

Abstract
Streptomyces regensis strain WC-3744 was identified as a potential phosphonic acid producer in a large-scale screen of microorganisms for the presence of the pepM gene, which encodes the key phosphonate biosynthetic enzyme phosphoenolpyruvate phosphonomutase. 31P NMR revealed the presence of several unidentified phosphonates in spent medium after growth of S. regensis. These compounds were purified and structurally characterized via extensive 1D and 2D NMR spectroscopic and mass spectrometric analyses. Three new phosphonic acid metabolites, whose structures were confirmed by comparison to chemically synthesized standards, were observed: (2-acetamidoethyl)phosphonic acid (1), (2-acetamido-1-hydroxyethyl)phosphonic (3), and a novel cyanohydrin-containing phosphonate, (cyano(hydroxy)methyl)phosphonic acid (4). The gene cluster responsible for synthesis of these molecules was also identified from the draft genome sequence of S. regensis, laying the groundwork for future investigations into the metabolic pathway leading to this unusual natural product.
Phosphonates and phosphinates are compounds containing highly stable C–P or C–P–C bonds, respectively. Owing to their structural similarity to ubiquitous phosphate esters and carboxylic acids, and the inherent stability of the C–P bond, phosphonates often display potent activities as enzyme inhibitors.1,2 Examples of naturally occurring phosphonates with established bioactivity include the industrially relevant herbicide phosphinothricin (PT);3 antimalarials FR-900098 and fosmidomycin;4 antimicrobials such as fosfomycin,5 dehydrophos,6 and the plumbemycins;7 the antifungal rhizocticins;8 and antihypertensive peptides K-49 and K-26.10
In an ongoing effort to discover new, bioactive natural product phosphonates our group has undertaken a gene-targeted screening approach11−13 to identify organisms with the biosynthetic capacity for phosphonate production. The basis of this approach lies in the fact that all known phosphonate biosynthetic pathways, with the exception of the angiotensin converting enzyme inhibitor K-26,14 share the same initiating reaction in which phosphoenolpyruvate (PEP) is isomerized by the product of the pepM gene, phosphoenolpyruvate phosphonomutase (PEP mutase), to phosphonopyruvate (PnPy)1,15 (Figure S1a). This commonality makes it possible to query the genome of any organism for the presence of pepM to determine if it harbors the genetic capacity to biosynthesize phosphonates. Furthermore, the C–P bond usually provides a diagnostic signal in phosphorus nuclear magnetic resonance (31P NMR) spectroscopy analyses,16 thus facilitating the rapid screening of crude biological samples for the presence or absence of phosphonates. Here, we report the isolation, structure elucidation, and biosynthetic gene cluster for three new phosphonates produced by one of the pepM positive strains identified in our screening program, Streptomyces regensis WC-3744.
Results and Discussion
S. regensis WC-3744 originally isolated from soil in La Pampa, Argentina, was obtained from the Agricultural Research Service (ARS) culture collection, USDA, Peoria, IL. The strain was grown on 30 L of agar-solidified International Streptomyces Project medium 4 (ISP4) for 10 days at 30 °C. The liquid fraction was recovered from the spent medium and analyzed by 31P NMR, revealing at least five signals in the range that is typical for phosphonic acids (Figure 1).
Figure 1.

31P NMR spectrum of WC-3744 crude extract. Signals from 1–4 are labeled. Compounds giving minor signals were not obtained in sufficient quantity for structure elucidation.
Coumpound 2 was shown to be 2-aminoethylphosphonic acid (2AEP), a common intermediate in numerous phosphonate biosynthetic pathways.1 This assignment was first suggested based on the 31P NMR chemical shift of 2 and further supported by its retention on AG 50W-X8 cation exchange resin presumably due to the primary amine. This proposal was verified after the AG 50W-X8 retentate was spiked with commercially available 2AEP and analyzed by 31P NMR spectroscopy, showing an increase in the signal at δ 20.5 ppm and no appearance of additional signals (Figure S2).
A substantially purified mixture of compounds 1 and 3 was obtained as a white powder; however, we were unable to further separate these highly similar compounds. A variety of data demonstrate that compound 1 is (2-acetamidoethyl)phosphonic acid (Figure 2). The molecular formula was established as C4H10NO4P by Fourier-transform ion cyclotron resonance mass spectrometry (FTICR-MS) (calculated m/z for C4H10NO4P [M – H]− 166.0274, experimental 166.0275; Figure S3). The 31P NMR spectrum contained one signal at δ 21.7 ppm (Table 1, Figures 1 and S4). The 1H NMR spectrum contained three signals at δ 3.26 (2H, m), 1.85 (3H, s), and 1.79 (2H, m) ppm (Table 1, Figure S5). The signals at δ 1.79 and 3.29 ppm showed correlations to P in the 1H–31P HMBC spectrum (Table 1, Figures 3 and S6). Protons correlated to carbons were determined by 1H–13C HSQC and 1H–13C HMBC spectra (Table 1, Figures 3 and S7, S8). The extracted 13C NMR spectra revealed four signals at δ 173.9, 34.4, 27.1, and 21.8 ppm (Table 1). These values are consistent with those reported for the same compound,17 in a study that showed 1 as an accumulated catabolic intermediate in Escherichia coli phn mutants that were fed 2AEP.17 To verify this structure, commercially available 2AEP was acetylated (Experimental Section), and 1H, 31P, 13C, 1H–31P HMBC, 1H–13C HSQC, and 1H–13C HMBC NMR spectra of the synthetic standard (Figures S9–S14) were in agreement with that of the natural product 1, as were the FTICR-MS spectra (Figure S15). The signal for the C-1 carbon was assigned by the appearance of a doublet at δ 27.1 ppm in the 13C NMR spectrum with a large coupling constant of J = 133.0 Hz due to splitting by the neighboring 31P (Table 1, Figure S11). The remaining carbons and their protons were assigned positions relative to C-1 based on their 1H–13C HSQC and 1H–13C HMBC correlations. To show that 1 was not modified during the purification process, synthetic standard 1 was spiked into S. regensis crude extract containing naturally produced 1 and analyzed by 31P NMR spectroscopy to show an increase in the signal at δ 21.7 ppm and no appearance of additional signals (Figure S16).
Figure 2.

Structures of phosphonates isolated from Streptomyces regensis WC-3744. 1: (2-acetamidoethyl)phosphonic acid (Ac2AEP); 2: (2-aminoethyl)phosphonic acid (2AEP); 3: (2-acetamido-1-hydroxyethyl)phosphonic acid (NAc1H2AEP); 4: (cyano(hydroxy)methyl)phosphonic acid (CHMP).
Table 1. NMR Spectroscopic Data for Compounds 1, 3, and 4 in D2O at 600 MHz (1H) and 150 MHz (13C).
| compound 1 |
compound 3 |
compound 4 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| position | δC, type (mult; J in Hz) | δH (mult; J in Hz) | HMBC | δC, type (mult; J in Hz) | δH (mult; J in Hz) | HMBC | δC, type (mult; J in Hz) | δH (mult; J in Hz) | HMBC |
| 1 | 27.1, CH2 (d; 133.0) | 1.79 (m) | 2, P | 68.6, CH (d; 148.5) | 3.62 (m) | 2, P | 59.4, CH (d; 136.5) | 4.31 (d; 16.0) | 2, P, N |
| 2 | 34.4, CH2 | 3.26 (m) | 1, 3, P | 42.4, CH2 | a 3.47, b 3.21 (m) | 1, 3, P | 120.7, CN | ||
| 3 | 173.9, qC | 174.3, qC | |||||||
| 4 | 21.8, CH3 | 1.85 (s) | 3 | 22.0, CH3 | 1.91 (s) | 3 | |||
| P | 21.7 (m) | 15.5 (m) | 7.6 (d; 16.0) | ||||||
| N | –137.9 | ||||||||
Figure 3.

Key HMBC correlations for 1, 3, and 4.
Compound 3 was shown to be (2-acetamido-1-hydroxyethyl)phosphonic acid (Figure 2). The molecular formula of 3 was established as C4H10NO5P by FTICR-MS (calculated m/z for C4H10NO5P [M – H]− 182.0223, experimental 182.0224; Figure S17). The 31P NMR spectrum contained one signal at δ 15.5 ppm (Table 1, Figures 1 and S4). The 1H NMR spectrum contained four signals at δ 3.62 (1H, m), 3.47 (1H, m), 3.21 (1H, m), and 1.91 (3H, s) ppm (Table 1, Figure S5).
The protons at δ 3.62, 3.47, and 3.21 ppm showed correlations to P in the 1H–31P HMBC spectrum (Table 1, Figures 3 and S6). As discussed for 1 above, protons were correlated to carbons by 1H–13C HSQC and 1H–13C HMBC spectra (Table 1, Figures 3 and S7, S8). The extracted 13C NMR spectra revealed four signals at δ 174.3, 68.6, 42.4, and 22.0 ppm (Table 1). These data suggest that 3 has the structure shown in Figure 2. To verify this structure, a synthetic standard was prepared by N-acetylation of rac-(2-amino-1-hydroxyethyl) phosphonate. Comparison of NMR and MS spectra of the purified natural product 3 and the synthetic standard confirmed its identity as (2-acetamido-1-hydroxyethyl)phosphonic acid (Figures S18–24). The signal associated with the C-1 position was assigned to the doublet at δ 68.6 ppm (CH, d, J = 146.5 Hz, C-1) in the 13C NMR spectrum (Table 1, Figure S20), which displayed a large coupling constant of J = 146.5 Hz due to splitting by the neighboring 31P. The remaining carbons and their attached protons were assigned positions relative to C-1 based on their 1H–13C HSQC and 1H–13C HMBC correlations. As with 1 above, synthetic standard 3 was also spiked into S. regensis crude extract containing naturally produced 3 and analyzed by 31P NMR spectroscopy to show an increase in the signal at δ 15.5 ppm with no appearance of additional signals (Figure S16).
Compound 4, (cyano(hydroxy)methyl)phosphonic acid, was obtained as a white powder, and its molecular formula was established as C2H4NO4P by FTICR-MS (calculated m/z for C2H4NO4P [M – H]− 135.9805, experimental 135.9805; Figure S25). The 31P NMR spectrum contained one signal at δ 7.6 ppm (Table 1, Figures 1 and S26). The 1H NMR spectrum contained only one signal, at δ 4.31 (1H, d, J = 16.0 Hz) ppm (Table 1, Figure S27). A 1H-coupled 31P NMR spectrum showed the signal at δ 7.6 ppm split into a doublet that shared the same large coupling constant (J = 16.0 Hz) as the proton at δ 4.31 ppm in the 1H NMR spectrum, suggesting assignment of the latter to H-1 (Table 1, Figure S28). Consistent with this, H-1 showed a correlation to P in the 1H–31P HMBC spectrum (Table 1, Figures 3 and S29). As discussed for compounds 1 and 3 above, protons were correlated to carbons by 1H–13C HSQC and 1H–13C HMBC spectra (Table 1, Figures 4 and S30, S31). The extracted 13C NMR spectra revealed two signals at δ 120.7 and 59.4 ppm (Table 1). The quaternary C atom was particularly interesting, as its signal at δ 120.7 ppm in the 13C NMR spectrum, which correlated only to H-1 in the 1H–13C HMBC spectrum (Figure S31), suggested the presence of a nitrile moiety. To further investigate the proposal of a nitrile at the C-2 position, a 15N isotopically labeled sample was generated by culturing S. regensis on ISP4 solid medium containing (15NH4)2SO4 as the sole nitrogen source. The spent media was then extracted as described above, and a 1H–15N HMBC spectrum was acquired (Table 1, Figures 3 and S32). In this spectrum the characteristic H-1 doublet at δ 4.31 ppm of 4 shared a cross-peak with a signal at δ −137.9 ppm in the 15N dimension, also characteristic of a nitrile functional group.18 Moreover, the proposed cyanohydrin phosphonate (Figure 2) had been previously synthesized,19 and the reported NMR data were in agreement with this study. Taken together, all NMR and MS spectra for 4 are in agreement with the proposed structure, which represents the first naturally occurring, cyanohydrin-containing phosphonate to be isolated and characterized. Among the over 150 000 known natural products, less than 0.1% contain a nitrile group and even less contain a C–P moiety.20 Thus the discovery of a phosphonate featuring a nitrile moiety is particularly fascinating, and future investigations into the biosynthesis of 4 are likely to reveal interesting enzymatic steps.
Figure 4.
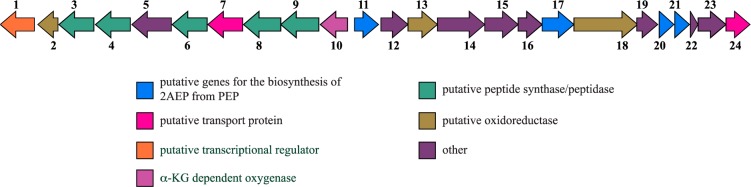
Organization of the phosphonate biosynthetic gene neighborhood from S. regensis. Open reading frames (ORFs) are labeled with numbers that correspond to Table 2. ORFs likely to be involved in 2AEP biosynthesis are colored blue. Other ORFs are colored according to their predicted functions concluded from BLAST and Phyre server analyses.
Metabolites 1, 3, and 4 were tested for bioactivity in both broth and disk diffusion assays against a panel of organisms including Gram-positive and Gram-negative bacteria as well as fungi. Inhibition of microbial growth among the tested strains by 1, 3, or 4 was not observed. Interestingly, however, 2-amino-1-hydroxyethylphosphonic acid (1H2AEP; i.e., 3 lacking the n-acetyl group)21 inhibited growth of an Escherichia coli strain, WM6242, which contains a phosphonate transport system (phnCDE) under control of the IPTG inducible promoter Ptac.13 The sensitivity of WM6242 to 1H2AEP was observed using a disk diffusion assay on Luria–Bertani agar and only when phosphonate transport was induced, suggesting that sensitivity is limited by transport (Figure S33). Considering that NAc1H2AEP (3) did not display activity against WM6242 with or without induction of phnCDE, one may postulate that N-acetylation of 1H2AEP may be a resistance mechanism employed by the producing strain when this potentially toxic intermediate accumulates to high levels in the cell.
The biosynthetic gene cluster (Figure 4, Table 2) that is probably responsible for production of 1–4 was identified by whole genome sequencing of S. regensis. The genome encodes a single pepM homologue as the first gene in a potential 14-gene operon. As described below, a divergent 10-gene operon is probably also involved in the synthesis of phosphonates by this organism. Both operons were analyzed using the Basic Local Alignment Search Tool (BLAST) program22 and Phyre23 to annotate predicted open reading frames (ORFs) (Table 2). In addition to pepM, the two operons encode numerous genes whose protein products have significant homology to characterized phosphonate biosynthetic enzymes (Figure 4, Table 2). These include orfs20 and 21 which encode the α and β subunits, respectively, of a predicted PnPy decarboxylase, which catalyzes the thiamine pyrophosphate (TPP)-dependent decarboxylation of PnPy to phosphonoacetaldehyde (PnAA),1,24 and a predicted 2AEP aminotransferase (orf17), which performs the pyridoxal 5′-phosphate (PLP)-dependent transamination of PnAA to afford 2AEP25 (Figure S1a). Together with PEP mutase, these enzymes would produce 2AEP as a pathway intermediate, a hypothesis consistent with our observation of 2AEP in extracts of S. regensis (Figures 1 and S2).
Table 2. Summary of Open Reading Frames in the Predicted Phosphonate Biosynthetic Gene Cluster from Steptomyces regensis Strain WC-3744.
| ORF | no. of amino acids | protein homologya | amino acid identity (%) |
|---|---|---|---|
| 1 | 410 | Streptomyces turgidiscabies car8, putative DNA-binding helix-turn-helix protein (zp_20876900.1) | 167/407 (41%) |
| 2 | 232 | Streptomyces rimosus subsp. rimosus ATCC 10970, putative FMN-binding oxidoreductase (ZP_20970515.1) | 118/201 (59%) |
| 3 | 415 | Streptomyces aureus, putative ATP-Grasp (AGH16374.1) | 158/405 (39%) |
| 4 | 416 | Streptomyces albus J1074, DabC ATP-Grasp (ZP_06590876) | 169/402 (42%) |
| 5 | 457 | Catenulispora acidiphila DSM 44928, putative condensation domain-containing protein (YP_003113992) | 143/459 (31%) |
| 6 | 423 | Streptomyces hygroscopicus subsp. jinggangensis 5008, putative ligase/carboxylase (AEY85881) | 154/392 (39%) |
| 7 | 420 | Streptomyces roseosporus NRRL 15998, putative major facilitator superfamily MFS_1 (ZP_06585169) | 168/407 (42%) |
| 8 | 448 | Thermacetogenium phaeum DSM 12270, PmbA/TldD putative peptidase (YP_006920355.1) | 72/215 (33%) |
| 9 | 448 | Thermacetogenium phaeum DSM 12270, PmbA/TldD putative peptidase (YP_006920356.1) | 146/363 (40%) |
| 10 | 318 | Streptomyces luridus, DhpA αKG-dependent oxygenase (ACZ13452.1) | 66/219 (30%) |
| 11 | 287 | Kitasatospora setae KM-6054, putative phosphoenolpyruvate mutase (YP_004904042) | 218/284 (77%) |
| 12 | 325 | Saccharomonospora cyanea NA-134, SbnA PLP-dependent transferase (ZP_09747688.1) | 169/315 (54%) |
| 13 | 350 | Saccharomonospora cyanea NA-134, SbnB NAD-dependent dehydrogenase (ZP_09747689.1) | 205/344 (60%) |
| 14 | 564 | Kitasatospora setae KM-6054, putative nonribosomal peptide synthetase adenylation domain (YP_004904044) | 311/552 (57%) |
| 15 | 396 | Kitasatospora setae KM-6054, hypothetical protein KSE_22690 (YP_004904045) | 231/373 (62%) |
| 16 | 291 | Streptomyces wedmorensis, FomA kinase (BAA32493) | 110/266 (42%) |
| 17 | 376 | Candidatus Nitrospira defluvii, 2-aminoethylphosphonate aminotransferase (YP_003796428) | 158/359 (45%) |
| 18 | 751 | Kitasatospora setae KM-6054, Rossmann-fold NAD(P)(+)-binding protein (YP_004904047) | 173/753 (23%) |
| 19 | 242 | Kitasatospora setae KM-6054, putative mycothiol-dependent maleylpyruvate isomerase (YP_004904048) | 152/224 (68%) |
| 20 | 181 | Bacillus subtilis subsp. Spizizenii ATCC 6633, putative phosphonopyruvate decarboxylase α subunit (ZP_06874616) | 82/167 (50%) |
| 21 | 183 | Bacillus subtilis subsp. Spizizenii ATCC 6633, putative phosphonopyruvate decarboxylase β subunit (ZP_06874617) | 87/177 (50%) |
| 22 | 87 | Azospirillum sp. B510, acyl carrier protein (YP_003453034) | 33/75 (44%) |
| 23 | 338 | Streptomyces wedmorensis, FomB kinase (BAA32494) | 192/331 (58%) |
| 24 | 285 | Streptomyces griseus subsp. griseus NBRC 13350, putative integral membrane protein (YP_001825847) | 177/284 (62%) |
Results obtained by BLAST analysis of putative open reading frames. Accession numbers are listed in parentheses.
Homologues of fomA and fomB (orfs16 and 23, respectively) from the biosynthetic gene cluster of the phosphonate antibiotic fosfomycin are also present in this neighborhood (Figure 4, Table 2). These kinases were previously shown to carry out the mono- and diphosphorylation of fosfomycin as a putative resistance mechanism,26 respectively. However, their roles in the biosynthesis or resistance related to the compounds reported here have yet to be investigated.
The first gene in the divergent operon, orf10, shares sequence similarity with the α-ketoglutarate-dependent oxygenases27 FrbJ and DhpA from the biosynthetic pathways of the phosphonates FR-900098 and dehydrophos, respectively.12,28 FrbJ has been shown to catalyze hydroxylation at the β-position of FR-900098 to yield the related phosphonate antibiotic FR-33289,28,29 while in dehydrophos biosynthesis DhpA catalyzes the α-hydroxylation of 2-hydroxyethylphosphonate (2HEP) to 1,2-dihydroxyethylphosphonate (1,2-DHEP).12 Considering the sequence similarity of the orf10 gene product to these two enzymes (Table 2) as well as the structures of 1–4, we propose that the product of orf10 likely installs the α-hydroxyl group present in 3 and 4 (Figure S1b).
Further upstream are orfs3, 4, and 6, which share significant sequence identity to amino acid ligases that contain characteristic ATP-grasp folds30 (Figure 4, Table 2). If involved in phosphonate biosynthesis, the presence of these putative peptide bond-forming enzymes would suggest that a peptide, possibly bearing one of 1–4 as a headgroup, may be the true end product of this gene cluster. This hypothesis would not be unprecedented, as many characterized peptide phosphonates including phosphinothricin tripeptide (PTT), the rhizocticins and plumbemycins, and dehydrophos gain entry into target cells via nonspecific peptide transport. Once inside the cell, these inactive peptides are hydrolyzed by endogenous peptidases to release the active phosphonate-containing headgroup.31 Under the culture conditions used in this study we did not isolate peptides containing any of 1–4 from extracts of S. regensis, although several very low abundance phosphonate-containing compounds can be observed in crude extracts of S. regensis by 31P NMR spectroscopy (Figure 1). This hypothesis may also explain why bioactivity was not observed for 1–4, as it is unlikely that these highly polar compounds are able to traverse the membranes of target cells without active transport.
Of particular interest are genes in this locus that may be involved in formation of the nitrile functional group on compound 4. As mentioned above, nitrile-containing natural products are quite rare, although they have been isolated from a variety of different organisms including arthropods,32 plants,33 fungi,34 and bacteria.20 To date, the most abundant and well-characterized nitrile-containing metabolites are the cyanogenic glycosides (CNGs), or α-hydroxynitrile glycosides, produced by various plant species. In these compounds, the nitrile is derived from an amino acid, in which the primary amine is first oxidized to an aldoxime, which then undergoes dehydration to afford the nitrile.20,35 While the biosynthesis of nitrile-containing natural products in plants has been studied extensively, the occurrence of bacterial secondary metabolites with nitrile groups appears to be much less common,20 although several different biosynthetic routes have been proposed in the literature. Biotransformation studies on borrelidin-blocked mutants implicated a PLP-dependent aminotransferase (BorJ) and a cytochrome P450 (BorI) in nitrile formation. In this proposed pathway a methyl group is oxidized to a formyl derivative in two steps catalyzed by BorI. An amine is then installed via the transamination activity of BorJ, which is both oxidized to an aldoxime and dehydrated to a nitrile by BorI in a scheme analogous to cyanogenic glycoside biosynthesis in plants.36 The cyanosporasides provide another example of a nitrile group that is likely formed via a primary amine; however after transamination this route appears to diverge from that proposed for borrelidin, as a flavin-dependent oxidoreductase with no significant sequence similarity to the cytochrome P450 BorI is implicated in converting the amine to a nitrile.37 Due to the observation of 2AEP (Figure 2) in extracts of S. regensis (Figures 1 and S2), we favor a pathway analogous to borrelidin and the cyanosporasides, in which the nitrile group of 4 is formed via oxidation of a primary amine to an aldoxime followed by elimination of water to yield the nitrile (Figure S1b). These reactions would likely be catalyzed by multiple oxidoreductases or a single multifunctional oxidoreductase as is proposed for borrelidin and the cyanosporasides.36,37 Following these observations, we have identified three putative oxidoreductases (orfs2, 13, and 18; Figure 4, Table 2) in the S. regensis pepM gene neighborhood as candidates for nitrile formation. None of these oxidoreductases, however, display significant sequence similarity to the cytochrome P450 (BorI) or the flavin-dependent oxidoreductase (CynN2/CyaN2) in the borrelidin and cyanosporaside gene clusters, respectively. This finding suggests that if nitrile formation does proceed via a primary amine, and one or more of these oxidoreductases are involved, the mechanism may differ from these two pathways. Future genetic and biochemical investigations will focus on establishing the involvement of these proteins in formation of the nitrile group.
In this study we have demonstrated the utility of a gene-targeted approach to facilitate the identification of organisms with the capacity to biosynthesize novel phosphonate natural products. Guided by 31P NMR spectroscopy, we have isolated three previously unknown phosphonate metabolites from S. regensis WC-3744, elucidated their structures using NMR and MS analyses, and identified the biosynthetic genes most likely responsible for their production. We have also set the stage for further characterization of this biosynthetic pathway, including investigations into nitrile formation. This work is an example of how such an approach can be applied to larger screening initiatives for efficient discovery of new phosphonate natural products along with their corresponding biosynthetic genes.
Experimental Section
General Experimental Procedures
NMR spectra were collected using an Agilent 600 MHz spectrometer with a 5 mm OneNMR probe, HCN PFG triple resonance probe, or ICG probe (Agilent). Samples were dissolved in 30–100% D2O. Mass spectrometry was performed using either a custom 11T linear ion trap FTICR-MS (LTQ-FT Ultra, Thermo Fisher Scientific) or an Agilent single quadrupole MSD/SL online with Agilent 1200 series HPLCs as previously described.38 Optical rotation was measured at room temperature on a Jasco P-1010 at 589 nm using a 10 mm path length cuvette and a 5 s integration time.
Cultivation, Extraction, and Purification
Streptomyces regensis strain WC-3744 was obtained from the ARS culture collection, USDA, Peoria, IL. An isolated colony was grown on solid International Streptomyces Project medium 2 (ISP2; Difco, Becton Dickinson Microbiology Systems, Sparks, MD, USA) and used to inoculate 5 mL of American Type Culture Collection medium 172 (ATCC 172). This starter culture was grown for three days at 30 °C on a drum roller and subsequently used to inoculate a 500 mL seed culture of ATCC medium 172 in a 2 L Erlenmeyer flask that was incubated for three days at 30 °C on a platform shaker rotating at 200 rpm. A 30 L amount of ISP4 (Difco, Becton Dickinson Microbiology Systems) solid medium, was then inoculated with 0.25 mL (per plate) of the seed culture and incubated for 10 days at 30 °C.
Extract was harvested by freezing plates at −80 °C, thawing, and pressing to collect the water-soluble fraction, which totaled 10 L. The sample was then concentrated by rotary evaporation to 500 mL, and the volume was brought back to 5 L with methanol (final concentration, 90%), where the phosphonates remained soluble, and the precipitate was removed by centrifugation. The sample was then dried by rotary evaporation and extracted a second time in 2.5 L of 90% methanol, and additional precipitate was removed by centrifugation. The soluble fraction was again dried by rotary evaporation, resuspended in 500 mL of H2O, and mixed in batch with activated charcoal (80 g; Sigma-Aldrich). The unbound fraction was collected and found to contain the phosphonate compounds. Multiple washes with H2O (totaling 1 L) were required to collect additional phosphonates weakly associated with the charcoal. No phosphonates were observed in subsequent methanol and hexanes wash fractions. The resulting solution (1.5 L) was concentrated to 200 mL by rotary evaporation, and the pH was adjusted to 3 with acetic acid. The sample was then fractionated over an Amberlite IRA67 weak base anion exchange column (80 g, 500–750 μm particle size, Supelco) equilibrated with 0.1% acetic acid. The column was washed successively with two volumes of 0.1% acetic acid and then H2O by gravity flow. Phosphonates and other bound material were then eluted with 500 mM NH4HCO3, acidified with acetic acid to remove excess bicarbonate, and dried by rotary evaporation to yield 10.5 g of material. The sample was then dissolved in 10 mL of 0.1% acetic acid and separated over a column of AG 50W-X8 cation exchange resin (10 g, 10–250 μm particle size, Bio-Rad) by gravity flow. Compounds 1, 3, and 4 were present in the flow-through and H2O wash, while 2AEP (2) remained bound due to its primary amine. The flow-through and H2O wash fractions were then combined and dried by lyophilization to yield 1.5 g of material, which was redissolved in 2.5 mL of H2O and fractionated over a Sephadex LH-20 size exclusion column (2.5 × 95 cm, 70 μm particle size, GE Healthcare). Compounds 1, 3, and 4 were recovered over the range of 165–200 mL, and, due to poor separation between phosphonates, all fractions containing phosphonates were pooled and dried to yield 125 mg of material.
The sample was then dissolved in 1 mL of 10 mM NH4HCO3, pH 9.2, containing 90% methanol, and compounds 1, 3, and 4 were purified using a 7.8 × 150 mm Cogent Diamond Hydride column (Microsolv, Eatontown, NJ, USA). The gradient used was as follows: 5 min at 100% solvent B1 (10 mM NH4HCO3 in 90% acetonitrile (ACN); A1 = 10 mM NH4HCO3) then a linear gradient to 40% B1 over 40 min, a hold at 40% B1 for 5 min, then a linear gradient back to initial conditions over 5 min and a re-equilibration time of 10 min; the flow rate was 4 mL/min. Fractions were monitored for the negative ions m/z 166 (1), m/z 182 (3), and m/z 136 (4) using an Agilent single quadrupole MSD (LC MSD/SL). While 4 was separated from 1 and 3 under these conditions, 1 and 3 coeluted. Attempts to establish conditions to separate these very similar compounds were unsuccessful. Fractions containing a signal at m/z 136 were pooled and lyophilized, as were those containing signals at m/z 166 and 182. These two samples were then dissolved in 0.1% formic acid and further purified with a final reverse-phase dimension using a 9.4 × 250 mm Synergi RP-Fusion column (Phenomenex, Torrence, CA, USA). The gradient used was as follows: 10 min at 100% solvent A2 (0.1% formic acid; B2 = 0.1% formic acid in ACN), then a linear gradient to 50% B2 over 10 min, a hold at 50% B2 for 5 min, then a linear gradient back to initial conditions over 5 min and a re-equilibration time of 10 min; the flow rate was 4 mL/min. Fractions were monitored using an Agilent single quadrupole MSD (LC MSD/SL). Fractions containing purified 4 (1 mg) and copurified 1 and 3 (3 mg) were pooled and lyophilized.
(2-Acetamidoethyl)phosphonic acid (1):
white powder; 1H NMR (600 MHz, D2O, δ, ppm, J/Hz) 3.26 (2H, m, H-2), 1.85 (3H, s, H-4), 1.79 (2H, m, H-1); 13C NMR (150 MHz, D2O, δ, ppm, JPC/Hz) 173.9 (qC, C-3), 34.4 (CH2, C-2), 27.1 (CH2, d, JPC = 133.0 Hz, C-1), 21.8 (CH3, C-4); FTICR-MS m/z 166.0275 [M – H]− (calcd for C4H9NO4P–, 166.0274).
(2-Acetamido-1-hydroxyethyl)phosphonic acid (3):
white powder; [α]22D +15.7 (c 0.7, H2O); 1H NMR (600 MHz, D2O, δ, ppm, J/Hz) 3.62 (1H, m, H-1), 3.47 (1H, m, H-2a), 3.21 (1H, m, H-2b), 1.91 (3H, s, H-4); 13C NMR (150 MHz, D2O, δ, ppm, JPC/Hz) 174.3 (qC, C-3), 68.6 (CH, d, JPC = 148.5 Hz, C-1), 42.4 (CH2, C-2), 22.0 (CH3, C-4); FTICR-MS m/z 182.0224 [M – H]− (calcd for C4H9NO5P–, 182.0223).
(Cyano(hydroxy)methyl)phosphonic acid (4):
white powder; [α]22D −180.0 (c 0.5, H2O); 1H NMR (600 MHz, D2O, δ, ppm, J/Hz) 4.31 (1H, d, J = 16.0 Hz, H-1); 13C NMR (150 MHz, D2O, δ, ppm, JPC/Hz) 120.7 (CN, C-2), 59.4 (CH, d, JPC = 136.5 Hz, C-1); FTICR-MS m/z 135.9805 [M – H]− (calcd for C2H3NO4P–, 135.9805).
Preparation of Standards 1 and 3
A 14.1 mg (0.1 M) portion of rac-1-hydroxy-2-aminoethylphosphonate (1H2AEP; Ryan Scientific, Inc., Mt Pleasant, SC, USA) or 12.4 mg (0.1 M) of 2-aminoethylphosphonate (2AEP; Sigma-Aldrich) and 4 mg (0.1 M) of NaOH were dissolved in 1 mL of H2O. To this solution was added an aliquot of 150 μL of acetic anhydride over a period of 20 min at 25 °C while stirring. During this addition the pH of the reaction mixture was maintained at 7.5 with NaOH. After the addition, the reaction mixture was stirred for 1 h and then acidified to pH 2.0 with HCl. To remove any remaining starting material, the reaction was passed over a 10 mL bed volume of AG 50W-X8 cation exchange resin (H+-form, 10–250 μm particle size, Bio-Rad). The flow-through was collected, lyophilized, and resuspended in 1 mL of 0.1% formic acid for final purification by semipreparative HPLC using a 9.4 × 250 mm Synergi RP-Fusion column (Phenomenex, Torrence, CA, USA). The gradient used was as follows: 10 min at 100% solvent A (0.1% formic acid; B2 = 0.1% formic acid in ACN), then a linear gradient to 50% B2 over 10 min, a hold at 50% B2 for 5 min, then a linear gradient back to initial conditions over 5 min and a re-equilibration time of 10 min; the flow rate was 4 mL/min. Fractions were monitored using an Agilent single quadrupole MSD (LC MSD/SL).
DNA Isolation and Sequencing
Genomic DNA was purified using the UltraClean Microbial DNA isolation kit (MoBio) and submitted for library preparation and sequencing to the University of Illinois at Urbana–Champaign Roy J. Carver Biotechnology Center. The library was prepared using the second-generation Nextera DNA sample prep kit (Epicenter) and run on an Illumina HiSeq platform using v3 chemistry. Assembly was performed using IDBA_UD.39 Gene prediction was performed using Prodigal v2.60,40 and annotations were assigned using results from BLAST22 and Phyre server23 analyses. The pepM biosynthetic gene cluster was found using known PEP mutase sequences as a BLAST query and verified through the EDKX5NS motif characteristic to all known proteins with this function.
Acknowledgments
Financial support from the National Institutes of Health (GM P01 GM077596) is gratefully acknowledged. NMR spectra were recorded on a 600 MHz instrument purchased with support from NIH Grant S10 RR028833. We also thank D. P. Labeda of the USDA for providing Streptomyces regensis WC-3744 from the ARS collection.
Supporting Information Available
Supplementary experimental procedures and Figures S1–S33. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The GenBank accession number for the Streptomyces regensis WC-3744 phosphonate biosynthetic gene cluster is KF594335.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Metcalf W.; van der Donk W. Annu. Rev. Biochem. 2009, 78, 65–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel R. Chem. Rev. 1977, 77, 349–367. [Google Scholar]; Hori T.; Horiguchi M.; Hayashi A. Occurence, identification and properties of phosphonic and phosphinic acids. In Biochemistry of Natural C-P Compounds; Japanese Association for Research on the Biochemistry of C-P Compounds: Shiga, Japan, 1984. [Google Scholar]
- Thompson C.; Seto H. Biotechnology 1995, 28, 197–222. [DOI] [PubMed] [Google Scholar]
- Jomaa H.; Wiesner J.; Sanderbrand S.; Altincicek B.; Weidemeyer C.; Hintz M.; Turbachova I.; Eberl M.; Zeidler J.; Lichtenthaler H. K.; Soldati D.; Beck E. Science 1999, 285, 1573–1576. [DOI] [PubMed] [Google Scholar]
- Hendlin D.; Stapley E. O.; Jackson M.; Wallick H.; Miller A. K.; Wolf F. J.; Miller T. W.; Chaiet L.; Kahan F. M.; Foltz E. L.; Woodruff H. B.; Mata J. M.; Hernandez S.; Mochales S. Science 1969, 166, 122–123. [DOI] [PubMed] [Google Scholar]
- Hunt A. H.; Elzey T. K. J. Antibiot. (Tokyo) 1988, 41, 802. [DOI] [PubMed] [Google Scholar]
- Park B. K.; Hirota A.; Sakai H. Agric. Biol. Chem. 1977, 41, 573–579. [Google Scholar]
- Michener H. D.; Snell N. Arch. Biochem. 1949, 22, 208–214. [PubMed] [Google Scholar]; Rapp C.; Jung G.; Kugler M.; Loeffler W. Liebigs Ann. Chem. 1988, 1988, 655–661. [Google Scholar]
- Koguchi T.; Yamada K.; Yamato M.; Okachi R.; Nakayama K.; Kase H. J. Antibiot. (Tokyo) 1986, 39, 364–371. [DOI] [PubMed] [Google Scholar]
- Yamato M.; Koguchi T.; Okachi R.; Yamada K.; Nakayama K.; Kase H.; Karasawa A.; Shuto K. J. Antibiot. (Tokyo) 1986, 39, 44–52. [DOI] [PubMed] [Google Scholar]
- Blodgett J. A.; Zhang J. K.; Metcalf W. W. Antimicrob. Agents Chemother. 2005, 49, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]; Peck S. C.; Gao J.; van der Donk W. A. Methods Enzymol. 2012, 516, 101–123. [DOI] [PubMed] [Google Scholar]
- Circello B. T.; Eliot A. C.; Lee J. H.; van der Donk W. A.; Metcalf W. W. Chem. Biol. 2010, 17, 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliot A.; Griffin B.; Thomas P.; Johannes T.; Kelleher N.; Zhao H.; Metcalf W. Chem. Biol. 2008, 15, 765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntai I.; Manier M. L.; Hachey D. L.; Bachmann B. O. Org. Lett. 2005, 7, 2763–2765. [DOI] [PubMed] [Google Scholar]
- Seidel H.; Freeman S.; Seto H.; Knowles J. Nature 1988, 335, 457–458. [DOI] [PubMed] [Google Scholar]; Bowman E.; McQueney M.; Barry R. J.; Dunaway-Marino D. J. Am. Chem. Soc. 1988, 110, 5575–5576. [Google Scholar]; Hidaka T.; Mori M.; Imai S.; Hara O.; Nagaoka K.; Seto H. J. Antibiot. (Tokyo) 1989, 42, 491–494. [DOI] [PubMed] [Google Scholar]
- Nielsen M. L.; Pustinger J. V.; Strobel J. J. Chem. Eng. Data 1964, 9, 167–170. [Google Scholar]
- Hove-Jensen B.; McSorley F. R.; Zechel D. L. PLoS One 2012, 7, e46416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witanowski M.; Stefaniak L.; Webb G. A.. Nitrogen NMR Spectroscopy. In Annual Reports on NMR Spectroscopy; Webb G. A., Ed.; Academic Press: New York, 1987; Vol. 18, pp 1–211. [Google Scholar]
- Oediger H.; Lieb F.; Disselnkotter H.. Phosphonohydroxyacetonitrile, a process for its preparation and its use as an intermediate product for the preparation of medicaments. U.S. Patent 4,370,280, 1983.
- Fleming F. Nat. Prod. Rep. 1999, 16, 597–606. [Google Scholar]
- Hammerschmidt F.; Völlenkle H. Liebigs Ann. Chem. 1989, 1989, 577–583. [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. J. Mol. Biol. 1990, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Kelley L. A.; Sternberg M. J. Nat. Protoc. 2009, 4, 363–371. [DOI] [PubMed] [Google Scholar]
- Warren W. Biochim. Biophys. Acta 1968, 156, 340–346. [DOI] [PubMed] [Google Scholar]; Horiguchi M. Biochim. Biophys. Acta 1972, 261, 102–113. [DOI] [PubMed] [Google Scholar]; Barry R.; Bowman E.; McQueney M.; Dunaway-Mariano D. Biochem. Biophys. Res. Commun. 1988, 153, 177–182. [DOI] [PubMed] [Google Scholar]
- Lacoste A. M.; Dumora C.; Balas L.; Hammerschmidt F.; Vercauteren J. Eur. J. Biochem. 1993, 215, 841–844. [DOI] [PubMed] [Google Scholar]; Chen C. C.; Zhang H.; Kim A. D.; Howard A.; Sheldrick G. M.; Mariano-Dunaway D.; Herzberg O. Biochemistry 2002, 41, 13162–13169. [DOI] [PubMed] [Google Scholar]; Kim A. D.; Baker A. S.; Dunaway-Mariano D.; Metcalf W. W.; Wanner B. L.; Martin B. M. J. Bacteriol. 2002, 184, 4134–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dumora C.; Lacoste A. M.; Cassaigne A. Eur. J. Biochem. 1983, 133, 119–125. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Kuzuyama T.; Seto H. Antimicrob. Agents Chemother. 2000, 44, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]; Woodyer R.; Shao Z.; Thomas P.; Kelleher N.; Blodgett J.; Metcalf W.; van der Donk W.; Zhao H. Chem. Biol. 2006, 13, 1171–1182. [DOI] [PubMed] [Google Scholar]
- Hausinger R. P. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 21–68. [DOI] [PubMed] [Google Scholar]
- DeSieno M. A.; van der Donk W. A.; Zhao H. Chem. Commun. (Cambridge) 2011, 47, 10025–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]; Johannes T. W.; DeSieno M. A.; Griffin B. M.; Thomas P. M.; Kelleher N. L.; Metcalf W. W.; Zhao H. Chem. Biol. 2010, 17, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuhara M.; Kuroda Y.; Goto T.; Okamoto M.; Terano H.; Kohsaka M.; Aoki H.; Imanaka H. J. Antibiot. (Tokyo) 1980, 33, 24–28. [DOI] [PubMed] [Google Scholar]
- Galperin M. Y.; Koonin E. V. Protein Sci. 1997, 6, 2639–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diddens H.; Zahner H.; Kraas E.; Gohring W.; Jung G. Eur. J. Biochem. 1976, 66, 11–23. [DOI] [PubMed] [Google Scholar]; Laber B.; Lindell S. D.; Pohlenz H. D. Arch. Microbiol. 1994, 161, 400–403. [DOI] [PubMed] [Google Scholar]; Circello B. T.; Miller C. G.; Lee J. H.; van der Donk W. A.; Metcalf W. W. Antimicrob. Agents Chemother. 2011, 55, 3357–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennesland B.Cyanide in Biology; Academic Press: London, 1981; p xiii. [Google Scholar]
- Mahadevan S. Annu. Rev. Plant Physiol. 1973, 24, 69–88. [Google Scholar]
- Legras J. L.; Chuzel G.; Arnaud A.; Galzy P. World J. Microbiol. Biotechnol. 1990, 6, 83–108. [DOI] [PubMed] [Google Scholar]
- Tapper B. A.; Reay P. F.. Cyanogenic Glycosides and Glucosinolates; Academic Press: London, 1973; Vol. 1. [Google Scholar]
- Olano C.; Moss S. J.; Brana A. F.; Sheridan R. M.; Math V.; Weston A. J.; Mendez C.; Leadlay P. F.; Wilkinson B.; Salas J. A. Mol. Microbiol. 2004, 52, 1745–1756. [DOI] [PubMed] [Google Scholar]
- Lane A. L.; Nam S. J.; Fukuda T.; Yamanaka K.; Kauffman C. A.; Jensen P. R.; Fenical W.; Moore B. S. J. Am. Chem. Soc. 2013, 135, 4171–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans B. S.; Zhao C.; Gao J.; Evans C. M.; Ju K. S.; Doroghazi J. R.; van der Donk W. A.; Kelleher N. L.; Metcalf W. W. Chem. Biol. 2013, 8, 908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Leung H. C.; Yiu S. M.; Chin F. Y. Bioinformatics 2012, 28, 1420–1428. [DOI] [PubMed] [Google Scholar]
- Hyatt D.; Chen G. L.; Locascio P. F.; Land M. L.; Larimer F. W.; Hauser L. J. BMC Bioinf. 2010, 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.