

Conspectus
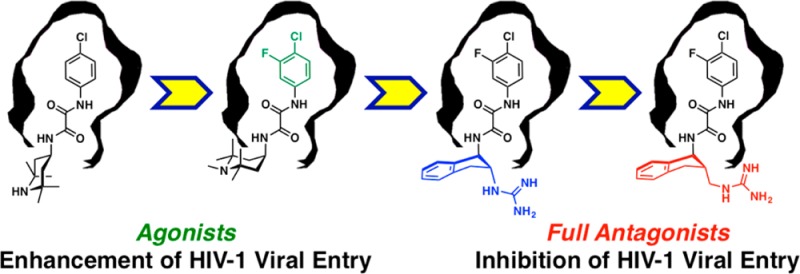
This Account provides an overview of a multidisciplinary consortium focused on structure-based strategies to devise small molecule antagonists of HIV-1 entry into human T-cells, which if successful would hold considerable promise for the development of prophylactic modalities to prevent HIV transmission and thereby alter the course of the AIDS pandemic.
Entry of the human immunodeficiency virus (HIV) into target T-cells entails an interaction between CD4 on the host T-cell and gp120, a component of the trimeric envelope glycoprotein spike on the virion surface. The resultant interaction initiates a series of conformational changes within the envelope spike that permits binding to a chemokine receptor, formation of the gp41 fusion complex, and cell entry. A hydrophobic cavity at the CD4–gp120 interface, defined by X-ray crystallography, provided an initial site for small molecule antagonist design. This site however has evolved to facilitate viral entry. As such, the binding of prospective small molecule inhibitors within this gp120 cavity can inadvertently trigger an allosteric entry signal.
Structural characterization of the CD4–gp120 interface, which provided the foundation for small molecule structure-based inhibitor design, will be presented first. An integrated approach combining biochemical, virological, structural, computational, and synthetic studies, along with a detailed analysis of ligand binding energetics, revealed that modestly active small molecule inhibitors of HIV entry can also promote viral entry into cells lacking the CD4 receptor protein; these competitive inhibitors were termed small molecule CD4 mimetics. Related congeners were subsequently identified with both improved binding affinity and more potent viral entry inhibition. Further assessment of the affinity-enhanced small molecule CD4 mimetics demonstrated that premature initiation of conformational change within the viral envelope spike, prior to cell encounter, can lead to irreversible deactivation of viral entry machinery. Related congeners, which bind the same gp120 site, possess different propensities to elicit the allosteric response that underlies the undesired enhancement of CD4-independent viral entry.
Subsequently, key hotspots in the CD4–gp120 interface were categorized using mutagenesis and isothermal titration calorimetry according to the capacity to increase binding affinity without triggering the allosteric signal. This analysis, combined with cocrystal structures of small molecule viral entry agonists with gp120, led to the development of fully functional antagonists of HIV-1 entry. Additional structure-based design exploiting two hotspots followed by synthesis has now yielded low micromolar inhibitors of viral entry.
Introduction
The acquired immunodeficiency syndrome (AIDS) derives from the infection and subsequent depletion of T lymphocytes, orchestrated by the human immunodeficiency viruses (HIV-1 and HIV-2).1,2 One potential tactic to intervene in the AIDS pandemic would be to block the viral entry process, exploiting a prophylactic microbicide or a therapeutic comprised of a small molecule viral entry inhibitor.3 To achieve this goal, a detailed understanding of the mechanism of the initial steps of the HIV entry cascade is required. This Account will provide an overview of an interdisciplinary research program to understand the molecular interactions that govern the initial virus–host cell recognition and entry events and in turn to develop small molecule probes that permit interrogation of the dynamic processes that underlie entry. Subsequent structure-based design and synthesis, in conjunction with thermodynamic characterization of hotspots for binding or allosteric activation within the interaction of the viral gp120 protein with the T-cell CD4 receptor protein has led to the development of small molecule antagonists of HIV-1 entry.
The HIV Entry Process: A Series of Coordinated Conformational Changes Drive Viral Entry
The first step of HIV entry into the host cells is mediated by a viral membrane glycoprotein assembly, organized as noncovalently associated trimers, collectively referred to as the envelope glycoprotein spike (Env; Figure 1).4,5 The glycoprotein momomer, initially produced as a single polypeptide (gp160), is post-translationally cleaved into glycoproteins 41 (gp41) and 120 (gp120). The transmembrane region of gp41 anchors the Env complex to the viral lipid bilayer, while the exposed Env surface is principally gp120. The Env trimer protein complex (gp1203/gp413) is the sole virus-specific protein present on the viral membrane and is the major target for neutralizing antibodies, vaccines, and small molecule entry inhibitors.
Figure 1.
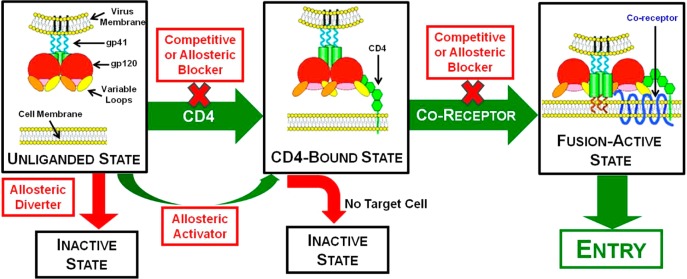
The HIV entry process (green arrows) illustrating strategies to disrupt the coordinated events that mediate viral entry (red boxes). Portions adapted from ref (5).
The primary human T-cell receptor for the Env is CD4, a membrane-associated glycoprotein present on the cell surface that binds gp120 with high affinity (Kd ≈ 4 nM).6 Binding between CD4 and gp120 leads to major conformational changes in the viral Env spike,7 revealing the binding epitope of a second host cell co-receptor (Figure 1).8 All HIV-1 strains utilize one of the transmembrane G-protein coupled chemokine receptors, CCR5 and CXCR4. Considerable progress has been achieved to develop CCR5 antagonists to block cellular HIV-1 penetration, with Pfizer’s maraviroc approved for treatment of individuals not responsive to standard highly active anti-retroviral therapy (HAART).9 A second approved entry inhibitor is enfuvirtide, a synthetic 36 amino acid peptide homologue of the HR2 subunit of gp41, currently employed in “salvage HAART” regimens.10
The viral fusion model, involving specific recognition events leading to unmasking of the co-receptor epitopes, is consistent with the triggered release of fusogenic conformations in other enveloped viruses and as such offers multiple opportunities for intervention (Figure 1).11 For HIV-1, cleavage of the gp160 precursor protein in conjunction with further post-translational modifications leads to a high potential energy state of the Env spike. Through a series of ligand-induced conformational transitions, the Env adopts lower energy states that deliver the driving force for viral entry.
Several tactics can be envisioned to interrupt the entry cascade including allosteric diversion of the initial unliganded state of the Env, premature allosteric activation of the Env to the CD4-bound state prior to encounter with transmembrane co-receptor, and direct blocking of either the CD4/gp120 or co-receptor/gp120 interactions. Although both “allosteric diversion” and receptor and co-receptor blockade are conceptually straightforward, “premature allosteric activation” and blocking CD4 binding with small molecule CD4 mimetics will be described in this Account.
At the outset, targeting the Env complex presented a minimum of three challenges: (1) The Env complex is conformationally dynamic and undergoes structural changes during viral entry. Although a variety of X-ray structures of gp120 in both ligand-bound and “unliganded” complexes were available, these structures reveal only portions of the unliganded Env. (2) The Env sequence differs among the three major HIV-1 subtypes and readily develops escape mutants. However, given the prerequisite of CD4 binding to initiate viral entry, the residues lining the gp120–CD4 binding site are highly conserved. (3) The surface of gp120, particularly the solvent-exposed outer domain, is heavily glycosylated,12 restricting access to large sections of the Env.13,14 Small molecule entry inhibitors have the advantage of potentially avoiding the “glycan shield”; thus, inhibitor development efforts recorded here focused on the CD4-binding site.
The gp120/CD4 Interaction: Thermodynamic and Structural Characterization
Prior to CD4–gp120 crystal structures, mutagenesis studies mapped the gp120 binding sites on CD4. These studies revealed that residues CD4Phe43 and CD4Arg59 comprise the centerpieces of the CD4–gp120 interaction: mutation of CD4Phe43 to alanine led to a 550-fold reduction in binding affinity for gp120,15 whereas mutation of CD4Arg59 to alanine reduced the affinity 9-fold.16
A significant milestone in HIV structural biology was achieved in 1998 when Kwong, Sodroski, and Hendrickson reported the 2.5 Å resolution X-ray crystal structure of a ternary complex composed of (1) two-domain (D1D2) CD4, (2) the deglycosylated core of gp120, and (3) the Fab of antibody 17b, which binds to a site that overlaps the co-receptor binding site on gp120 (Figure 2).17,18 Approximately 65% of the gp120 protein was preserved in the core; a total of 19 and 52 residues were deleted from the C- and N-termini, respectively, and 67 residues of the V1/V2 variable loops and 32 residues from the V3 variable loop were replaced with the tripeptide Gly-Ala-Gly to facilitate crystallization. Notwithstanding these deletions, substitutions, and removal of over 90% of the carbohydrates, this gp120 core interacted with both CD4 and a variety of CD4-binding site antibodies at or near wild-type affinity.
Figure 2.
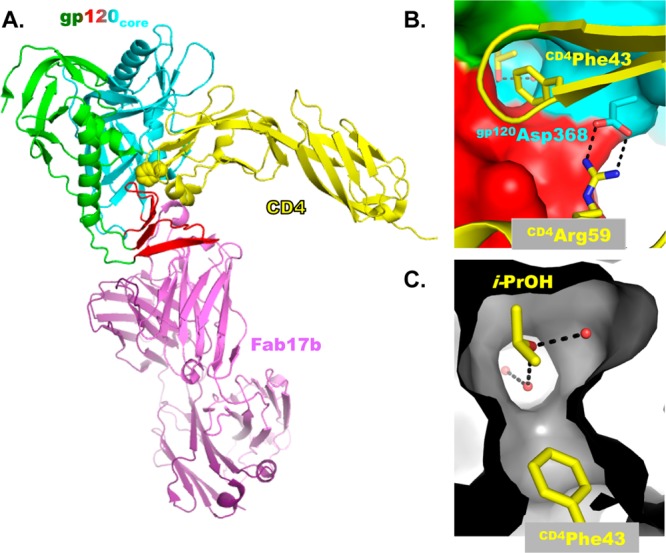
(A) The X-ray crystal structure of the CD4/gp120core/Fab17b complex (PDB 1G9M). The gp120core is color coded by domain: outer domain, cyan; inner domain, green; bridging sheet domain, red. CD4 is shown in yellow, with the CD4Phe43 side chain in spheres. The 17b Fab is shown in purple. (B) The pivotal residues, CD4Phe43 and CD4Arg59, identified by CD4 mutagenesis. (C) The interior of the Phe43 cavity, with i-PrOH from the crystallization media and crystallographic water molecules (red spheres) shown.
Analysis of the gp120 core led to the definition of three domains: (1) the inner domain, (2) the outer domain, and (3) the bridging sheet domain. The inner domain, proximal to the trimeric axis of the Env spike, contains the N- and C-termini that interact with gp41. The bridging sheet domain comprises an antiparallel four-stranded β-sheet, composed of the V1/V2 loop stem emanating from the inner domain and the β20/21 hairpin. During viral entry, CD4-induced conformational changes occur within the inner and bridging sheet domains and propagate to gp41.19 Finally, the outer domain, distal from the trimer axis and forming the majority of the solvent exposed surface of the Env spike, is heavily glycosylated and conformationally invariant.
A unique feature of the CD4–gp120 interface is the large spherical, water-accessible hydrophobic cavity formed at the intersection of the gp120 domains (Figure 2C). When CD4 binds to gp120, the CD4Phe43 phenyl ring extends midway into the cavity and “seals” the entrance. From the perspective of structure-based inhibitor design, the serendipitous cocrystallization of an isopropyl alcohol in the Phe43 cavity (Figure 2C) demonstrates that the cavity can accept small molecule fragments.18
At the outset of this program, site-directed mutagenesis was employed to incorporate a reactive cysteine residue at position 43 to afford D1D2-F43C-CD4 (Figure 3).20 This construct was selectively alkylated with a library of diverse α-bromoacetamides to furnish D1D2-CD4 conjugates that could deliver structural probes precisely to the Phe43 cavity.
Figure 3.
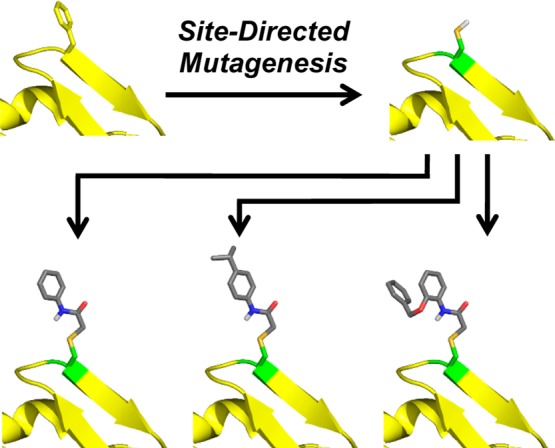
Design of the D1D2-F43C-CD4 protein (green residue). Alkylation with α-bromoacetamides allows probing of the Phe43 cavity. Structural representations are hypothetical.
Interactions between the Phe43 cavity and the probes were characterized by X-ray crystallography.21,22 Unexpectedly, the Phe43 cavity exhibits significant plasticity, adapting to accommodate increasingly large fragments. The structures suggested that residues lining the Phe43 cavity might be involved in transduction of conformational changes, upon cavity occupation, to more remote portions of gp120. Thus, binding within and proximal to the Phe43 cavity hotspot was reasoned to be associated with eliciting an allosteric response.
In a recent analysis employing alanine-scanning mutagenesis within CD4, isothermal titration calorimentry (ITC) was employed to characterize the energetic contributions to the CD4–gp120 interactions.23 The results revealed that the residues within the binding interface contribute differently to affinity and to the conformational structuring that leads to co-receptor binding. Residues CD4Phe43 and CD4Arg59 were of particular interest. The results demonstrate that CD4Phe43 contributes significantly to both the binding affinity and allosteric activation of gp120, whereas the interaction between CD4Arg59 and gp120Asp368 contributes much more to affinity than allosteric activation. Additional residues within gp120 strongly associated with binding (cf. gp120Met426) were also identified. We therefore viewed designing interactions between small molecule ligands and gp120 “binding hotspot” residues to be critical for the successful development of viral entry antagonists that do not promote an allosteric response.23
Although the cocrystal structure of CD4/gp120/Fab17b provided a foundation for rational design and synthesis of gp120-directed inhibitors, the initial small molecules were designed using the CD4-bound conformation of gp120. This dilemma was resolved in part with the structure of an extended, unliganded gp120 core, by altering the truncations to the V1/V2 and V3 loops and incorporating additional N-terminal residues (Figure 4).24 Remarkably, the CD4-bound and unliganded structures of the gp120 core proved quite similar. More recently, cryo-ET and single-particle cryo-EM of various trimer constructs revealed the overall architecture of the Env trimer in both the unliganded and bound states.25−27
Figure 4.
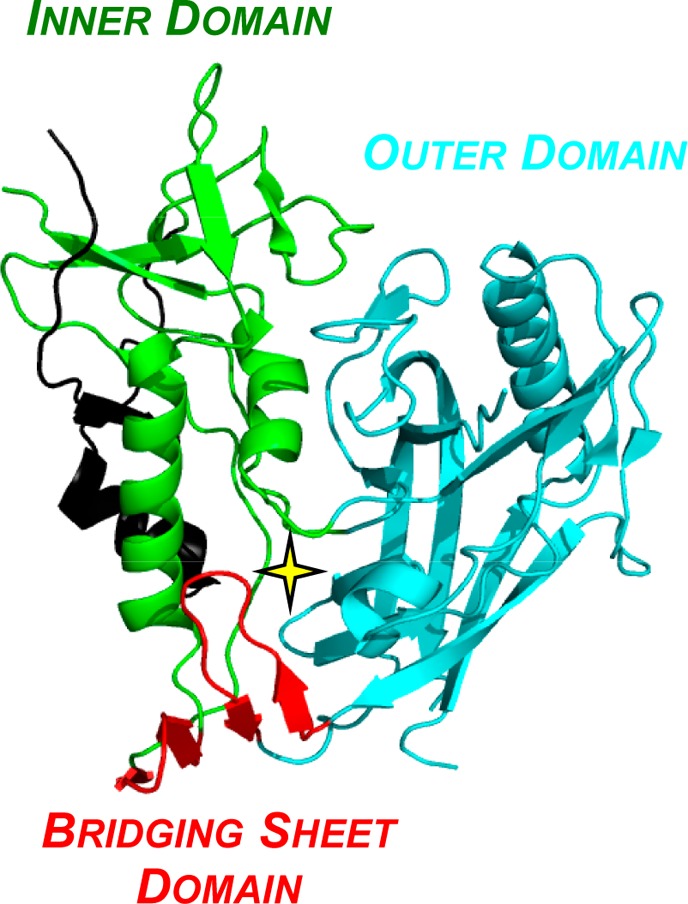
The crystal structure of the unliganded “extended” gp120 core, with the star denoting the location of the Phe43 cavity (additional N-terminal residues, relative to Figure 2A, are shown in black).
Small Molecule Inhibitors of the CD4–gp120 Interaction28
A major effort at Bristol-Myers Squibb led to a series of small molecule HIV-1 viral entry inhibitors. Central here was BMS-378806 (Figure 5; EC50 = 3–62 nM).29 When formulated as a microbicide for prophylaxis, in conjunction with both a co-receptor and gp41-directed entry antagonist, Moore in collaboration with BMS, demonstrated that macaque monkeys were protected from vaginal challenge by a human/simian immunodeficiency hybrid virus that exploits the CCR5 co-receptor.30 The BMS team reported that BMS-378806 binds within the Phe43 cavity and directly inhibits the CD4–gp120 interaction. Data from our laboratories however support an alternative mode of action, wherein BMS-378806 binds to the unliganded state of gp120 and prevents the requisite conformational transitions to expose the gp41 heptad repeat region 1 (HR1).31,32
Figure 5.
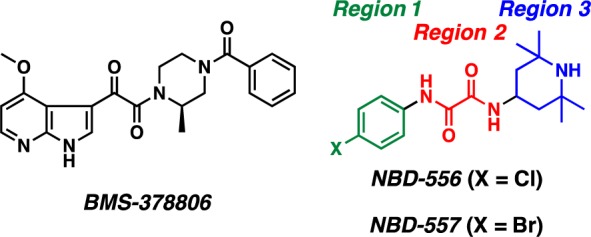
Small molecule inhibitors of HIV-1 viral entry.
In 2005, in what must be considered a major discovery, Debnath and co-workers reported the identification of two small molecule HIV-1 viral entry inhibitors, NBD-556 and NBD-557 (Figure 5), via high-throughput screening.33 While these small molecules inhibit HIV entry into cells expressing the CD4 protein, we were intrigued to discover that they also enhance viral entry into cells that lack the CD4 receptor.34 The activation of HIV-1 entry led us to focus on defining this mechanism. The subsequent understanding provided the cornerstone that led to rational, structure-based conversion of the initial NBD small molecule viral entry agonists to full functional antagonists.
The NBD Compounds: Small Molecule CD4 Mimetics
First, a description of the methods employed to define viral inhibition and the binding affinity of small molecules is required. Functional evaluation entails cell-based assays to measure (1) the inhibition of HIV-1 viral entry into cells expressing both the CD4 and CCR5 receptors (i.e., CD4+CCR5+ Cf2Th cells), expressed as an IC50 value and (2) the propensity of the small molecule to enhance CD4-independent viral entry into cells lacking CD4 (i.e., CD4–CCR5+ Cf2Th cells), expressed relative to enhancement of HIV-1 entry induced by NBD-556.35 It is important to note that the levels of CCR5 expressed on these target cells are significantly higher than those on human T-cells. Congeners that either inhibit HIV-1 viral entry into CD4+CCR5+ cells or enhance viral entry into CD4–CCR5+ cells were subjected to direct binding analysis to full-length gp120, employing isothermal titration calorimetry (ITC).
For NBD-556, ITC revealed a thermodynamic signature of gp120 binding similar to that of the CD4–gp120 interaction. The interaction between soluble CD4 and full-length, monomeric gp120 is characterized by a high affinity (Kd = 4 nM), wherein a highly favorable ΔH is observed; this favorable enthalpy change is however compensated by a significant unfavorable entropic contribution (Figure 6).6 The binding of NBD-556 to gp120 also revealed a favorable ΔH that is partially opposed by an unfavorable ΔS (Figure 6), consistent with NBD-556 binding to gp120 that induces conformational ordering within the protein.34 The similar thermodynamic signatures of sCD4 and NBD-556 binding are remarkable, given the large difference in both ligand size and interaction surface area. This thermodynamic signature is in marked contrast to that observed for the binding of BMS-378806 to gp120, and suggests viral entry inhibition via a different mechanism.
Figure 6.
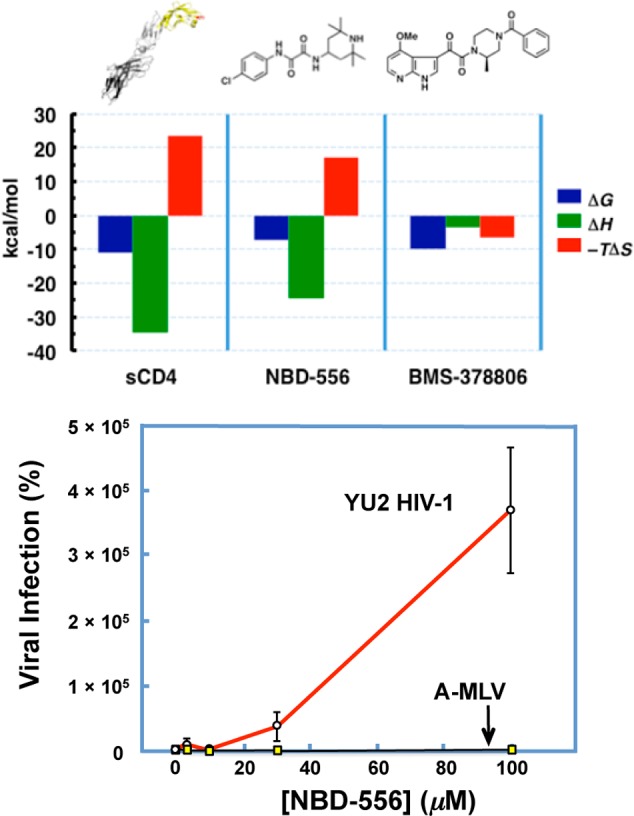
(top) The thermodynamic signatures of sCD4, NBD-556, and BMS-378806 binding to monomeric gp120. (bottom) The selective enhancement of CD4-independent entry of YU2 HIV-1 into CD4–CCR5+ target cells versus the control virus A-MLV.
Further evidence of NBD-556 functional mimicry of CD4 was obtained with cell-based, single-round viral entry assays, utilizing cells that lack the CD4 receptor (CD4–) but express the co-receptor (CCR5+) necessary to mediate entry. When these CD4–CCR5+ cells were incubated with NBD-556 and a pseudovirus expressing the Env from YU2HIV-1, an enhancement of entry was observed (Figure 6).34 This remarkable effect was specific to HIV-1, since the viral entry of the unrelated pseudovirus expressing the Env of amphotropic murine leukemia virus (A-MLV) was not enhanced. Thus, in a functional viral entry assay utilizing the full Env trimer and CD4–CCR5+ cells, NBD-556 acts as a surrogate for CD4 and promotes entry. Based on both the similarity of the thermodynamic signatures for gp120 binding and the enhancement of entry in the absence of cell-associated CD4, we termed NBD-556 a small molecule CD4 mimetic.(34)
Docking studies employing the CD4-bound conformation of gp120 suggested that NBD-556 binds within the Phe43 cavity.35 The modeling results also predict that the para-chlorophenyl ring (Region 1; Figure 5) binds ∼6.5 Å more deeply within the cavity than that of the CD4Phe43 side chain in the native CD4/gp120 interaction (Figure 2). Moreover, when the Phe43 “cavity filling mutant” S375W-gp120 was tested, no binding between NBD-556 and S375W-gp120 mutant was observed (Figure 7). Taken together, these results suggested that congeners of NBD-556 with enhanced affinity for gp120 might compete more efficiently with CD4 for binding to gp120 and in turn, if appropriately designed, prevent the downstream allosteric events in the entry cascade that are initiated by CD4 binding at the Phe43 cavity allosteric hotspot.
Figure 7.
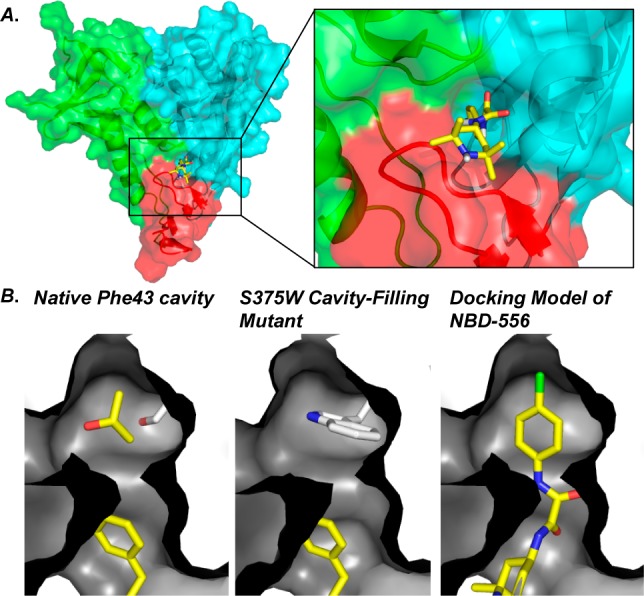
(A) The docking model of NBD-556 to gp120 in the CD4 bound conformation. (B) Region 1 of NBD-556 binds deeply within the cavity. NBD-556 does not bind the cavity filling S375W-gp120 mutant.
To explore binding within the Phe43 cavity, we first examined region 1 congeners. Biological evaluation revealed that alterations to the aromatic ring had a significant effect on affinity for gp120 and the degree of CD4 mimicry. This led to the identification of JRC-II-191 possessing a m-fluorine on NBD-556, which possessed both a higher affinity for gp120 (Kd = 0.76 μM) and a 2-fold enhancement of viral entry into CD4–CCR5+ T-cells relative to NBD-556.35 Pleasingly, modest inhibition of HIV entry (IC50 = 54.4 μM) into CD4+CCR5+ target T-cells was also observed. These observations led us to ask: How can a small molecule mimetic of CD4 both enhance viral entry in the absence of CD4 and modestly inhibit entry when CD4 is present on the target cell?
To answer this question, a magnetically controlled assay was developed to regulate temporally the association of target cells with HIV-1 virions activated either by soluble CD4 or JRC-II-191. This study revealed that sCD4 and JRC-II-191 activate the Env to mediate viral fusion to cells lacking CD4. This activated state of the Env is, however, transient and leads to irreversible changes in the Env conformation that inactivate the virus, demonstrating for the first time that premature allosteric activation (vide supra) by a small molecule comprises a means to inhibit viral entry (Figure 8).36
Figure 8.

CD4-independent HIV-1 viral entry (CD4–CCR5+ target cells, bottom) can be induced by CD4-mimetic JRC-II-191. If however the activated state upon ligand binding does not encounter a target cell, the Env irreversibly assumes a fusion-inactive state.36
We next turned to optimizing regions 2 and 3 of the JRC-II-191 scaffold. While changes to region 2 were not tolerated, a second binding hotspot in the native protein–protein interaction at gp120Asp368, located within the vestibule of the Phe43 cavity and proximal to region 3, contributes significantly to binding affinity, without induction of conformational transitions in the native protein–protein interaction. We thus sought to generate a gp120Asp368 interaction with the designed small molecule CD4 mimetics to increase binding affinity without promoting enhancement of CD4-independent viral entry.
A virtual screening program led to identification of a new series of scaffolds designed to recapitulate the native CD4Arg59–gp120Asp368 ionic interaction (Figure 9).37 In particular, we searched for commercially available amines that contained a hydrogen bond donor for gp120Asp368 that could also be readily coupled to a common region 1 and 2 precursor. A modest improvement was observed when the piperidine ring was methylated to furnish TS-II-224 (IC50 = 48.8 ± 3.6 μM; Kd = 0.30 μM). Congener MAE-II-120 also revealed an improved IC50 (38.5 ± 10.1 μM), despite a modest loss in affinity (Kd = 0.60 μM). Both TS-II-224 and MAE-II-120 however enhance CD4-independent viral entry into CD4–CCR5+ cells, with a ∼2 fold increase relative to JRC-II-191. In contrast, MAE-II-116 had an acceptable affinity for gp120 (Kd = 2.1 μM), but remained a weak inhibitor of entry that, importantly, did not enhance CD4-independent viral entry into CD4–CCR5+ cells. This combination of SAR and thermodynamic analysis, employing a collection of small molecule congeners, demonstrated that small molecule inhibitors can bind to the same site of gp120 with similar affinities yet elicit different allosteric responses.38 The results of these analyses in turn supported the important hypothesis that structural alterations to region 3 might modulate the observed allosteric activation. Unfortunately, a large number of the NBD congeners that were designed and synthesized during the virtual screening program proved inactive.
Figure 9.

Region 3 congeners designed by virtual screening and synthesis.
Undaunted, guided by a collection of X-ray cocrystal structures of the small-molecule CD4 mimetics bound to an extended gp120 core, we achieved significant advancement. Specifically, the cocrystal structure of NBD-556 bound to the extended gp120 core demonstrated that the region II oxalamide moiety formed two hydrogen bonds with backbone carbonyls of residues on opposite sides of the Phe43 cavity.24 The subsequent cocrystal structure of TS-II-224 bound to this extended gp120 core was obtained (Figure 10).39 In neither structure however were interactions observed between region 3 and the gp120Asp368 hotspot.
Figure 10.

(A) The cocrystal structure of TS-II-224/gp120E (Clade A/E, Strain 93TH057, H375S). The N-terminal extension is shown in pale green; the CD4 binding footprint is shown in pale domain colors (PDB 4DKO). (B) The interactions of TS-II-224 within the Phe43 cavity (left) and orientation with respect to gp120Asp368 (right). (C) Virtual screening led to amino indane (+)-AWS-I-50.
We therefore refined the virtual screening program. Analysis of the TS-II-224 cocrystal structure (Figure 10A,B) revealed that substituents such as an amino group located at the C4 position of the piperidine ring would be directed toward the gp120Asp368 side chain. A hypothetical gem-diamine was therefore designed as a new region 3 scaffold (Figure 10C). This prototype structure, while not chemically stable, was employed as a virtual screening search query. These efforts suggested amino indanol (+)-TK-II-52 (Figure 9) might comprise a promising scaffold; however, upon synthesis (+)-TK-II-52 proved to be a weak, nonselective inhibitor of HIV-1 entry. To increase interactions with gp120Asp368, the hydroxyl was converted to the amine.39 While we were pleased to find that (+)-AWS-I-50 possessed a Kd = 1.9 μM, nonselective entry inhibition and enhancement of CD4-independent HIV-1 entry continued.
The cocrystal structure of (+)-AWS-I-50 bound to the extended gp120 core revealed that, as with the earlier NBD congeners, the amine was oriented away from the gp120Asp368 hotspot (Figure 11). However, comparison with the CD4-bound gp120 structure indicated that the indane ring effectively mimics the CD4 β-turn positioned over the Phe43 cavity of gp120. Clearly we had yet to test the hypothesis that simultaneous engagement of the gp120Asp368 and Phe43 cavity hotspots would lead to functional antagonists of viral entry.
Figure 11.

(left) The 1.8 Å resolution X-ray cocrystal structure of (+)-AWS-I-50/gp120E (PDB 4DKP) overlaid with the CD4 β-turn and CD4Arg59 (white). (right) Interactions between (+)-AWS-I-50 and the Phe43 cavity of gp120.
Encouraged by the affinity of (+)-AWS-I-50 for gp120, as well as the CD4 β-turn mimicry by the indane scaffold, an effort was made to replicate the CD4Arg59 side chain interaction via incorporation of a guanidinium group. Significant improvements in both affinity and functional entry inhibition were observed upon evaluation of guanidinium congener (+)-DMJ-I-228 (Figure 12; Kd = 250 nM). Moreover, in the cell-based assay, HIV-1 entry inhibition was observed (IC50 = 22.9 μM) with excellent selectivity. Equally significant, when (+)-DMJ-I-228 was tested against a panel of 42 primary HIV-1 isolates, 57% of the isolates tested display an IC50 < 10 μM, with a mean IC50 = 7.9 μM. In contrast, the original lead compound NBD-556 has a mean IC50 = 29.3 μM, while it possesses an IC50 < 10 μM against only 12% of the viral isolates examined.39
Figure 12.
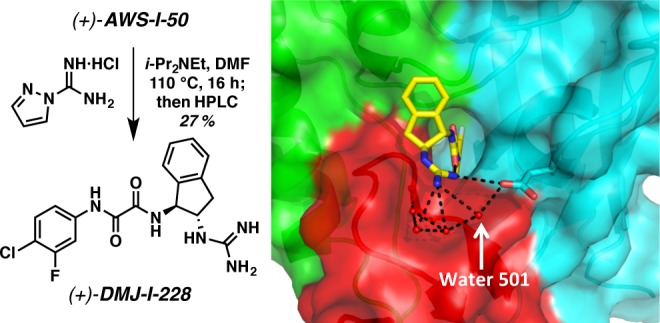
The conversion of amino indane (+)-AWS-I-50 to dual hotspot inhibitor (+)-DMJ-I-228. The 1.89 Å resolution (+)-DMJ-I-228/gp120E cocrystal structure (PDB 4DKQ). A network of crystallographic water molecules (water 501 labeled) mediates interactions between (+)-DMJ-I-228, gp120Asp368, and the bridging sheet.
Of critical importance, however, (+)-DMJ-I-228 did not enhance CD4-independent viral entry, unlike the previous small molecule CD4 mimetics.(39) When the thermodynamic signature of (+)-DMJ-I-228 binding to gp120 was determined, a significant reduction in the entropic penalty was observed, relative to NBD-556, suggesting that (+)-DMJ-I-228 does not stabilize gp120 conformationally to the same degree as the earlier small molecule CD4 mimetics.39
To derive a structural understanding, the high-resolution crystal structure of the (+)-DMJ-I-228/gp120 complex was completed, demonstrating interactions with the two well-conserved gp120 hotspots, namely, the Phe43 cavity and gp120Asp368 (Figure 12). We currently hypothesize that the acquisition of interactions with the affinity hotspot gp120Asp368 is at least partially responsible for obtaining a fully functional antagonist of viral entry that does not promote CD4-independent viral entry relative to NBD-556. The development of (+)-DMJ-I-228 thus demonstrated for the first time the feasibility of converting an agonist of HIV-1 entry to an antagonist, achieved by an integrated understanding of ligand interactions with hotspots that are associated with driving binding affinity, and not allosteric transduction, in the native protein–protein interaction.39
The (+)-DMJ-I-228/gp120 cocrystal structure was subsequently exploited in structure-based design and synthesis by addition of a methylene spacer between the indane core and the guanidinium functionality to improve both the binding affinity and HIV-1 entry inhibition; (+)-DMJ-II-121 (Figure 13) exhibited an enhancement in affinity (KD = 110 nM). Moreover, a 10-fold improvement in viral entry inhibition (IC50 = 2.3 ± 0.05 μM) was observed.40
Figure 13.
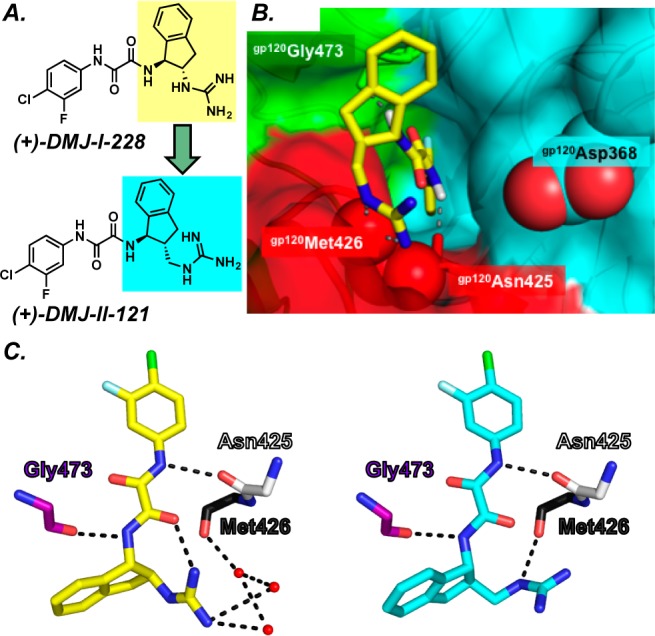
(A) Modification of the guanidinium indane core to afford (+)-DMJ-II-121. B. The (+)-DMJ-II-121/gp120E X-ray cocrystal structure demonstrates that (+)-DMJ-II-121 interacts with the Phe43 cavity and gp120Met426 hotspots. (C) The interactions between (+)-DMJ-I-228 (yellow) and (+)-DMJ-II-121 (cyan) with main chain carbonyls of gp120.
The cocrystal structure of the (+)-DMJ-II-121/gp120(Clade A/E) complex revealed that instead of the anticipated improved interactions with the gp120Asp368 hotspot, the extended guanidinium moiety displaces crystallographic water molecules observed in the (+)-DMJ-I-228 structure, thus forming a direct hydrogen bond to bridging sheet residue gp120Met426.40 Given that our first-generation dual hotspot antagonist (+)-DMJ-I-228 interacts with gp120Met426 via a water-mediated hydrogen bond, the direct interaction between (+)-DMJ-II-121 and the backbone bone carbonyl of gp120Met426 clearly leads to an improvement in activity. We take these results as very exciting, given that gp120Met426 was previously defined as a binding hotspot (vide supra).23 Importantly, interactions between the antagonist ligand and a backbone carbonyl are less prone to resistance mutations.
Summary
The development of (+)-DMJ-I-228 and (+)-DMJ-II-121 demonstrates that HIV-1 entry inhibitors that bind the conformationally dynamic Env trimer and exhibit fully functional antagonist activity in viral infectivity assays can be achieved. Such inhibitors were obtained through interdisciplinary research, involving structure-based ligand design based on alanine-scanning mutagenesis, thermodynamic profiling, and cocrystal structures with the gp120 core, in conjunction with synthesis and detailed biological evaluation. Studies continue with the hope of developing a viable tactic for the preventive intervention of the AIDS pandemic.
Acknowledgments
The authors thank all previous co-workers who have helped advance this project. The research presented in this Account has been primarily supported for over 15 years through an NIH Program Project (P01 GM 56550).
Biographies
Joel R. Courter received his Ph.D. in Chemistry from the University of Pennsylvania in 2012, working in the laboratories of Amos B. Smith, III.
Navid Madani is a Research Scientist in the Department of Cancer Immunology and AIDS at the Dana-Farber Cancer Institute (DFCI).
Joseph Sodroski is a Professor of Microbiology and Immunology at DFCI, Harvard Medical School, and in the Department of Immunology and Infectious Diseases at the Harvard School of Public Health. He is also an associate member of the Ragon Institute.
Arne Schön is a Research Scientist in the Department of Biology at Johns Hopkins University, specializing in biothermodynamics.
Ernesto Freire is the Henry Walters Professor of Biology at Johns Hopkins University.
Peter D. Kwong is the chief of the Structural Biology Section, National Institute of Allergy and Infectious Disease, National Institutes of Health.
Wayne A. Hendrickson is currently a University Professor at Columbia; Chief Life Scientist, Photon Sciences Directorate, Brookhaven National Laboratory; and the Scientific Director of the New York Structural Biology Center.
Irwin M. Chaiken is a Professor of Biochemistry and Molecular Biology at the Drexel University College of Medicine.
Judith M. LaLonde is a Research Associate in the Chemistry Department at Bryn Mawr College.
Amos B. Smith, III is the Rhodes-Thompson Professor of Chemistry at Penn and Founding Editor-in-Chief of Organic Letters.
During the review of this Account structures of an Env trimer construct (termed the BG505 SOSIP.664 gp140 trimer) obtained by X-ray crystallography41 and cryo-electron microscopy42 were reported.
The authors declare no competing financial interest.
This paper was published ASAP on February 6, 2014 with errors in references 41 and 42. The corrected version was reposted on February 20, 2014.
Funding Statement
National Institutes of Health, United States
References
- Barré-Sinoussi F.; Chermann J. C.; Rey F.; Nugeyre M. T.; Chamaret S.; Gruest J.; Dauguet C.; Axler-Blin C.; Vezinet-Brun F.; Rouzioux C.; Rozenbaum W.; Montagnier L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune-deficiency syndrome (AIDS). Science 1983, 220, 868–871. [DOI] [PubMed] [Google Scholar]
- Gallo R. C.; Salahuddin S. Z.; Popovic M.; Shearer G. M.; Kaplan M.; Haynes B. F.; Palker T. J.; Redfield R.; Oleske J.; Safai B.; White G.; Foster P.; Markham P. D. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [DOI] [PubMed] [Google Scholar]
- Mertenskoetter T.; Kaptur P. E. Update on microbicide research and development - seeking new HIV prevention tools for women. Eur. J. Med. Res. 2011, 16, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilton J. C.; Doms R. W. Entry inhibitors in the treatment of HIV-1 infection. Antiviral Res. 2010, 85, 91–100. [DOI] [PubMed] [Google Scholar]
- Wilen C. B.; Tilton J. C.; Doms R. W. HIV: Cell binding and entry. Cold Spring Harbor Perspect. Med. 2012, 2, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myszka D. G.; Sweet R. W.; Hensley P.; Brigham-Burke M.; Kwong P. D.; Hendrickson W. A.; Wyatt R.; Sodroski J.; Doyle M. L. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 9026–9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattentau Q. J.; Moore J. P. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J. Exp. Med. 1991, 174, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragic T.; Litwin V.; Allaway G. P.; Martin S. R.; Huang Y. X.; Nagashima K. A.; Cayanan C.; Maddon P. J.; Koup R. A.; Moore J. P.; Paxton W. A. HIV-1 entry into CD4(+) cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [DOI] [PubMed] [Google Scholar]
- MacArthur R. D.; Novak R. M. Maraviroc: The first of a new class of antiretroviral agents. Clin. Infect. Dis. 2008, 47, 236–241. [DOI] [PubMed] [Google Scholar]
- Matthews T.; Salgo M.; Greenberg M.; Chung J.; DeMasi R.; Bolognesi D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discovery 2004, 3, 215–225. [DOI] [PubMed] [Google Scholar]
- Wyatt R.; Sodroski J. The HIV-1 envelope glycoproteins: Fusogens, antigens, and immunogens. Science 1998, 280, 1884–1888. [DOI] [PubMed] [Google Scholar]
- Kwong P. D.; Mascola J. R.; Nabel G. J. Rational design of vaccines to elicit broadly neutralizing antibodies to HIV-1. Cold Spring Harbor Perspect. Biol. 2012, 4, a007278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M. HIV envelope: Challenges and opportunities for development of entry inhibitors. Trends Microbiol. 2011, 19, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira C.; Gomes J. R. B.; Gomes P.; Maurel F. Viral surface glycoproteins, gp120 and gp41, as potential drug targets against HIV-1: Brief overview one quarter of a century past the approval of zidovudine, the first anti-retroviral drug. Eur. J. Med. Chem. 2011, 46, 979–992. [DOI] [PubMed] [Google Scholar]
- Moebius U.; Clayton L. K.; Abraham S.; Harrison S. C.; Reinherz E. L. The human immunodeficiency virus-gp120 binding-site on CD4 - Delineation by quantitative equilibrium and kinetic binding-studies of mutants in conjunction with a high-resolution CD4 atomic-structure. J. Exp. Med. 1992, 176, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshevsky U.; Helseth E.; Furman C.; Li J.; Haseltine W.; Sodroski J. Identification of individual human-immunodeficiency-virus type-1 gp120 amino-acids important for CD4 receptor-binding. J. Virol. 1990, 64, 5701–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong P. D.; Wyatt R.; Majeed S.; Robinson J.; Sweet R. W.; Sodroski J.; Hendrickson W. A. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure 2000, 8, 1329–1339. [DOI] [PubMed] [Google Scholar]
- Kwong P. D.; Wyatt R.; Robinson J.; Sweet R. W.; Sodroski J.; Hendrickson W. A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong P. D.; Doyle M. L.; Casper D. J.; Cicala C.; Leavitt S. A.; Majeed S.; Steenbeke T. D.; Venturi M.; Chaiken I.; Fung M.; Katinger H.; Parren P.; Robinson J.; Van Ryk D.; Wang L. P.; Burton D. R.; Freire E.; Wyatt R.; Sodroski J.; Hendrickson W. A.; Arthos J. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 2002, 420, 678–682. [DOI] [PubMed] [Google Scholar]
- Xie H.; Ng D.; Savinov S. N.; Dey B.; Kwong P. D.; Wyatt R.; Smith A. B. III; Hendrickson W. A. Structure–activity relationships in the binding of chemically derivatized CD4 to gp120 from human immunodeficiency virus. J. Med. Chem. 2007, 50, 4898–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H.Inhibiting the binding of HIV to its initial cellular receptor: Design of chemical derivatives of human CD4 and characterization of their interactions with HIV gp120. Ph.D. Thesis, Columbia University, 2006. [Google Scholar]
- Hendrickson W. A.; Xie H.; Smith A. B.; Ng D.. Chemically modified CD4 peptides comprising D1D2 domain epitope to inhibit HIV gp120-CD4 binding and prevent HIV and HIV-1 infection. WO 2007075414 A22007.
- Liu Y.; Schön A.; Freire E. Optimization of CD4/gp120 inhibitors by thermodynamic-guided alanine-scanning mutagenesis. Chem. Biol. Drug Des. 2013, 81, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Kwon Y.; Finzi A.; Wu X.; Dogo-Isonagie C.; Lee L. K.; Moore L. R.; Schmidt S. D.; Stuckey J.; Yang Y.; Zhou T.; Zhu J.; Vicic D. A.; Debnath A. K.; Shapiro L.; Bewley C. A.; Mascola J. R.; Sodroski J. G.; Kwong P. D. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 5663–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y.; Wang L.; Gu C.; Herschhorn A.; Xiang S.-H.; Haim H.; Yang X.; Sodroski J. Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat. Struct. Mol. Biol. 2012, 19, 893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y.; Wang L.; Gu C.; Herschhorn A.; Désormeaux A.; Finzi A.; Xiang S.-H.; Sodroski J. G. Molecular architecture of the uncleaved HIV-1 envelope glycoprotein trimer. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 12438–12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E. E. H.; Borgnia M. J.; Kuybeda O.; Schauder D. M.; Bartesaghi A.; Frank G. A.; Sapiro G.; Milne J. L. S.; Subramaniam S. Structural mechanism of trimeric HIV-1 envelope glycoprotein activation. PLoS Pathog. 2012, 8, e1002797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews, see ref (5) and; Debnath A. K.Rational design of HIV-1 entry inhibitors. In In Silico Models for Drug Discovery; Kortagere S., Ed.; Methods in Molecular Biology; Springer: New York, 2013; Vol. 993; pp 185–204. [DOI] [PubMed] [Google Scholar]
- Lin P. F.; Blair W.; Wang T.; Spicer T.; Guo Q.; Zhou N. N.; Gong Y. F.; Wang H. G. H.; Rose R.; Yamanaka G.; Robinson B.; Li C. B.; Fridell R.; Deminie C.; Demers G.; Yang Z.; Zadjura L.; Meanwell N.; Colonno R. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 11013–11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veazey R. S.; Klasse P. J.; Schader S. M.; Hu Q. X.; Ketas T. J.; Lu M.; Marx P. A.; Dufour J.; Colonno R. J.; Shattock R. J.; Springer M. S.; Moore J. P. Protection of macaques from vaginal SHIV challenge by vaginally delivered inhibitors of virus-cell fusion. Nature 2005, 438, 99–102. [DOI] [PubMed] [Google Scholar]
- Si Z. H.; Madani N.; Cox J. M.; Chruma J. J.; Klein J. C.; Schön A.; Phan N.; Wang L.; Biorn A. C.; Cocklin S.; Chaiken I.; Freire E.; Smith A. B.; Sodroski J. G. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5036–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani N.; Perdigoto A. L.; Srinivasan K.; Cox J. M.; Chruma J. J.; LaLonde J.; Head M.; Smith A. B.; Sodroski J. G. Localized changes in the gp120 envelope glycoprotein confer resistance to human immunodeficiency virus entry inhibitors BMS-806 and #155. J. Virol. 2004, 78, 3742–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Ma L. Y.; Jiang S. B.; Lu H.; Liu S. W.; He Y. X.; Strick N.; Neamati N.; Debnath A. K. N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339, 213–225. [DOI] [PubMed] [Google Scholar]
- Schön A.; Madani N.; Klein J. C.; Hubicki A.; Ng D.; Yang X.; Smith A. B. III; Sodroski J.; Freire E. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry 2006, 45, 10973–10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani N.; Schön A.; Princiotto A. M.; LaLonde J. M.; Courter J. R.; Soeta T.; Ng D.; Wang L.; Brower E. T.; Xiang S.-H.; Do Kwon Y.; Huang C.-C.; Wyatt R.; Kwong P. D.; Freire E.; Smith A. B. III; Sodroski J. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure 2008, 16, 1689–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim H.; Si Z.; Madani N.; Wang L.; Courter J. R.; Princiotto A.; Kassa A.; DeGrace M.; McGee-Estrada K.; Mefford M.; Gabuzda D.; Smith A. B. III; Sodroski J. Soluble CD4 and CD4-mimetic compounds inhibit HIV-1 infection by induction of a short-lived activated state. PLoS Pathog. 2009, 5, e1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLonde J. M.; Elban M. A.; Courter J. R.; Sugawara A.; Soeta T.; Madani N.; Princiotto A. M.; Do Kwon Y.; Kwong P. D.; Schön A.; Freire E.; Sodroski J.; Smith A. B. III Design, synthesis and biological evaluation of small molecule inhibitors of CD4-gp120 binding based on virtual screening. Bioorg. Med. Chem. 2011, 19, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön A.; Madani N.; Smith A. B.; Lalonde J. M.; Freire E. Some binding-related drug properties are dependent on thermodynamic signature. Chem. Biol. Drug Des. 2011, 77, 161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLonde J. M.; Kwon Y. D.; Jones D. M.; Sun A. W.; Courter J. R.; Soeta T.; Kobayashi T.; Princiotto A. M.; Wu X.; Schön A.; Freire E.; Kwong P. D.; Mascola J. R.; Sodroski J.; Madani N.; Smith A. B. III Structure-based design, synthesis, and characterization of dual hotspot small-molecule HIV-1 entry inhibitors. J. Med. Chem. 2012, 55, 4382–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLonde J. M.; Le-Khac M.; Jones D. M.; Courter J. R.; Park J.; Schön A.; Princiotto A. M.; Wu X.; Mascola J. R.; Freire E.; Sodroski J.; Madani N.; Hendrickson W. A.; Smith A. B. III Structure-based design and synthesis of an HIV-1 entry inhibitor exploiting X-ray and thermodynamic characterization. ACS Med. Chem. Lett. 2013, 4, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien J.-P.; Cupo A.; Sok D.; Stanfield R. L.; Lyumkis D.; Deller M. C.; Klasse P.-J.; Burton D. R.; Sanders R. W.; Moore J. P.; Ward A. B.; Wilson I. A. Crystal Structure of a Soluble Cleaved HIV-1 Envelope Trimer. Science 2013, 342, 1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyumkis D.; Julien J.-P.; de Val N.; Cupo A.; Potter C. S.; Klasse P.-J.; Burton D. R.; Sanders R. W.; Moore J. P.; Carragher B.; Wilson I. A.; Ward A. B. Cryo-EM Structure of a Fully Glycosylated Soluble Cleaved HIV-1 Envelope Trimer. Science 2013, 342, 1484–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]