

Abstract
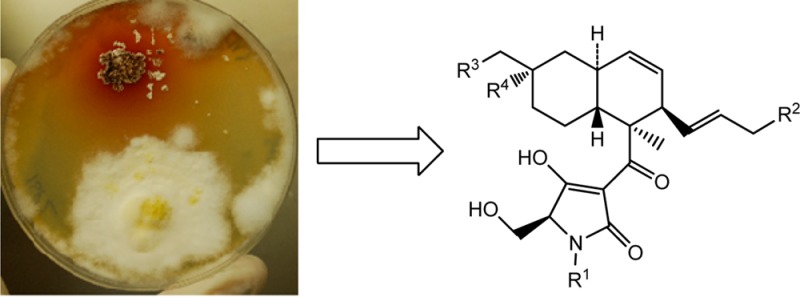
Coculture of the fungus Fusarium pallidoroseum with the bacterium Saccharopolyspora erythraea was found to produce three new decalin-type tetramic acid analogues related to equisetin. The structures were determined by spectroscopic methods. The absolute configurations were established by circular dichroism spectroscopy and comparing the data with those of equisetin.
Natural products continue to play an integral role in the discovery and development of drugs used to treat various diseases. From genomic sequencing in the early 2000s of the actinomycetes Streptomyces avermitilis(1) and S. coelicolor(2) it became apparent that there were 10 to 20-plus putative (cryptic) secondary metabolite clusters in the genomes of these microbes. In the middle 2000s, from investigations of the genomes of certain Aspergillii, the Keller group reported that in contrast to the then current dogma, putative secondary metabolite biosynthetic clusters in the Aspergillii were on single chromosomes, rather than being spread among a number, as is the case in fungal primary metabolism.3,4 Thus over the past few years, a number of investigators have been attempting to activate these cryptic biosynthetic clusters by a variety of means, including activation by exogenous epigenetic modifiers such as demethylase or histone acetylase/deacetylase activators/inhibitors.5
Using exogenous molecules does work, but the levels required are purely empirical in nature and costs become significant as fermentation volume increases. One very useful approach to overcoming the increasing difficulty in discovering new active structures by activation of the cryptic clusters is to mimic the competitive microbial ecosystems employed in the natural environment through mixed culture of two or more organisms known to produce bioactive compounds. The resulting interactions may lead to induced production of previously unreported secondary metabolites or an improved titer of known and at times unidentified low level analogues, as we and others have demonstrated.6−13
As part of our ongoing study of metabolites produced by cocultures, we analyzed the mixed culture of Fusarium pallidoroseum with Saccharopolyspora erythraea. The genus Fusarium includes a large group of filamentous fungi widely distributed in soil and in association with plants. It produces a broad array of active metabolites including the HDAC inhibitor apicidin,14 the immunosuppressor cyclosporine A,15 and the antibiotic and insecticidal beauvericin,16 just to name a few examples. S. erythaea is a Gram-positive bacterium producer of the antibiotic erythromycin A.17
Another metabolite isolated from a number of species of Fusarium is equisetin,18−23 a tetramic acid-containing analogue with antibiotic,18,21 cytotoxic,24 and phytotoxic25 activities and potent inhibitor of mitochondrial ATPases26 and HIV-1 integrase.19,27 Equisetin is active against several genera of Gram-positive bacteria, including Bacillus subtilis and Staphylococcus aureus, and inhibits the growth of the acid-fast bacteria Mycobacterium phlei.(18,21) Several close analogues of equisetin have been reported, including its C-5′ epimer epi-equisetin,21 its enantiomer phomasetin,19 ophiosetin,28 and trichosetin.29

The LCMS chromatogram of the extract from the coculture of S. erythraea with F. pallidoroseum showed the presence of four peaks that were not observed in either of the controls (Figure 1, peaks 1–4). After further analysis it was also evident that although the peak labeled as 5 was present in the chromatogram of the extract of the Fusarium control, its area on the coculture was 30 times larger. These peaks exhibited absorbance bands in the UV spectrum with maxima at 250 and 290 nm. This UV profile was very similar to that reported for the known Fusarium metabolite equisetin.20
Figure 1.

Chromatogram in positive ion mode (m/z 350–450 uma) of the extracts of Fusarium pallidoroseum (top), Saccharopolyspora erythraea (bottom), and coculture (middle) showing the new peaks observed only in the coculture (1–3 are new metabolites, 4 is ophiosetin, and 5 is equisetin).
Scale-up of this coculture allowed us to isolate those compounds produced only in the mixed fermentation. The analogues were identified as three new metabolites and two known compounds identified as equisetin18 and ophiosetin28 by comparison with reported data. Ophiosetin has been reported from the fungus Elaphocordyceps ophioglossoides(28) but had never been reported from the genus Fusarium or Saccharopolyspora.
The molecular weight of compound 1 was determined as 375. The UV–vis spectrum showed two maxima at 250 and 287 nm, which were consistent with the spectrum reported for equisetin.20 The signals in the 1H NMR spectrum (DMSO-d6) of compound 1 were broad, supporting the possibility that it was an equisetin analogue since it is known that several tautomers occur with tetramic acid analogues.30 Despite the broad signals, it was possible to assign all of the signals in the spectrum. The 1H NMR spectrum showed two signals integrating for 3H each that were assigned to methyl groups [one singlet at δH 1.30 and one doublet at δH 1.48 (J = 4.8 Hz)]. The spectrum also showed signals for an additional nine protons resonating between 0.7 and 2 ppm, six protons between 3.2 and 3.7 ppm, one D2O exchangeable proton (δH 4.36), and four vinylic protons. 2D NMR experiments including COSY, HSQC, and HMBC allowed us to assign all of the signals in the decalin core. The HSQC correlations between δC 67.0 and δH 3.20 and 3.23 and between δC 62.8 and δH 3.47 and 3.63 suggested that four of the signals between 3.1 and 3.8 ppm corresponded to two methylene groups carrying hydroxyl groups. The HMBC correlation between one of these methylene groups (δH 3.22, H-16) and δC 37.0 (C-7), δC 41.4 (C-8), and δC 30.4 (C-9) and between H-7a (δH 0.76), H-7b (δH 1.86), H-8 (δH 1.46), and H-9a (δH 0.99) and C-16 (δC 67.0) indicated that the CH3-16 present in equisetin was oxidized to a primary alcohol. The second primary alcohol was assigned to C-6′ on the tetramic acid part of the molecule by HMBC correlations between H-6′b (δH 3.63) and C-4′ (δC 191.2) and C-5′ (δC 62.5). Thus analysis of the NMR data suggested that compound 1 had a structure very similar to ophiosetin. Comparison of the NMR data obtained in DMSO-d6 of both molecules indicated that the main difference was the absence of the N-methyl group in the new analogue. The relative configuration of the decalin skeleton of compound 1 was determined by NOESY and tROESY experiments. tROESY correlations between H-6 (δH 1.77) and H-8 (δH 1.46) and CH3-12 (δH 1.30) and NOESY correlation between H-3 (δH 3.62) and CH3-12 (δH 1.30) indicated a syn relationship among these protons. The absence of correlation between H-6 and H-11 supported a trans fusion of the two six-membered rings in the decalin skeleton. NOESY correlations between H-11 (δH 1.54) and H-13 (δH 5.14) suggested a syn relationship but on the other side of the molecule’s plane. The optical rotation ([α]25D −334) was the same sign as equisetin. Comparison of the CD spectra obtained for compound 1 [λmax (Δε) 232 (−3.3), 250 (−2.3), 285 (−8.4), 323 (+0.7) nm] and equisetin allowed us to establish that the absolute configuration was the same as the one reported for equisetin. Compound 1 was named N-demethylophiosetin.
The molecular weight of compound 2 was established as 389, which corresponds to the same molecular weight as ophiosetin. However, careful analysis of the NMR data and comparison with an authentic sample indicated that compound 2 was not ophiosetin. Its 1H NMR spectrum showed three singlets (δH 1.08, 1.24, and 2.77) and one doublet (δH 1.46) integrating for three protons each. Three of the methyl groups were assigned by 2D NMR correlations to the methyl groups C-12, C-15, and N-CH3; however the singlet at highest field (δH 1.08) was not observed in the 1H NMR spectrum of ophiosetin. HMBC correlation between this signal (δH 1.08) and C-7 (δC 46.4), C-8 (δC 68.1), and C-9 (δC 40.1) suggested that this methyl group was on C-8. The low-field chemical shift for C-8 (δC 68.1) in the 13C NMR spectrum together with HMBC correlations between one D2O exchangeable proton (δH 3.91) and C-8 (δC 68.1) and C-16 (δC 31.8) indicated the presence of a hydroxyl group on C-8. To the best of our knowledge this is the first report of an equisetin-type analogue bearing a hydroxyl on C-8. This substitution had been reported in tanzawaic acid31 and in betaenones,32 both metabolites containing a decalin core. Comparison of the NMR data with those reported for tanzawaic acid for CH2-7 (δH 1.04 and 1.59, δC 46.4) C-8 (δC 68.1), CH2-9 (δH 1.30 and 1.50, δC 40.1), and CH3-16 (δH 1.08, δC 31.9) confirmed the presence of the hydroxyl group at C-8. The coupling constant H-13/H-14 of 15.4 Hz indicated that the configuration of the double bond of the side chain was E. The relative configuration of the decalin ring was determined by tROESY experiments. Correlations between CH3-12 (δH 1.24) and H-3 (δH 4.09) and H-6 (δH 2.10) and between H-6 (δH 2.10) and OH-8 (δH 3.91) indicated a syn relationship among all these protons, and correlations between H-11 (δH 1.43) and H-13 (δH 5.14) supported these two protons being in a syn relationship. The absence of correlation between H-6 and H-11 suggested that these two protons were on opposite sides of the molecule’s plane, indicating a trans fusion of the two rings of the decalin skeleton. The optical rotation ([α]25D −153) and the CD spectrum [λmax (Δε) 228 (−4.0), 256 (−2.5), 292 (−11.5), 324 (+2.5) nm] were the same sign as that determined for equisetin, indicating the same absolute configuration. We proposed the name pallidorosetin A for compound 2.
Compound 3 was more polar than the other isolated metabolites, as indicated by its shorter retention time on reversed-phase chromatography. Its molecular weight was determined to be 405, indicating that it contains one oxygen more than ophiosetin. The absence of the doublet assigned to CH3-15 in the 1H NMR spectra of all the previous analogues, together with a COSY correlation between H-15 (δH 3.74) and a vinylic proton on the lateral chain (H-14, δH 5.22) and the signal for one of the D2O exchangeable protons (δH 4.38), suggested the presence of a hydroxyl group on the side chain, accounting for the extra oxygen in the molecular formula. HMBC correlations between H-15 (δH 3.74) and C-13 (δC 132.9) and C-14 (δC 130.5) and between H-13 (δH 5.30) and H-14 (δH 5.22) and C-15 (δC 62.1) confirmed the position of the new hydroxyl group on C-15. Two pairs of signals were assigned to CH-5′ (δH 3.19, δC 65.3 and δH 3.42, δC 72.8) and two to CH2-6′ (δH 3.54 and 3.67; δC 62.2 and δH 3.29 and 3.36; δC 63.4). These data suggested that compound 3 was a mixture of epimers at C-5′.
A similar mixture of epimers at C-5′ had been described previously for equisetin,19 phomasetin,19 and the melophlins.33 For equisetin and phomasetin the epimerization at C-5′ was also reported in pyridine.21 The coupling constant JH-13/H-14 = 15.4 Hz indicated that the configuration of the double bond of the side chain was E. The relative configuration of compound 3 was determined by analysis of a tROESY experiment. The lack of correlation between H-6 and H-11 together with the correlation between H-11 (δH 1.48) and H-13 (δH 5.30) and between CH3-12 (δH 1.24) and H-6 (δH 1.67) and H-3 (δH 4.16) indicated trans fusion of the two six-membered rings on the decalin skeleton and the relative configuration of C-2 and C-3. Correlations between H-16 (δH 3.21) and H-11 (δH 1.48) with H-9a (δH 0.95) and H-7a (δH 0.71) indicated a syn relationship among these protons, while correlation between H-8 (δH 1.44) and H-7b (δH 1.82) and H-9b (δH 1.69) also indicated a syn relationship but on the other side of the molecule’s plane. The optical rotation obtained for compound 5 ([α]D −142) had the same sign as those observed for all previous analogues, and its CD spectrum was also qualitatively similar [λmax (Δε) 237 (−1.6), 250 (−1.6), 293 (−4.4), 323 (+1.0) nm], confirming that the absolute configuration for all the centers, except for C-5′, which could not be determined, was the same as the one reported for equisetin. We propose the name pallidorosetin B for compound 3.
The spectral data for equisetin and ophiosetin were in agreement with those reported in the literature.
All the isolated analogues were tested using a Kirby-Bauer disk diffusion assay34 against the Gram-positive bacteria Staphylococcus erythraea and S. aureus. Only equisetin was active (minimum inhibitory concentrations were <1.25 μg against S. aureus and 2.5 μg against S. erythraea). The inhibition of S. aureus is in agreement with the data previously reported.18 Hydroxylation of C-16 was reported as detrimental for the antimicrobial activity based on the comparison of activity observed between equisetin and ophiosetin.23 Compounds 1 and 3 also bear a hydroxyl group on C-16 and were both inactive against the bacteria screened. Compound 2, which has a hydroxyl group on C-8 instead of C-16, also was inactive. All the inactive analogues are more polar than equisetin, which could suggest that the antibacterial activity has a negative correlation with the polarity of the metabolite. Analogues containing a tetramic acid moiety were proposed to have antibacterial properties by inhibiting the formation of biofilms in Gram-positive bacteria.35 It is possible that Fusarium increases the production of equisetin-type analogues as a defense mechanism against Saccharopolyspora. Mixed cultures of Fusarium and other Gram-positive bacteria (Bacillus and Staphylococcus) cultured in our laboratory also increased production of equisetin (data not shown). Interestingly, the tetramic acid trichosetin29 was isolated from a coculture of the fungus Trichoderma harzianum with the callus of Catharanthus roseus and not produced by either of the organisms alone.
The cytotoxicity of all the isolated metabolites was assessed in the NCI-60 cell line screen.36,37 Equisetin (NSC 772378) exhibited low μM GI50 (Supporting Information) against all the cell lines and GI50 = 144 nM against the leukemia cell line CCRF-CEM. The rest of the compounds were inactive (GI50 > 20 μM).
Experimental Section
General Experimental Procedures
UV spectra were acquired in spectroscopy grade MeOH using an Agilent 8453 UV–vis spectrophotometer; NMR spectra were recorded on Bruker Avance III 600 MHz (Bruker Biospin) and Varian Inova 500 MHz NMR spectrometers. Measured chemical shifts were referenced to residual solvent (δH 2.50 and δC 39.5 in DMSO-d6). LCMS analyses and HRESIMS measurements were performed with an XTerra MS C18 (2.1 × 50 mm, 3.5 μm, Waters) column on a Waters Acquity UPLC system coupled to a Waters LCT Premier TOF mass spectrometer, a Waters Acquity PDA detector, and a Sedex 75 evaporative light scattering detector. Matrex C18 adsorbent (Varian) was used for flash chromatography. Preparative-scale purification was performed with a Dynamax 60 Å C18 column (19 × 300 mm, Varian) on an HPLC system consisting of a Waters 600 pump, a Waters 3100 mass detector, a Waters 996 photodiode array detector, and a Shimadzu ELSD-LT2 evaporative light scattering detector.
Material
Both microorganisms were purchased from the American Type Culture Collection (ATCC). Fusarium pallidoroseum (ATCC 74289) was maintained on Difco potato dextrose agar at room temperature. Saccharopolyspora erythraea (ATCC 31772) was maintained on International Streptomyces Project Medium 2 (ISP2) agar, consisting of malt extract, 10 g/L; dextrose, 4 g/L; and yeast extract, 4 g/L, at 27 °C.
Fermentation and Isolation
S. erythraea and F. pallidoroseum were grown on agar for two weeks and one week, respectively, before inoculation into 100 mL of ISP2 seed medium in 500 mL Erlenmeyer flasks. Both organisms were incubated at room temperature for 24 h on a rotary shaker (150 rpm), after which the seeds were used to inoculate 1 L Erlenmeyer flasks containing 200 mL of ISP2 broth. The inoculum per 1 L flask was 20 mL of S. erythraea and 1 mL of F. pallidoroseum with a ratio of packed cell volume to media of <0.5 mL/15 mL and 1 mL/15 mL, respectively. Fermentation was carried out for 4 days at 27 °C on a rotary shaker (200 rpm). Single control cultures of S. erythraea and F. pallidoroseum were grown under identical conditions.
The full volume (3.5 L) of culture filtrate was concentrated to 300 mL in vacuo (<40 °C) and partitioned three times with equal volumes of ethyl acetate. Evaporation of the pooled organic layers yielded 1.45 g of an oily brown crude extract. The extract was applied to a 10 mL SPE cartridge packed with 1.5 g of C18 silica. The cartridge was eluted in a stepwise fashion with increasing percentages of MeOH in H2O. Fractions containing the compounds of interest were pooled and lyophilized, yielding 52.4 mg of enriched extract. Further purification was performed with a stepwise gradient (H2O/AcN w/0.1% F.A.; 0–20 min 65:35, 20–60 min 60:40; 60–80 min 55:45; 10 mL/min) on a preparative-scale reversed-phase HPLC system. Analysis and pooling of the resulting fractions afforded ophiosetin (2.53 mg), N-demethylophiosetin (1) (2.86 mg), pallidorosetin A (2) (1.59 mg), pallidorosetin B (3) (1.06 mg), and equisetin (5.07 mg).
N-Demethylophiosetin (1):
[α]25D −334 (c 0.025, CHCl3); CD (c 0.1 mM, MeOH) λmax (Δε) 232 (−3.3), 250 (−2.3), 285 (−8.4), 323 (+0.7) nm; UV (MeOH) λmax (log ε) 250 (3.73), 287 nm (3.92); 1H NMR (500 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) see Table 1; HRESIMS m/z 376.2108 [M + H]+ (calc for C21H30NO5, 376.2124).
Table 1. 1H (500 MHz) and 13C (150 MHz) NMR Data for Compounds 1–3 in DMSO-d6.
|
1 |
2 |
3 |
||||
|---|---|---|---|---|---|---|
| position | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC,a type | δH (J in Hz) |
| 1 | 199.5, C | 198.1, C | 197.7, C | |||
| 2 | 49.6, C | 48.6, C | 49.9, C | |||
| 3 | 43.9, CH | 3.62b | 42.2, CH | 4.09 m | 41.8, CH | 4.16 br s |
| 4 | 127.6, CH | 5.36 ddd (10.2, 4.7, 2.1) | 128.3, CH | 5.34 ddd (10.0, 5.2, 2.6) | 128.2, CH | 5.38 br s |
| 5 | 130.5, CH | 5.42 d (10.2) | 130.0, CH | 5.27 d (10.0) | 130.4, CH | 5.38 br s |
| 6 | 38.4, CH | 1.77b | 33.3, CH | 2.10 t (10.7) | 38.5, CH | 1.67 m |
| 7 | 37.0, CH2 | 0.76 q (12.0) | 46.4, CH2 | 1.04 d (11.5) | 37.1, CH2 | 0.71 q (11.8) |
| 1.86 d (12.0) | 1.59 d (11.5) | 1.82 d (11.8) | ||||
| 8 | 41.4, CH | 1.46* | 68.1, C | 3.91 s (OH) | 41.2, CH | 1.44 m |
| 9 | 30.4, CH2 | 0.99 q (12.0) | 40.1, CH2 | 1.30 m | 30.5, CH2 | 0.95 br d (13.8) |
| 1.75 m | 1.50 d (12.9) | 1.69 m | ||||
| 10 | 27.7, CH2 | 0.89 q (12.0) | 22.9, CH2 | 1.10 m | 27.2, CH2 | 0.73b |
| 1.96 br s | 1.61 br d (11.9) | 1.92 br d (10.0) | ||||
| 11 | 40.6, CH | 1.54 m | 40.4, CH | 1.43 m | 40.4, CH | 1.48 m |
| 12 | 14.4, CH3 | 1.30 s | 14.7, CH3 | 1.24 s | 14.6, CH3 | 1.24 s |
| 13 | 132.6, CH | 5.14 m | 134.3, CH | 5.14 dd (15.4,6.9) | 132.9, CH | 5.30 dd (15.4, 7.0) |
| 14 | 125.8, CH | 5.14 m | 123.1, CH | 5.07 dq (15.4, 6.0) | 130.5, CH | 5.22 dt (15.4, 5.4) |
| 15 | 18.3, CH3 | 1.48 d (4.8) | 18.1, CH3 | 1.46 d (5.6) | 62.1, CH2 | 3.74 br s |
| 16 | 67.0, CH2 | 3.20 d (11.6) | 31.8, CH3 | 1.08 s | 67.0, CH2 | 3.21 br d (4.2) |
| 3.23 d (11.6) | ||||||
| 2′ | 178.6, C | 175.0, C | 175.4, C | |||
| 3′ | 101.2, C | 101.5, C | ND | |||
| 4′ | 191.2, C | 188.5, C | 189.2, C | |||
| 5′ | 62.5, CH | 3.64 m | 64.7, CH | 3.21 br s | 65.3, CH (A) | 3.19b (A) |
| 72.8, CH (B) | 3.42 dd (10.9, 5.7) (B) | |||||
| 6′ | 62.8, CH2a | 3.47 dd (11.1, 5.0) | 61.8, CH2 | 3.54 dd (10.5, 5.1) | 62.2, CH2 (B) | 3.36b (a) |
| 3.63 dd (11.1, 2.8) | 3.66 dd (10.5, 2.3) | 3.29 dd (10.8, 5.7) (b) | ||||
| 63.4, CH2 (A) | 3.67 dd (10.8, 3.0) (a) | |||||
| 3.54 dd (10.8, 5.0) (b) | ||||||
| NCH3 | 27.0, CH3 | 2.77 s | 27.6, CH3 | 2.77 s | ||
| OH-16 | 4.36 br s | 4.32 br s | ||||
| OH-15 | 4.38 br s | |||||
δC determined by 2D experiments,
Superposed with another signal; δH determined by analysis of 2D experiments.
Pallidorosetin A (2):
[α]D −153 (c 0.025, CHCl3); CD (c 0.1 mM, MeOH) λmax (Δε) 228 (−4.0), 256 (−2.5), 292 (−11.5), 324 (+2.5) nm; UV (MeOH) λmax (log ε) 251 (3.98), 292 nm (3.99); 1H NMR (500 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) see Table 1; HRESIMS m/z 390.2262 [M + H]+ (calc for C22H32NO5, 390.2280).
Pallidorosetin B (3):
[α]D −142 (c 0.025, CHCl3); CD (c 0.1 mM, MeOH) λmax (Δε) 237 (−1.6), 250 (−1.6), 293 (−4.4), 323 (+1.0) nm; UV (MeOH) λmax (log ε) 250 (3.76), 290 nm (3.73); 1H NMR (500 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) see Table 1; HRESIMS m/z 388.2115 [M – H2O + H]+ (calc for C22H30NO5, 388.2124).
Equisetin:
[α]D −258 (c 0.025, CHCl3); CD (c 0.1 mM, MeOH) λmax (Δε) 234 (−4.5), 255 (−1.7), 292 (−8.3), 326 (+0.6) nm.
Ophiosetin:
[α]D −241 (c 0.025, CHCl3); CD (c 0.1 mM, MeOH) λmax (Δε) 234 (−4.2), 256 (−2.2), 292 (−7.7), 328 (+0.3) nm.
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported [in part] by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute. We gratefully acknowledge T. DeLloyd for assistance with fermentations, Q. Van and A. Castro Ruiz for running NMR experiments, S. Tarasov and M. Dyba (Structural Biophysics Laboratory) for assistance with the CD determinations, and L. Cartner for help running the polarimeter.
Supporting Information Available
1H NMR, 13C NMR, COSY, gHSQC, gHMBC, NOESY, TROESY, and CD spectra for compounds 1–3 together with dose response NCI-60 screen data for equisetin. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- O̅mura S.; Ikeda H.; Ishikawa J.; Hanamoto A.; Takahashi C.; Shinose M.; Takahashi Y.; Horikawa H.; Nakazawa H.; Osonoe T.; Kikuchi H.; Shiba T.; Sakaki Y.; Hattori M. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 12215–12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley S. D.; Chater K. F.; Cerdeño-Tárraga A.-M.; Challis G. L.; Thomson N. R.; James K. D.; Harris D. E.; Quail M. A.; Kieser H.; Harper D.; Bateman A.; Brown S.; Chandra G.; Chen C. W.; Collins M.; Cronin A.; Fraser A.; Goble A.; Hidalgo J.; Hornsby T.; Howarth S.; Huang C.-H.; Kieser T.; Larke L.; Murphy L.; Oliver K.; O’Neil S.; Rabbinowitsch E.; Rajandream M.-A.; Rutherford K.; Rutter S.; Seeger K.; Saunders D.; Sharp S.; Squares R.; Squares S.; Taylor K.; Warren T.; Wietzorrek A.; Woodward J.; Barrell B. G.; Parkhill J.; Hopwood D. A. Nature 2002, 417, 141–147. [DOI] [PubMed] [Google Scholar]
- Bok J. W.; Hoffmeister D.; Maggio-Hall L. A.; Murillo R.; Glasner J. D.; Keller N. P. Chem. Biol. 2006, 13, 31–37. [DOI] [PubMed] [Google Scholar]
- Lim F. Y.; Sanchez J. F.; Wang C. C. C.; Keller N. P. Methods Enzymol. 2012, 517, 303–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichewicz R. H. Nat. Prod. Rep. 2010, 27, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueto M.; Jensen P.; Kauffman C.; Fenical W.; Lobkovsky E.; Clardy J. J. Nat. Prod. 2001, 64, 1444–1446. [DOI] [PubMed] [Google Scholar]
- Oh D.-C.; Jensen P. R.; Kauffman C. A.; Fenical W. Bioorg. Med. Chem. 2005, 13, 5267–5273. [DOI] [PubMed] [Google Scholar]
- Zhu F.; Lin Y. Chin. Sci. Bull. 2006, 51, 1426–1430. [Google Scholar]
- Oh D.-C.; Kauffman C. A.; Jensen P. R.; Fenical W. J. Nat. Prod. 2007, 70, 515–520. [DOI] [PubMed] [Google Scholar]
- Park H. B.; Kwon H. C.; Lee C.-H.; Yang H. O. J. Nat. Prod. 2009, 72, 248–252. [DOI] [PubMed] [Google Scholar]
- Zuck K. M.; Shipley S.; Newman D. J. J. Nat. Prod. 2011, 74, 1653–1657. [DOI] [PubMed] [Google Scholar]
- Nonaka K.; Abe T.; Iwatsuki M.; Mori M.; Yamamoto T.; Shiomi K.; O̅mura S.; Masuma R. J. Antibiot. 2011, 64, 769–774. [DOI] [PubMed] [Google Scholar]
- Wang J.-P.; Lin W.; Wray V.; Lai D.; Proksch P. Tetrahedron Lett. 2013, 44, 2492–2496. [Google Scholar]
- Han J.-W.; Ahn S. H.; Park S. H.; Wang S. Y.; Bae G.-U.; Seo D.-W.; Kwon H.-K.; Hong S.; Lee H. Y.; Lee Y.-W.; Lee H.-W. Cancer Res. 2000, 60, 6068–6074. [PubMed] [Google Scholar]
- Ferraccioli G. F.; Tomietto P.; De Santis M. Ann. N.Y. Acad. Sci. 2005, 1051, 658–665. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Xu L. Molecules 2012, 17, 2367–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirst H. A. In Milestones in Drug Therapy. Macrolide Antibiotics; Schönfeld W., Kirst H. A., Eds.; Birkhäuser Verlag: Basel, 2002; Chapter 1, pp 1–14. [Google Scholar]
- Burmeister H. R.; Bennett G. A.; Vesonder R. F.; Hesseltine C. W. Antimicrob. Agents Chemother. 1974, 5, 634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. B.; Zink D. L.; Goetz M. A.; Dombrowski A. W.; Polishook J. D.; Hazuda D. J. Tetrahedron Lett. 1998, 39, 2243–2246. [Google Scholar]
- Vesonder R. F.; Tjarks L. W.; Rohwedder W. K.; Burmeister H. R.; Laugal J. A. J. Antibiot. 1979, 32, 759–761. [DOI] [PubMed] [Google Scholar]
- Phillips N. J.; Goodwin J. T.; Fraiman A.; Cole R. J.; Lynn D. G. J. Am. Chem. Soc. 1989, 111, 8223–8231. [Google Scholar]
- Turos E.; Audia J. E.; Danishefsky S. J. J. Am. Chem. Soc. 1989, 111, 8231–8236. [Google Scholar]
- Burmeister H. Patent USPTO 3,959,468; May 25, 1976.
- Quek N. C. H.The Characterization of TA-289, A Novel Antifungal from Fusarium sp. Master’s Thesis, Victoria University of Wellington, New Zealand, 2011. [Google Scholar]
- Wheeler M. H.; Stipanovic R. D.; Puckhaber L. S. Mycol. Res. 1999, 103, 967–973. [Google Scholar]
- König T.; Kapus A.; Sardaki B. J. Bioenerg. Biomembr. 1993, 25, 537–545. [DOI] [PubMed] [Google Scholar]
- Hazuda D.; Uncapher Blau C.; Felock P.; Hastings J.; Pramanik B.; Wolfe A.; Bushman F.; Farnet C.; Goetz M.; Williams M.; Silverman K.; Lingham R.; Singh S. Antiviral Chem. Chemother. 1999, 10, 63–70. [DOI] [PubMed] [Google Scholar]
- Putri S. P.; Kinoshita H.; Ihara F.; Igarashi Y.; Nihira T. J. Antibiot. 2010, 63, 195–198. [DOI] [PubMed] [Google Scholar]
- Marfori E. C.; Kajiyama S.; Fukusaki E.; Kobayashi A. Z. Naturforsch. 2002, 57c, 465–470. [DOI] [PubMed] [Google Scholar]
- Schobert R.; Schlenk A. Bioorg. Med. Chem. 2008, 16, 4203–4221. [DOI] [PubMed] [Google Scholar]
- Malmstrøm J.; Christophersen C.; Frisvad J. C. Phytochemistry 2000, 54, 301–309. [DOI] [PubMed] [Google Scholar]
- Brauers G.; Edrada R. A.; Ebel R.; Proksch P.; Wray V.; Berg A.; Gräfe U.; Schächtele C.; Totzke F.; Finkenzeller G.; Marme D.; Kraus J.; Münchbach M.; Michel M.; Bringmann G.; Schaumann K. J. Nat. Prod. 2000, 63, 739–745. [DOI] [PubMed] [Google Scholar]
- Wang C.-Y.; Wang B.-G.; Wiryowidagdo S.; Wray V.; van Soest R.; Steube K. G.; Guan H.-S.; Proksch P.; Ebel R. J. Nat. Prod. 2003, 66, 51–56. [DOI] [PubMed] [Google Scholar]
- Bauer A. W.; Kirby W. M. M.; Sherris J. C.; Turck M. Am. J. Clin. Pathol. 1996, 45, 493–496. [PubMed] [Google Scholar]
- Kaufmann G. F.; Sartorio R.; Lee S.-H.; Rogers C. J.; Meijler M. M.; Moss J. A.; Clapham B.; Brogan A. P.; Dickerson T. J.; Janda K. D.; Brenner S. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudiero D. A.; Shoemaker R. H.; Paul K. D.; Monks A.; Tierney S.; Nofziger T. H.; Currens M. J.; Seniff D.; Boyds M. R. Cancer Res. 1988, 48, 4827–4833. [PubMed] [Google Scholar]
- Shoemaker R. H. Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.