

Abstract
Gemcitabine is a potent anticancer drug approved for the treatment of pancreatic, non-small-cell lung, breast, and ovarian cancers. The major deficiencies of current gemcitabine therapy, however, are its rapid metabolic inactivation and narrow therapeutic window. Herein, we employed polyethylene glycol-b-distearoylphosphatidylethanolamine (PEG-DSPE)/tocopheryl polyethylene glycol 1000 succinate (TPGS) mixed micelles as a delivery system, to improve the pharmacokinetic characteristics of gemcitabine and enhance its antitumor efficacy. By conjugating stearic acid to gemcitabine and subsequently encapsulating stearoyl gemcitabine (GemC18) within PEG-DSPE/TPGS mixed micelles, the deamination of gemcitabine was delayed in vitro and in vivo. Importantly, compared to free gemcitabine, GemC18-loaded micelles pronouncedly prolonged the circulation time of gemcitabine and elevated its concentration in the tumor by 3-fold, resulting in superior antitumor efficacy in mice bearing human pancreatic cancer BxPC-3 xenografts. Our findings demonstrate the promise of PEG-DSPE/TPGS mixed micelles as a nanocarrier system for the delivery of gemcitabine to achieve safer and more efficacious therapeutic outcomes.
Keywords: gemcitabine, cytidine deamination, polymeric micelles, pharmacokinetics, antitumor efficacy
Introduction
As a deoxycytidine analogue that interferes with DNA synthesis, gemcitabine (2′,2′-difluorocytidine, dFdC) is a potent anticancer drug against an unusually broad spectrum of solid tumors.1 It is an FDA-approved first-line therapy for advanced or metastatic pancreatic cancer as a single agent, a first-line therapy for advanced or metastatic non-small-cell lung cancer in combination with cisplatin, a first-line therapy for metastatic breast cancer in combination with paclitaxel, and a second-line therapy for advanced ovarian cancer in combination with carboplatin. In addition, a large number of gemcitabine-based therapies combined with cytotoxins or molecularly targeted agents are currently being evaluated in clinical trials for the treatment of many common cancer types.2−6
The major deficiencies of gemcitabine therapy, however, are its rapid metabolic inactivation and narrow therapeutic window. The standard gemcitabine regimen is to administer the drug via 30 min intravenous infusion at a weekly dose of 1000–1250 mg/m2. During circulation, gemcitabine is extensively deaminated to the inactive metabolite 2′,2′-difluorouridine (dFdU) by cytidine deaminase, which is abundantly expressed in leukocytes and normal tissues.7 The rapidly declining gemcitabine concentration in plasma necessitates the administration of large doses of the drug in cancer patients. However, the clinical benefits of gemcitabine are limited and short-lived with the median survival extended merely for a few months.8−11 This is largely attributable to insufficient drug accumulation and activation in the tumor cells. On the other hand, the very high initial gemcitabine concentration in plasma immediately following intravenous administration commonly causes severe myelosuppression and toxicities in well-perfused organs including liver, lung, and kidney, which prohibit more frequent administration of the drug than once-weekly dosing in cancer patients. Moreover, the combination of gemcitabine with cytotoxins such as cisplatin, carboplatin, and paclitaxel further exacerbates the hematological toxicities of gemcitabine.8−11
There is therefore an urgent need to improve the therapeutic outcomes of gemcitabine so that the full potential of gemcitabine-based therapies could be realized. One promising approach to improve the pharmacokinetic characteristics of gemcitabine and enhance its anticancer effectiveness is to utilize nanosized drug delivery systems.12 The primary advantages of such nanosystems are 2-fold: (1) to prolong the circulation time of the drug in the bloodstream by protecting the drug from enzymatic inactivation and restricting the drug distribution mainly within the circulation; and (2) to augment the drug accumulation in the tumor owing to the enhanced permeability and retention (EPR) effect,13 a well-known phenomenon responsible for the preferential extravasation of nanoparticles into solid tumors. Liposomes have demonstrated promise in entrapping and delivering gemcitabine, greatly reducing deamination, prolonging the circulation time of gemcitabine, and leading to more potent tumor growth arrest in mice than free gemcitabine.14,15 However, while the liposomal gemcitabine improves the drug accumulation in the tumor tissue, the prolonged presence of liposomal gemcitabine in the circulation also causes massive drug retention in the liver and spleen at 12–24 h, which is likely to elicit long-term toxicities in these organs.14 Conjugation of gemcitabine with highly lipophilic moieties has been investigated to increase the lipophilicity of gemcitabine in order to promote its incorporation into the nanoparticulate delivery systems.16 Squalenoylation of gemcitabine by conjugating its 4-amino group with squalene, a natural lipid and a precursor of cholesterol synthesis, yields a lipophilic prodrug that gets incorporated into liposomes17 or supramolecular vesicular aggregates,18,19 or self-organizes to form nanoassemblies when dispersed in water.20−23 Although the nanoformulations of squalenoyl gemcitabine improve the metabolic stability and potentiate the antitumor efficacy of gemcitabine, they again cause rapid and highly elevated drug uptake in the reticuloendothelial system (RES) including lung, liver, and spleen.19,22 Stearoyl gemcitabine (GemC18), a conjugate of stearic acid with gemcitabine at the 4-(N)-position, displays resistance against metabolic deamination and releases gemcitabine in the presence of cathepsin B.24 When incorporated within solid lipid nanoparticles engineered from lecithin/glycerol monostearate in water emulsion, the stearoyl gemcitabine nanoparticles (GemC18-NPs) demonstrate superior antitumor efficacy compared to free gemcitabine.25 The coupling of epidermal growth factor (EGF) as a targeting ligand onto GemC18-NPs further enhances the in vivo efficacy against EGF receptor-overexpressing tumors.26 However, GemC18-NPs also accumulate in healthy organs including heart, lung, liver, spleen, and kidney.
Self-assembled from biodegradable amphiphilic block polymers, polymeric micelles are considered to be promising drug delivery vehicles for lipophilic drugs.27 The unique core–shell structure of a micelle is afforded by the hydrophobic interactions among the hydrophobic blocks surrounded by the hydrophilic blocks extending into the aqueous milieu. While polyethylene glycol (PEG) is the most commonly used hydrophilic block because of its excellent biocompatibility and “stealth” property, the composition of the hydrophobic block can be tailored to achieve stable encapsulation of lipophilic molecules without the inclusion of any organic solvent. We have recently shown that PEG-b-distearoylphosphatidylethanolamine (PEG-DSPE)/tocopheryl polyethylene glycol 1000 succinate (TPGS) mixed micelles provide a protective shield for the entrapped molecules from elimination and increase drug delivery to the tumor without raising the drug levels in normal organs.28
In the current study, we explored the feasibility of employing PEG-DSPE/TPGS mixed micelles as a delivery system for gemcitabine (Figure 1A). We demonstrated that by conjugating stearic acid to gemcitabine and subsequently encapsulating GemC18 within PEG-DSPE/TPGS mixed micelles, the deamination of gemcitabine to dFdU was attenuated. Importantly, GemC18-loaded micelles drastically elevated gemcitabine concentration in the tumor, resulting in enhanced antitumor efficacy in the human pancreatic tumor xenograft mouse model.
Figure 1.

(A) The structural scheme of GemC18-loaded PEG-DSPE/TPGS mixed micelles. (B) The hydrodynamic diameter of GemC18-loaded PEG-DSPE/TPGS mixed micelles. (C) The release kinetics of free gemcitabine, free GemC18, and GemC18-loaded PEG-DSPE/TPGS mixed micelles as a function of dialysis time. The line represents the respective best-fit regression line for each data set. (D) Cytotoxicity of free gemcitabine, free GemC18, and GemC18-loaded PEG-DSPE/TPGS mixed micelles in human pancreatic cancer BxPC-3 cells for 72 h. All results show representative data obtained from at least 3 independent experiments and are reported as the means + SD (n ≥ 3).
Experimental Section
Materials
Gemcitabine (free base) was purchased from LC Laboratories (Woburn, MA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG-DSPE) was purchased from Corden Pharma (Cambridge, MA). d-α-Tocopheryl polyethylene glycol 1000 succinate (TPGS) was from Eastman Chemical Company (Kingsport, TN). 1-Hydroxybenzotriazole hydrate (HOBt) was purchased from AK Scientific (Union City, CA). Potassium hydroxide (KOH), di-tert-butyl dicarbonate (Boc2O), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI), stearic acid, and trifluoroacetic acid (TFA) were all purchased from Alfa Aesar (Ward Hill, MA). Human recombinant cytidine deaminase was from ProSpec-Tany TechniGene (Israel). Cathepsin B was from Sigma-Aldrich (St. Louis, MO). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), and 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) were from Biotium (Hayward, CA).
Cell Culture
Human pancreatic cancer BxPC-3 cells (American Type Culture Collection, Manassas, VA) were grown in RPMI medium, supplemented with 10% fetal bovine serum, 100 U/m of penicillin, and 100 μg/mL of streptomycin (all from Invitrogen, Carlsbad, CA). The cells were maintained at 37 °C with 5% CO2 in a humidified incubator.
Synthesis of GemC18
The synthesis of GemC18 was carried out according to a reported method29 with minor modifications, as shown in Scheme 1. Gemcitabine 1 (500 mg, 1.90 mmol) was dissolved in 50 mL of 1 M KOH aqueous solution and kept stirring for 1 h. A solution of Boc2O (6.22 g, 28.5 mmol) in 40 mL of dioxane was added dropwise over 1 h. The mixture was stirred at room temperature for 4 h and was then extracted by ethyl acetate. The organic layer was washed 3 times with brine, dried over anhydrous Na2SO4, and concentrated by rotary evaporation under vacuum. The residue was further purified by column chromatography with hexane–acetone (8:1) to give 3′,5′-O-bis(tert-butoxycarbonyl)gemcitabine (2) as a white solid (818 mg, 93% yield). The 1H NMR spectrum (600 MHz, CDCl3) showed the following resonances: δ = 7.43 (s, 1 H), 6.41 (s, 1 H), 5.92 (s, 1 H), 5.25 (m, 1 H), 4.42–4.21 (m, 3 H), 1.51 (m, 9 H), 1.37 (m, 9 H).
Scheme 1. Synthetic Route of GemC18.

(a) KOH, Boc2O, dioxane, H2O, rt, 4 h; (b) stearic acid, EDCI, HOBt, DCM, rt, overnight; (c) TFA, dioxane, reflux, 2 h.
A mixture of stearic acid (540 mg, 1.90 mmol), EDCI (670 mg, 3.50 mmol), and HOBt (600 mg, 3.5 mmol) in 20 mL of dry dichloromethane was stirred for 0.5 h. A solution of 2 (800 mg, 1.73 mmol) in 10 mL of dichloromethane was added, and then the resulting mixture was stirred at room temperature overnight. The organic layer was washed 3 times with brine, dried over anhydrous Na2SO4, and concentrated by rotary evaporation under vacuum. The residue was purified by the column chromatography with hexane–acetone (20:1) to give 3′,5′-O-bis(tert-butoxycarbonyl)-4-N-stearoylgemcitabine (3) as a white solid (1.05 g, 83% yield). 1H NMR (600 MHz, CDCl3): δ = 8.12 (s, 1 H), 7.41 (s, 1 H), 6.51 (s, 1 H), 5.28 (m, 1 H), 4.49–4.27 (m, 3 H), 2.53 (m, 2 H), 1.70 (m, 2 H), 1.57 (s, 9 H), 1.41 (s, 9 H), 1.38–1.21 (m, 28 H), 0.85 (t, 3H).
A mixture of 3 (1 g, 1.37 mmol) and TFA (100 μL) in 15 mL of dioxane was heated, stirred, and refluxed for 2 h. Solvent and excess reagents were evaporated under reduced pressure. The resulting syrup was purified by the column chromatography with dichlormethane–methanol (30:1) to yield 4-(N)-stearoylgemcitabine (GemC18) as a white solid (630 mg, 87% yield). 1H NMR (600 MHz, DMSO): δ = 9.22 (s, 1H), 8.27 (s, 1H), 7.31 (s, 1H), 7.12 (s, 1H), 6.28 (m, 1H), 5.20–5.13 (m, 1H), 4.76–4.51 (m, 2H), 3.39 (t, 2H), 2.50 (m, 2H), 2.35–2.18 (m, 28H), 1.80 (t, 3H).
Micelle Preparation
GemC18-loaded micelles were prepared using a solvent evaporation method as described previously.30 Briefly, GemC18 dissolved in acetone, together with PEG-DSPE and TPGS dissolved in chloroform, was rotor-evaporated at room temperature in a round-bottom flask to form a homogeneous thin drug–polymer film, which was further dried under vacuum overnight to remove any residual solvent. The film was then hydrated with HEPES-buffered saline (HBS, 10 mM, pH 7.4) with vigorous vortexing for 5 min at room temperature. The mixture was then centrifuged at 12000g for 5 min and filtered through a 0.2 μm membrane to remove any undissolved drugs/polymers. The loading concentrations of GemC18 in the micelles, defined as the amount of the drugs in the resulting micellar solution per unit volume of HBS, were quantified by HPLC as described below. The encapsulation efficiency of GemC18 was calculated as the percentage of the incorporated vs the input GemC18. Since the aqueous solubility of GemC18 at room temperature was below 10 nM, free GemC18 dissolved in HBS was negligible. To study cellular uptake of PEG-DSPE/TPGS mixed micelles, equal amounts of DiO and/or DiI were loaded into the micelles following the same procedures as described above.
Characterization of GemC18-Loaded Micelles
The micelle sample was diluted in HBS (10 mM, pH 7.4). The hydrodynamic diameter and zeta potential of the micelles were evaluated by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments, U.K.). The CONTIN approach (number distribution) was used for curve fitting. The storage stability of GemC18-loaded micelles was assessed by monitoring the drug concentration and the micelle size when incubation at 4 or 37 °C for up to 5 weeks.
In Vitro Release Study
To evaluate the release kinetics of GemC18 from the drug-loaded micelles, the release study was studied using a dialysis method.30 Briefly, GemC18-loaded micelles, free gemcitabine, or free GemC18 was loaded into a dialysis cassette (Thermo Scientific, Rockford, IL) with a 20 kDa MWCO, which was placed in a sink of 500 mL of phosphate-buffered saline (PBS, 20 mM, pH 7.4) at 37 °C that was refreshed at each sampling time point. Due to its extremely poor aqueous solubility (<10 nM), free GemC18 was first dissolved in Tween 80/ethanol (1:4, v/v)24 to obtain a stock solution of 6 mM, and then the solution was diluted with HEPES-buffered saline to a final concentration of 100 μM before being inserted into a dialysis cassette. Under this condition, GemC18 remained soluble for at least 24 h and there was no aggregation/precipitation of GemC18 at any time point during the release study. At predetermined time points, a sample (15–25 μL) was collected from each cassette. The concentration of GemC18 or gemcitabine was determined by HPLC as described below. The size and zeta potential of the micellar solution within the dialysis cassette were monitored throughout the release study, and no alteration was observed. To derive the first-order release rate constant (k), the drug concentration in the dialysis cassette (Ct) as a function of release time (t) was fitted to the equation Ct/C0 = e–kt, wherein C0 is the initial drug concentration.30 The best-fit nonlinear regression was obtained by Sigma Plot (San Jose, CA), and the release half-life (t1/2,release) was calculated by 0.693/k.
Cell Proliferation Assay
Stock solutions of free gemcitabine and free GemC18 (1 mM) were prepared in PBS and DMSO, respectively. BxPC-3 cells were seeded in 96-well plates and treated with 10–150 nM gemcitabine, GemC18, or GemC18-loaded micelles. All formulations remained in solution during the entire duration of the study. After 72 h, cells were fixed with 1% glutaraldehyde, stained with 0.1% crystal violet, dissolved in 10% acetic acid, and analyzed for absorbance at 595 nm using FLUOstar Omega plate reader (BMG LABTECH, Germany).31 The relative cell viability was calculated as the percentage of absorbance of the treated vs the untreated wells.
Cellular Uptake
BxPC-3 cells (1.5 × 105/well) were seeded in a 12-well plate and cultured overnight. To study the uptake of PEG-DSPE/TPGS mixed micelles, the cells were treated with equal concentration of DiI, DiO, or DiI/DiO-loaded micelles for 3 h. After removal of the culture medium, cells were washed 3 times with cold PBS and then trypsinized and resuspended in 300 μL of PBS before being subjected to flow cytometry analysis. Cell-associated fluorescence was analyzed using a BD Accuri C6 Flow Cytometer System (BD Biosciences, San Jose, CA). Fluorescence signals were acquired with the excitation wavelength of 488 nm. The spectral filter of 530 ± 15 nm was used to detect DiO, and 585 ± 20 nm was used to detect DiI. All signals were obtained with the same gain and offset. Data collection involved 10,000 counts per sample. Data were analyzed using the FlowJo 9.3.1 software (Tree Star, Inc., Ashland, OR) and expressed as the geometric mean of the entire cell population.
Metabolic Stability
Stock solutions of free gemcitabine and free GemC18 (1 mM) were prepared in PBS and DMSO, respectively. Gemcitabine, GemC18, and GemC18-loaded micelles (50 μM) were incubated in 50 mM sodium acetate buffer (pH 6.5) containing 0.5 mM magnesium chloride, 1 mM EDTA, 200 mM sodium chloride, in the presence of cytidine deaminase (15 μg/mL) and cathepsin B (5 U/mL) at 37 °C. All formulations remained in solution during the entire duration of the study. At predetermined time points, namely, 15, 30, 60, and 120 min, a sample of the mixture was collected and 5 μL of glacial acetic acid was added to quench the enzymatic activities. The samples were then precipitated with 100 μL of acetonitrile. The dried supernatant samples were reconstituted and quantified by HPLC as described below.
Tumor Xenograft Mouse Model
BxPC-3 cells (2 × 106 in 0.1 mL of matrigel/RMPI mixture) were implanted subcutaneously in each flank of 5–6-week-old female athymic nude mice (nu/nu, Charles River, Wilmington, MA). The tumor size was measured using a caliper, and the tumor volume was calculated as 1/2 × length × width2.
Pharmacokinetic Study
When the tumor volumes reached 100–300 mm3, the mice were randomized into three different treatment groups (3 mice per group). Each group was intravenously administrated with 25 mg/kg gemcitabine equivalent dose of free gemcitabine dissolved in PBS, free GemC18 dissolved in Tween 80/ethanol:PBS (1:4, v/v),24 or GemC18-loaded micelles. At 3, 15, 30, 45, 60, and 90 min postinjection, whole blood was collected via retro-orbital bleeding. The concentrations of GemC18, gemcitabine, and/or gemcitabine deamination metabolite dFdU were quantified by HPLC as described below. Noncompartmental analysis was performed to obtain pharmacokinetic parameters including the elimination half-life (t1/2,e), the total body clearance (CLT), and the apparent volume of distribution (Vd,ss) using WinNonlin software (version 5.1, Pharsight, Sunnyvale, CA). To assess gemcitabine concentration in different tissues, normal organs (liver, spleen, kidney, lung, and heart) and tumor tissues were harvested at 60 min and were processed and analyzed by HPLC as described below.
Antitumor Efficacy Study
Once the BxPC-3 xenografts reached 50–100 mm3, the mice were randomized into 3 treatment groups (4 mice per group): untreated control group, free gemcitabine group, and GemC18-loaded micelle group. The mice were treated with 10 mg/kg gemcitabine equivalent dose as free gemcitabine dissolved in PBS or GemC18-loaded micelles, twice weekly for 2 consecutive weeks.
Statistical Analysis
All data were presented as mean ± SD or SE. Data from different groups were compared using Student’s t-test. A p value of less than 0.01 was considered to be statistically significant.
HPLC Methodology
The HPLC system consisted of a Waters 2695 separations module, a Waters 996 photodiode array detector, and an Empower software system (Milford, MA). The standard curves for GemC18, or gemcitabine and its metabolite in PBS buffer, plasma, and tissue homogenates were established by spiking the samples with the drug stocks (dissolved in DMSO) with a linear range of 1–100 μM. Plasma and tissue homogenate samples were treated with glacial acetic acid to terminate possible deamination and were precipitated with acetonitrile. Following the centrifugation at 12000g for 5 min, the supernatants were evaporated overnight. The dried samples were reconstituted in the mobile phase, and the supernatant was injected into the HPLC system.
Due to its high lipophilicity, GemC18 was quantified separately from gemcitabine and dFdU. For the quantification of GemC18, a Phenomenex (Torrance, CA) Luna C18 column (5 μm, 150 × 4.6 mm) was used with an isocratic mobile phase of 15% (v/v) sodium phosphate buffer (10 mM, pH 7.0, supplemented with 10 mM triethylamine) and 85% (v/v) methanol. α-Naphthoflavone (2 μM) was used as an internal standard. The flow rate was 1.0 mL/min with a column temperature of 40 °C. The detection wavelengths for GemC18 and α-naphthoflavone were 252 and 281 nm, respectively. GemC18 was eluted at 18 min, α-naphthoflavone at 4 min. The recovery rate of GemC18 in plasma was 88%.
An ion-pairing methodology was developed to simultaneously resolve gemcitabine and dFdU onto a Luna C18 column (5 μm, 250 × 4.6 mm), and 5-methylcytidine (10 μM) was used as an internal standard. To analyze drug concentrations in aqueous buffer and plasma samples, a gradient mobile phase system A [octanesulfonic acid (5 mM) in sodium phosphate buffer (10 mM, pH 2.9, supplemented with 10 mM triethylamine)] and B [methanol] was used: 7% B for 12 min, increased and maintained at 25% B for 13 min, increased and maintained at 50% B for 5 min, then increased and maintained at 95% B for 5 min, finally gradually reduced to initial 7% B and maintained for 10 min. The flow rate was 0.8 mL/min with a column temperature of 40 °C. Gemcitabine was eluted at 26 min, dFdU at 12 min, and 5-methylcytidine at 21 min. The detection wavelengths for gemcitabine, dFdU, and 5-methylcytidine were 275 nm, 259 nm, and 288 nm, respectively. The recovery rates of gemcitabine and dFdU in plasma were 87% and 91%, respectively.
To quantify gemcitabine in the tissue homogenates, an isocratic mobile phase of 93% (v/v) octanesulfonic acid (5 mM) in sodium phosphate buffer (10 mM, pH 2.9, supplemented with 10 mM triethylamine) and 7% (v/v) methanol was used. The flow rate was 0.8 mL/min with a column temperature of 40 °C. Gemcitabine and 5-methylcytidine were eluted at 78 and 46 min, respectively. The recovery rates of gemcitabine in the homogenates of tumor, liver, spleen, kidney, lung, and heart tissues were 73%, 73%, 87%, 85%, 82%, and 78%, respectively.
Results
Synthesis and Characterization of GemC18-Loaded Micelles
We have recently demonstrated that PEG-DSPE/TPGS mixed micelles can efficiently load hydrophobic drugs and serve as drug carriers in vivo.28 To explore this micellar system for the delivery of gemcitabine, we first conjugated stearic acid to gemcitabine at the 4-(N)-position to increase the lipophilicity of the drug. Briefly, the 3′ and 5′ hydroxyl groups of gemcitabine were protected by Boc anhydride in alkaline condition to afford 2 (Scheme 1). Stearic acid was then conjugated to the amino group of compound 2 in the presence of EDCI and HOBt. The removal of Boc groups in compound 3 by TFA furnished GemC18 with a total yield of 67%. Structures of the synthesized compounds were confirmed by their respective 1H NMR spectra. We hypothesized that the modification of gemcitabine with an acyl chain identical to that in DSPE moiety would promote hydrophobic interactions, confer good compatibility, and enable stable incorporation of GemC18 into the hydrophobic core of PEG-DSPE/TPGS mixed micelles. We found that GemC18-loaded PEG-DSPE/TPGS mixed micelles were formed readily with encapsulation efficiency above 95%. The average hydrodynamic diameter of GemC18-loaded PEG-DSPE/TPGS mixed micelles was 11.8 nm (Figure 1B) with a surface charge of −23 mV and a polydispersity index of 0.38. GemC18 could be loaded into PEG-DSPE/TPGS mixed micelles at a maximum loading concentration of 10 mM, whereas the molar ratio of GemC18:PEG-DSPE:TPGS was 1:2:4. At 4 °C GemC18-loaded micelles remained stable in aqueous buffer for at least 5 weeks with less than 10% decrease in the loading concentration and negligible change in size. At 37 °C, GemC18-loaded micelles were stable in aqueous buffer for 48 h without noticeable alteration in the loading concentration and size. In plasma, GemC18-loaded micelles were stable for 4 h at 37 °C with less than 2% loss in GemC18 loading. Following 24 h incubation, the loading concentration of GemC18 decreased by 16%.
In Vitro Release Study
To function as a drug carrier system for GemC18 and improve its delivery to the tumor, it is a prerequisite that PEG-DSPE/TPGS mixed micelles can retain GemC18 for extended period of time in order to promote preferential drug accumulation into the tumor via the EPR effect. The release kinetics of GemC18-loaded micelles was examined in comparison with that of free gemcitabine and free GemC18 by monitoring the release of drug molecules from a dialysis cassette into a sink. As shown in Figure 1C, the release of free gemcitabine across the dialysis membrane was rapid with a release half-life (t1/2,release) of about 15 min (see also Table 1). Free GemC18 was released more slowly compared to free gemcitabine with 50% GemC18 released into the sink in approximately 2 h. Strictly speaking, GemC18 solubilized in Tween 80 was not in free form due to its incorporation into the Tween 80 micelles, which deters the diffusion of free GemC18 within the dialysis cassette. The observed release half-life of GemC18 herein was therefore an overestimation for that of free GemC18. In contrast, when GemC18 was loaded into the PEG-DSPE/TPGS mixed micelles, the release of GemC18 was significantly diminished with a t1/2,release of about 12 h. The much reduced release rate constant of GemC18-loaded micelles compared to that of free GemC18 strongly implies that the liberation of GemC18 from PEG-DSPE/TPGS mixed micelles is the rate-limiting step during the entire drug release process into the sink. These results indicate that PEG-DSPE/TPGS mixed micelles are able to entrap GemC18 for prolonged period of time, and function as carriers to deliver GemC18.
Table 1. The Release Kinetics of GemC18 from PEG-DSPE/TPGS Mixed Micelles, Free GemC18, and Free Gemcitabine.
| release rate constant (h–1) | t1/2,release (h) | goodness-of-fit (r2) | |
|---|---|---|---|
| GemC18-loaded micelles | 0.055 | 12.60 | 0.997 |
| free GemC18 | 0.328 | 2.11 | 0.970 |
| free gemcitabine | 2.920 | 0.24 | 0.990 |
Cell Proliferation Assay
As a prodrug, GemC18 needs to be hydrolyzed to gemcitabine to result in cytotoxicity. To evaluate the activity of GemC18-loaded micelles, a proliferation assay was performed in human pancreatic cancer BxPC-3 cells. As shown in Figure 1D, the EC50 of gemcitabine was 25 nM; while for GemC18, the EC50 was increased to 40 nM; when loaded into the micelles, the EC50 of micellar GemC18 was further increased to 65 nM. There was no significant loss in cell viability caused by the empty micelles across the entire tested concentration range. The reduced cytotoxicity could be accounted by the slow conversion of GemC18 into gemcitabine, and by the entrapment into the micelles that further shields GemC18 from hydrolyzing into gemcitabine.
Cellular Uptake of PEG-DSPE/TPGS Mixed Micelles
Fluorescence resonance energy transfer (FRET) occurs when a donor dye and an acceptor dye are present within the range of Förster distance; the emission fluorescence from the excited donor dye is used as the excitation energy for the acceptor dye, resulting in the emission of the acceptor fluorescence.32 As a FRET pair of hydrophobic dyes, DiO (donor, Ex/Em 488/501 nm) and DiI (acceptor, Ex/Em 501/565 nm) can be enclosed within the hydrophobic core of the micelles, to monitor the structural integrity of micelles. For the micelles loaded with both DiO and DiI, when they are excited at wavelength of 488 nm, the emission energy generated from DiO can be transferred efficiently to adjacent DiI, which subsequently gets excited and emits fluorescence at wavelength 565 nm.33 By contrast, when the micelles disintegrate or the dye molecules are released from the micellar core, no emission fluorescence from DiI can be observed due to the lack of excitation energy. The cellular uptake of PEG-DSPE/TPGS mixed micelles was studied by directly determining the fluorescence signals of DiO-, DiI-, or DiO/DiI-loaded micelles in BxPC-3 cells. The washing steps appeared to be sufficient to remove micelles adhering to the cell surface, and all fluorescence signals could be assigned to the intracellular dye molecules, since further repeated washing did not reduce the fluorescence intensity. As shown in Figure 2A, when excited at 488 nm, the cells incubated with either DiO- or DiO/DiI-loaded micelles displayed strong green fluorescence signal at 530 ± 15 nm; whereas only the cells incubated with DiO/DiI-dual-loaded micelles emitted red fluorescent signal at 585 ± 20 nm. Because the emission of red fluorescence by DiI requires excitation energy transferred from adjacent DiO, these results strongly suggest that PEG-DSPE/TPGS mixed micelles are internalized into the tumor cells with both DiI and DiO enclosed inside the micellar core. As expected, no red fluorescence signal was detected from the cells treated with DiI-loaded micelles.
Figure 2.

Cellular uptake of PEG-DSPE/TPGS mixed micelles in BxPC-3 cells. Cells were incubated with DiO/DiI-dual-loaded PEG-DSPE/TPGS mixed micelles for 3 h, in comparison to DiO- or DiI-loaded micelles or the untreated controls. All micelles were loaded with equal concentration of DiO and/or DiI. The intracellular fluorescence intensity was analyzed by a flow cytometer, as displayed in dot plots (A) and histogram plots (B, C). The insets show the median fluorescence intensity (MFI) of each cell population. All results show representative data obtained from 3 independent experiments. The excitation wavelength was set at 488 nm, the FL1 channel had a spectral filter of 530 ± 15 nm to detect the fluorescence emission from DiO, and the FL2 channel with a spectral filter of 585 ± 20 nm to detect the fluorescence emission from DiI.
The histogram analysis of the fluorescence intensity in the cells following the incubation with DiI-, DiO-, or DiI/DiO-loaded PEG-DSPE micelles was in agreement with the above results. While the green fluorescence intensity in the cells incubated with DiI-loaded micelles was identical to that of the untreated control cells, the fluorescence intensity in the cells treated with DiO-loaded micelles was higher than that of cells treated with DiO/DiI-dual-loaded micelles (Figure 2B). This is attributable to the energy transfer from DiO to DiI when both dyes are entrapped within Förster distance inside the micellar core, resulting in the reduced emission intensity by DiO. Importantly, the red fluorescence intensity in the cells incubated with DiI/DiO-dual-loaded micelles was at least 2-fold above the background noise observed in the cells treated with DiI- or DiO-loaded micelles (Figure 2C). Moreover, as highly lipophilic dyes, both DiI and DiO molecules have a stearoyl moiety, which imparts good compatibility and retention of the dye molecules within the PEG-DSPE/TPGS micellar core and allows them to serve as the payload markers for GemC18. Together, these results strongly imply that PEG-DSPE/TPGS mixed micelles can retain the structural integrity and be taken up by the tumor cells along with the payload.
Metabolic Study in the Presence of Cathepsin B and Cytidine Deaminase
GemC18 is converted to gemcitabine by amidases such as cathepsin B, which is subsequently susceptible to deamination by cytidine deaminase. To mimic the in vivo setting where these metabolic enzymes usually work in concert, we studied the metabolic stability of free gemcitabine, free GemC18, and GemC18-loaded micelles in the presence of both cathepsin B and cytidine deaminase. We found that GemC18-loaded micelles were completely resistant to cathepsin B and cytidine deaminase; there was no decrease in GemC18 or formation of dFdU (Figures 3A and 3B) during the 2 h incubation period. Under identical conditions, free gemcitabine was rapidly metabolized with 80% conversion to dFdU by 1 h. Free GemC18 was metabolized and converted to dFdU at a slower rate than free gemcitabine, with about 20% conversion to dFdU at 2 h. These results indicate that PEG-DSPE/TPGS micelles protect GemC18 from enzymatic conversions, which deters the metabolic inactivation of gemcitabine into dFdU.
Figure 3.
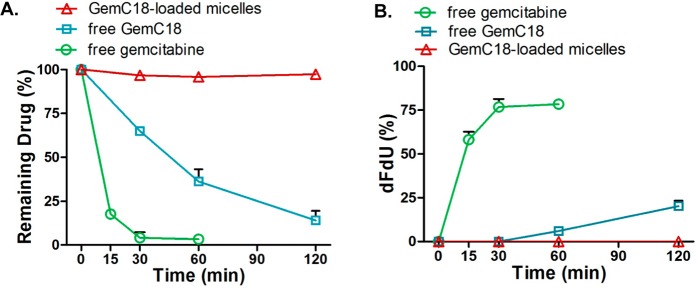
Metabolic stability of GemC18-loaded PEG-DSPE/TPGS mixed micelles in comparison to free gemcitabine and free GemC18. Micellar GemC18, free GemC18, or free gemcitabine (100 μM) was incubated in a buffer (pH 6.5) containing cathepsin B (5 U/mL) and cytidine deaminase (15 μg/mL) at 37 °C. (A) The decrease in GemC18 or gemcitabine as a function of the incubation time. (B) The formation of dFdU as a function of the incubation time. All results are reported as the means + SD (n = 3).
Pharmacokinetic Study in Mice Bearing BxPC-3 Xenografts
To evaluate the therapeutic potential of GemC18-loaded PEG-DSPE/TPGS mixed micelles, we first examined the pharmacokinetics of GemC18-loaded micelles in comparison to free GemC18 and free gemcitabine. Owing to its sparse aqueous solubility, GemC18 was solubilized in the Tween 80 formulation, which entrapped GemC18 within the Tween 80 micelles. The pharmacokinetic profile of GemC18 in the Tween 80 formulation is a close approximation of that of free GemC18, because the Tween 80 micelles are known to disintegrate rapidly in the circulation.34 As shown in Figure 4A, intravenous administration of GemC18-loaded micelles resulted in 2–5-fold higher plasma concentration of GemC18 than that of free prodrug. The volume of distribution at the steady state (Vd,ss) of GemC18-loaded micelles was markedly lower than that of free GemC18 (Table 2), indicating that the distribution of the micellar prodrug is much more restricted to the circulation. The elimination half-life (t1/2,e) of micellar GemC18 was similar to that of free GemC18, suggesting that the elimination of the micellar prodrug is rate-limited by the removal of the free prodrug. The total body clearance (CLT) of micellar GemC18 was about one-third of free GemC18, causing a more than 3-fold increase in the systemic exposure (AUC) of the prodrug. These results suggest that when GemC18-loaded micelles are administered intravenously, a significant proportion of GemC18 remains associated with PEG-DSPE/TPGS mixed micelles while circulating in the bloodstream.
Figure 4.

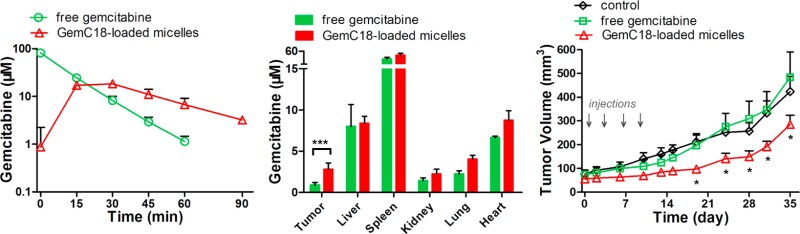
Pharmacokinetics of GemC18-loaded PEG-DSPE/TPGS mixed micelles in nude mice bearing human pancreatic cancer BxPC-3 xenografts. The mice were iv administered 25 mg/kg equivalent gemcitabine as either free gemcitabine dissolved in PBS, free GemC18 dissolved in Tween 80/ethanol:PBS (1:4, v/v), or GemC18-loaded micelles. Each data point was the mean + SD, n = 3 mice per group. (A) GemC18 concentrations in plasma. (B) Gemcitabine concentrations in plasma. (C) dFdU concentrations in plasma. (D) Gemcitabine concentrations in the tumor and normal organs (***, p = 0.0002; *, p = 0.005).
Table 2. Pharmacokinetic Parameters of GemC18-loaded PEG-DSPE/TPGS Mixed Micelles and Free GemC18 in Mice (Mean ± SD, n = 3).
| GemC18-loaded micelles | free GemC18 | |
|---|---|---|
| t1/2,ea (min) | 26.5 ± 6.5 | 21.0 ± 1.6 |
| AUCb (μM·min) | 10821.3 ± 1190.8 | 3500.7 ± 301.7 |
| CLTc (mL/kg/min) | 8.0 ± 0.2 | 26.7 ± 2.3 |
| Vd,ssd (mL/kg) | 185.3 ± 37.8 | 386.7 ± 32.3 |
t1/2,e: terminal elimination half-life.
AUC: area under the plasma drug concentration versus time curve.
CLT: total clearance.
Vd,ss: apparent volume of distribution at the steady-state.
In addition to the prodrug, we also quantified the concentrations of gemcitabine and its inactive metabolite dFdU in plasma. As shown in Figure 4B and Table 3, gemcitabine was formed gradually following micellar GemC18 administration, with a time to reach maximum (Tmax) of 15 min and t1/2,e of 27 min. By contrast, intravenous administration of an identical dose of free gemcitabine resulted in high initial plasma concentration ensued by a rapid decline with t1/2,e of 10 min. The metabolism of gemcitabine to dFdU occurred immediately in the case of free gemcitabine administration, with a substantial amount of dFdU already formed within the initial 3 min (Figure 4C). Meanwhile, following the administration of micellar GemC18, the deamination of gemcitabine proceeded at a much slower rate (Figure 4C). This is consistent with the in vitro finding that the biotransformation of GemC18 requires the involvement of amidases such as cathepsin B as well as cytidine deaminase while the micellar nanocarriers provide extra protection for GemC18 from the enzymatic attacks.
Table 3. Pharmacokinetic Parameters of Gemcitabine Released from GemC18-Loaded PEG-DSPE/TPGS Mixed Micelles and Free Gemcitabine in Mice (Mean ± SD, n = 3).
| GemC18-loaded micelles | free gemcitabine | |
|---|---|---|
| t1/2,e (min) | 26.6 ± 4.6 | 10.1 ± 0.4 |
| AUC (μM·min) | 995.9 ± 111.7 | 1130.0 ± 39.8 |
| CLT (mL/kg/min) | 84.0 ± 4.0 | |
| Vd,ss (mL/kg) | 1062.7 ± 131.5 |
Next, the distribution of gemcitabine into both normal organs and the tumor tissues was also analyzed. We found that, compared to free gemcitabine, GemC18-loaded micelles elevated gemcitabine concentration in the tumor by over 3-fold (p = 0.0002) at 1 h, whereas the drug concentration in normal organs was increased by less than 100% (Figure 4D). The increased gemcitabine level in the tumor following the treatment of GemC18-loaded micelles is likely driven by the preferential extravasation of the micellar prodrug into the tumor, combined with the more sustained gemcitabine concentration in the circulation. Collectively, these results indicate that GemC18-loaded micelles prolong the circulation time and tumor accumulation of gemcitabine as well as slow down its metabolic inactivation.
Antitumor Efficacy Study in Mice Bearing BxPC-3 Xenografts
Encouraged by the improved pharmacokinetics of gemcitabine resulting from the micellar delivery approach, we next examined the in vivo antitumor efficacy of GemC18-loaded micelles in BxPC-3 tumor bearing mice. Although gemcitabine is known to be efficacious at weekly 50 mg/kg in mice, we chose a treatment regimen of 10 mg/kg equivalent dose twice weekly in order to observe possible potentiation in the efficacy by the micellar formulation. As shown in Figures 5A and 5B, GemC18-loaded micelles potently suppressed tumor growth. Starting on day 19, the average tumor sizes of the mice receiving micellar GemC18 were significantly smaller than those of the untreated or free drug treated mice (p < 0.01). On the contrary, free gemcitabine was unable to arrest the tumor growth for the tumor volume was similar to that found in the untreated controls. Composed of biodegradable and biocompatible polymers, the empty PEG-DSPE/TPGS mixed micelles did not exhibit antitumor efficacy.28 There was no significant decrease in the body weight of the treated mice (Figure 5C), an indication of no overt toxicity of either free gemcitabine or micellar GemC18. These results clearly indicate that the anticancer efficacy of GemC18-loaded micelles is superior to that of free gemcitabine.
Figure 5.

GemC18-loaded PEG-DSPE/TPGS mixed micelles potentiate the antitumor efficacy of gemcitabine in nude mice bearing BxPC-3 xenografts. Mice were randomized, and treatment was initiated on day 0. The mice were dosed intravenously with 10 mg/kg equivalent gemcitabine either as free gemcitabine dissolved in PBS or as GemC18-loaded micelles on days 0, 3, 7, and 10. The untreated mice served as controls. (A) Tumor growth curves in mice. Each data point was the mean + SD (n = 4 mice per group; *, p < 0.01). (B) The average body weight of mice remained constant in all groups throughout the study.
Discussion
Gemcitabine is among the select few nucleoside analogues that exhibit potent anticancer activity against a broad spectrum of solid tumors. To exert cytotoxicity, gemcitabine first needs to be transported across the cell membrane, mainly via human equilibrating nucleoside transporter 1 (hENT1).35 Once inside the cytoplasm, gemcitabine undergoes sequential phosphorylations and gives rise to gemcitabine diphosphate and triphosphate, the active forms of gemcitabine, which disrupt DNA synthesis and cause apoptosis.1 In the cell culture setting, in the tumor cells with wild-type hENT1 and deoxycytidine kinase, gemcitabine is highly cytotoxic with EC50 in the low nanomolar range.36 The challenge of gemcitabine therapy in vivo, however, arises from the instantaneous inactivation of gemcitabine by cytidine deaminase, which occurs at a much faster rate while circulating than the intracellular activation of gemcitabine in the tumor cells.37 Consequently, even being administered at the maximally tolerated doses, the plasma concentration of gemcitabine quickly declines below the minimal effective concentration, leading to the suboptimal drug accumulation and activation in the tumor. To enhance the antitumor efficacy of gemcitabine therapy, it is therefore of critical importance to attenuate metabolic inactivation of gemcitabine as well as to enhance the drug accumulation in the tumor.
Herein, PEG-DSPE/TPGS mixed micelles were explored as a nanocarrier system for the delivery of gemcitabine. The critical micelle concentrations (CMC) of PEG-DSPE and TPGS are within the 10–6–10–5 M range, reflecting the high thermodynamic stability of the mixed micelles in aqueous solution and upon dilution.39 We have previously shown that, at a 1:2 molar ratio, PEG-DSPE and TPGS self-orient to form mixed micelles with thermodynamically stable construct and excellent loading capacity for hydrophobic drugs.30,38 Furthermore, by employing indocyanine green, a near-infrared dye that could be efficiently loaded within PEG-DSPE/TPGS mixed micelles, a significant proportion of these nanocarriers remained intact in the circulation for at least 4–6 h, which protected the loaded molecules from elimination and preferentially accumulated in the tumor.28 In the present study, GemC18 was found to be stably incorporated into PEG-DSPE/TPGS mixed micelles at therapeutically relevant concentration. Though the modification of gemcitabine at the 4-(N)-position blocked the deamination site, the combined presence of cathepsin B and cytidine deaminase could still readily convert the prodrug into the inactive metabolite dFdU. By contrast, micellar GemC18 was found to be much less prone to the biotransformation by cathepsin B and cytidine deaminase, which greatly deterred the formation of dFdU in vitro. Accordingly, compared to free gemcitabine, the intravenous administration of micellar GemC18 yielded a more sustained level of gemcitabine, which was accompanied by the retarded conversion to dFdU in plasma.
In addition to serving as a sustained-release formulation for gemcitabine, another important advantage of micellar GemC18 was its ability to directly deliver the payload into the tumor tissue via the EPR effect. With an average diameter around 12 nm, PEG-DSPE/TPGS mixed micelles were large enough to avoid the glomerular filtration, yet small enough to evade the macrophage engulfment in the RES and extravasate across the angiogenic tumor endothelium. As a result, GemC18-loaded PEG-DSPE/TPGS mixed micelles boosted gemcitabine concentration in the tumor by over 3-fold without drastically increasing the drug level in normal organs. This is in sharp contrast with the tissue distribution patterns of gemcitabine-loaded liposomes (∼200 nm),14 squalenoyl gemcitabine-loaded supramolecular vesicular aggregates (∼100 nm),19 squalenoyl gemcitabine nanoassemblies (∼130 nm),22 or C18Gem solid nanoparticles (∼170–200 nm),25 which increase gemcitabine level not only in the tumor but also in the RES such as liver, lung, and spleen by 2–5-fold. With a much smaller size, the drug-loaded PEG-DSPE/TPGS mixed micelles are also expected to diffuse more freely within the tumor, which is beneficial to exert the anticancer efficacy.
Upon arrival at the tumor site, micellar GemC18 could have access to the tumor cells via two possible routes: (1) GemC18 could first be released from the micelles prior to its permeation across the tumor cell membrane; (2) micellar GemC18 could get internalized into the tumor cells by endocytosis, and then release GemC18 in the cytoplasm. Although the first route may dominate, both scenarios are likely to coexist, which potentially bypass hENT1-mediated drug transport. In fact, by using FRET imaging probes, our flow cytometry data clearly indicate that PEG-DSPE/TPGS mixed micelles are able to be internalized into the tumor cells along with the payload. In the tumor tissue, the released GemC18 is expected to be readily hydrolyzed to gemcitabine by amidases such as cathepsin B. It has been shown that cathepsin B, localized inside the endosomes and lysosomes, on the cell membrane surface and in the extracellular matrix, is overexpressed in various tumor types.40
Finally, our work has demonstrated that GemC18-loaded PEG-DSPE/TPGS mixed micelles can significantly enhance the in vivo efficacy of gemcitabine, similar to what was observed with liposomal gemcitabine,14,15 squalenoyl gemcitabine-based nanoformulations,17−23 and GemC18 nanoparticles.25,26 The prolonged circulation of gemcitabine combined with the augmented delivery of micellar GemC18 to the tumor by the EPR effect is responsible for this favorable therapeutic outcome. Both PEG-DSPE and TPGS are FDA-approved pharmaceutical excipients, which makes the current micellar delivery system highly translatable to the clinic. Our findings raise the exciting possibility of optimizing the current therapy regimen of gemcitabine by administering the drug at a much lower dose with higher frequency to maximize the anticancer efficacy and minimize the debilitating toxicities toward normal organs.
Acknowledgments
This work was supported by National Institutes of Health Grant R15 CA152860 to C.T.
Author Contributions
‡ Y.W. and W.F. contributed equally to the work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Mini E.; Nobili S.; Caciagli B.; Landini I.; Mazzei T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17, v7–v12. [DOI] [PubMed] [Google Scholar]
- Michaelson M. D.; Zhu A. X.; Ryan D. P.; McDermott D. F; Shapiro G. I.; Tye L.; Chen I.; Stephenson P.; Ruiz-Garcia A.; Schwarzberg A. B. Sunitinib in combination with gemcitabine for advanced solid tumours: a phase I dose-finding study. Br. J. Cancer 2013, 108, 1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Hurt C. N.; Bridgewater J.; Falk S.; Cummins S.; Wasan H.; Jephcott C.; Roy R.; Radhakrishna G.; McDonald A.; Ray R.; Joseph G.; Staffurth J.; Abrams R. A.; Griffiths G.; Maughan T. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): a multicentre, randomised, phase 2 trial. Lancet Oncol. 2013, 14, 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balar A. V.; Apolo A. B.; Ostrovnaya I.; Mironov S.; Iasonos A.; Trout A.; Regazzi A. M.; Garcia-Grossman I. R.; Gallagher D. J.; Milowsky M. I.; Bajorin D. F. Phase II study of gemcitabine, carboplatin, and bevacizumab in patients with advanced unresectable or metastatic urothelial cancer. J. Clin. Oncol. 2013, 31, 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay V. M.; Middleton M. R.; Protheroe A. S.; Tolcher A.; Dieras V.; Sessa C.; Bahleda R.; Blay J. Y.; LoRusso P.; Mery-Mignard D.; Soria J. C. Phase I study of humanized monoclonal antibody AVE1642 directed against the type 1 insulin-like growth factor receptor (IGF-1R), administered in combination with anticancer therapies to patients with advanced solid tumors. Ann. Oncol. 2013, 24, 784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérol M.; Chouaid C.; Pérol D.; Barlési F.; Gervais R.; Westeel V.; Crequit J.; Léna H.; Vergnenègre A.; Zalcman G.; Monnet I.; Le Caer H.; Fournel P.; Falchero L.; Poudenx M.; Vaylet F.; Ségura-Ferlay C.; Devouassoux-Shisheboran M.; Taron M.; Milleron B. Randomized, phase III study of gemcitabine or erlotinib maintenance therapy versus observation, with predefined second-line treatment, after cisplatin-gemcitabine induction chemotherapy in advanced non–small-cell lung cancer. J. Clin. Oncol. 2012, 30, 3516–3524. [DOI] [PubMed] [Google Scholar]
- Ho D. H. W. Distribution of kinase and deaminase of 1-β-D-arabinofuranosylcytosine in tissues of man and mouse. Cancer Res. 1973, 33, 2816–2820. [PubMed] [Google Scholar]
- Burris H. A.; Moore M. J.; Andersen J.; Green M. R.; Rothenberg M. L.; Modiano M. R.; Cripps M. C.; Portenoy R. K.; Storniolo A. M.; Tarassoff P.; Nelson R.; Dorr F. A.; Stephens C. D.; Von Hoff D. D. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [DOI] [PubMed] [Google Scholar]
- Sandler A. B.; Nemunaitis J.; Denham C.; Von Pawel J.; Cormier Y.; Gatzemeier U.; Mattson K.; Manegold Ch.; Palmer M. C.; Gregor A.; Nguyen B.; Niyikiza C.; Einhorn L. H. Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non-small-cell lung cancer. J. Clin. Oncol. 2000, 18, 122–130. [DOI] [PubMed] [Google Scholar]
- Albain K. S.; Nag S. M.; Calderillo-Ruiz G.; Jordaan J. P.; Llombart A. C.; Pluzanska A.; Rolski J.; Melemed A. S.; Reyes-Vidal J. M.; Sekhon J. S.; Simms L.; O’Shaughnessy J. Gemcitabine plus paclitaxel versus paclitaxel monotherapy in patients with metastatic breast cancer and prior anthracycline treatment. J. Clin. Oncol. 2008, 26, 3950–3957. [DOI] [PubMed] [Google Scholar]
- Pfisterer J.; Plante M.; Vergote I.; du Bois A.; Hirte H.; Lacave A. J.; Wagner U.; Stähle A.; Stuart G.; Kimmig R.; Olbricht S.; Le T.; Emerich J.; Kuhn W.; Bentley J.; Jackisch C.; Lück H. J.; Rochon J.; Zimmermann A. H.; Eisenhauer E. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J. Clin. Oncol. 2006, 24, 4699–4707. [DOI] [PubMed] [Google Scholar]
- Celia C.; Cosco D.; Paolino D.; Fresta M. Gemcitabine-loaded innovative nanocarriers vs GEMZAR: biodistribution, pharmacokinetics features and in vivo antitumor activity. Expert Opin. Drug Delivery 2011, 8, 1609–1629. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Nakamura H.; Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Delivery Rev. 2013, 65, 71–79. [DOI] [PubMed] [Google Scholar]
- Paolino D.; Cosco D.; Racanicchi L.; Trapasso E.; Celia C.; Iannone M.; Puxeddu E.; Costante G.; Filetti S.; Russo D.; Fresta M. Gemcitabine-loaded PEGylated unilamellar liposomes vs GEMZAR®: biodistribution, pharmacokinetic features and in vivo antitumor activity. J. Controlled Release 2010, 144, 144–150. [DOI] [PubMed] [Google Scholar]
- Cosco D.; Bulotta A.; Ventura M.; Celia C.; Calimeri T.; Paolino D.; Costa N.; Neri P.; Tagliaferri P.; Tassone P.; Fresta M. In vivo activity of gemcitabine-loaded PEGylated small unilamellar liposomes against pancreatic cancer. Cancer Chemother. Pharmacol. 2009, 64, 1009–1020. [DOI] [PubMed] [Google Scholar]
- Moysan E.; Bastiat G.; Benoit J.-P. Gemcitabine versus modified gemcitabine: a review of several promising chemical Modifications. Mol. Pharmaceutics 2013, 10, 430–444. [DOI] [PubMed] [Google Scholar]
- Pili B.; Reddy H.; Bourgaux C.; Lepêtre-Mouelhi S.; Desmaële D.; Courveur P. Liposomal squalenoyl-gemcitabine: formulation, characterization and anticancer activity evaluation. Nanoscale 2010, 2, 1521–1526. [DOI] [PubMed] [Google Scholar]
- Licciardi M.; Polino D.; Celia C.; Giammona G.; Cavallaro G.; Fresta M. Folate-targeted supramolecular vesicular aggregates based on polyaspartyl-hydrazide copolymers for the selective delivery of antitumor drugs. Biomaterials 2010, 31, 7340–7354. [DOI] [PubMed] [Google Scholar]
- Polino D.; Licciardi M.; Celia C.; Giammona G.; Fresta M.; Cavallaro G. Folate-targeted supramolecular vesicular aggregates as a new frontier for effective anticancer treatment in in vivo model. Eur. J. Pharm. Biopharm. 2012, 82, 94–102. [DOI] [PubMed] [Google Scholar]
- Couvreur P.; Stella B.; Reddy L. H.; Hillaireau H.; Dubernet C.; Desmaële D.; Lepêtre-Mouelhi S.; Rocco F.; Dereuddre-Bosquet N.; Clayette P.; Rosilio V.; Marsaud V.; Renoir J. M.; Cattel L. Squalenoyl nanomedicines as a potential therapeutics. Nano Lett. 2006, 6, 2544–2548. [DOI] [PubMed] [Google Scholar]
- Reddy L. H.; Dubernet C.; Mouelhi S. L.; Marque P. E.; Desmaele D.; Couvreur P. A new nanomedicine of gemcitabine displays enhanced anticancer activity in sensitive and resistant leukemia types. J. Controlled Release 2007, 124, 20–27. [DOI] [PubMed] [Google Scholar]
- Reddy L. H.; Khoury H.; Paci A.; Deroussent A.; Ferreira H.; Dubernet C.; Declèves X.; Besnard M.; Chacun H.; Lepêtre-Mouelhi S.; Desmaële D.; Rousseau B.; Laugier C.; Cintrat J. C.; Vassal G.; Couvreur P. Squalenoylation favorably modifies the in vivo pharmacokinetics and biodistribution of gemcitabine in mice. Drug Metab. Dispos. 2008, 36, 1570–1577. [DOI] [PubMed] [Google Scholar]
- Réjiba S.; Reddy L. H.; Bigand C.; Parmentier C.; Couvreur P.; Hajri A. Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine 2011, 7, 841–849. [DOI] [PubMed] [Google Scholar]
- Immordino M. L.; Brusa P.; Rocco F.; Arpicco S.; Ceruti M.; Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J. Controlled Release 2004, 100, 331–346. [DOI] [PubMed] [Google Scholar]
- Sloat B. R.; Sandoval M. A.; Li D.; Chung W. G.; Lansakara-P D. S.; Proteau P. J.; Kiguchi K.; DiGiovanni J.; Cui Z. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int. J. Pharm. 2011, 409, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval M. A.; Sloat B. R.; Lansakara-P D. S.; Kumar A.; Rodriguez B. L.; Kiguchi K.; DiGiovanni J.; Cui Z. EGFR-targeted stearoyl gemcitabine nanoparticles show enhanced anti-tumor activity. J. Controlled Release 2012, 157, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhail A. S.; Allen C. Block copolymer micelles for delivery of cancer therapy: transport at the whole body, tissue and cellular levels. J. Controlled Release 2009, 138, 214–223. [DOI] [PubMed] [Google Scholar]
- Katragadda U.; Fan W.; Wang Y.; Teng Q.; Tan C. Combined delivery of paclitaxel and tanespimycin via micellar nanocarriers: pharmacokinetics, efficacy and metabolomic analysis. PloS One 2013, 8, e58619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z. W; Gallo J. M. Selective Protection of 2′,2′-Difluorodeoxycytidine (Gemcitabine). J. Org. Chem. 1999, 64, 8319–8322. [DOI] [PubMed] [Google Scholar]
- Chandran T.; Katragadda U.; Teng Q.; Tan C. Design and evaluation of micellar nanocarriers for 17-allyamino-17-demethoxygeldanamycin (17-AAG). Int. J. Pharm. 2010, 92, 170–177. [DOI] [PubMed] [Google Scholar]
- Roforth M. M.; Tan C. Combination of rapamycin and 17-allylamino-17-demethoxygeldanamycin abrogates Akt activation and potentiates mTOR blockade in breast cancer cells. Anticancer Drugs 2008, 19, 681–688. [DOI] [PubMed] [Google Scholar]
- Berney C.; Danuser G. FRET or no FRET: a quantitative comparison. Biophys. J. 2003, 84, 3992–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Lansakara-P D. S.; Li X.; Cui Z. Lysosomal delivery of a lipophilic gemcitabine prodrug using novel acid-sensitive micelles improved its antitumor activity. Bioconjugate Chem. 2012, 23, 966–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tellingen O.; Beijnen J. H.; Verweij J.; Scherrenburg E. J.; Nooijen W. J.; Sparreboom A. Rapid esterase-sensitive breakdown of polysorbate 80 and its impact on the plasma pharmacokinetics of docetaxel and metabolite in mice. Clin. Cancer Res. 1999, 5, 2918–2924. [PubMed] [Google Scholar]
- Mackey J. R.; Mani R. S.; Selner M.; Mowles D.; Young J. D.; Belt J. A.; Crawford C. R.; Cass C. E. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357. [PubMed] [Google Scholar]
- Achiwa H.; Oguri T.; Sato S.; Maeda H.; Niimi T.; Ueda R. Determinants of sensitivity and resistance to gemcitabine: the roles of human equilibrative nucleoside transporter 1 and deoxycytidine kinase in non-small cell lung cancer. Cancer Sci 2004, 95, 753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbruzzese J. L.; Grunewald R.; Weeks E. A.; Gravel D.; Adams T.; Nowak B.; Mineishi S.; Tarassoff P.; Satterlee W.; Raber M. N. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J. Clin. Oncol. 1991, 9, 491–498. [DOI] [PubMed] [Google Scholar]
- Katragadda U.; Teng Q.; Rayaprolu B. M.; Chandran T.; Tan C. Multi-drug delivery to tumor cells via micellar nanocarriers. Int. J. Pharm. 2011, 419, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu L.; Elbayoumi T. A.; Torchilin V. P. Mixed micelles made of poly(ethylene glycol)-phosphatidylethanolamine conjugate and D-α-tocopheryl polyethylene glycol 1000 succinate as pharmaceutical nanocarriers for camptothecin. Int. J. Pharm. 2005, 306, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed M. M.; Sloane B. F. Cysteine cathepsins: multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [DOI] [PubMed] [Google Scholar]