

Abstract
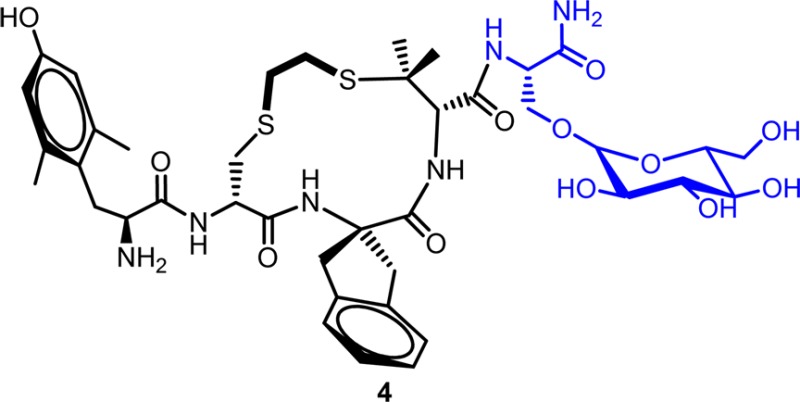
We have previously described a cyclic tetrapeptide, 1, that displays μ opioid receptor (MOPr) agonist and δ opioid receptor (DOPr) antagonist activity, a profile associated with a reduced incidence of opioid tolerance and dependence. Like many peptides, 1 has poor bioavailability. We describe here an analogue of 1 with an added C-terminal β-glucosylserine residue, Ser(β-Glc)NH2, a modification that has previously been shown to improve bioavailability of opioid peptides. The resulting peptide, 4, exhibits full antinociceptive efficacy in the mouse warm water tail withdrawal assay after intraperitoneal administration with potency similar to that of morphine. Further, 4 does not give rise to acute tolerance and thus represents a promising lead for the development of opioid analgesics with reduced side effects.
Introduction
The growing recognition that multifunctional ligands simultaneously acting at multiple targets may yield a more desirable drug profile than selectively targeted drugs has opened a new approach for the development of therapeutics.1−3 For opioid analgesics this is exemplified by the observation that coadministration of a μ opioid receptor (MOPr) agonist with a δ opioid receptor (DOPr) antagonist retains MOPr-mediated analgesia but reduces development of tolerance and dependence,4−6 features that hamper the clinical use of opioid analgesics. For pharmacokinetic simplicity it is preferable to incorporate both activities into a single compound, and the development of bifunctional opioid ligands has thus become a topic of increasing interest. Peptide,7,8 peptidomimetic,9−11 and alkaloid12 structures have been reported that display a MOPr agonist/DOPr antagonist (MOPr(ag)/DOPr(ant)) profile. The best studied of these are Schiller’s peptide DIPPψNH2 8 and Balboni’s peptidomimetic, UFP-505.9,10 Consistent with the expectations for a compound with a MOPr(ag)/DOPr(ant) profile, DIPPψNH2 was reported to produce reduced tolerance compared to morphine and no dependence after intracerebroventricular (icv) infusion;8 however, its therapeutic potential is compromised by its poor blood–brain barrier (BBB) penetration.13 UFP-505, on the other hand, did give rise to the development of tolerance after icv administration and displayed significant toxicity (G. Balboni, personal communication). We have previously described a cyclic tetrapeptide KSK-103 (1, Figure 1) that shows an improved in vitro profile compared with DIPPψNH2 and UFP-505; of the three ligands, only 1 demonstrated equal high affinity for MOPr and DOPr, much lower affinity for the κ opioid receptor (KOPr), and high efficacy and potency at MOPr with no stimulation of DOPr.14 However, like the previously reported ligands and like most peptides, 1 has poor bioavailability.
Figure 1.

Structure of lead MOPr (ag)/DOPr (ant) peptide 1 (KSK103).
Several approaches have been developed to increase stability and peptide penetration of biological membranes.15−17 In particular, Polt and co-workers have shown that in many cases glycosylation of opioid peptides affords improved metabolic stability and CNS activity after peripheral administration.17−21 We report here the observation that side chain glycosylation of a C-terminal SerNH2 extension of 1 results in a peptide that retains the desirable in vitro profile of 1 while displaying centrally mediated antinociception after intraperitoneal (ip) administration. Further, the resulting glycosylated peptide does not give rise to acute tolerance and thus represents a lead toward the development of opioid analgesics with lessened side effects.
Results
In Vitro Profile of Analogues of 1
Our approach toward peptide glycosylation, following that of Polt and co-workers, was via the side chain hydroxyl moiety of a serine residue. Accordingly, we first examined the effect of C-terminal extension of 1 with an unmodified serine residue to determine its possible effect on the in vitro profile of the lead peptide. As presented in Table 1, which summarizes opioid receptor binding affinities and efficacies relative to standard full agonists (the latter as stimulation of [35S]GTPγS binding), compounds 2 and 3, the C-terminal Ser carboxylic acid and carboxamide extension of 1, respectively, display in vitro profiles generally analogous to that of 1: similar MOPr and DOPr affinities, with reduced KOPr affinity; partial agonist activity at MOPr but no stimulation of DOPr or KOPr. While 3 displayed lower maximum stimulation in the [35S]GTPγS binding assay than compound 2 (17% vs 61% of control DAMGO), its higher potency and binding affinity led us to choose 3 for glycosylation with β-d-glucose. As seen in Table 1, the resulting glycosylated analogue, 4, exhibits a very similar in vitro profile to the original lead peptide 1. The only significant difference is the somewhat lower potency of 4 at MOPr (EC50 of 36.9 nM vs 4.7 nM). Like 1, 4 was confirmed to be an antagonist at DOPr by examining its effect on stimulation of [35S]GTPγS binding by the DOPr agonist DPDPE. The Ke (equilibrium dissociation constant, a measure of antagonist affinity) observed for 4 (6.1 nM, Table 1) is very similar to that previously reported for 1 (4.4 nM).14 The promising in vitro profile of 4 coupled with the anticipated bioavailability improvement resulting from glycosylation led us to examine the in vivo, antinociceptive activity of this analogue.
Table 1. Binding and Efficacy Results for 1 and Analoguesa.
|
Ki (nM) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| sequence | MOPr | DOPr | KOPr | efficacy (% of control), MOPr | EC50 (nM), MOPr | efficacy (% of control), DOPr | Ke (nM), DOPr | efficacy (% of control), KOPr | |
| 1b | Dmt-c(SEtS)[DCys-Aci-DPen]OH | 2.4 ± 0.7 | 2.3 ± 0.5 | 776 ± 149 | 59 ± 11 | 4.7 ± 0.7 | dns | 4.4 ± 1.4 | dns |
| 2 | Dmt-c(SEtS)[DCys-Aic-DPen]SerOH | 10.3 ± 2.3 | 4.5 ± 1.0 | 7100 ± 1260 | 61.4 ± 8.7 | 35.0 ± 12 | dns | dns | |
| 3 | Dmt-c(SEtS)[DCys-Aic-DPen]SerNH2 | 0.53 ± 0.07 | 0.93 ± 0.11 | 93.4 ± 1.4 | 17.3 ± 1.2 | 3.4 ± 1.0 | dns | dns | |
| 4 | Dmt-c(SEtS)[DCys-Aic-DPen]Ser(Glc)NH2 | 1.74 ± 0.2 | 2.43 ± 0.34 | 420 ± 25 | 41.3 ± 2.8 | 36.9 ± 8.4 | dns | 6.1 ± 1.5 | dns |
Binding affinities (Ki) were obtained by competitive displacement of radiolabeled [3H]diprenorphine. Efficacy data were obtained using [35S]GTPγS binding assay. Efficacy is presented as percent maximal stimulation relative to standard agonists DAMGO (MOPr), DPDPE (DOPr), and U69,593 (KOPr) at 10 μM. DOPr equilibrium dissociation constant, Ke, was determined to confirm the antagonist activity of 1 and 4. All values are expressed as the mean ± SEM of three separate assays performed in duplicate. dns = does not stimulate.
From ref (14).
In Vivo Antinociception Activity of 4
The antinociceptive activity of 4 after ip administration was assessed in the mouse warm water tail withdrawal (WWTW) assay. The results are presented in Figure 2 as latency to tail withdrawal. Figure 2 shows that 4 exhibits a dose dependent antinociceptive response, achieving approximately 80% maximal effect (relative to the 20 s cutoff used) at 10 mg/kg, the highest dose used in this experiment. The time course for the antinociceptive action of 4 was examined at a higher dose of 32 mg/kg, as shown in Figure 3. As seen there, 4 displays a maximal antinociceptive effect between approximately 30 and 60 min after administration. The effect then diminishes, approaching baseline after 150 min. By comparison, morphine displays similar antinociceptive potency, with an approximately 2-fold longer duration of action.11 Figure 4 shows that the observed antinociceptive effect of 4 is mediated by opioid receptors; pretreatment with the nonselective opioid antagonist naltrexone (3.2 mg/kg, ip) completely blocks antinociception by 10 mg/kg 4.
Figure 2.

Antinociception as a function of dose observed for 4 in mouse warm water tail withdrawal assay following ip administration.
Figure 3.

Antinociception (mouse WWTW assay) time course following 32 mg/kg ip dose of 4.
Figure 4.

Latency to tail withdrawal observed for 4 (10 mg/kg, ip) with (right) and without (left) pretreatment with the opioid antagonist naltrexone.
We next examined whether acute tolerance develops to the antinociceptive effect of 4. For these experiments, mice were treated with a single dose of compound (either 4 or fentanyl), and when these initial antinociceptive effects dissipated, a second dose of compound was given to determine if previous exposure to each compound produced acute tolerance to the antinociceptive effects. The left-hand side of Figure 5 compares the antinociceptive time course after ip administration of 10 mg/kg 4 to that of vehicle. As seen in Figure 5, at this dose 4 reaches its maximum effect after 30 min, then diminishes to baseline at t = 90 min. By contrast no antinociception is observed in the vehicle treated mice. At t = 90 min both groups of mice were treated with 10 mg/kg 4, ip, and monitoring of the antinociceptive time course continued (right side of Figure 5). For this latter half of the experiment no difference was observed in the antinociceptive effects between mice previously treated with 4 or vehicle; therefore, previous exposure to 4 failed to produce acute tolerance. This was quantified by measuring the area under the curve (AUC) of the antinociceptive effects of 10 mg/kg 4 at t = 90 min (vehicle-treated AUC of 718 (±100) vs 4 AUC of 740 (±114)). No significant difference was observed in the antinociceptive effects of 4 in vehicle-pretreated and 4-pretreated mice (t = 0.14, p = 0.89). This is in contrast to the results observed for the potent MOPr agonist fentanyl, shown in Figure 6. The left side of Figure 6 compares the antinociceptive time course following ip administration of 0.3 mg/kg fentanyl (a dose determined to be equiefficacious to 10 mg/kg 4) to that of vehicle-treated mice. The time course observed for fentanyl was quite similar to that of 4 (Figure 5). After the antinociceptive effect of fentanyl had returned to baseline, both groups were then injected with 0.3 mg/kg fentanyl (right side of Figure 6). In contrast to the behavior displayed by 4, acute tolerance to fentanyl is clearly observed; the antinociception observed in the previously fentanyl treated group was greatly attenuated (by ∼50%) compared with the vehicle-pretreated mice. Acute tolerance was quantified by measuring AUC of the antinociceptive effects of 0.3 mg/kg fentanyl measured at t = 120 min (vehicle-treated AUC of 740 (±182) vs fentanyl-pretreated AUC of 236 (±66)). The antinociceptive effects of fentanyl were smaller in magnitude and/or shorter in duration as measured in fentanyl-pretreated mice compared with drug-naive mice (t = 2.6, p = 0.04).
Figure 5.

Determination of acute tolerance to antinociceptive effect (mouse WWTW assay) of 4. Mice were treated with 10 mg/kg 4 (closed squares) or saline (open circles), and the time course of the antinociceptive response was determined. At t = 90 min (indicated by dashed vertical line) both groups of mice were injected with 10 mg/kg 4 and antinociceptive response was again examined.
Figure 6.

Determination of acute tolerance to antinociceptive effect (mouse WWTW assay) of fentanyl. Mice were treated with 0.3 mg/kg fentanyl (closed squares) or saline (open circles), and the time course of the antinociceptive response was determined. At t = 120 min (indicated by dashed vertical line) both groups of mice were injected with 0.3 mg/kg fentanyl, and antinociceptive response was again examined.
Discussion
Although compound 1 displays an ideal MOPr (ag)/DOPr (ant) in vitro profile, like many peptides, it has poor bioavailability. The challenge then was to modify 1 in such a way that its desirable in vitro profile was maintained while its bioavailability was significantly improved. Our earlier proposed models for the docking of 1 to MOPr and DOPr14 suggested that C-terminal elongation would not signficantly affect binding to MOPr and DOPr, as the C-terminus points out toward solvent. Further, early work by Roques and colleagues had shown that extension of pentapeptide enkephalin analogues by a Ser or Thr residue maintained opioid binding character,22 and Polt and co-workers demonstrated that glycosylation of these Ser- and Thr-extended opioids could greatly improve bioavailability without jeopardizing the in vitro profile of the parent peptide.18 Thus, the observation that C-terminal extension of 1 by Ser (2 and 3) and subsequent glycosylation (4) had only modest effect on the in vitro activity was expected.
The in vivo antinociceptive activity of 4 further demonstrates the utility of peptide glycosylation. As seen in Figures 2 and 3, 4 displays effective antinociception after ip injection with a potency similar to that of morphine and with approximately half of morphine’s duration of action.11 Most significant is the finding (Figure 5) that acute tolerance to 4 was not observed, in contrast with the results seen for fentanyl (Figure 6), a widely used opiate analgesic with a similar duration of action. Acute tolerance observed with fentanyl in the present study is likely to be due to reversible changes at the receptor level,23 such as desensitization and internalization, compared to the more permanent events occurring after chronic administration. The role of acute processes in long-term tolerance is not confirmed, although morphine tolerance does appear to be associated with desensitization of the MOPr.24 If this holds true, the present results imply that the beneficial effects of the δ antagonist component of MOPr (ag)/DOPr (ant) compounds are effective early in the process of tolerance development. These results suggest that 4, or a related analogue with MOPr (ag)/DOPr (ant) activity, may be effective clinically as a safer opioid analgesic. Follow-up studies assessing the development of chronic tolerance and dependence after prolonged administration of 4 are in progress.
Experimental Section
Chemistry
All reagents and solvents were purchased from commercial sources (Sigma Aldrich (St. Louis, MO) or Fisher Scientific (Hudson, NH), unless otherwise noted) and used without further purification. Peptide synthesis reagents, amino acids, and Rink resin were purchased from Advanced Chem Tech (Louisville, KY) except for Fmoc-2-aminoindan-2-carboxylic acid (Aic), which was purchased from Chem Impex (Wood Dale, IL). Wang resins were purchased from Nova Biochem, EMD (Gibbstown, NJ). Fmoc-Ser-(β-Glc(Ac)4)-OH (the glycosylated serine building block) was synthesized using microwave accelerated glycosylation of Fmoc-Ser-OBn with β-glucose peracetate and indium(III) bromide as the promoter, followed by the removal of the benzyl protecting group via hydrogenolysis, according to previously published protocols and confirmed by NMR and LCMS.25
Peptide Synthesis
Peptides were synthesized on solid support (0.2 mM scale on a resin with a substitution ∼0.6 mmol/g), using fluorenylmethyloxycarbonyl (Fmoc) chemistry on a Discover S class CEM microwave using Synergy software. Deprotection of the Fmoc protecting group was performed using either a 20% solution (v/v) of piperidine in N-methyl-2-pyrrolidone (NMP) or 5% piperazine in 0.1 M HOBt-Cl in NMP. Double coupling was performed for the addition of each amino acid: using the microwave for the first coupling and at room temperature for 3 h on a Labmate shaker (Advanced ChemTech, Louisville, KY) for the second coupling, with 3 equiv of the protected amino acid, 0.4 M O-(7-azabenzotriazol-1-yl)-N,N,N′-N′-tetramethyluronium hexafluorophosphate (HATU) and 1-hydroxy-7-azabenzotriazole (HOAt) in dimethylformamide (DMF), and either 1 M diisopropylethylamine (DIEA) in NMP or neat collidine.
After each double coupling the resin was washed three times with NMP. Following the double coupling, unreacted amino groups were acetylated in the microwave (under the same conditions as for coupling) using a solution of 0.5 M acetic anhydride, 0.125 M DIEA, and 0.015 M HOBT-Cl in NMP. After the removal of the final Fmoc group, the resin was washed three times with NMP, then three times with methylene chloride (DCM) and dried under vacuum.
All peptides were cleaved from the resin and side chain protecting groups removed by treatment at room temperature for 2 h with a cleavage cocktail consisting of 9.5 mL of trifluoroacetic (TFA) acid, 0.25 mL of water, and 0.25 mL of triisopropylsilane (TIS) and filtered to remove the resin. The filtrate was concentrated in vacuo, and peptides were precipitated using cold diethyl ether. The filtered crude material was then purified using a Waters semipreparative HPLC (Waters Corporation, Milford, MA) with a Vydac protein and peptide C18 column (10 μm particle size, 10 mm × 150 mm), using a linear gradient of 10% solvent B (0.1% TFA acid in acetonitrile) in solvent A (0.1% TFA acid in water) to 60% solvent B in solvent A, at a rate of 1% per minute (flow rate 10 mL/min). The molecular weight of all peptides was confirmed using ESI-MS performed on an Agilent Technologies LCMS system using a 1200 series LC instrument and 6130 quadrupole LCMS instrument (Agilent Technologies, Santa Clara, CA). The purity of all peptides was determined using a Waters Alliance 2690 analytical HPLC system (Waters Corporation, Milford, MA) and Vydac protein and peptide C18 reverse phase column (5 μm particle size, 5 mm × 220 mm), using a linear gradient of 0–70% solvent B in solvent A at a rate of 1% per minute, with a flow rate of 1 mL/min. Linear peptides were at least 95% pure as determined by HPLC monitored at 230 nm.
Dithioether Cyclization of Linear Peptides
A DMF solution of the linear peptide (15 mg/40 mL) containing 5 mol equiv of 1,2-dibromoethane was added dropwise to a round-bottom flask containing 10 mol equiv of potassium tert-butoxide in 100 mL of anhydrous DMF saturated with argon, on ice. The mixture was stirred for 2 h under argon, on ice, and then quenched to pH 3.5 with glacial acetic acid. Solvents were removed in vacuo, and the crude cyclized peptides were purified using the same conditions as for linear precursors. All final peptides were at least 98% pure as measured by HPLC monitored at 230 nm, and their molecular weights were confirmed by ESI-MS.
Synthesis of 2, Dmt-c(SEtS)[DCys-Aci-DPen]SerOH
Compound 2 was synthesized on a 0.2 mmol scale starting with the Ser(t-Bu)OH-preloaded Wang resin at ∼0.8 mmol/g substitution. The synthesis proceeded with double coupling for each amino acid as described above. To avoid racemization, collidine was used as the base for the couplings and piperazine for Fmoc deprotection for d-Pen and d-Cys; for all other amino acids DIEA and piperidine were used as the base for the couplings and Fmoc deprotection, respectively. The resulting linear peptide was cleaved from the resin, purified by HPLC, cyclized and repurified by HPLC, as described above.
Synthesis of 3, Dmt-c(SEtS)[DCys-Aci-DPen]SerNH2
Compound 3 was synthesized following the same protocol used for the synthesis of 2 but employing a Rink resin, not preloaded, ∼0.6 mmol/g substitution.
Synthesis of 4, Dmt-c(SEtS)[DCys-Aci-DPen]Ser(Glc)NH2
Compound 4 was synthesized following the same protocol as for the synthesis of 3; however, the acetates protecting the carbohydrate were removed prior to peptide cleavage from the resin. Acetate removal was effected by treating the peptide-resin twice with 80% hydrazine monohydrate in methanol with stirring under a stream of nitrogen for 30 min and then for another hour using fresh hydrazine solution, following previously reported protocols.26
Pharmacology
All tissue culture reagents were purchased from Gibco Life Sciences (Grand Island, NY, USA). Radioactive compounds were purchased from Perkin-Elmer (Waltham, MA, USA).
Cell Lines and Membrane Preparations
C6-rat glioma cells stably transfected with a rat μ (C6-MOPr) or rat δ (C6-DOPr) opioid receptor27 and Chinese hamster ovary (CHO) cells stably expressing a human κ (CHO-KOPr) opioid receptor28 were used for all in vitro assays. Cells were cultured and membranes prepared as previously described.29
Radioligand Binding Assays
Radioligand binding assays were performed as previously described.29 In brief, assays were performed using competitive displacement of 0.2 nM [3H]diprenorphine (250 μCi, 1.85 TBq/mmol) by the test compound from membrane preparations containing opioid receptors. The assay mixture, containing membrane suspension (20 μg of protein/well) in 50 mM Tris-HCl buffer (pH 7.4), [3H]diprenorphine, and various concentrations of test peptide, was incubated at room temperature for 1 h to allow binding to reach equilibrium. The samples were filtered through Whatman GF/C filters and washed three times with cold 50 mM Tris-HCl buffer (pH 7.4). The radioactivity retained on dried filters was determined by liquid scintillation counting after saturation with EcoLume liquid scintillation cocktail in a Wallac 1450 MicroBeta (Perkin-Elmer, Waltham MA, USA). Nonspecific binding was determined using 10 μM naloxone. Ki values were calculated using nonlinear regression analysis to fit a logistic equation to the competition data using GraphPad Prism, version 5.01, for Windows. The results presented are the mean ± standard error from at least three separate assays performed in duplicate.
Stimulation of [35S]GTPγS Binding
Agonist stimulation of [35S]guanosine 5′-O-[γ-thio]triphosphate ([35S]GTPγS, 1250 Ci, 46.2 TBq/mmol) binding was measured as described previously.30 Briefly, membranes (10–20 μg of protein/well) were incubated for 1 h at room temperature in GTPγS buffer (50 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl2, pH 7.4) containing 0.1 nM [35S]GTPγS, 30 μM guanosine diphosphate (GDP), and varying concentrations of test peptides. Peptide stimulation of [35S]GTPγS was compared with 10 μM standard compounds [d-Ala2,N-MePhe4,Gly-ol]enkephalin (DAMGO) at MOPr, d-Pen2,5-enkephalin (DPDPE) at DOPr, or U69,593 at KOPr. The reaction was terminated by rapidly filtering through GF/C filters and washing 10 times with cold GTPγS buffer. Retained radioactivity was measured as described above. The results are presented as the mean ± standard error from at least three separate assays performed in duplicate; maximal stimulation and EC50 values were determined using nonlinear regression analysis with GraphPad Prism, version 5.01, for Windows. Antagonist affinities for peptides 1 and 4 at DOPr were determined as Ke values using a single concentration of test peptide according to the formula
Animals
Adult male and female C57BL/6 mice (bred in-house) weighing between 20 and 35 g at 8–16 weeks old were used for the current experiments. Mice were group-housed and had free access to food and water at all times. Experiments were conducted in the housing room, which was maintained on a 12 h light/dark cycle (with lights on at 0700). Each mouse was used only once, and experiments were conducted between 10 a.m. and 4 p.m. Studies were performed in accordance with the University of Michigan Committee on the Use and Care of Animals and the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011 publication).
Antinociception
All compounds were dissolved in sterile saline and administered by ip injection in a volume of 10 mL/kg body weight. Antinociceptive effects were evaluated in the warm water tail withdrawal (WWTW) assay. Tail withdrawal latencies were determined by briefly placing a mouse into a plastic, cylindrical restrainer and putting 2–3 cm of the tail tip into a water bath maintained at 50 °C. The latency to withdraw the tail from the water or rapidly flick the tail back and forth was recorded with a maximum cutoff time of 20 s. If the mouse did not remove its tail by the cutoff time, the experimenter removed its tail from the water to prevent tissue damage.
Acute antinociceptive effects were determined using a cumulative dosing procedure. Each animal received an injection of saline ip, and then 30 min later, baseline withdrawal latencies (3–6 s) were recorded. Following baseline determinations, increasing, cumulative doses of the test compound were given ip at 30 min intervals. At 30 min after each injection, the tail withdrawal latency was measured as described above. To evaluate the time course of the antinociceptive effects of 4, a single injection of 32 mg/kg 4 was administered ip after baseline measurements and tail withdrawal latencies were evaluated at 15, 30, 45, 60, 90, 120, and 150 min after injection. For antagonism studies, a pretreatment of saline or 3.2 mg/kg naltrexone was administered 30 min before 10 mg/kg 4 and withdrawal latencies were measured 30 min after the pretreatment and 30 min after the injection of 4.
For acute tolerance studies, separate groups of mice were treated (ip) with saline, 10 mg/kg 4, or 0.3 mg/kg fentanyl after baseline withdrawal latency was determined, and the time course of these initial antinociceptive effects was measured at 15, 30, 45, 60, and 90 min (and at 120 min in fentanyl-treated mice) after injection. Fentanyl was selected as a positive control because it was determined that 0.3 mg/kg fentanyl had a similar effect and duration of action compared with 4 in preliminary studies. At 90–120 min after the initial injection, all mice were injected with 10 mg/kg 4 or 0.3 mg/kg fentanyl, and withdrawal latencies were measured 15, 30, 60, and 90 min after injection. Tolerance was evaluated by comparing the antinociceptive effects of 4 or fentanyl in drug-naive versus drug-treated mice. For statistical comparisons, the AUC over baseline withdrawal latency for the antinociceptive effects of 4 or fentanyl was calculated for each mouse and AUCs were averaged within treatment group. Unpaired Student’s t tests were performed to determine if the AUCs were statistically different in drug-naive versus drug-treated mice (GraphPad Prism, La Jolla, CA).
Acknowledgments
This work was funded by NIH Grants DA003910 (H.I.M.) and MH083754 (J.R.T.). J.P.A. was supported by NIH Predoctoral Training Grants DA007281 and GM007767. V.P. and L.Y. were supported by the Ruth L. Kirschtein National Service Award NIH Training Grant DA007267.
Glossary
Abbreviations Used
- MOPr
μ opioid receptor
- DOPr
δ opioid receptor
- KOPr
κ opioid receptor
- Aic
2-aminoindane, 2-carboxylic acid
- Pen
penicillamine
- cSEtS
cyclization through an ethylene dithioether linkage
- Dmt
2′,6′-dimethyl-l-tyrosine
- Ser(Glc)
β-glucosyl-l-serine
- CHO
Chinese hamster ovary
- GTPγS
guanosine 5′-O-[γ-thio]triphosphate
- WWTW
warm water tail withdrawal
- icv
intracerebroventricular
- BBB
blood–brain barrier
- ip
intraperitoneal
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Morphy R.; Kay C.; Rankovic Z. From magic bullets to designed multiple ligands. Drug Discovery Today 2004, 9, 641–651. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. Designing multiple ligands—medicinal chemistry strategies and challenges. Curr. Pharm. Des. 2009, 15, 587–600. [DOI] [PubMed] [Google Scholar]
- Abdelhamid E. E.; Sultana M.; Portoghese P. S.; Takemori A. E. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991, 258, 299–303. [PubMed] [Google Scholar]
- Fundytus M. E.; Schiller P. W.; Shapiro M.; Weltrowska G.; Coderre T. J. Attenuation of morphine tolerance and dependence with the highly selective delta-opioid receptor antagonist TIPP[ψ]. Eur. J. Pharmacol. 1995, 286, 105–108. [DOI] [PubMed] [Google Scholar]
- Hepburn M. J.; Little P. J.; Gingras J.; Kuhn C. M. Differential effects of naltrindole on morphine-induced tolerance and physical dependence in rats. J. Pharmacol. Exp. Ther. 1997, 281, 1350–1356. [PubMed] [Google Scholar]
- Purington L. C.; Pogozheva I. D.; Traynor J. R.; Mosberg H. I. Pentapeptides displaying mu opioid receptor agonist and delta opioid receptor partial agonist/antagonist properties. J. Med. Chem. 2009, 52, 7724–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller P. W.; Fundytus M. E.; Merovitz L.; Weltrowska G.; Nguyen T. M.; Lemieux C.; Chung N. N.; Coderre T. J. The opioid mu agonist/delta antagonist DIPP-NH(2)[Psi] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 1999, 42, 3520–3526. [DOI] [PubMed] [Google Scholar]
- Balboni G.; Guerrini R.; Salvadori S.; Bianchi C.; Rizzi D.; Bryant S. D.; Lazarus L. H. Evaluation of the Dmt-Tic pharmacophore: conversion of a potent delta-opioid receptor antagonist into a potent delta agonist and ligands with mixed properties. J. Med. Chem. 2002, 45, 713–720. [DOI] [PubMed] [Google Scholar]
- Balboni G.; Salvadori S.; Trapella C.; Knapp B. I.; Bidlack J. M.; Lazarus L. H.; Peng X.; Neumeyer J. L. Evolution of the bifunctional lead μ agonist/δ antagonist containing the Dmt-Tic opioid pharmacophore. ACS Chem. Neurosci. 2010, 1, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosberg H. I.; Yeomans L.; Harland A. A.; Bender A. M.; Sobczyk-Kojiro K.; Anand J. P.; Clark M. J.; Jutkiewicz E. M.; Traynor J. R. Opioid peptidomimetics: leads for the design of bioavailable mixed efficacy μ opioid receptor (MOR) agonist/δ opioid receptor (DOR) antagonist ligands. J. Med. Chem. 2013, 56, 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiebel A.-C.; Lee Y. S.; Bilsky E.; Giuvelis D.; Deschamps J. R.; Parrish D. A.; Aceto M. D.; May E. L.; Harris L. S.; Coop A.; Dersch C. M.; Partilla J. S.; Rothman R. B.; Cheng K.; Jacobson A. E.; Rice K. C. Probes for narcotic receptor mediated phenomena. 34. Synthesis and structure–activity relationships of a potent mu-agonist delta-antagonist and an exceedingly potent antinociceptive in the enantiomeric C9-substituted 5-(3-hydroxyphenyl)-N-phenylethylmorphan series. J. Med. Chem. 2007, 50, 3765–3776. [DOI] [PubMed] [Google Scholar]
- Schiller P. W. Bi- or multifunctional opioid peptide drugs. Life. Sci. 2009, 86, 698–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purington L. C.; Sobczyk-Kojiro K.; Pogozheva I. D.; Traynor J. R.; Mosberg H. I. Development and in vitro characterization of a novel bifunctional μ-agonist/δ-antagonist opioid tetrapeptide. ACS Chem. Biol. 2011, 6, 1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egleton R. D.; Davis T. P. Development of neuropeptide drugs that cross the blood–brain barrier. NeuroRx 2005, 2, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Andaloussi S.; Holm T.; Langel U. Cell-penetrating peptides: mechanisms and applications. Curr. Pharm. Des. 2005, 11, 3597–3611. [DOI] [PubMed] [Google Scholar]
- Li Y.; Lefever M. R.; Muthu D.; Bidlack J. M.; Bilsky E. J.; Polt R. Opioid glycopeptide analgesics derived from endogenous enkephalins and endorphins. Future Med. Chem. 2012, 4, 205–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmagbari N. O.; Egleton R. D.; Palian M. M.; Lowery J. J.; Schmid W. R.; Davis P.; Navratilova E.; Dhanasekaran M.; Keyari C. M.; Yamamura H. I.; Porreca F.; Hruby V. J.; Polt R.; Bilsky E. J. Antinociceptive structure–activity studies with enkephalin-based opioid glycopeptides. J. Pharmacol. Exp. Ther. 2004, 311, 290–297. [DOI] [PubMed] [Google Scholar]
- Keyari C. M.; Knapp B. I.; Bidlack J. M.; Lowey J.; Bilsky E. J.; Polt R. Glycosyl-enkephalins: synthesis and binding at the mu, delta & kappa opioid receptors. Antinociception in mice. Adv. Exp. Med. Biol. 2009, 611, 495–496. [DOI] [PubMed] [Google Scholar]
- Lowery J. J.; Raymond T. J.; Giuvelis D.; Bidlack J. M.; Polt R.; Bilsky E. J. In vivo characterization of MMP-2200, a mixed δ/μ opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011, 336, 767–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polt R.; Dhanasekaran M.; Keyari C. M. Glycosylated neuropeptides: a new vista for neuropsychopharmacology?. Med. Res. Rev. 2005, 25, 557–585. [DOI] [PubMed] [Google Scholar]
- Gacel G.; Daugé V.; Breuzé P.; Delay-Goyet P.; Roques B. P. Development of conformationally constrained linear peptides exhibiting a high affinity and pronounced selectivity for delta opioid receptors. J. Med. Chem. 1988, 31, 1891–1897. [DOI] [PubMed] [Google Scholar]
- Gintzler A. R.; Chakrabarti S. The ambiguities of opioid tolerance mechanisms: barriers to pain therapeutics or new pain therapeutic possibilities. J. Pharmacol. Exp. Ther. 2008, 325, 709–713. [DOI] [PubMed] [Google Scholar]
- Williams J. T.; Ingram S. L.; Henderson G.; Chavkin C.; von Zastrow M.; Schulz S.; Koch T.; Evans C. J.; Christie M. J. Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 2013, 65, 223–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefever M. R.; Szabò L. Z.; Anglin B.; Ferracane M.; Hogan J.; Cooney L.; Polt R. Glycosylation of α-amino acids by sugar acetate donors with InBr3. Minimally competent Lewis acids. Carbohydr. Res. 2012, 351, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S. A.; Pratt M. R.; Hruby V. J.; Polt R. Solid-phase synthesis of O-linked glycopeptide analogues of enkephalin. J. Org. Chem. 2001, 66, 2327–2342. [DOI] [PubMed] [Google Scholar]
- Lee K. O.; Akil H.; Woods J. H.; Traynor J. R. Differential binding properties of oripavines at cloned mu- and delta-opioid receptors. Eur. J. Pharmacol. 1999, 378, 323–330. [DOI] [PubMed] [Google Scholar]
- Husbands S. M.; Neilan C. L.; Broadbear J.; Grundt P.; Breeden S.; Aceto M. D.; Woods J. H.; Lewis J. W.; Traynor J. R. BU74, a complex oripavine derivative with potent kappa opioid receptor agonism and delayed opioid antagonism. Eur. J. Pharmacol. 2005, 509, 117–125. [DOI] [PubMed] [Google Scholar]
- Anand J. P.; Purington L. C.; Pogozheva I. D.; Traynor J. R.; Mosberg H. I. Modulation of opioid receptor ligand affinity and efficacy using active and inactive state receptor models. Chem. Biol. Drug Des. 2012, 80, 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor J. R.; Nahorski S. R. Modulation by mu-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995, 47, 848–854. [DOI] [PubMed] [Google Scholar]