with an equimolar amount of [LtBuCo(μ-H)]2 (Figures S-6–S-8, Supporting Information). Thus, this dimer also exhibits PIECS, though the shifts are not as pronounced as in the iron species described above. Heating the mixture of [LtBuCo(μ-H)]2 and [LtBuCo(μ-D)]2 to 80 °C for 12 h gave no change in the NMR spectrum, indicating that there is no significant exchange of hydrides between complexes in this time frame.29 This contrasts with the iron analogues (Table 1) and is consistent with the idea that the cobalt(II) hydride dimer does not break up in solution.
Discussion
Characterization of High-Spin Iron Hydride Complexes, Including PIECS in 1H NMR Spectra
The Mössbauer spectra of 1 and 3 have similar isomer shifts, δ = 0.59 and 0.51 mm/s, which are consistent with the values observed in other high-spin iron(II) β-diketiminate complexes.23,24 High-spin, tetrahedral iron(II) sites in iron-sulfide clusters have similar isomer shifts (δ = 0.6–0.7 mm/s).30 Low-spin octahedral iron(II) sites have very different isomer shifts in the range of δ = 0.3–0.45 mm/s.30b The intermediate-spin (S = 1) iron(II) hydride complex [Fe(dppe)2H]+ has δ = 0.23 mm/s.31 The much higher isomer shifts in 1 and 3 strongly support the assignment of 1 and 3 having high-spin Fe(II) subsites and are consistent with the paramagnetic shifts in the 1H NMR spectra. We note that our assignment of the Mössbauer spectrum of 3 here replaces an incorrect assignment we gave in an earlier paper;3 the previous spectrum actually corresponds to LMeFe(arene), which can be formed during the synthesis of 3 when arenes are present.
The 1H NMR spectra of 1 and 3 were found to exhibit significant PIECS, as all the resonances shifted upon deuterium substitution. The term PIECS was coined by Heintz and Theopold22 and has been reported in a number of complexes.21,32 For example, deuteration of the hydrides in (Cp″)4Cr4(μ3–H)4 (Cp″ = η5-C5Me4Et)22 gives changes in the chemical shifts of the Cp″ protons, though they are far removed from the bridging hydride ligands. Most explanations for PIECS are based on the shorter bond lengths to D versus H.33 The Heintz/Theopold study is particularly relevant to our complexes because of the presence of bridging hydrides: shorter M–D bonds could decrease the M–M distance and the exchange coupling, which in turn would influence the magnetic susceptibility and thus the chemical shift of the protons.11 However, other explanations have been advanced for other cases of PIECS: for example, differential M–H/M–D bond energies could influence the ligand-field splitting slightly, which in turn could influence the paramagnetic shift.21a,21c However, we saw no evidence for any sizable change in the ligand field of iron upon deuteration, because the quadrupole splittings were the same within error between 3 and 3-D2, and only slightly different (ΔΔEQ = 0.12 mm/s) between 1 and 1-D2.
The PIECS in the 1H NMR spectra of 1 and 3 varied from a negligible change in some resonances up to 5.7 ppm in others. Importantly, PIECS was observed only for the dimeric form of 1, as the monomer LtBuFe(H/D) had the same chemical shifts in the 1H NMR spectrum for both isotopologues. This observation supports the hypothesis that the PIECS is connected to changes in the Fe–Fe distance.
A significant finding of this study is that variable-temperature 1H NMR spectroscopy, together with PIECS, was essential for defining the correct 1H NMR assignments of 1 and 3.2,4,14 Compound 3 was particularly vexing, because almost half of the seven expected resonances were masked. One resonance was hidden under the residual solvent, another apparent resonance was actually two resonances with the same chemical shift, and one was broadened into the baseline at room temperature and was broad even at elevated temperatures. However, variable-temperature studies enabled us to finally assign the resonances for 3 with confidence.
Examination of the variable-temperature 1H NMR data for 3 also revealed a fluxional process in solution with a barrier of ΔG‡ = 10.5 kcal/mol at 0 °C. We tentatively assign the low-temperature structure to be similar to the solid-state structure,4,14 in D2 symmetry with the diketiminate planes perpendicular to one another. In this case, the barrier would correspond to the energy required to reach the D2h symmetric conformation in which the N2C3Fe planes are coplanar (Scheme 1) and through which two D2 isomers of different chirality can interconvert.
Scheme 1.

Meanwhile, PIECS was vital to the assignment of all resonances in the 1H NMR spectrum of 1, which is complicated by the presence of both monomer and dimer.2 In addition, the crystallographic symmetry of the dimer in 1 is lower than that in 3, because crowding gives a boat conformation of the N2C3Fe rings that lowers the symmetry to idealized C2. We observed 18 resonances in dimeric 1, which is somewhat less than the 21 predicted from C2 symmetry in solution, implying that three of the resonances are lost to overlap or broadness. However, the number of peaks is significantly larger than in 3, indicating that the increased steric bulk of the LtBu ligand in 1 (from a buttressing effect)3,25 prevented the fluxional process that was observed in 3. The 1H NMR spectra imply that the hydride ligands are oriented such that there is no plane of symmetry relating the two LtBu ligands. Overall, these studies show the usefulness of variable-temperature 1H NMR spectroscopy and PIECS to decipher paramagnetic spectra and solution structure despite multiple overlapping resonances.
Intermolecular Hydride Exchange
Compounds 1 and 3 undergo intermolecular hydride exchange with their deuterated isotopologues. Dissolving 1 and 1-D2 in C6D6 produced an equilibrium mixture containing dimeric 1, 1-D, and 1-D2, along with monomeric LtBuFeH and LtBuFeD. In the iron(II) complexes, formation of 1-D most likely results from equilibrium between monomeric and dimeric 1, which is slow on the NMR time scale but rapid on the chemical manipulation time scale. (The rate of dissociation has been estimated to be 5 × 10–4 s–1 at 288 K, based on kinetics studies of the reaction of 1 with alkynes.2) Consistent with this rate regime, equilibrium between the isotopologues of 3 is reached within 45 min at room temperature, as judged by 1H NMR spectroscopy.
The hydride isotope exchange between 3 and 3-D2 cannot follow this process because 3 does not interconvert with monomer in solution, as previously shown using kinetics studies on the reaction with boranes.14 Two possible mechanisms for hydride isotope exchange between 3 and 3-D2 are shown in Scheme 2. The first is a concerted process, while the second requires rate-limiting cleavage of one of the bridging hydrides to give a single terminal hydride. The hydride ligands in the terminal position could attack another dimer to give exchange. Partial breaking of the dimer of 3 to give a terminal hydride, as in Scheme 2b, was previously found to be the most reasonable mechanism for the reaction of 3 with trialkylboranes,14 and so we consider this to be the most likely possibility that is consistent with the combined studies on 3. It is also notable that the cobalt analogue, [LtBuCo(μ-H)]2, did not undergo H/D exchange even with heating to 80 °C. Its low reactivity in general may be attributable to its inability to open one of the bridges, as in Scheme 2b for 3, or both bridges, as in 1.
Scheme 2. Possible Mechanisms for Hydride Ligand Exchange between Isotopologues in 3.

Dissociation of 3 into monomers is inconsistent with earlier kinetics studies on the reaction of boranes with 3.
Hydride Exchange with H2 and D2
The exchange of hydride complexes with H2 and D2 has been studied in detail in the literature.16 It is well-established in mononuclear iron-hydride complexes.34 In each of these cases, an open coordination site is required to bind H2 in an η2 binding mode. The oxidative addition of the H2 is not necessary, because there can be direct H transfer from coordinated H2 to the hydride without changing the oxidation state at the metal.
It is interesting that the inability of 3 to form monomeric LMeFeH in solution did not hinder hydride ligand exchange with D2. Equilibrium was established in less than a minute after treatment of 3 with D2. Though we cannot determine the mechanism unambiguously, we can advance two possible mechanisms. First, the “opened” form of the dimer has an open coordination site on one iron that could coordinate H2 or D2 to make a transient side-on D2 complex that is well-situated to exchange with the hydride on the other metal (Scheme 3a). Another potential mechanism involves oxidative addition of H2, either to one metal (giving a transient iron(IV) on one side) or to both metals (giving a diiron(III) species). The latter possibility is shown in Scheme 3b.
Scheme 3. Proposed Mechanisms for Hydride Ligand Exchange with D2 in 3.

It is also relevant that compound 1 does not undergo facile hydride exchange with D2, despite its ability to form monomer in solution. This required over 2 d at room temperature under 1 atm of D2 and 1 d under 8 atm of D2. The hindered reaction of 1 with D2 supports the contention that H2/D2 exchange in these hydride species does not proceed through a transient monomer. It is possible that the reaction of 1 with D2 proceeded via the monomer at a significantly slower rate. Another possibility is that the exchange goes through the dimeric form of 1, but that the bulkier ligands hinder its ability to access the reactive conformation.
Dinuclear iron sites with bridging hydride ligands have been studied extensively as [FeFe]-hydrogenase models.35 These diiron hydride complexes have been reported to undergo H/D hydride ligand exchange with D2/H2 via photolysis.36 In these systems, photolysis opens a coordination site by dissociating CO or cleaving a hydride bridge, and the H/D hydride exchange requires days (which contrasts with exchange in the 3/3-D2 system that occurs in seconds).36 H/D hydride ligand exchange has also been reported using D2/H2O for hydrogenases,37 diiron complexes,36a,38 other metal complexes,39 along with D+ as a deuterium source.40 In addition, nitrogenase can exchange D from D2O into H2, but only does so in the presence of N2.17−19 We suggest that the mechanisms advanced above for hydride/D2 exchange in 3 should be considered in nitrogenase: specifically, bridging hydride species may play key roles in H2/D2 exchange in the FeMoco cluster.
Conclusions
D2 was utilized as a deuterium source to enable the isolation and characterization of the low-coordinate iron deuteride complexes [LtBuFe(μ-D)]2 and [LMeFe(μ-D)]2. The Mössbauer spectra of these hydride complexes indicated that the metal centers are high-spin iron(II). The 1H NMR spectra of the hydride isotopologues exhibited PIECS. This effect was observed only in the dimeric complexes, implicating the slightly smaller size of the M2(μ-D)2 core as the main cause of PIECS.
PIECS also enabled the correct 1H NMR assignments of the hydride complexes, as well as the study of intermolecular hydride exchange. The exchange of hydrides between hydride complexes of the largest supporting ligand is likely to occur through dissociation of the dimers into monomers. However, exchange of the hydrides with added H2 occurs most rapidly with the smaller supporting ligand, implicating diiron(II) hydrides as the key intermediates. More generally, these studies show that 1H NMR spectroscopy can be a powerful tool for the study of paramagnetic iron hydride complexes: not despite the paramagnetism, but because of the paramagnetism through the PIECS effect.
Experimental Section
General Considerations
All manipulations were performed under a nitrogen atmosphere (or argon atmosphere where specified) by Schlenk techniques or in an M. Braun glovebox maintained at or below 1 ppm of O2 and H2O. Glassware was dried at 150 °C overnight, and Celite was dried overnight at 200 °C under vacuum. Pentane, hexane, benzene, diethyl ether, and toluene were purified by passage through activated alumina and Q5 columns from Glass Contour Co. (Laguna Beach, CA). THF was distilled under N2 from a potassium benzophenone ketyl solution. All solvents were degassed by removing a small amount of solvent under reduced pressure prior to argon glovebox entry. All solvents were stored over 3 Å molecular sieves. Benzene-d6 was dried and stored over flame-activated alumina. Toluene-d8 and THF-d8 were vacuum-transferred from sodium benzophenone ketyl solutions and were stored over 3 Å molecular sieves. Before use, an aliquot of each solvent was tested with a drop of sodium benzophenone ketyl in THF solution. Ultrahigh-purity H2 was purchased from Air Products, and D2 (99.8% D) was purchased from Sigma-Aldrich or Cambridge Isotope Laboratories. LtBuFeCl,25a potassium graphite,15 [LtBuFe(μ-H)]2,5 [LtBuCo(μ-H)]2,8 and LMeH41 were prepared by published procedures. LMeK was prepared using the published procedure,42 except Et2O was used as the solvent instead of toluene.
1H NMR data were recorded on a Bruker Avance spectrometer at 500 MHz. All resonances in the 1H NMR spectra are referenced to residual protiated solvents: benzene (7.16 ppm), toluene (2.09 ppm), or THF (3.58 or 1.73 ppm). Resonances were singlets unless otherwise noted. The NMR probe temperature was calibrated using either ethylene glycol or methanol.43 IR data were recorded on a Shimadzu FTIR spectrophotometer (FTIR-8400S) using a KBr pellet. UV–vis spectra were recorded on a Cary 50 spectrophotometer using Schlenk-adapted quartz cuvettes with a 1 mm optical path length. GC-MS was performed using a Shimadzu QP2010 system with electron impact ionization. Solution magnetic susceptibilities were determined by the Evans method.44 Elemental analyses were obtained from the CENTC Elemental Analysis Facility at the University of Rochester. Microanalysis samples were weighed with a PerkinElmer Model AD-6 Autobalance in a VAC Atmospheres glovebox under argon, and their compositions were determined with a PerkinElmer 2400 Series II Analyzer.
1H NMR Data for 1
1H NMR (C6D6, 25 °C): see Figure 1. Dimeric [LtBuFe(μ-H)]2: δ 67.3, 21.8, 19.8, 14.3 (18H, tBu), 12.0 (18H, tBu), 5.4, −2.6, −7.5, −9.7, −10.6, −14.5, −15.4, −22.0, −27.8, −31.6, −37.2, −51.8, −57.5 ppm. Resonances in the dimers could not be assigned to specific proton environments, because of overlap that prevented accurate integration. Monomeric LtBuFeH: δ 115 (1H, α-H), 41.7 (18H, tBu), 11.7 (4H, iPr-CH or aryl m-H), −26.5 (12H, iPr CH3), −109 (4H, iPr-CH or aryl m-H), −113 (2H, aryl p-H), −122 (12H, iPr CH3) ppm.
Improved Synthesis of 1-D2 from D2
The synthesis of [LtBuFe(μ-D)]2 relied on the same procedure as the synthesis of 1 from H2.5 After 16 h, the headspace gases were removed, and fresh D2 was added; this process was repeated twice. 1-D2 was isolated in 51% yield. 1H NMR (C6D6, 25 °C): [LtBuFe(μ-D)]2: δ 73.0, 23.0, 21.0, 14.3 (18H, tBu), 13.0 (18H, tBu), 6.4, −1.7, −3.9, −8.0, −10.6, −16.9, −17.5, −25.1, −29.6, −33.0, −43.0, −50.2, −55.0 ppm. LtBuFeD: δ 115 (1H, α-H), 41.7 (18H, tBu), 11.7 (4H, iPr-CH or aryl m-H), −26.5 (12H, iPr CH3), −109 (4H, iPr-CH or aryl m-H), −113 (2H, aryl p-H), −122 (12H, iPr CH3) ppm. The monodeuterated hydride dimer, 1-D, was also observed in solution as described above. 1H NMR (C6D6, 25 °C): δ 70.2, 22.4, 20.5, 14.3 (18H, tBu), 12.5 (18H, tBu), 5.9, −2.1, −6.6, −7.7, −10.6, −15.7, −16.5, −23.6, −28.7, −32.3, −40.1, −51.0, −56.2 ppm.
Synthesis of LMeFeBr(THF) (2)
LMeK (3.215 g, 7.039 mmol) was added to a flask with a Teflon pin closure and was dissolved in THF (75 mL) to give a light yellow solution. Anhydrous FeBr2 (1.532 g, 7.104 mmol, 1.01 equiv) was added to the solution, which produced a red reaction mixture. The flask was sealed, and the mixture was heated at 70 °C for 16 h. The reaction mixture turned yellow in color upon heating. The reaction mixture was cooled to room temperature and filtered through Celite. The yellow solution was concentrated to 20 mL, and pentane (100 mL) was added to precipitate additional insoluble material (presumably KBr), which was removed by filtration through Celite. The yellow solution was concentrated to 5 mL, which resulted in the formation of a large amount of yellow crystalline solid. The supernatant was decanted, and the crystalline yellow solid was washed with pentane (12 mL). The solid was dried under reduced pressure to give 2.282 g of product. Additional product (1.328 g) was collected from subsequent crystallizations of the supernatant by layering with pentane and cooling to −45 °C. The total yield was 3.610 g (82.0%). 1H NMR (THF-d8, 22 °C): δ 18.6 (4H, aryl m-H), 4.9 (12H, iPr CH3), −8.7 (12H, iPr CH3), −12.3 (br, 4H, iPr-CH), −39.9 (2H, aryl p-H), −67.3 (6H, backbone CH3), −78.7 (1H, α-H) ppm. μeff (THF-d8, 22 °C) 5.5(1) μB. IR (KBr): 3058 (w), 2964 (s), 2928 (s), 1529 (s), 1459 (s), 1437 (s), 1388 (vs), 1316 (s), 1261 (m), 1176 (m), 1100 (m), 1057 (w), 1022 (m), 935 (m), 899 (w), 872 (m), 855 (m), 795 (s), 758 (s) cm–1. UV–vis (THF): 333 (21.2 mM–1cm–1), 433 (sh, ∼0.9 mM–1cm–1) nm. Anal. Calcd for C33H49N2FeBrO: C, 63.36; H, 7.91; N, 4.48. Found: C, 63.13; H, 8.10; N, 4.31%.
Modified Synthesis of 3
In an Ar-filled glovebox, LMeFeBr(THF) (703 mg, 1.12 mmol) was dissolved in THF (20 mL) to give a yellow solution, which was added to a small three-neck round-bottom flask with a stir bar. On a vacuum line, H2 (1 atm) was added to a bulb (297.5 mL, 12.4 mmol, 11 equiv), and the bulb was brought into the glovebox. KC8 (182 mg, 1.35 mmol, 1.2 equiv) was added to a solid addition tube. The three-neck flask was attached to the volume bulb, a vacuum adapter, and the solid addition piece. The reaction apparatus was degassed under reduced pressure until a small amount of THF had been removed. Then, the apparatus was backfilled with H2 by slowly opening the stopcock of the volume bulb. KC8 was added to the stirring solution by inverting the solid addition piece, which immediately produced a dark green reaction mixture. The mixture was stirred and turned brown in color after 20 min. After 3 h, the mixture was filtered through Celite, and the volatile components were removed under reduced pressure. The resulting brown residue was dissolved in toluene (35 mL) and was filtered through Celite to remove additional insoluble material. Toluene was removed under reduced pressure to give a brown powder, which was washed with cold pentane (−45 °C, 10 mL). The solid was dried under reduced pressure to give 263 mg of brown powder. The pentane wash was concentrated to 3 mL and was layered with hexamethyldisiloxane (4 mL). Cooling to −45 °C yielded an additional 35 mg of product. The total yield was 298 mg (56.1%). 1H NMR (C6D6, 25 °C): δ 13.0 (12H + 8H, backbone CH3 and aryl m-H), 7.1 (24H, iPr CH3), −24.0 (4H, aryl p-H), −24.8 (br, 24H, iPr CH3), −55.9 (2H, α-H) ppm. The iPr-CH protons were not observed at this temperature (see text). The deuterated isotopologue of 3, 3-D2, was synthesized using the same method with D2, in 58.1% yield.
Details of High Pressure Gas Addition Apparatus
A Wilmad 522-PV-7 pressure NMR tube with a 5 mm outer diameter (OD) and a maximum pressure rating of 200 psi was used for all high-pressure gas experiments. The tube comes equipped with a Wilmad PV-ANV valve that is capable of accepting a Swagelok 1/8″ tubing nut and ferrule. Poly(tetrafluoroethylene) (PTFE) tubing (OD = 1/8″) and Swagelok 1/8″ tubing nuts and ferrules were used for all the connections. PTFE tubing was used to connect the gas regulator to a T-shaped splitter, which provided two paths. One path connected through PTFE tubing a high-pressure gas gauge and the PV-ANV valve on the NMR tube. The second path connected the PTFE tubing to a Swagelok valve, which was connected to a metal O-ring joint. The other part of the O-ring joint was equipped with a metal-to-glass flange that had a glass 14/20 female joint. This allowed the apparatus to be attached to the Schlenk line for evacuation.
Mössbauer Spectroscopy
Mössbauer data were recorded on a spectrometer with alternating constant acceleration. The minimum experimental line width was 0.24 mm/s (full width at half-height). The sample temperature was maintained constant in an Oxford Instruments Variox cryostat. The γ-ray source was ca. 0.6 GBq 57Co/Rh. Isomer shifts are quoted relative to iron metal at 300 K. The zero-field spectra were simulated by using Lorentzian doublets.
Acknowledgments
This work was supported by the National Institutes of Health (GM065313). We thank Bernd Mienert for the collection of Mössbauer data.
Supporting Information Available
Additional spectral and crystallographic information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Recent Advances in Hydride Chemistry; Peruzzini M.; Poli R., Eds.; Elsevier: New York, 2001. [Google Scholar]
- Smith J. M.; Lachicotte R. J.; Holland P. L. J. Am. Chem. Soc. 2003, 125, 15752. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Sadique A. R.; Smith J. M.; Dugan T. R.; Cowley R. E.; Brennessel W. W.; Flaschenriem C. J.; Bill E.; Cundari T. R.; Holland P. L. J. Am. Chem. Soc. 2008, 130, 6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vela J.; Smith J. M.; Yu Y.; Ketterer N. A.; Flaschenriem C. J.; Lachicotte R. J.; Holland P. L. J. Am. Chem. Soc. 2005, 127, 7857. [DOI] [PubMed] [Google Scholar]
- Dugan T. R.; Holland P. L. J. Organomet. Chem. 2009, 694, 2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. M.; Bill E.; Brennessel W. W.; Holland P. L. Science 2011, 334, 780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland P. L. Acc. Chem. Res. 2008, 41, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High-spin diketiminate-cobalt-hydride complexes:; a Ding K.; Brennessel W. W.; Holland P. L. J. Am. Chem. Soc. 2009, 131, 10804. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dugan T. R.; Goldberg J. M.; Brennessel W. W.; Holland P. L. Organometallics 2012, 31, 1349. [Google Scholar]
- Related high-spin diketiminate-nickel-hydride complexes:; a Pfirrmann S.; Limberg C.; Ziemer B. Dalton Trans. 2008, 6689. [DOI] [PubMed] [Google Scholar]; b Pfirrmann S.; Yao S.; Ziemer B.; Stosser R.; Driess M.; Limberg C. Organometallics 2009, 28, 6855. [Google Scholar]; c Gehring H.; Metzinger R.; Herwig C.; Intemann J.; Harder S.; Limberg C. Chem.—Eur. J. 2013, 19, 1629. [DOI] [PubMed] [Google Scholar]
- Upon reducing the complex to the iron(I) oxidation state, the bridging hydrides became terminal, and it was possible to crystallographically characterize three-coordinate iron-hydride species:Chiang K. P.; Scarborough C. C.; Horitani M.; Lees N. S.; Ding K.; Dugan T. R.; Brennessel W. W.; Bill E.; Hoffman B. M.; Holland P. L. Angew. Chem., Int. Ed. 2012, 51, 3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a NMR of Paramagnetic Molecules, La Mar G. N.; Horrocks W. D.; Holm R. H. Eds.; Academic Press: New York, 1973. [Google Scholar]; b Ming L.-J.Nuclear Magnetic Resonance of Paramagnetic Metal Centers in Proteins and Synthetic Complexes. In Physical Methods in Bioinorganic Chemistry; Que L., Ed.; University Science Books: Sausalito, CA, 2000; p 375. [Google Scholar]
- Limberg has recently reported interesting shifts in the hydride signal of a nickel-hydride complex with a low-lying paramagnetic excited state:Pfirrmann S.; Limberg C.; Herwig C.; Knispel C.; Braun B.; Bill E.; Stösser R. J. Am. Chem. Soc. 2010, 132, 13684. [DOI] [PubMed] [Google Scholar]
- Electron–nuclear double-resonance (ENDOR) spectroscopy can detect NMR transitions in hydrides using EPR. See:; a Brecht M.; van Gastel M.; Buhrke T.; Friedrich B.; Lubitz W. J. Am. Chem. Soc. 2003, 125, 13075. [DOI] [PubMed] [Google Scholar]; b Igarashi R. Y.; Laryukhin M.; Dos Santos P. C.; Lee H. I.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2005, 127, 6231. [DOI] [PubMed] [Google Scholar]; c Kinney R. A.; Hetterscheid D. G. H.; Hanna B. S.; Schrock R. R.; Hoffman B. M. Inorg. Chem. 2010, 49, 704. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jablonskyte A.; Wright J. A.; Fairhurst S. A.; Peck J. N. T.; Ibrahim S. K.; Oganesyan V. S.; Pickett C. J. J. Am. Chem. Soc. 2011, 133, 18606. [DOI] [PubMed] [Google Scholar]; e Pandelia M.-E.; Infossi P.; Stein M.; Giudici-Orticoni M.-T.; Lubitz W. Chem. Commun. 2012, 48, 823. [DOI] [PubMed] [Google Scholar]; f Kinney R. A.; Saouma C. T.; Peters J. C.; Hoffman B. M. J. Am. Chem. Soc. 2012, 134, 12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Brennessel W. W.; Holland P. L. Organometallics 2007, 26, 3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. M.; Sadique A. R.; Cundari T. R.; Rodgers K. R.; Lukat-Rodgers G.; Lachicotte R. J.; Flaschenriem C. J.; Vela J.; Holland P. L. J. Am. Chem. Soc. 2006, 128, 756. [DOI] [PubMed] [Google Scholar]
- Selected reviews and recent examples:; a Maseras F.; Lledós A.; Clot E.; Eisenstein O. Chem. Rev. 2000, 100, 601. [DOI] [PubMed] [Google Scholar]; b Pons V.; Conway S. L. J.; Green M. L. H.; Green J. C.; Herbert B. J.; Heinekey D. M. Inorg. Chem. 2004, 43, 3475. [DOI] [PubMed] [Google Scholar]; c Trovitch R. J.; Lobkovsky E.; Chirik P. J. Inorg. Chem. 2006, 45, 7252. [DOI] [PubMed] [Google Scholar]; d Weller A. S.; McIndoe J. S. Eur. J. Inorg. Chem. 2007, 4411. [Google Scholar]
- Burgess B. K.; Lowe D. J. Chem. Rev. 1996, 96, 2983. [DOI] [PubMed] [Google Scholar]
- a Leigh G. J. Eur. J. Biochem. 1995, 229, 14. [PubMed] [Google Scholar]; b Sellmann D.; Fürsattel A.; Sutter J. Coord. Chem. Rev. 2000, 200–202, 545. [Google Scholar]; c Hoffman B. M.; Lukoyanov D.; Dean D. R.; Seefeldt L. C. Acc. Chem. Res. 2013, 46, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.-Y.; Khadka N.; Lukoyanov D.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iron-hydride complexes are important because the binding site of N2 to the FeMoco of nitrogenases is most likely to be iron.; a Seefeldt L. C.; Dance I. G.; Dean D. R. Biochemistry 2004, 43, 1401. [DOI] [PubMed] [Google Scholar]; b Igarashi R. Y.; Dos Santos P. C.; Niehaus W. G.; Dance I. G.; Dean D. R.; Seefeldt L. C. J. Biol. Chem. 2004, 279, 34770. [DOI] [PubMed] [Google Scholar]; c Hoffman B. M.; Dean D. R.; Seefeldt L. C. Acc. Chem. Res. 2009, 42, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hebendanz N.; Köhler F. H.; Scherbaum F.; Schlesinger B. Magn. Reson. Chem. 1989, 27, 798. [Google Scholar]; b Theopold K. H.; Silvestre J.; Byrne E. K.; Richeson D. S. Organometallics 1989, 8, 2001. [Google Scholar]; c Medforth C. J.; Shiau F.-Y.; La Mar G. N.; Smith K. M. J. Chem. Soc. Chem. Commun. 1991, 590. [Google Scholar]; d Evans B.; Smith K. N.; La Mar G. N.; Viscio D. B. J. Am. Chem. Soc. 1977, 99, 7070. [DOI] [PubMed] [Google Scholar]; e Horn R. R.; Everett G. W. Jr. J. Am. Chem. Soc. 1971, 93, 7173. [Google Scholar]; f Johnson A.; Everett G. W. Jr. J. Am. Chem. Soc. 1972, 94, 1419. [Google Scholar]; g Johnson A.; Everett G. W. Jr. J. Am. Chem. Soc. 1972, 94, 6397. [Google Scholar]; h Wieder N. L.; Gallagher M.; Carroll P. J.; Berry D. H. J. Am. Chem. Soc. 2010, 132, 4107. [DOI] [PubMed] [Google Scholar]; i Hetterscheid D. G. H.; Hanna B. S.; Schrock R. R. Inorg. Chem. 2009, 48, 8569. [DOI] [PMC free article] [PubMed] [Google Scholar]; j There can also be influences on 31P NMR spectra: Beck R.; Shoshani M.; Johnson S. A. Angew. Chem., Int. Ed. Engl. 2012, 51, 11753. [DOI] [PubMed] [Google Scholar]
- Heintz R. A.; Neiss T. G.; Theopold K. H. Angew. Chem., Int. Ed. Engl. 1994, 33, 2326. [Google Scholar]
- Eckert N. A.; Stoian S.; Smith J. M.; Bominaar E. L.; Münck E.; Holland P. L. J. Am. Chem. Soc. 2005, 127, 9344. [DOI] [PubMed] [Google Scholar]
- a Andres H.; Bominaar E. L.; Smith J. M.; Eckert N. A.; Holland P. L.; Münck E. J. Am. Chem. Soc. 2002, 124, 3012. [DOI] [PubMed] [Google Scholar]; b Vela J.; Stoian S.; Flaschenriem C. J.; Münck E.; Holland P. L. J. Am. Chem. Soc. 2004, 126, 4522. [DOI] [PubMed] [Google Scholar]; c Cowley R. E.; Bill E.; Neese F.; Brennessel W. W.; Holland P. L. Inorg. Chem. 2009, 48, 4828. [DOI] [PubMed] [Google Scholar]; d Cowley R. E.; Elhaïk J.; Eckert N. A.; Brennessel W. W.; Bill E.; Holland P. L. J. Am. Chem. Soc. 2008, 130, 6074. [DOI] [PubMed] [Google Scholar]; e Stoian S. A.; Smith J. M.; Holland P. L.; Munck E.; Bominaar E. L. Inorg. Chem. 2008, 47, 8687. [DOI] [PubMed] [Google Scholar]; f Eckert N. A.; Vaddadi S.; Stoian S.; Lachicotte R. J.; Cundari T. R.; Holland P. L. Angew. Chem., Int. Ed. 2006, 45, 6868. [DOI] [PubMed] [Google Scholar]
- a Smith J. M.; Lachicotte R. J.; Holland P. L. Chem. Commun. 2001, 1542. [DOI] [PubMed] [Google Scholar]; b Eckert N. A.; Smith J. M.; Lachicotte R. J.; Holland P. L. Inorg. Chem. 2004, 43, 3306. [DOI] [PubMed] [Google Scholar]; c Stubbert B. D.; Holland P. L.; Adhikari D.; Mindiola D. J. Inorg. Synth. 2010, 35, 38. [Google Scholar]
- Tonzetich Z. J.; Heroguel F.; Do L. H.; Lippard S. J. Inorg. Chem. 2011, 50, 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- It is particularly important to protect the reaction mixture from arenes. Even traces of benzene in the vapor phase in the glove box led to undesired formation of LMeFe(C6H6). This benzene complex was previously reported:Smith J. M.; Sadique A. R.; Cundari T. R.; Rodgers K. R.; Lukat-Rodgers G.; Lachicotte R. J.; Flaschenriem C. J.; Vela J.; Holland P. L. J. Am. Chem. Soc. 2006, 128, 756. [DOI] [PubMed] [Google Scholar]
- a Legzdins P.; Phillips E. C.; Yee V. C.; Trotter J.; Einstein F. W. B.; Jones R. H. Organometallics 1991, 10, 986. [Google Scholar]; b Thomas W. A. Annu. Rev. NMR Spectrosc. 1968, 1, 43. [Google Scholar]
- In an independent experiment, a mixture of Et3SiH and Et3SiD was used to make a statistical mixture of isotopologues of [LtBuCo(μ-H)]2, and the peaks for the singly deuterated isotopologue were distinguishable by 1H NMR spectroscopy. See Supporting Information.
- a Beinert H.; Holm R. H.; Münck E. Science 1997, 277, 653. [DOI] [PubMed] [Google Scholar]; b Münck E.Aspects of 57Fe Mössbauer Spectroscopy. In Physical Methods in Bioinorganic Chemistry; Que L., Ed.; University Science Books: Sausalito, CA, 2000; p 287. [Google Scholar]
- Franke O.; Wiesler B. E.; Lehnert N.; Peters G.; Burger P.; Tuczek F. Z. Anorg. Allg. Chem. 2006, 632, 1247. [Google Scholar]
- This phenomenon is distinct from IPR (isotopic perturbation of resonance) effects in diamagnetic complexes. For an example of IPR in a hydride-H2 complex:Heinekey D. M.; Oldham W. J. J. Am. Chem. Soc. 1994, 116, 3137. [Google Scholar]
- Bartell L. S.; Roskos R. R. J. Chem. Phys. 1966, 44, 457. [Google Scholar]
- See reference (14)c and references therein.
- a Darensbourg M. Y.; Lyon E. J.; Smee J. J. Coord. Chem. Rev. 2000, 206–207, 533. [Google Scholar]; b Tard C.; Pickett C. J. Chem. Rev. 2009, 109, 2245. [DOI] [PubMed] [Google Scholar]
- a Zhao X.; Georgakaki I. P.; Miller M. L.; Yarbrough J. C.; Darensbourg M. Y. J. Am. Chem. Soc. 2001, 123, 9710. [DOI] [PubMed] [Google Scholar]; b Zhao X.; Georgakaki I. P.; Miller M. L.; Mejia-Rodriguez R.; Chiang C.-Y.; Darensbourg M. Y. Inorg. Chem. 2002, 41, 3917. [DOI] [PubMed] [Google Scholar]
- Vignais P. M. Coord. Chem. Rev. 2005, 249, 1677. [Google Scholar]
- Georgakaki I. P.; Miller M. L.; Darensbourg M. Y. Inorg. Chem. 2003, 42, 2489. [DOI] [PubMed] [Google Scholar]
- a Carriker J. L.; Wagenknecht P. S.; Hosseini M. A.; Fleming P. E. J. Mol. Catal. A: Chem. 2007, 267, 218. [Google Scholar]; b Kovács G.; Nádasdi L.; Joó F.; Laurenczy G. C. R. Acad. Sci., Ser. IIc: Chim. 2000, 3, 601. [Google Scholar]; c Kovács G.; Nádasdi L.; Laurenczy G.; Joó F. Green Chem. 2003, 5, 213. [Google Scholar]; d Kovács G.; Schubert G.; Joó F.; Pápai I. Organometallics 2005, 24, 3059. [Google Scholar]
- Wang N.; Wang M.; Liu J.; Chen L.; Sun L. Inorg. Chem. 2009, 48, 11551. [DOI] [PubMed] [Google Scholar]
- Budzelaar P. H. M.; Moonen N. N. P.; De Gelder R.; Smits J. M. M.; Gal A. W. Eur. J. Inorg. Chem. 2000, 753. [DOI] [PubMed] [Google Scholar]
- Clegg W.; Cope E. K.; Edwards A. J.; Mair F. S. Inorg. Chem. 1998, 37, 2317. [DOI] [PubMed] [Google Scholar]
- a Kaplan M. L.; Bovey F. A.; Cheng H. N. Anal. Chem. 1975, 47, 1703. [Google Scholar]; b Ammann C.; Meier P.; Merbach A. E. J. Magn. Reson. 1982, 46, 319. [Google Scholar]
- a Baker M. V.; Field L. D.; Hambley T. W. Inorg. Chem. 1988, 27, 2872. [Google Scholar]; b Schubert E. M. J. Chem. Educ. 1992, 69, 62. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.