

Abstract
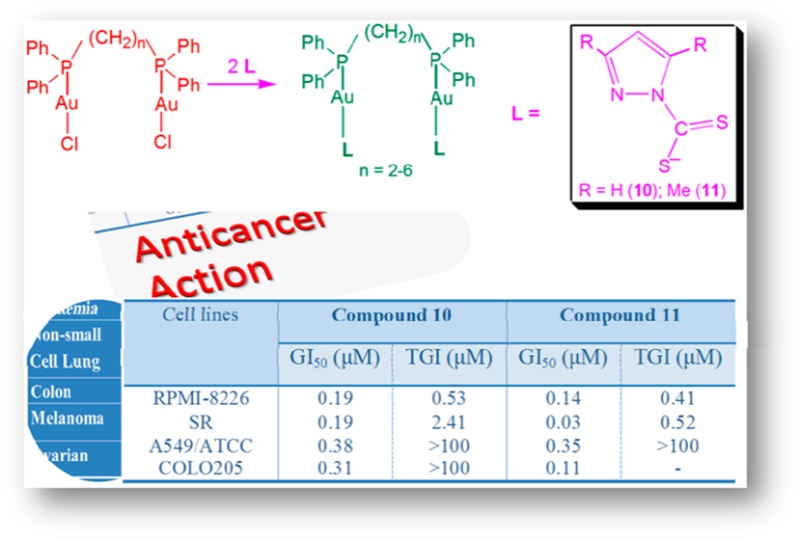
The reactions of potassium salts of the dithiocarbamates L {where L = pyrazolyldithiocarbamate (L1), 3,5-dimethylpyrazolyldithiocarbamate (L2), or indazolyldithiocarbamate (L3)} with the gold precursors [AuCl(PPh3)], [Au2Cl2(dppe)], [Au2Cl2(dppp)], or [Au2Cl2(dpph)] lead to the new gold(I) complexes [AuL(PPh3)] (1–3), [Au2L2(dppe)] (4–6), [(Au2L2)(dppp)] (7–9), and [Au2(L)2(dpph)] (10–12) {where dppe = 1,2-bis(diphenylphosphino)ethane, dppp = 1,3-bis(diphenylphosphino)propane, and dpph = 1,6-bis(diphenylphosphino)hexane}. These gold compounds were characterized by a combination of NMR and infrared spectroscopy, microanalysis, and mass spectrometry; and in selected cases by single-crystal X-ray crystallography. Compounds 4–6, which have dppe ligands, are unstable in solution for prolonged periods, with 4 readily transforming to the Au18 cluster [Au18S8(dppe)6]Cl2 (4a) in dichloromethane. Compounds 1–3 and 7–12 are all active against human cervical epithelioid carcinoma (HeLa) cells, but the most active compounds are 10 and 11, with IC50 values of 0.51 μM and 0.14 μM, respectively. Compounds 10 and 11 are more selective toward HeLa cells than they are toward normal cells, with selectivities of 25.0 and 70.5, respectively. Further tests, utilizing the 60-cell-line Developmental Therapeutics Program at the National Cancer Institute (U.S.A.), showed 10 and 11 to be active against nine other types of cancers.
Short abstract
Phosphinogold(I) complexes, featuring heterocycle dithiocarbamate ligands, have been synthesized and characterized. These complexes are active against several cancer cells, with the most active compound able to act against ten cancer cells in vitro and with GI50 as low as 0.03 μM against leukemia SR cell line.
Introduction
Gold compounds in the oxidation states of +1 and +3 continue to be of interest in medicinal chemistry because their efficacies toward certain diseases can be fine-tuned.1,2 For instance, a number of gold(III) dithiocarbamates have been reported as anticancer agents by Fregona and co-workers.3 This includes the classics [Au(DMDT)X2] and [Au(ESDT)X2] (where X = Cl, Br; DMDT = N,N-dimethyldithiocarbamate; ESDT = ethylsarcosinedithiocarbamate), which were shown to be 1–4 times more active than cis-platin and particularly potent against the lymphoproliferative-type HL-60 cell line.3,4 Various modes of action have been proposed for these compounds, from weak DNA binding to proteasome inhibition.5−7 A second generation of gold(III) dithiocarbamate (dtc) anticancer compounds includes the gold(III)-dipeptidedithiocarbamato derivatives AuX2(dtc-Sar-AA-O(t-Bu))] (X = Cl, Br; Sar = Sarcosine; AA = Gly, α-aminoisobutyric acid, and l-Phe)8 and several [AuX2(pdtc)] (X = Cl, Br; pdtc = oligopeptidedithiocarbamate) compounds, which are active against four human tumor cell lines at lower IC50 values than is cis-platin.9
Gold(I) thiolate compounds have also been of interest as anticancer agents since the first reports on the antiarthritic agent auranofin,1,10,11 and other gold(I) thiolates12−14 were shown to possess anticancer properties. These include the report by Tiekink and co-workers that triorganophosphinogold(I) dithiocarbamates of general formula [(R3P)Au(S2CNR2)] were active against seven human cancer cell lines.13 Of these, [(Et3P)Au(S2CNEt2)] (Et = ethyl) was the most active against the IGROV ovarian cell line (ID50 = 12 ng/mL).13
An early report by Mirabelli et al.,14 supported by others,12,13 on various gold(I) compounds of structural formula R3P–Au–X (R = ethyl; X = thioglucose (SR)) has demonstrated that the presence of a P–Au–S motif in these compounds enhances their anticancer activity. Notably, phosphinogold(I) thiolate complexes of type [Au(PR3)(SR)] are more active than gold(I) thiolates of type [Au(SR)], suggesting that the presence of phosphine ligands increases the lipophilicity and membrane permeability of the phosphinogold(I) complexes that make them active.12
It has been established that gold compounds, such as auranofin, act against cancer cells via the mitochondria by inhibiting thioredoxin reductase.11−18 In doing so, gold(I) binds the C-terminal of the redox-active selenocysteine, leading to cytotoxic effects.18−20 For these reasons, several phosphinogold(I) thiolate complexes have been prepared and investigated for their anticancer activities. Examples include the hydrophilic tetrakis((tris(hydroxymethyl)phosphino)gold(I) complex, which was reported to be cytotoxic toward HCT-15 tumor cells.21 Other examples are [Au(PPh3)(sppa)] (sppa = sulfanylpropenoate),22 [Au(PEt3)(K3TSC)] (K3TSC = vitamin K3 derivative),23 and the gold(I) 7-azacoumarin complex,24 which exhibit activity against HeLa and A2780cis cells, [(AuOmS)2(Ph2P(CH2)2PPh2)] (OmS = lupinylsulfide), exhibiting activity against ovarian carcinomas,25 and [Et3PAu(S2CNEt2)] and [Ph3PAu(S2CNC2C4H8)], which exhibit activity against breast, ovarian, and colon cancer.12,13a This strongly suggests that one strategy to make gold(I) anticancer drugs is to prepare molecules that have a phosphine and a sulfur-containing ligand bound to gold(I) to have the P–Au–S motif.
For phosphinogold(I) complexes, the nature of the phosphine ligand appears to be important in regulating their anticancer behavior. For example a report on the anticancer properties of dinuclear di(phosphino)alkane gold(I) chloride by Mirabeli et al.26 suggested two key factors determine the activity of these gold(I) compounds. The first is to have phenyl groups on the phosphorus, and the second is the effect of CH2 linkers, with the compound with two CH2 linkers being more active than compounds with three, four, five, or six CH2 linkers. However, a recent report by Raubenheimer and co-workers indicates that dinuclear di(phosphino)alkane gold(I) azole complexes with longer CH2 linkers are highly active for a number of cancer cell lines, and at very low dosage.27 Clearly, from these two reports, the nature of the di(phosphino)alkanegold(I) and the ancillary ligand bound to the gold(I) is important in determining the anticancer activity of these compounds.
We report here new phosphinogold(I) dithiocarbamate complexes, using triphenylphosphine, 1,2-bis(diphenylphosphino)ethane (dppe), 1,3-bis(diphenylphosphino)-propane (dppp), 1,6-bis(diphenylphosphino)hexane (dpph), and the pyrazol-1-yl- and indazol-1-yl-based dithiocarbamate ligands (dtcs) to test the hypothesis that combining alkane chain length in the diphosphino ligand and using dithiocarbamate as the thiolate can lead to new gold anticancer compounds. Our choice of pyrazoles and indazole as the backbone of the dithiocarbamate is based on their medicinal properties.28,29 This approach to prepare phosphinogold(I) dithiocarbamato compounds has resulted in compounds that have excellent anticancer activities against several cancer cell lines. Several reports on the chemistry of gold(I) dithiocarbamate compounds,30−39 gold(I) thiosemicarbazones, and gold(I) thiosemicarbazone complexes40,41 have appeared in the literature. Some of these compounds have anticancer activity that has been attributed to the synergistic effect of the metals and the thiolate ligands, but none of these compounds has phosphine ligands.
We have further shown for the first time that dithiocarbamate can act as a sulfur transfer reagent to form the high-nuclearity gold cluster [Au18S8(dppe)6]2+ from [Au(L1)(dppe)], a previously known cluster that was erroneously reported as neutral [Au18S8(dppe)6]·H2O.42 The heavier congener of this cluster, [Au18Se8(dppe)6]2+, is known.43
Results and Discussion
Triphenylphosphinogold(I) Dithiocarbamato Complexes (1–3)
The gold(I) complexes 1–3 were prepared from the reaction of [AuCl(PPh3)] and the dtc ligands L1–L3 (Scheme 1) and were isolated as orange-yellow solids. The complexes were characterized by a combination of spectroscopy, mass spectrometry, and microanalysis; in the case of 3, single-crystal X-ray crystallography was also used. Complex 1 displays a typical 1H NMR spectrum for these compounds, which shows dtc protons for 4-pz and 5-pz proton peaks at 6.27 and 7.63 ppm, respectively. The 31P{1H} NMR spectra of complexes 1–3 displayed broad signals between 29.9 and 36.3 ppm.
Scheme 1. Preparations of (Monophosphino)gold(I) Dithiocarbamate Complexes 1–3.

Crystal data, together with the data collection and refinement parameters, are presented in Table 1. Compound 3 exhibits the expected near-linear geometry about the Au(I) center with the P1–Au1–S1 angle spanning 175.36(2)°. A molecular drawing of 3 is shown in Figure 1. The Au–P distance of 2.2533(6) Å is in excellent agreement with the average value of 2.26(2) Å obtained by averaging 628 Au–PPh3 distances from 501 complexes reported to the Cambridge Structural Database (CSD).44 The Au–S distance in 3 (2.3272(6) Å) is slightly longer than the Au–P distance, but is somewhat shorter than the average distance of 2.38(16) Å computed based on 89 Au–S distances for 58 dithiocarbamate complexes reported to the CSD. However, the difference with the latter is not statistically significant as Au–S bond lengths fall in a broad range. In closely related compounds these distances are as follows: [Au(S2CNEt2)(PPh3)], Au–S (2.338(4) Å) and Au–P (2.251(3) Å)33; [Au(S2CNC10H20O4)(PPh3)] (where C10H20O4 = aza-15-crown-5), Au–S (2.339(3) Å) and Au–P (2.253(3) Å).31 The C–S distances are different, with the C1–S1 distance to the coordinated S atom being longer (single bond, 1.732(3) Å) than the C1–S2 bond that clearly shows multiple character (1.663(3) Å).
Table 1. Crystallographic Data for Compounds 3, 4a, and 12.
| 3 | 4a | 12 | |
|---|---|---|---|
| empirical formula fw | C26H20AuN2PS2 652.50 | C156H144Au18Cl2P12S8 6263.13 | C46H42Au2N4P2S4 1234.95 |
| temp (K) | 100(2) | 105(2) | 150(2) |
| wavelength (Å) | 0.710 73 | 0.710 73 | 0.774 90a |
| cryst syst | monoclinic | monoclinic | monoclinic |
| space group | P21/n | P21/n | P21/n |
| a (Å) | 9.1382(12) | 17.6854(7) | 13.499(11) |
| b (Å) | 22.487(3) | 34.4361(14) | 11.560(19) |
| c (Å) | 11.3760(14) | 29.1894(12) | 15.396(15) |
| β (deg) | 91.541(2) | 92.4570(10) | 108.60(6) |
| volume (Å3) | 2336.8(5) | 17760.5(12) | 2277(5) |
| Z | 4 | 4 | 2 |
| density (calc) (Mg/m3) | 1.855 | 2.342 | 1.801 |
| abs. coeff. (mm–1) | 6.560 | 15.077 | 8.334 |
| F(000) | 1264 | 11376 | 1196 |
| final R indices (R1) | 0.0587 | 0.0473 | 0.0404 |
| reflections collected | 36 929 | 188 093 | 31 033 |
| completeness to θ | 99.3% | 99.8% | 99.2% |
| goodness-of-fit on F2 | 1.19 | 1.02 | 1.02 |
| final R indices [I > 2σ(I)] | R1 = 0.0210, wR2 = 0.0542 | R1 = 0.0473, wR2 = 0.1258 | R1 = 0.0404, wR2 = 0.0984 |
| R indices (all data) | R1 = 0.0216, wR2 = 0.0544 | R1 = 0.0677, wR2 = 0.1347 | R1 = 0.0566, wR2 = 0.1071 |
| larg. diff. peak hole (e.Å–3) | 1.15 and −0.88 | 2.14 and −1.44 | 1.45 and −0.86 |
Using synchrotron radiation tuned to λ = 0.7749 Å.
Figure 1.
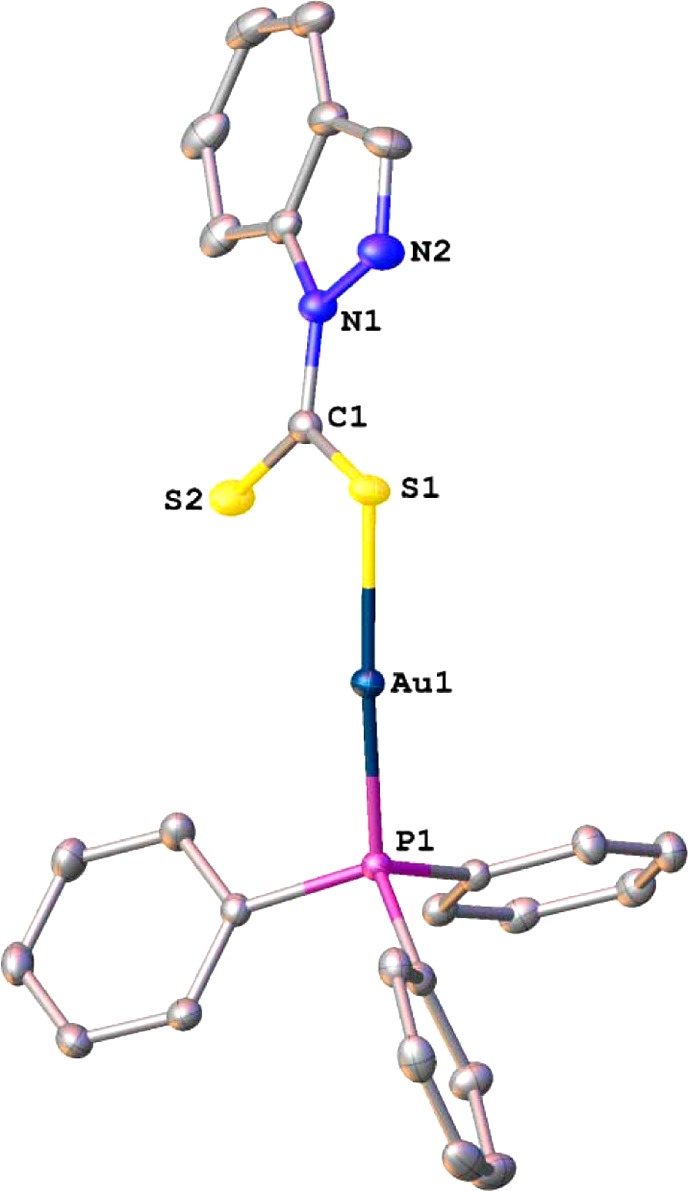
Molecular structure of 3 drawn with 30% probability ellipsoids. H atoms are omitted for clarity. Selected bond lengths [Å] and angles [deg]: Au1–P1, 2.2533(6); Au1–S1, 2.3272(6); P1–C1, 1.811(2); N1–C1, 1.399(3); S2–C1, 1.663(3); P1–Au1–S1, 175.36(2); C1– S1–Au1, 100.04(9); C9–P1–Au1, 118.20(8); S2–C1–S1, 124.85(15).
(Diphosphino)ethanegold(I) Dithiocarbamato Complexes (4–6)
Complexes 4–6 (Scheme 2) were prepared from the reaction of [Au2Cl2(dppe)] with 2 equiv of L1–L3 and were isolated as flaky yellow solids in moderate yields. In the 1H NMR spectra of 4–6, the CH2 protons of the di(phosphino)alkane ligand appeared upfield (1.8–2.9 ppm) as second-order multiplets, and the 13C{1H} NMR spectra showed peaks at ca. 215.0 and ca. 129.2–133.5 ppm, which were assigned to C(C=S) and the phenyl carbons, respectively; the 31P{1H} NMR spectra showed broad singlets between 29.0 and 33.0 ppm. The spectroscopic data for 4–6 are similar to the data for the bis(diphenylphosphino)digold(I) cations with azotate neutral heterocycles [μ-(dppe)Au2(pzH)2]2ClO4 and [μ-dppmAu2(pzH)2]2ClO4 (pzH = pyrazole).45
Scheme 2. Preparations of (Diphosphino)alkylgold(I) Dithiocarbamato Complexes 4–12.

Attempts to grow crystals of compound 4 were unsuccessful. On prolonged standing, a solution of 4 in a mixture of dichloromethane and diethylether at room temperature led to the isolation of the chloride salt of the cationic gold(I). cluster [Au18S8(dppe)6]2+ (4a), whose molecular structure was established by single crystal X-ray crystallography (Table 1, Figure 2). The molecular structure of 4a contains 18 gold atoms, 8 sulfur atoms, and 6 dppe units, with 2 chloride counterions and some unidentified solvents of crystallization (see Supporting Information for more details). It is isomorphous with the Se analogue [Au18Se8(dppe)6]2+.43 The geometry of 4a is similar to that of the selenium analogue [Au18Se8(dppe)6]2+.43 At the core of the cluster is a Au6S2 cubane composed of atoms Au1–Au6, S1, and S2 in such a fashion that each face contains three Au and one S atoms; the S atoms are positioned across the body diagonal of the cubane. The three Au atoms of each face are capped with a Au2S triangle. Each S atom is μ3-bridging: atoms S1 and S2 coordinate to three Au atoms of the cubane, whereas the other S atoms ligate one Au atom from the cubane and two Au atoms from a capping Au2S triangle. Each of the Au atoms in the capping triangles is ligated by one P atom of the six bidentate dppe ligands. The Au–Au distances vary between 2.9147(7) and 3.2838(8) Å; the Au–S bond length falls within a 2.328(4)–2.374(3) Å range, and the 12 Au–P distances average 2.260(7) Å, a value similar to that in compound 3. Other high nuclearity gold(I) sulfide complexes with bridging diphosphines have been reported.46−48 As for the neutral hydrated cluster [Au18S8(dppe)6]·H2O,32 its formulation implies a mixed-valent Au0–AuI compound, contrary to our findings and unlike the related cluster [Au18Se8(dppe)6]2+.43,48 The formation of 4a is unique because dithiocarbamate as a sulfur source is clearly unusual in forming high-nuclearity clusters. The formation of 4a from 4 is likely to occur via a C–S bond cleavage, triggered by the heterocycle attached to the CS2 in the dithiocarbamate. This is supported by [Au(dithiocarbamate)(dppe)] complexes that are stable in solution and have solid-state structures where the dithiocarbamate ligands remain intact, for example, [Au2(S2CNEt2)2(dppe)],49 [Au2(S2CNEt2)2{μ-(PPh2)2C=CH2}],49 and[Au(S2CNC10H20O4)(dppe)] (where C10H20O4 = aza-15-crown-5).31 The dppe backbone and aurophilicity of the two Au atoms in 4 must have aided the cluster formation since increase in the alkyl chain length in the diphosphine gave stable diphosphinogold(I) dithiocarbamate complexes (vide infra).
Figure 2.

Molecular structure of 4a. H atoms are omitted for clarity. Selected bond lengths [Å] and angles [deg]: Au1–P1, 2.2533(6); Au1–S2, 2.343(3); Au1–S3, 2.368(4); Au1–Au2, 2.9263(7); Au1–Au17, 2.9452(7); Au1–Au7, 3.1395(7); Au7–P10, 2.268(4); S2–Au1–S3, 175.80(12); Au10–Au2–Au3, 77.409(18); S2–Au1–Au2, 92.05(8); S1–Au2–Au1, 109.71(8); P8–Au9–S4, 109.71(8).
(Diphosphino) Propyl and Hexyl Gold(I) Dithiocarbamato Complexes (7–12)
To avoid the cluster formation observed during crystallization of 4, the −CH2– linker count within the diphosphines was increased to three in dppp and six in dpph. Their respective diphosphinogold(I) dithiocarbamates (7–12) were synthesized from the reaction between [Au2Cl2(dppp)] or [Au2Cl2(dpph)] and 2 equiv of L1, L2, or L3 (Scheme 2). The 1H and 31P{1H} NMR spectra of 7–12 showed similar patterns as those of 4–6, with the CH2 protons in the backbone of the diphosphine ligands appearing upfield in the region of 1.49–2.93 ppm and singlets for phosphorus between 29.0 and 33.0 ppm. These complexes were stable in solution for several weeks, with no signs of decomposition. ESI-MS of 9 (m/z = 1190.88, Figure S1, Supporting Information) showed molecular ions that correspond to a binuclear complex with a molecular ion peak at m/z = 1190.88, and the crystal structure of 12 (Figure 3) further indicates that the length of the alkyl linker in the diphosphine ligand is important in preventing compounds 7–12 from transforming into clusters.
Figure 3.
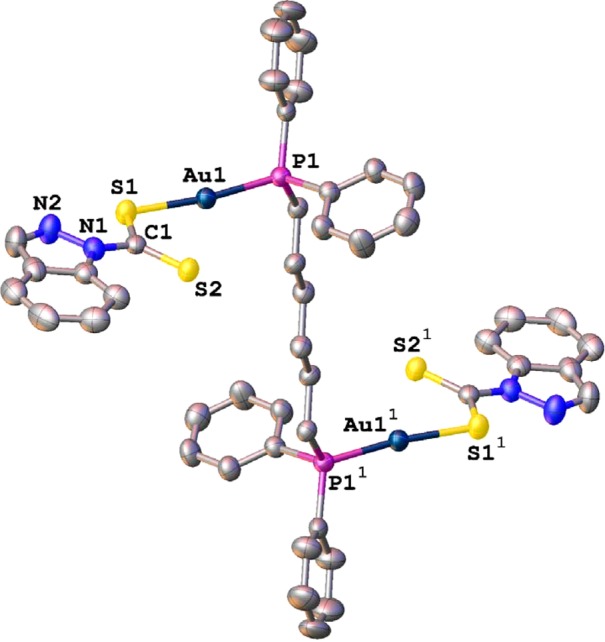
Molecular structure of 12 drawn with 50% probability ellipsoids. H atoms are omitted for clarity. Selected bond length [Å] and angles [deg]: Au1–P1, 2.255(2); Au1–S1,2.312(3); P1–C9, 1.818(6); N1–C8, 1.390(8); S1–C1, 1.720(7); S2–C1, 1.656(6); P1–Au1–S1, 173.41(6); C1–S1–Au1, 100.9(2); C15–P1–Au1, 112.6(8); S2–C1–S1, 124.1(4). Selected symmetry-related atoms are labeled with a superscript.
Crystal data, together with the data collection and refinement parameters, are presented in Table 1. The molecular drawing of 12 is shown in Figure 3. The dinuclear complex resides on a crystallographic inversion center, and only one-half of it is symmetry independent; the most important distances are discussed. The symmetry-independent half of complex 12 can be considered a congener of complex 3. In complex 12 the coordination environment about the Au atoms is nearly linear, with the P1–Au1–S1 angle spanning 173.41(6)°; the Au1–S1 (2.312(3) Å) and Au1–P1 (2.255(2) Å) distances are typical and in good agreement with the ones observed in complex 3. The single C1–S1 distance to the ligating atom S1 (1.720(7) Å) is longer than the formally double C1=S2 bond of 1.656(6) Å, but both values closely match the corresponding values in complex 3. In the lattice, pairs of clusters are packed with an inversion center at the center of the Au–S···Au–S parallelepiped. Within each parallelepiped, the nonbonding Au···Au distance measures 3.986(3) Å, and the Au···S distance is 3.598(4) Å. Both distances exceed the sum of the van der Waals radii of the involved elements; thus, there are no aurophilic interactions in the lattice. The structural findings for complex 12 are similar to those reported by Uson et al.50 and Cookson et al.51 for the gold(I) complexes [Au(PPh3)(S2C-aza-15-crown-5)] and [Au(C5H5NS)2], respectively. The other bonding distances and angles are in the same range and correspond with the expected values. Crystal data, together with the data collection and refinement parameters, are presented in Table 1.
Biological Activity
Fifteen compounds (L1–L3, 1–12) were initially screened for their ability to inhibit cell growth on human cervical epitheloid carcinoma (HeLa) cells in vitro. All data were acquired in triplicate, and the final values were recorded as averages. The dose values that caused 50% inhibition of cell growth (IC50) are listed in Table 2. To establish the activities of phosphine gold(I) complexes it was important to first establish the activities of the ligands (L1–L3) to determine whether activities of the metal complexes could be due to the presence of the dithiocarbamate ligands. All the free ligands were inactive against HeLa cells.
Table 2. Growth Inhibition Data of Complexes 1–3, 7–9, and 10–12 against HeLa Cells and Corresponding Tumor Specificities.
| drug | HeLa IC50 (μM) | lymph (resting) IC50 (μM) | lymph (stimulated) IC50 (μM) | tumor specificity (TS) | |
|---|---|---|---|---|---|
| PPh3 derivatives | 1 | 2.56 ± 0.12 | 9.05 ± 3.50 | 8.55 ± 3.37 | 3.4 |
| 2 | 2.63 ± 0.10 | 9.53 ± 3.18 | 8.63 ± 3.77 | 3.5 | |
| 3 | 2.56 ± 0.17 | 13.98 ± 10.50 | 5.08 ± 2.48 | 3.7 | |
| dppp derivatives | 7 | 3.07 ± 0.48 | 7.05 ± 2.02 | 5.02 ± 1.72 | 1.9 |
| 8 | 6.96 ± 0.51 | 6.13 ± 2.92 | 2.94 ± 0.91 | 0.7 | |
| 9 | 3.38 ± 0.30 | 18.48 ± 3.88 | 8.16 ± 4.10 | 3.9 | |
| dpph derivatives | 10 | 0.51 ± 0.08 | 18.89 ± 5.66 | 6.68 ± 3.40 | 25.0 |
| 11 | 0.14 ± 0.01 | 18.23 ± 15.90 | 1.38 ± 0.39 | 70.5 | |
| 12 | 4.04 ± 0.49 | 37.74 ± 12.60 | 30.25 ± 11.06 | 8.4 | |
| cis-platin | 0.45 ± 0.09 |
The gold(I) complexes were grouped into three sets according to ligand type, PPh3 (1–3), dppp (7–9), and dpph (10–12), for testing. Complexes 4–6 (the dppe set) were not tested because of the instability of 4 in solution discussed earlier. Although complexes 1–3, 7–9, and 12 had quite good IC50 values (2.2–7.0 μM), their tumor specificity (TS) values were very low (Table 2), and they were less active than cis-platin (0.476 μM). The low TS factors suggest that these compounds were toxic to both tumor and normal cells and were thus not further investigated. Compounds 10 and 11 had IC50 values of 0.51 μM and 0.14 μM, respectively, compared to 0.45 μM for cis-platin. These two gold compounds (10 and 11) displayed very good TS values of 25.0 and 70.5, respectively (Table 2). Our findings are comparable to and in some cases much better than activities reported for phosphine gold(I) thiolate complexes,12,13,23 which further buttresses the conception that a P–Au–S motif enhances the therapeutic effect of phosphine gold(I) thiolate compounds as anticancer agents. Complexes 10 and 11 were further investigated against a panel of 60 cancer cell lines of the Developmental Therapeutics Program (DTP) at the National Cancer Institute (U.S.A.), where the concentration that inhibits growth of cells by 50% (GI50), the concentration that causes total tumor growth inhibition (TGI), and the least concentration required to kill 50% of tumor cells (LC50) were determined. The 60 cell lines were organized into subpanels, representing various histologies, for example, nonsmall cell lung-, colon-, breast-, ovarian-, renal-, prostrate-, and CNS cancers, leukemia, and melanoma. Because of the extensive data, we have highlighted only the most important findings in Tables 3 and 4, but more data are provided as Supporting Information (Figures S2 and S3; Tables S1 and S2). Complexes 10 and 11 were, in general, active against all of the 60 cell lines in the subpanels mentioned above, with GI50 values in the range of 0.03–100 μM.
Table 3. Selected DTP Antitumor Inhibition Results of Complex 10.
| type of cancer | panel/cell line | GI50 (μM) | TGI (μM) | LC50 (μM) |
|---|---|---|---|---|
| leukemia | RPMI-8226 | 0.19 | 0.53 | >100 |
| SR | 0.19 | 2.41 | >100 | |
| nonsmall cell lung | A549/ATCC | 0.38 | >100 | >100 |
| colon | COLO205 | 0.31 | >100 | >100 |
| KM12 | 0.43 | 1.73 | >100 | |
| melanoma | UACC-62 | 0.38 | >100 | >100 |
| ovarian | OVCAR-3 | 0.70 | 14.20 | >100 |
| renal | RXF393 | 0.45 | >100 |
Table 4. Selected DTP Antitumor Inhibition Results of Complex 11.
| type of cancer | panel/cell line | GI50 (μM) | TGI (μM) | LC50 (μM) |
|---|---|---|---|---|
| leukemia | RPMI-8226 | 0.14 | 0.41 | >100 |
| SR | 0.03 | 0.52 | 90 | |
| nonsmall cell lung | A549/ATCC | 0.35 | >100 | >100 |
| colon | COLO205 | 0.11 | >100 | |
| HCC-2998 | 0.25 | 0.80 | 2.99 | |
| melanoma | UACC-62 | 0.25 | >100 | |
| ovarian | OVCAR-8 | 0.60 | >100 | >100 |
| renal | A498 | 0.38 | >100 |
Compound 10 showed good activity against all cell lines tested but was particularly highly active against the leukemia RPMI-8226 and SR cell lines, with a GI50 value of 0.19 μM for each cell line and TGI values of 0.53 and 2.41 μM for these cell lines, respectively (Table 3). This indicates the inhibitory property of 10 at very low molar concentrations. However, what was interesting is that the LC50 for 10 was greater than 100 μM for most cell lines, suggesting minimal cytotoxic properties of 10. It, therefore, implies that this complex is cytostatic. Similar results have been reported in literature, where a compound is cystostatic/antiproliferative and not necessarily cytotoxic.52
We observed a similar activity pattern for complex 11 against all the cell lines, but 11 was even more potent than 10. For instance, the GI50 values of 11 against the leukemia cell lines RPMI-8226 and SR were 0.14 μM and 0.03 μM, respectively. In fact, the best activity registered for all the compounds tested was that of 11 against the SR cell line (0.03 μM) (Table 4). The TGI concentrations recorded were similarly quite low, RPMI-8226 (0.41 μM) and SR (0.52 μM), which corroborates the cytostatic properties of 11. Of interest is that compound 11, unlike 10, showed very good cytotoxic activity against the HCC-2998 cell line (LC50 = 2.99 μM). Compound 11 also showed activity against the colon cancer COLO205 and HCC-2998 cell lines, with GI50 of 0.11 μM and 0.25 μM, respectively. In general, complexes 10 and 11 recorded high cytostatic median values (GI50 = −6.25; TGI = −4.51) and cytotoxic median value (LC50 = −4.05) against the 60 cell lines (Table 5, Figures S5 and S7, Supporting Information), which in the DTP program indicates high activity across most of the panel of cell lines.
Table 5. Cytostatic and Cytotoxic Median Data of 10 and 11.
| GI50 |
TGI |
LC50 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| drug | MG-MID | Δ | range | MG-MID | Δ | range | MG-MID | Δ | range |
| 10 | –5.93 | 0.8 | 2.73 | –4.36 | 1.92 | 2.28 | –4.02 | 0.69 | 0.71 |
| 11 | –6.25 | 1.22 | 1.96 | –4.51 | 1.87 | 2.38 | –4.05 | 1.47 | 1.52 |
Following on the in vitro results, compound 11 was further tested in vivo in hollow fiber studies, but the activity data suggested that the compound could not be delivered to the targets. We are therefore investigating the use of various drug delivery vehicles, including the use of β– and γ–cyclodextrin, to deliver this drug in vivo.
Conclusions
We have prepared the first examples of phosphinogold(I) dithiocarbamates derived from heterocycles. The stability of these phosphinogold(I) dithiocarbamate complexes depends on the nature of the phosphine ligand used. Triphenylphosphino and diphenylphosphinoalkyl ligands with alkyl chains longer than ethyl produce stable gold dithocarbamates in solution, but the diphenylphosphinoethanegold(I) dithiocarbamates are unstable and were found to transform to a Au18 cluster. All the phosphinogold(I) dithiocarbamates that are stable in solution are active against HeLa cancer cells, suggesting the importance of the P–Au–S moiety in conferring activity to the compounds. Compounds with hexyl chain were found to be the most active and extremely selective; in particular compounds 10 and 11 were 25.0 and 70.5 times more selective for HeLa cells than normal cells. Bis(diphosphines) alkanes with longer CH2 linkers appear to hold better promise as anticancer agents than their shorter CH2-linker counterparts, similar to the observation by Horvath et al.27 We also found compounds 10 and 11 to have excellent activities for nine other cancer cell lines in vitro. Of the nine cancer cell lines tested, the best activity against RPMI-8226 was found for 10 (GI50 = 0.19 μM), while the best activity against SR cells (GI50 = 0.03 μM) was for 11. Although activities for 10 and 11 in vivo were not so great, we believe finding drug delivery vehicles to transport these two compounds would improve their activities in vivo. We are therefore investigating the use of various delivery vehicles for these compounds.
Experimental Section
Materials and Instrumentation
All manipulations were performed under a dry, deoxygenated nitrogen atmosphere using Schlenk techniques. All commercially available chemicals were used as received. Pyrazol-1-yldithiocarbamate (L1), 3,5-dimethylpyrazol-1-yldithiocarbamate (L2), and indazol-1-yldithiocarbamate (L3) were synthesized according to literature methods.53,54 Gold starting materials [Au2Cl2(dppe)], [Au2Cl2(dppp)], and [Au2Cl2(dpph)] {dppe = 1,2-bis(diphenyl-phosphino)ethane; dppp = 1,3-bis(diphenylphosphino)propane; and dpph = 1,6-bis(diphenyl-phosphino)hexane} were synthesized according to the literature procedures.55 Infrared (IR) spectra were recorded as KBr pellets on a Bruker Tensor27 spectrophotometer. 1H, 13C{1H}, and 31P{1H} NMR spectra were recorded on a Varian 2000 spectrometer (1H, 300 MHz; 13C, 75.4 MHz; and 31P, 121.5 MHz) in CDCl3 or D2O at room temperature. Elemental analysis was performed on a Fisons elemental analyzer at the University of Cape Town, South Africa. ESI-MS spectra were recorded on a Waters API Quattro Micro spectrometer at the University of Stellenbosch, South Africa. The mass spectra were collected using 3.0 s cyclical scans and applying the sample cone voltage of 15 V at the source block temperature of 100 °C. Desolvation temperature was 350 °C at desolvation cone gas flow rate of 350 L/h.
Synthesis of Triphenylphosphinegold(I) Complexes
Pyrazolyl-1-dithiocarbamato-triphenylphosphinogold(I) (1)
Complex 1 was prepared by dissolving L1 (0.05 g, 0.3 mmol) in deionized water (10 mL), after which a solution of [AuCl(PPh3)] (0.15 g, 0.3 mmol) in dichloromethane (10 mL) was added. The resultant biphasic mixture was vigorously stirred at room temperature for 30 min, during which time the color of the organic layer changed from colorless to red. The aqueous and organic layers were separated, and the organic layer was dried over anhydrous MgSO4. The solvent was removed from the organic extract in vacuo to afford an orange-yellow solid. Yield = 0.17 g (93%). 1H NMR (CDCl3): δ 7.63 (s, 1H, 5-pz); 7.57 (s, 1H, 3-pz); 7.49 (m, 9H, PPh3); 7.35 (m, 6H, PPh3); 6.27 (s, 1H, 4-pz). 13C{1H} NMR (CDCl3): δ 213.4 (C(C=S)); 144.0 (C(5-pz); 134.2–129.1 (phenyl region); 140.8 (C(3-pz)); 106.1 (C(4-pz)). IR (KBr, cm–1): υC=N = 1620, υC–S = 1121, υC=S = 884. 31P{1H} NMR (CDCl3): δ 35.2 (PPh3). Anal. Calc. for C24H22AuN2PS2: C 43.86, H 3.01, N 4.65. Found: C 43.53, H 3.05, N 4.62%.
Compounds 2 and 3 were prepared using the procedure described for 1 above, using the reagents indicated for each compound.
3,5-dimethylpyrazolyl-1-dithiocarbamato-triphenylphosphinogold(I) (2)
L2 (0.1g, 0.47 mmol), [AuCl(PPh3)] (0.24 g, 0.47 mmol). Yield = 0.17 g (57%). 1H NMR (CDCl3): δ 7.44, 7.29 (m, 15H, PPh3); 6.12, 5.82 (s, 1H, 4-pz); 2.29 (m, 6H, 3, 5-pz). 13C{1H} NMR (CDCl3): δ 215.5 (C(C=S)); 149.7 (C(5-pz); 134.1–128.7 (phenyl region); 142.1 (C(3-pz)); 112.4, 104.1 (C(5-pz)); 13.9 (C(CH3, 5-pz); 12.31 (C(CH3, 3-pz). IR (KBr, cm–1): υC=N = 1580, υC–S = 1154, υC=S = 854. 31P{1H} NMR (CDCl3): δ 33.8 (PPh3). Anal. Calc. for C24H22AuN2PS2: C 45.72, H 3.52, N 4.44. Found: C 45.89, H 3.89, N 3.29%.
Indazolyl-1-dithiocarbamato-triphenylphosphinogold(I) (3)
L3 (0.1 g, 0.4 mmol), [AuCl(PPh3)] (0.2 g, 0.4 mmol). Yield = 0.15 g (58%). 1H NMR (CDCl3): δ 9.21 (d, 1H, 4JHH = 9.0 Hz, Ha); 8.17 (s, 1H, Hb); 7.73 (d, 1H, 3JHH = 8.4 Hz, He); 7.61, 7.50 (m, 15H, PPh3); (7.58 (1H, Hd); 7.35 (t, 1H, 4JHH = 15 Hz, Hc). 13C{1H} NMR (CDCl3): δ 214.2 (C(C=S)); 139.5 (C(5C-pz); 134.7–129.5 (phenyl region); 129.5 (C(3C-pz)); 126.9 (C(7C-Ph)); 124.6 (C(4C-Ph); 121.4 (C(8C, 9C-Ph)); 118.7 (C(6C-Ph)). IR (KBr, cm–1): υC=N = 1601, υC–S = 1148, υC=S = 905. 31P{1H} NMR (CDCl3): δ 36.8 (PPh3). Anal. Calc. for C26H20AuN2PS2: C 47.86, H 3.09, N 4.29. Found: C 47.41, H 3.04, N 3.85%.
Synthesis of Dinuclear Di(phosphino)alkylgold(I) Complexes
Bis-(pyrazolyl-1-dithiocarbamato)-bis-(diphenylphosphino)ethane Dinuclear Gold(I) (4) and [Au18S8(dppe)6]Cl2 (4a)
To a solution of [Au2Cl2(dppe)] (0.08 g, 0.09 mmol) in CH2Cl2 (10 mL) was added L1 (0.03 g, 0.19 mmol) previously dissolved in water (10 mL). The resultant mixture was stirred at room temperature for 20 min during which time the CH2Cl2 layer changed color from a white suspension to a clear orange solution, while the yellowish water layer became clear. The two layers were separated and the organic fraction dried over anhydrous MgSO4. The organic fraction was evaporated in vacuo to obtain 4 as an orange solid. Yield = 0.07 g (73%). 1H NMR (CDCl3): δ 8.78 (d, 2H, 3JHH = 2.7 Hz, 5-pz); 7.80 (s, 2H, 3-pz); 7.78 (m, 8H, (Ph)2P(CH2)2P(Ph)2); 7.49 (m, 12H, (Ph)2P(CH2)2P(Ph)2); 6.43 (s, 2H, 4-pz); 2.87 (bs, 4H, CH2, (Ph)2P(CH2)2P(Ph)2). 13C{1H} NMR (CDCl3): δ 214.2 (C(C=S)); 149.7 (C(5-pz); 142.0 (C(3-pz)); 134.1–128.7 (phenyl region); 111.4 (C(4-pz)); 24.6 (C(Ph2P(CH2)2PPh2). IR (KBr, cm–1): υC=N = 1590, υC–S = 1190, υC=S = 889. 31P{1H} NMR: (CDCl3): δ 36.1 (Ph2P(CH2)2PPh2). Anal. Calc. for C34H30Au2N4P2S4: C 37.85, H 2.80, N 5.14, S 11.89. Found: C 38.18, H 3.01, N 4.57, S 11.73%.
Attempts to crystallize 4a in a solution of dichloromethane/ether led to the formation of the gold cluster, [Au18S8(dppe)6]Cl2, (4a). Anal. Calc. for C26H20AuCl2N2PS2: C 29.71, H 2.32, N 4.10. Found: C 29.80, H 2.00, N 3.80%.
Compounds 5–12 were prepared using the procedure described for 4 above, using the appropriate starting materials.
Bis-(3,5-dimethylpyrazolyl-1-dithiocarbamato)-bis-(diphenylphosphino)ethane Dinuclear Gold(I) (5)
L2 (0.04 g, 0.19 mmol), [Au2Cl2(dppe)] (0.1 g, 0.09 mmol). Yield = 0.09 g (88%). 1H NMR (CDCl3): δ 7.64 (m, 8H, Ph2P(CH2)2PPh2); 7.35 (m, 12H, Ph2P(CH2)2PPh2); 6.09 (s, 2H, 4-pz); 2.69 (bs, 4H, Ph2P(CH2)2PPh2); 2.26 (d, 12H, CH3, 3,5-pz). 13C{1H} NMR (CDCl3): δ 216.5 (C(C=S)); 152.7 (C(5-pz); 143.1 (C(3-pz)); 135.1–128.6 (phenyl region); 112.6 (C(4-pz)); 14.0 (C(5-pPz); 12.4 (C(3-pz)); 25.7 (C(Ph2P(CH2)2PPh2). IR (KBr, cm–1): υC=N = 1603, υC–S = 1263, υC=S = 989. 31P{1H} NMR: (CDCl3): δ 35.7 (Ph2P(CH2)2PPh2). Anal. Calc. for C38H38Au2N4P2S4: C 40.22, H 3.37, N 4.94, S 11.30. Found: C 39.89, H 3.59, N 4.67, S 11.15%.
Bis-(indazolyl-1-dithiocarbamato)-bis-(diphenylphosphino)ethane Dinuclear Gold(I) (6)
L3 (0.04 g, 0.19 mmol), [Au2Cl2(dppe)] (0.08 g, 0.09 mmol). Yield = 0.07 g (63%). 1H NMR (CDCl3): δ 9.16 (s, 1H, Ha); 8.06 (s, 1H, Hb); 7.78 (m, 8H, (Ph)2P(CH2)2P(Ph)2); 7.49 (m, 12H, (Ph)2P(CH2)2P(Ph)2); 7.67 (d, 1H, 3JHH = 8.4 Hz, He); 7.71 (1H, Hd); 7.43 (t, 1H, 4JHH = 15 Hz, Hc); 2.87 (bs, 4H, CH2, (Ph)2P(CH2)2P(Ph)2). 13C{1H} NMR (CDCl2): δ 213.9 (C(C=S)); 142.1 (C(5-Pz); 139.0 (C(3-Pz)); 134.1–129.7 (phenyl region); 126.1 (C(7C-Ph)); 123.1 (C(4C-Ph)); 122.2 (C(8C-Ph, 9C-Ph)); 117.5 (C(6C-Ph) 25.2 (C(Ph2P(CH2)2PPh2). IR (KBr, cm–1): υC=N = 1612, υC–S = 1137, υC=S = 915. 31P{1H} NMR: (CDCl3): δ 34.9 (Ph2P(CH2)2PPh2). Anal. Calc. for C42H34Au2N4P2S4: C 42.79, H 2.91, N 4.75, S 10.88. Found: C 43.24, H 2.82, N 4.65, S 10.57%.
Bis(pyrazol-1-yldithiocarbamato)-bis-(diphenylphosphino)propane Dinuclear Gold(I) (7)
L1 (0.08 g, 0.46 mmol), [Au2Cl2(dppp)] (0.2 g, 0.23 mmol). Yield = 0.15 g (59%). 1H NMR (CDCl3): δ 8.73 (s, 2H, 3JHH = 2.7 Hz, 5-pz); 7.73 (m, 8H, Ph2P(CH2)3PPh2); 7.51 (s, 4H, 3JHH = 1.8 Hz, 3-pz); 7.41 (m, 12H, Ph2P(CH2)3PPh2); 6.41 (s, 2H, 3JHH = 2.7 Hz, 4-pz); 2.93 (m, 2H, Ph2PCH2CH2CH2PPh2); 1.99 (m, 4H, Ph2PCH2CH2CH2PPh2). 13C{1H} NMR (CDCl3): δ 218.9 (C(C=S)); 147.5 (C(5-pz); 140.8 (C(3-pz)); 133.2–129.2 (phenyl region); 106.2 (C(4-pz)); 28.5 (2C, 1JC–P = 62.7 Hz, P–CH2, Ph2PCH2CH2CH2PPh2); 21.4 (C, Ph2PCH2CH2CH2PPh2). IR (KBr, cm–1): υC=N = 1628, υC–S = 1088, υC=S = 891. 31P{1H} NMR (CDCl3): δ 32.8. Anal. Calc. for C35H32Au2N4P2S4: C 38.47, H 2.95, N 5.13, S 11.74. Found: C 38.99, H 3.08, N 5.09, S 11.45%.
Bis(3,5-dimethylpyrazol-1-yldithiocarbamato)-bis-(diphenylphosphino)propane Dinuclear Gold(I) (8)
L2 (0.1 g, 0.46 mmol), [Au2Cl2(dppp)] (0.2 g, 0.23 mmol). Yield = 0.13 g (50%). 1H NMR (CDCl3): δ 7.66 (m, 8H, Ph2P(CH2)3PPh2); 7.34 (m, 12H, Ph2P(CH2)3PPh2); 5.85 (s, 2H, 4-pz); 2.91 (m, 2H, Ph2PCH2CH2CH2PPh2); 1.88 (m, 4H, Ph2PCH2CH2CH2PPh2); 2.24, 2.26 (d, 12H, CH3, 3,5-pz). 13C{1H} NMR (CDCl3): δ 215.9 (C(C=S)); 153.3 (C(5-pz); 144.2 (C(3-pz)); 133.4–129.2 (phenyl region); 112.5 (C(4-pz)); 27.9 (2C, 1JC–P = 58.4 Hz, P–CH2, Ph2PCH2CH2CH2PPh2); 20.2 (C, Ph2PCH2CH2CH2PPh2); 13.8 (C(CH3, 5-pz); 13.2 (C(CH3, 3-pz). IR (KBr, cm–1): υC=N = 1653, υC–S = 1099, υC=S = 957. 31P{1H} NMR (CDCl3): δ 29.2. Anal. Calc. for C39H40Au2N4P2S4: C 40.77, H 3.51, N 4.88, S 11.16. Found: C 40.99, H 3.74, N 4.79, S 11.10%.
Bis(indazol-1-yldithiocarbamato)-bis-(diphenylphosphino)propane Dinuclear Gold(I) (9)
L3 (0.04 g, 0.22 mmol), [Au2Cl2(dppp)] (0.1 g, 0.11 mmol). Yield = 0.07 g (54%). 1H NMR (CDCl3): δ 9.13 (s, 1H, Ha); 8.16 (s, 1H, Hb); 7.73 (1H, Hd); 7.69 (d, 1H, 3JHH = 8.0 Hz, He); 7.66 (m, 8H, Ph2P(CH2)3PPh2); 7.45 (t, 1H, 4JHH = 16.8 Hz, Hc); 7.42 (m, 12H,Ph2P(CH2)3PPh2); 2.89 (m, 2H, Ph2PCH2CH2CH2PPh2); 1.92 (m, 4H, Ph2PCH2CH2CH2PPh2). 13C{1H} NMR (CDCl3): δ 213.8 (C(C=S)); 140.3 (C(5C-pz); 139.1 (C(3C-pz)); 133.5–129.2 (phenyl region); 127.9 (C(7C-Ph)); 124.2 (C(4C-Ph)); 121.1 (C(8C-Ph, 9C-Ph)); 118.3 (C(6C-Ph)); 28.6 (2C, 1JC–P = 64.2 Hz, P–CH2, Ph2PCH2CH2CH2PPh2); 19.8 (Ph2PCH2CH2CH2PPh2). IR (KBr, cm–1): υC=N = 1606, υC–S = 1150, υC=S = 993. 31P{1H} NMR (CDCl3): δ 29.2. ESI-MS: m/z 1190.9 [Au2(L3)2(dppp)]+ (5%). Anal. Calc. for C43H36Au2N4P2S4: C 43.29, H 3.04, N 4.70, S 10.75. Found: C 43.23, H 3.05, N 4.62, S 10.56%.
Synthesis of Dinuclear Bis(pyrazol-1-yldithiocarbamato)-bis-(diphenylphosphino)hexane Gold(I) (10)
L1 (0.02 g, 0.11 mmol), [Au2Cl2(dpph)] (0.05 g, 0.05 mmol). Yield = 0.04 g (70%). 1H NMR (CDCl3): δ 8.78 (d, 2H, 3JHH = 2.7 Hz, 5-pz); 7.80 (s, 2H, 3-pz); 7.78 (m, 8H, (Ph)2P(CH2)6P(Ph)2); 7.49 (m, 12H, (Ph)2P(CH2)6P(Ph)2); 6.43 (s, 2H, 4-pz); 2.87 (bs, 4H, Ph2P(CH2)2(CH2)2(CH2)2PPh2); 1.49 (bs, 8H, Ph2P(CH2)2(CH2)2(CH2)2PPh2). 13C{1H} NMR (CDCl3): δ 213.8 (C(C=S)); 140.3 (C(5C-pz); 139.1 (C(3C-pz)); 133.5–129.2 (phenyl region); 127.9 (C(7C-Ph)); 124.2 (C(4C-Ph)); 121.1 (C(8C-Ph, 9C-Ph)); 118.3 (C(6C-Ph)); 30.1 (4C, PPh2(CH2)2(CH2)2(CH2)2PPh2); 25.9 (2C, Ph2P(CH2)2(CH2)2(CH2)2PPh2). IR (KBr, cm–1): υC=N = 1620, υC–S = 1108, υC=S = 960. 31P{1H} NMR (CDCl3): δ 32.3. Anal. Calc. for C38H38Au2N4P2S4: C 40.22, H 3.37, N 4.94, S 11.30. Found: C 40.17, H 3.45, N 4.36, S 10.28%.
Synthesis of Dinuclear Bis(3,5-dimethylpyrazol-1-yldithiocarbamato)-bis-(diphenylphosphino)hexane Gold(I) (11)
L2 (0.09 g, 0.42 mmol), [Au2Cl2(dpph)] (0.2 g, 0.2 mmol). Yield = 0.12 g (50%). 1H NMR (CDCl3): δ 7.67 (m, 8H, Ph2P(CH2)6PPh2); 7.40 (m, 12H, Ph2P(CH2)6PPh2); 6.00 (s, 2H, 4-pz); 2.76 (bs, 4H, Ph2P(CH2)2(CH2)2(CH2)2PPh2); 2.18, 2.27 (d, 12H, CH3, 3,5-pz); 1.60, 1.52 (bs, 8H, Ph2P(CH2)2(CH2)2(CH2)2PPh2). 13C{1H} NMR (CDCl3): δ 215.9 (C(C=S)); 153.3 (C(5-pz); 144.2 (C(3-pz)); 133.4–129.2 (phenyl region); 112.5 (C(4-pz)); 31.2 (4C, PPh2(CH2)2(CH2)2(CH2)2PPh2); 26.7 (2C, Ph2P(CH2)2(CH2)2(CH2)2PPh2); 13.8 (C(5-pz); 13.2 (C(3-pz). IR (KBr, cm–1): υC=N = 1653, υC–S = 1099, υC=S = 957. 31P{1H} NMR (CDCl3): δ 32.7. Anal. Calc. for C42H46Au2N4P2S4: C 42.36, H 3.89, N 4.70, S 10.77. Found: C 42.12, H 3.84, N 4.62, S 10.56%.
Synthesis of Dinuclear Bis(indazol-1-yldithiocarbamato)-bis-(diphenylphosphino)hexane Gold(I) (12)
L3 (0.1 g, 0.44 mmol), [Au2Cl2(dpph)] (0.2 g, 0.22 mmol). Yield = 0.14 g (52%). 1H NMR (CDCl3): δ 9.18 (d, 1H, 4JHH = 8.4 Hz, (Ha)); 8.10 (s, 1H, Hb); 7.49 (d, 1H, 3JHH = 8.7 Hz, He); 7.24 (t, 1H, 4JHH = 15.0 Hz, Hd); 7.32 (t, 1H, 4JHH = 15.0 Hz, Hc); 7.66 (m, 8H, Ph2P(CH2)6PPh2); 7.42 (m, 12H, Ph2P(CH2)6PPh2); 2.39 (m, 4H, Ph2P(CH2)2(CH2)2(CH2)2PPh2); 1.57 (m, 8H, Ph2P(CH2)2(CH2)2(CH2)2PPh2). 13C{1H} NMR (CDCl3): δ 213.8 (C(C=S)); 140.3 (C(5C-pz); 139.1 (C(3C-pz)); 133.5–129.2 (phenyl region); 127.9 (C(7C-Ph)); 124.2 (C(4C-Ph)); 121.1 (C(8C-Ph, 9C-Ph)); 118.3 (C(6C-Ph)); 28.9 (4C, PPh2(CH2)2(CH2)2(CH2)2PPh2); 25.6 (2C, Ph2P(CH2)2(CH2)2(CH2)2PPh2). IR (KBr, cm–1): υC=N = 1657, υC–S = 1099, υC=S = 994. 31P{1H} NMR CDCl3): δ 31.8. Anal. Calc. for C46H42Au2N4P2S4: C 44.74, H 3.43, N 4.54, S 10.39. Found: C 44.37, H 3.46, N 4.50, S 10.32%.
Crystal Structure Determination
Crystals of 3 and 4a were mounted in oil on a glass fiber, and data collection was performed on a Bruker CCD-1000 diffractometer with Mo Kα (λ = 0.71073 Å) radiation at a diffractometer-to-crystal distance of 4.9 cm. The reflections were successfully indexed by an automated indexing routine built in the SMART program. The data were corrected for Lorentz and polarization effects. Intensity data for 12 were collected at 150 K on a D8 goniostat equipped with a Bruker APEXII CCD detector at beamline 11.3.1 at the Advanced Light Source (Lawrence Berkeley National Laboratory) using synchrotron radiation tuned to λ = 0.7749 Å. The data frames were collected using the program APEX2 and processed using the program SAINT routine within APEX2. Option SQUEEZE of program PLATON56 was used to correct the diffraction data for diffuse scattering effects and to identify the solvate molecule.
The absorption correction for the all three complexes was based on fitting a function to the empirical transmission surface as sampled by multiple equivalent measurements. A successful solution by the direct methods provided all non-hydrogen atoms from the E-map. All non-hydrogen atoms were refined with anisotropic displacement coefficients. All hydrogen atoms were included in the structure factor calculation at idealized positions and were allowed to ride on the neighboring atoms with relative isotropic displacement coefficients.57
Testing of Compounds for Anticancer Activity
Biological Reagents and Instrumentation
All commercial reagents were used as received. Phosphate-buffered saline (PBS), Eagle’s RPMI-1640 medium, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) kit, phytohemagglutinin-protein form (PHA-P) and the 96-well flat-bottomed culture plates were all purchased from BD Biosciences Ltd. Cis-platin was purchased from Sigma Aldrich. Eagle’s medium with 0.1 mM nonessential amino acids was prepared by adding 2 mM l-glutamine, 1.0 mM sodium pyruvate, and 5% bovine fetal calf serum to the pure Eagle’s RPMI-1640 medium. A total of fifteen compounds (L1–L3, 1–3, and 7–12) were screened for their anticancer activities. Human cervix epithelial carcinoma (HeLa) cells and human lymphocytes (PBMCs), from preservative-free heparinised peripheral blood, were obtained from the Department of Pharmacology and Pretoria Medical Hospital, University of Pretoria, South Africa. The absorbance values were recorded on a Whittaker Microplate Reader 2001 spectrophotometer at 570 nm and the reference wavelength of 630 nm.
Two complexes (10 and 11) were further tested against the 60-cell-line panel at the DTP using their internal procedures.58,59 From this study, three important (cytostatic and cytotoxic) properties are evident, namely, cytostatic values that include molar concentrations of drug required for 50% growth inhibition (GI50), cytostatic values that include molar concentrations of drug required for total growth inhibition (TGI), and cytostatic values that include molar concentrations of drug required to kill 50% of the cell population (LC50).
Cell Culture and Drug Treatment
HeLa cells were cultured in Eagle’s medium with 0.1 mM nonessential amino acids, 2 mM l-glutamine, 1.0 mM sodium pyruvate, and 5% bovine fetal calf serum at 37 °C in an atmosphere of 5% CO2. Cells were placed in 96-well sterile plates, at a density of 1 × 104 cells/well in 100 μL of medium, and incubated for 1 h. Subsequently, the ligands or gold compounds were added, with concentrations ranging from 0 to 100 μM. Cytotoxicity was determined by using MTT to stain treated HeLa cells after 7 d, according to literature methods.60 MTT dye is reduced by living cells to yield a soluble formazan product that can be assayed colorimetrically.60 A 20 μL volume of freshly prepared MTT (5 mg/mL) was added to each well, and the cells were incubated for another 4 h. Cell survival was evaluated by measuring absorbance at 570 nm, using a Whittaker Microplate Reader 2001. All experiments were performed in triplicate.
The inhibition of the growth of normal cells by the complexes tested was also measured by employing human lymphocytes (PBMC) cells. The same procedure described above was used, except that the treated PBMC cells were incubated for 3 d as opposed to 7 d for HeLa cells. The aim of testing these compounds on normal cells was to determine whether the compounds could target the cancerous (HeLa) cells specifically and not the normal cells. Lymphocytes were divided into two groups, namely, (i) normal cells that were stimulated using PHA-P so as to increase their proliferation rate (stimulated lymphocytes) and (ii) unstimulated normal cells (resting lymphocytes).
Acknowledgments
We acknowledge financial support for this work from Harmony Gold under project AuTEK, run by Mintek (South Africa), and University of Johannesburg. Crystallographic data were collected through the SCrALS (Service Crystallography at Advanced Light Source) program at the Small-Crystal Crystallography Beamline 11.3.1 at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. The ALS is supported by the U.S. Department of Energy, Office of Energy Sciences Materials Sciences Division, under contract DE-AC02-05CH11231. The authors would also like to thank the National Cancer Institute (U.S.A.) for the anticancer data generated under their DTP program and Dr. Frederik H. Kriel of Mintek who arranged for samples to be tested at the NCI.
Supporting Information Available
Crystallographic information data (CIF); ESI-MS of 9; biological data for compounds 11 and 12. This material is available free of charge via the Internet at http://pubs.acs.org. The deposition numbers CCDC 680273, 949434, and 920397 contain the supplementary crystallographic data for compounds 3, 4a, and 12, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Author Present Address
¶ School of Chemistry and Physics, University of KwaZulu-Natal, Westville Campus, Private Bag X54001, Durban, 4000, South Africa.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bindoli A.; Rigobello M. P.; Scutari G.; Gabbiani C.; Casini A.; Messori L. Coord. Chem. Rev. 2009, 253, 1692. [Google Scholar]
- Tiekink E. R. T. Crit. Rev. Oncol./Hematol. 2002, 42, 225. [DOI] [PubMed] [Google Scholar]
- Ronconi L.; Giovagnini L.; Marzano C.; Bettio F.; Graziani R.; Pilloni G.; Fregona D. Inorg. Chem. 2005, 44, 1867. [DOI] [PubMed] [Google Scholar]
- Tiekink E. R. T. Inflammopharmacology 2008, 16, 138. [DOI] [PubMed] [Google Scholar]
- Buac D.; Schmitt S.; Ventro G.; Kona F. R.; Dou P. Q Mini-Rev. Med. Chem. 2012, 12, 1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saggioro D.; Rigobello M. P.; Paloschi L.; Folda A.; Moggach S. A.; Parsons S.; Ronconi L.; Fregona D.; Bindoli A. Chem. Biol. 2007, 14, 1128. [DOI] [PubMed] [Google Scholar]
- Casini A.; Messori L. Curr. Top. Med. Chem. 2011, 11, 2647. [DOI] [PubMed] [Google Scholar]
- Kouodom M. N.; Ronconi L.; Celegato M.; Nardon C.; Marchio L.; Dou Q. P.; Aldinucci D.; Formaggio F.; Fregona D. J. Med. Chem. 2012, 55, 2212. [DOI] [PubMed] [Google Scholar]
- Kouodom M. N.; Boscutti G.; Celegato M.; Crisma M.; Sitran S.; Aldinucci D.; Formaggio F.; Ronconi L.; Fregona D. J. Inorg. Biochem. 2012, 117, 248. [DOI] [PubMed] [Google Scholar]
- a Cox A. G.; Brown K. K.; Arner E. S.; Hampton M. B. Biochem. Pharmacol. 2008, 76, 1097. [DOI] [PubMed] [Google Scholar]; b Rigobello M. P.; Scutari G.; Boscolo R.; Bindoli A. Br. J. Pharmacol. 2002, 136, 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Simon T. M.; Kunishima D. H.; Vibert G. J.; Lorber A. Cancer 1979, 44, 1965. [DOI] [PubMed] [Google Scholar]
- a Ott I. Coord. Chem. Rev. 2009, 253, 1670. [Google Scholar]
- Gandin V.; Fernandes A. P.; Rigobello M. P.; Dani B.; Sorrentino F.; Tisato F.; Bjornstedt M.; Bindoli A.; Sturaro A.; Rella R.; Marzano C. Biochem. Pharmacol. 2010, 79, 90. [DOI] [PubMed] [Google Scholar]
- a de Vos D.; Ho S. Y.; Tiekink E. R. T. Bioinorg. Chem. Appl. 2004, 2, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tiekink E. R. T. Bioinorg. Chem. Appl. 2003, 1, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabelli C. K.; Johnson R. K.; Hill D. T.; Faucette L. F.; Girard G. R.; Kuo G. Y.; Sung C.; Crooke S. T. J. Med. Chem. 1986, 29, 218. [DOI] [PubMed] [Google Scholar]
- McKeage M. J.; Maharaj L.; Berners-Price S. J. Coord. Chem. Rev. 2002, 232, 127. [Google Scholar]
- Rigobello M. P.; Scutari G.; Folda A.; Bindoli A. Biochem. Pharmacol. 2004, 67, 689. [DOI] [PubMed] [Google Scholar]
- Barnard P. J.; Berners-Price S. J. Coord. Chem. Rev. 2007, 251, 1889. [Google Scholar]
- Saccoccia F.; Angelucci F.; Boumis G.; Brunori M.; Miele A. E.; Williams D. L.; Bellelli A. J. Inorg. Biochem. 2012, 108, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigobello M. P.; Messori L.; Marcon G.; Cinellu M. A.; Bragadin M.; Folda A.; Scutari G.; Bindoli A. J. Inorg. Biochem. 2004, 98, 1634. [DOI] [PubMed] [Google Scholar]
- Urig S.; Fritz-Wolf K.; Réau R.; Herold-Monde C.; Tóth K.; Davioud-Charvet E.; Becker K. Angew. Chem., Int. Ed. 2006, 45, 1881. [DOI] [PubMed] [Google Scholar]
- Pillarsetty N.; Katti K. K.; Hoffman T. J.; Volkert W. A.; Katti K. V.; Kamei H.; Koide T. J. Med. Chem. 2003, 46, 1130. [DOI] [PubMed] [Google Scholar]
- Barreiro E.; Casas J. S.; Couce M. D.; Sanchez A.; Sanchez-Gonzalez A.; Sordo J.; Varela J. M.; Lopez E. M. V. J. Inorg. Biochem. 2008, 102, 184. [DOI] [PubMed] [Google Scholar]
- Casas J. S.; Castellano E. E.; Couce M. D.; Ellena J.; Sanchez A.; Sordo J.; Taboada C. J. Inorg. Biochem. 2006, 100, 1858. [DOI] [PubMed] [Google Scholar]
- Casas J. S.; Castellano E. E.; Couce M. D.; Crespo O.; Ellena J.; Laguna A.; Sanchez A.; Sordo J.; Taboada C. Inorg. Chem. 2007, 46, 6236. [DOI] [PubMed] [Google Scholar]
- Cagnoli M.; Alama A.; Barbieri F.; Novelli F.; Bruzzo C.; Speratore F. Anti-Cancer Drugs 1998, 9, 603. [DOI] [PubMed] [Google Scholar]
- Mirabelli C. K.; Hill D. T.; Faucette L. F.; McCabe F. L.; Girard G. R.; Byran D. B.; Sutton B. M.; Bartus J. O.; Crooke S. T.; Johnson R. K. J. Med. Chem. 1987, 30, 2181. [DOI] [PubMed] [Google Scholar]
- Horvath U. E. I.; Dobrzańska L.; Strasser C. E.; Bouwer (neé Potgieter) W.; Joone G.; van Rensburg C. E. J.; Cronje S.; Raubenheimer H. G. J. Inorg. Biochem. 2012, 111, 80. [DOI] [PubMed] [Google Scholar]
- Keter F. K.; Darkwa J. BioMetals 2012, 25, 9. [DOI] [PubMed] [Google Scholar]
- Thangadurai A.; Minu M.; Wakode S.; Agrawal S.; Narasimhan B. Med. Chem. Res. 2012, 21, 1509. [Google Scholar]
- a Knight E. R.; Leung N. H.; Thompson A. L.; Hogarth G.; Wilton-Ely J. D. E. T. Inorg. Chem. 2009, 48, 3866. [DOI] [PubMed] [Google Scholar]; b Naeem S.; Delaude L.; White A. J. P.; Wilton-Ely J. D. E. T. Inorg. Chem 2010, 49, 1784. [DOI] [PubMed] [Google Scholar]; c Oliver K.; White A. J. P.; Hogarth G.; Wilton-Ely J. D. E. T. Dalton Trans. 2011, 40, 5852. [DOI] [PubMed] [Google Scholar]
- Arias J.; Bardaji M.; Espinet P. Inorg. Chem. 2008, 47, 1597. [DOI] [PubMed] [Google Scholar]
- Fernandez E. J.; Lopez-de-Luzuriaga J. M.; Monge M.; Olmos E.; Gimeno M. C.; Laguna A.; Jones P. G. Inorg. Chem. 1998, 37, 5532. [DOI] [PubMed] [Google Scholar]
- Wijnhoven J. G.; Bosman W. J. P.; Beurskens P. T. J. J. Cryst. Mol. Struct. 1972, 2, 7. [Google Scholar]
- Guzei I. A.; Spencer L. C.; Lillywhite S.; Darkwa J. Acta Crystallogr., Sect. E: Struct. Rep. Online 2011, 67, m1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscutti G.; Marchio L.; Ronconi L.; Fregona D. Chem.—Eur. J. 2013, 19, 13428. [DOI] [PubMed] [Google Scholar]
- a Nagy E. M.; Ronconi L.; Nardon C.; Fregona D. Mini-Rev. Med. Chem. 2012, 12, 1216. [DOI] [PubMed] [Google Scholar]; b Hogarth G. Mini-Rev. Med. Chem. 2012, 12, 1205. [DOI] [PubMed] [Google Scholar]
- Marya A.; Mamba S.; Yang X.-H.; Darkwa J.; Kumar P.; Narain R. Bioconjugate Chem. 2013, 24, 979. [DOI] [PubMed] [Google Scholar]
- Jamaludin N. S.; Goh Z.-J.; Cheah Y. K.; Ang K.-P.; Sim J. H.; Khoo C. H.; Fairuz Z. A.; Halim B. A. S. N.; Ng S. W.; Seng H.-L.; Tiekink E. R. T. Eur. J. Med. Chem. 2013, 67, 127. [DOI] [PubMed] [Google Scholar]
- Boscutti Giulia Feltrin L.; Lorenzon D.; Sitran S.; Aldinucci D.; Ronconi L.; Fregona D. Inorg. Chim. Acta 2012, 393, 304. [Google Scholar]
- Beraldo H.; Gambino D. Mini-Rev. Med. Chem. 2004, 4, 159. [Google Scholar]
- Lessa J. A.; Guerra J. C.; de Miranda L. F.; Romeiro C. F. D.; Da Silva J. G.; Mendes I. C.; Speziali N. L.; Souza-Fagundes E. M.; Beraldo H. J. Inorg. Biochem. 2011, 105, 1729. [DOI] [PubMed] [Google Scholar]
- Bates P. A.; Waters J. M. Acta Crystallogr. 1987, A43, C194. [Google Scholar]
- a Fenske D.; Langetepe T.; Kappes M. M.; Hampe O.; Weis P. Angew. Chem., Int. Ed. 2000, 39, 1857. [DOI] [PubMed] [Google Scholar]; b Sevillano P.; Fuhr O.; Hampe O.; Lebedkin S.; Neiss C.; Ahlrichs R.; Fenske D.; Kappes M. M. Eur. J. Inorg. Chem. 2007, 5163.and references therein.. [DOI] [PubMed] [Google Scholar]
- Allen F. H. Acta Crystallogr. 2002, B58, 380. [DOI] [PubMed] [Google Scholar]
- Bachechi F.; Burini A.; Galassi R.; Pietroni B. R.; Severini M. J. Organomet. Chem. 1999, 575, 269. [Google Scholar]
- Chen J.; Mohamed A. A.; Abdou H. E.; Bauer J. A. K.; Fackler J. P. Jr; Bruce A. E.; Bruce M. R. M. Chem. Commun. 2005, 1575. [DOI] [PubMed] [Google Scholar]
- a Yam V. W.-W.; Cheng E. C.-C.; Cheung K. K. Angew Chem., Int. Ed. 1999, 38, 197. [Google Scholar]; b Yam V. W.-W.; Cheng E. C.-C.; Zhou Z.-Y. Angew. Chem., Int. Ed. 2000, 39, 1683. [DOI] [PubMed] [Google Scholar]; c Yam V. W.-W.; Cheng E. C.-C.; Zhu N. New J. Chem. 2002, 26, 279. [Google Scholar]
- For an overview of multinuclear gold(I) complexes, see:Yam V. W.-W.; Cheng E. C.-C. Angew. Chem., Int. Ed. 2000, 39, 4240. [DOI] [PubMed] [Google Scholar]
- Fernandez E. J.; Gimeno M. C.; Jones P. G.; Laguna A.; Lopez-de-Luzuriaga J. M.; Monge M.; Olmos M. E. Inorg. Chem. 1998, 37, 5532. [DOI] [PubMed] [Google Scholar]
- Uson R.; Laguna A.; Laguna M.; Jimenez J.; Gomez M. P.; Sainz A.; Jones P. G. J. Chem. Soc., Dalton Trans. 1990, 3457. [Google Scholar]
- Cookson P. D.; Tiekink E. R. T. J. Chem. Soc., Dalton Trans. 1993, 259. [Google Scholar]
- Fonteh P. N.; Keter F. K.; Meyer D. J. Inorg. Biochem. 2011, 105, 1173. [DOI] [PubMed] [Google Scholar]
- Trofimenko S. J. J. Org. Chem. 1968, 33, 890. [Google Scholar]
- Keter F. K.; Nell M. J.; Guzei I. A.; Omondi B.; Darkwa J. J. Chem. Res. 2009, 322. [Google Scholar]
- Bruce I.; Nicholson B. K.; Bin Shawkataly O. Inorg. Synth. 1989, 324. [Google Scholar]
- Spek A. L. Acta Crystallogr. 1990, A46, C34. [Google Scholar]
- Bruker-AXS SADABS V.2.05, SAINT V.6.22, SHELXTL V.6.10 & SMART 5.622 Software Reference Manuals; Bruker-AXS: Madison, WI, 2000. [Google Scholar]
- a Paull D. K.; Shoemaker R. H.; Hodes L.; Monks A.; Scudiero D. A.; Rubinstein L.; Plowman J.; Boyd M. R. J. Natl. Cancer Inst. 1989, 81, 1088. [DOI] [PubMed] [Google Scholar]; b Boyd M. R., DeVita V. T., Hellmann S., Rpsemberg S. A., Ed. Cancer: Principles and Practices of Oncology Updates, Vol. 3; Phildelphia, PA, 1989, p 1. [Google Scholar]
- Monks A.; Scudiero D.; Skehan P.; Shoemaker R.; Paull K.; Vistica D.; Hose C.; Langley J.; Cronise P.; Vaigro-Wolff A.; Gray-Goodrich M.; Campbell H.; Mayo J.; Boyd M. J. Natl. Cancer Inst. 1991, 83, 757. [DOI] [PubMed] [Google Scholar]
- Mossman T. J. Immunol. Methods 1983, 65, 55.6606682 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.