

Abstract
Isoxazole ring formation was examined as a potential Cu-free alternative click reaction to CuI-catalyzed alkyne/azide cycloaddition. The isoxazole reaction was explored at macroscopic and radiotracer concentrations with the fac-[MI(CO)3]+ (M = Re, 99mTc) core for use as a noncoordinating linker strategy between covalently linked molecules. Two click assembly methods (click, then chelate and chelate, then click) were examined to determine the feasibility of isoxazole ring formation with either alkyne-functionalized tridentate chelates or their respective fac-[MI(CO)3]+ complexes with a model nitrile oxide generator. Macroscale experiments, alkyne-functionalized chelates, or Re complexes indicate facile formation of the isoxazole ring. 99mTc experiments demonstrate efficient radiolabeling with click, then chelate; however, the chelate, then click approach led to faster product formation, but lower yields compared to the Re analogues.
Short abstract
Isoxazole ring formation was examined as a potential Cu-free alternative click scheme to CuAAC reactions at macroscopic and radiotracer concentrations with fac-[MI(CO)3]+ (M = Re, 99mTc) for use as a noncoordinating linker between covalently linked molecules. Two click assembly methods (click, then chelate and chelate, then click) were examined to determine the feasibility of isoxazole ring formation with either alkyne-functionalized tridentate chelates or their respective fac-[MI(CO)3]+ complexes with a model nitrile oxide generator.
The CuI-catalyzed azide/alkyne cycloaddition (CuAAC) reaction to yield 1,2,3-triazole has become analogous with the phrase “click chemistry” despite limitations (i.e., toxicity, transchelation) from residual Cu that require additional purification steps for biological applications.1 To circumvent the use of a CuI catalyst, Cu-free reactions have been developed with sterically strained cyclooctynes to perform an analogous cycloaddition reaction.2−4 However, these reactive alkynes consist of bulky, lipophilic, and high-molecular-weight molecules that may adversely impact the pharmacokinetic behavior, particularly with small molecules and peptide drugs. Alternative approaches to CuAAC are being explored with other reactive group pairs that maintain specificity, eliminate a metal catalyst, and avoid bulky functional groups incorporated into the assembled or click product.5−7
In recent years, the CuAAC reaction has gained particular prominence as a rapid assembly technique for radiopharmaceutical preparation through the pairing of fast reaction times with the short half-lives of diagnostic and therapeutic radionuclides to incorporate these probes into biological targeting molecules.8−11 In particular, the CuAAC reaction has been extensively utilized with group VII congeners, diagnostic 99mTc (t1/2 = 6.0 h; γ = 140 keV), and radiotherapeutic 186/188Re (β–, 1.071 and 2.118 MeV). Several CuAAC strategies have been developed for the organometallic fac-[MI(OH2)3(CO)3]+ (M = Re, 99mTc) precursor. Schibli and Mindt pioneered an innovative chelate building strategy (click to chelate) to incorporate 1,2,3-triazole as part of a chelate system in peptides/small molecules for targeted in vivo delivery when subsequently complexed with fac-[MI(CO)3]+.8,12−18 While our group has demonstrated the potency of the CuAAC reaction to couple fac-[MI(CO)3]+ complexes under mild conditions (15 min, 37 °C) using 1,2,3-triazole as a linker between the metal and targeting vector, uncoordinated 1,2,3-triazole can impact the coordination mode and stability of the complex.12,17
Since their structural determination by Claisen and Stock,19 isoxazoles have been widely used in therapeutics (i.e., anticancer, immune suppressor, cardiovascular) as agonists and antagonists because of their hydrolytic stability, π-stacking, and hydrogen-bonding capacities.20 Similar in molecular shape, volume, and electron distribution, isoxazoles present a potential alternative for 1,2,3-triazoles. Analogous to CuAAC, isoxazoles are prepared from an alkyne and nitrile oxide in good-to-excellent yields without the presence of Cu.21−23 Nitrile oxide is generated from a reactive oxime or from in situ activation of an oxime with chloramine-T or N-chlorosuccinimide.24,25
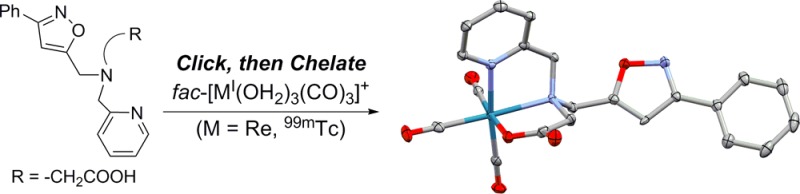
In this work, the isoxazole 1,3-dipolar cycloaddition was explored as a linker-based click reaction with fac-[MI(CO)3]+. The isoxazole orientation was designed to exclude N-coordination of the isoxazole ring to the metal center, unlike N-coordinated bidentate bis(isoxazole) chelates with fac-[ReI(CO)3]+.22 Two strategies, click, then chelate and chelate, then click, were utilized to investigate the versatility of the cycloaddition reaction between phenylchlorooxime and a terminal alkyne tethered to a tridentate chelate. In the click, then chelate method, the isoxazole click reaction was conducted prior to chelation with fac-[MI(CO)3]+, while alkyne-tethered fac-[MI(CO)3]+ complexes were utilized in the chelate, then click method.
The general preparation of the ligands is depicted in Scheme 1. Several of the precursor compounds (1–4) were synthesized as previously reported.12,17,25 Formation of the isoxazole linkers (5 and 6) was carried out at room temperature with 2 equiv of 4 in the presence of a weak base in moderate-to-good yields (45 and 50%, respectively). 1H NMR confirmed cycloaddition by the disappearance of the alkyne triplet and the appearance of the isoxazole proton (6.63 and 6.79 ppm, respectively) and additional aromatic resonances that correlated with the 13C NMR data. Deprotection of 6 was carried out through base hydrolysis to produce 7 in reasonable yields (49%) after isolation. The synthesis of 7 was confirmed by 1H NMR analysis, demonstrating loss of the methyl ester signal of 6 at 3.73 ppm and loss of CH3 in mass spectrometry (MS) analysis. 7 could also be directly prepared from 3 in significantly lower yields (<10%).
Scheme 1. Synthetic Strategy for Generating Isoxazole Ligands and fac-[MI(CO)3]+ Complexes.

Methyl ester deprotection of 2 and 6 with LiOH/methanol (1:3) was conducted prior to complexation with fac-[MI(OH2)3(CO)3]+.
The possible flexidentate nature of the ligands was examined using two synthetic approaches (click, then chelate and chelate, then click) at macroscopic concentrations to inspect speciation of the fac-[ReI(CO)3]+ products based on assembly of the ligand (Scheme 1). The click, then chelate strategy was centered on formation of the full ligands prior to complexation with the metal centers. Reaction of the ligands (5 and 7) with fac-[ReI(OH2)3(CO)3]+ yielded one product each (8 and 9) regardless of the pH conditions. Complexation of the ligand was carried out at room temperature (5) and 40 °C (7) for 12 h, producing 8 and 9 in excellent-to-moderate yields (75 and 59%, respectively). 1H NMR analysis of 8 and 9 exhibited a downfield shift in all resonances relative to the free ligand. In 8, the adjacent pyridine CH2’s exhibit a symmetric AB quartet splitting, indicating that both pyridines are coordinated to the metal center.17 The CH2 adjacent to the isoxazole ring was observed as a singlet at 5.14 ppm, indicating that isoxazole is not involved in coordination with the metal. In 9, similar shifts and splitting patterns were also observed upon metal complexation. However, the asymmetric nature of the chelate impacted the splitting patterns of the CH2’s adjacent to the tertiary amine. The three different CH2’s exhibited AB quartet splitting patterns, indicating a unique magnetic environment for each proton as expected because of the asymmetric nature of the chelate.12 The ambiguity of the NMR splitting patterns does not clearly elucidate the coordination environment of the metal center; consequently, X-ray crystallography was used to determine the structure. Single crystals of 9 were grown by slow evaporation of a dichloromethane/methanol (2:1) mixture for X-ray diffraction analysis (Figure 1).26 Re bond distances (Å) and angles (deg) are analogous to those of previously reported complexes with this chelate (Table S1 in the Supporting Information, SI).12 The structure clearly confirms noncoordination of the isoxazole linker with the Re center. IR analysis gave the expected CO peaks for fac-[ReI(CO)3]+ complexes12,17 and showed a shift in the IR of the isoxazole ring peak (from 1620 cm–1 in 5 to 1611 cm–1 in 8 and from 1620 cm–1 in 7 to 1609 cm–1 in 9). MS analyses of 8 and 9 were consistent with the anticipated structures and Re splitting pattern at m/z 627.2 and 594.2, respectively.
Figure 1.
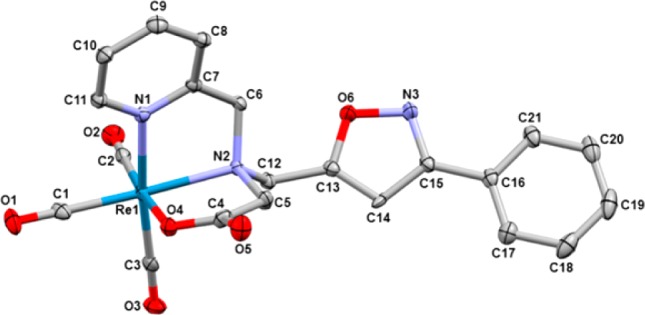
X-ray structure of 9 with H atoms omitted for clarity. Ellipsoids are drawn at 30% probability. Selected bond lengths (Å): Re1–O4 2.125(4), Re1–N4 2.172(4), Re1–N2 2.233(4), Re1–C2 1.909(5), Re1–C3 1.915(5), Re1–C1 1.914(6). Selected bond angles (deg): O4–Re1–N1 78.4(1), O4–Re1–N2 78.6(1), O4–Re1–C2 174.7(2), O4–Re1–C3 96.8(2), O4–Re1–C1 94.7(2), N1–Re1–N2 77.1(1), N1–Re1–C2 97.7(2), N1–Re1–C3 173.4(2), N1–Re1–C1 96.4(2), N2–Re1–C2 97.1(2), N2–Re1–C3 97.5(2), N2–Re1–C1 171.4(2), C2–Re1–C3 86.7(2), C2–Re1–C1 89.3(2), C2–Re1–C1 88.6(2).
The chelate, then click approach was examined to determine if the metal center would have any steric or electrochemical interactions that would perturbate isoxazole formation. An increased reactivity in CuAAC reactions performed with 10 and 11 was proposed because of the presence of fac-[MI(CO)3]+; similar effects due to the metal center may also impact the isoxazole reaction.12,17 In both cycloaddition reactions with 4 in the present study, the corresponding Re complexes (8 and 9) were formed in moderate-to-good yields after isolation. Analytical characterization data of 8 and 9 prepared by the chelate, then click approach from 10 and 11 corresponded to the data obtained via the click, then chelate method from 5 and 7, showing that both strategies successfully produced the final Re complexes. The two synthetic approaches (click, then chelate and chelate, then click) were also examined at radiotracer levels. In the click, then chelate approach, assessment of the speciation and complexation of 5 and 7 with fac-[99mTcI(OH2)3(CO)3]+ was achieved by varying the ligand concentrations (10–3–10–5 M) at constant temperature and reaction time (70 °C, 30 min). The chromatograms of 8A and 9A exhibited a single peak that correlated with the tR of the corresponding Re analogues (Figure 2). Compounds 5 and 7 displayed labeling efficiencies similar to those of previously reported systems.12,17 Evaluation of the stability of the metal complexes and isoxazole linker were examined using cysteine (1 mM) and histidine (1 mM) in a phosphate buffer (10 mM, pH 7.4). The challenge assays showed >99% stability of 8A and 9A through 24 h, with both amino acids indicating minimal transchelation or coordination rearrangement correlating with Re stability studies (Table S2 in the SI).
Figure 2.
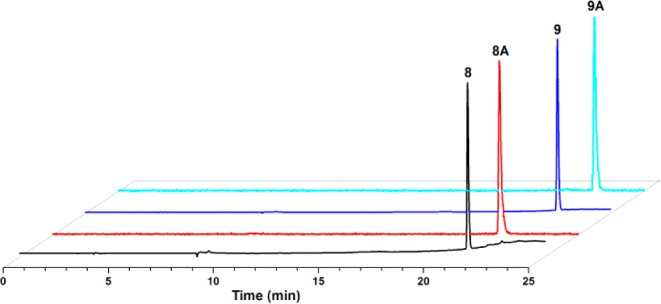
Normalized high-performance liquid chromatography chromatograms of purified 8 and 9 (UV, 254 nm, Re) and 8A and 9A (NaI, γ, 99mTc).
Evaluation of the chelate, then click method gave unanticipated results with 99mTc compared to the Re analogues. The reaction of 10A with 4 rapidly produced 8A with tR similar to that of 8 in ∼60% yield after 2.5 min at room temperature. In efforts to improve yields, reaction times were extended to 1 h, but yields decreased to <25%. A new peak was observed with tR similar to that of 10A; however, upon isolation, this species failed to react after subsequent exposure to Cu-free or CuAAC reaction conditions. Similar behavior was also observed in the reaction of 11A with 4 to generate 9A but with decreased yields of 9% (2.5 min) and 2% (1 h). Dimer formation of 4 or cycloaddition intermediate formation27 might explain the decreased reactivity of 10A and 11A. Alternatively, reductive isoxazole ring N–O bond cleavage in 8A and 9A may have occurred, as has been shown previously with transition-metal carbonyls,28 resulting in significantly lower yields. The variability between the Re and 99mTc reactivities in the chelate, then click approach may also be attributed to the intrinsic properties of the metals within group VII as well as radiotracer versus macroscopic concentrations.
In conclusion, by replacement of the azide dipole with nitrile oxide, it is possible to achieve CuAAC speed and selectivity without the drawbacks of Cu toxicity. The potential issue that arises is that of dipole instability, which can lead to dimerization or nucleophile trapping.29 It is possible to eliminate the probability of multiple coordination modes of the 1,2,3-triazole ring while still maintaining selective reactivity between the alkyne and oxime. Although conditions for isoxazole formation in the present study are not optimal for ideal click reactions, future work to improve yields and maintain biocompatibility would enhance the utility of this click strategy for applications with fac-[MI(CO)3]+. While the chelate, then click approach for the cycloaddition reaction was not as favorable for 99mTc as for Re, the click, then chelate approach is still a viable approach for generating SPECT imaging agents with 99mTc.
Acknowledgments
The authors thank the Office of Science, U.S. Department of Energy, Radiochemistry and Radiochemistry Instrumentation Program (Grant DE-FG02-08ER64672), NIH/NIGMS (Institutional Award T32-GM008336), and Washington State University for financial assistance. We greatly appreciate Dr. Mary Dyszlewski at Covidien for providing the IsoLink kits.
Supporting Information Available
Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Valko M.; Morris H.; Cronin M. T. D. Curr. Med. Chem. 2005, 12, 1161–1208. [DOI] [PubMed] [Google Scholar]
- Chang P. V.; Prescher J. A.; Sletten E. M.; Baskin J. M.; Miller I. A.; Agard N. J.; Lo A.; Bertozzi C. R. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debets M. F.; Van Berkel S. S.; Dommerholt J.; Dirks A. J.; Rutjes F. P. J. T.; Van Delft F. L. Acc. Chem. Res. 2011, 44, 805–815. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Acc. Chem. Res. 2011, 44, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- Zeng D. X.; Zeglis B. M.; Lewis J. S.; Anderson C. J. J. Nucl. Med. 2013, 54, 829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahta M.; Burke T. R. ChemMedChem 2011, 6, 1363–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly T.; Kasten B. B.; Hayes T. R.; Benny P. D.. Recent Advances in Re/Tc Radiopharmaceutical Design Utilizing Orthogonal and Metal Template Based Click Reactions. In Advances in Chemistry Research; Taylor J. C., Ed.; Nova Publishers: New York, 2013; Vol. 18, pp 93–141. [Google Scholar]
- Wangler C.; Schirrmacher R.; Bartenstein P.; Wangler B. Curr. Med. Chem. 2010, 17, 1092–1116. [DOI] [PubMed] [Google Scholar]
- Mamat C.; Ramenda T.; Wuest F. R. Mini-Rev. Org. Chem. 2009, 6, 21–34. [Google Scholar]
- Meldal M.; Tornøe C. W. Chem. Rev. 2008, 108, 2952–3015. [DOI] [PubMed] [Google Scholar]
- Bottorff S. C.; Moore A. L.; Wemple A. R.; Bucar D. K.; MacGillivray L. R.; Benny P. D. Inorg. Chem. 2013, 52, 2939–2950. [DOI] [PubMed] [Google Scholar]
- Schibli R.; La Bella R.; Alberto R.; Garcia-Garayoa E.; Ortner K.; Abram U.; Schubiger P. A. Bioconjugate Chem. 2000, 11, 345–351. [DOI] [PubMed] [Google Scholar]
- Struthers H.; Spingler B.; Mindt T. L.; Schibli R. Chem.—Eur. J. 2008, 14, 6173–6183. [DOI] [PubMed] [Google Scholar]
- Mindt T. L.; Muller C.; Stuker F.; Salazar J.-F.; Hohn A.; Mueggler T.; Rudin M.; Schibli R. Bioconjugate Chem. 2009, 20, 1940–1949. [DOI] [PubMed] [Google Scholar]
- Kluba C. A.; Mindt T. L. Molecules 2013, 18, 3206–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore A. L.; Bucar D.-K.; MacGillivray L. R.; Benny P. D. Dalton Trans. 2010, 39, 1926–1928. [DOI] [PubMed] [Google Scholar]
- Zeglis B. M.; Houghton J. L.; Evans M. J.; Viola-Villegas N.; Lewis J. S.. Inorg. Chem. DOI: 10.1021/ic401607z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claisen L.; Stock R. Ber. Dtsch. Chem. Ges. 1891, 24, 130–138. [Google Scholar]
- Pevarello P.; Amici R.; Brasca M. G.; Villa M.; Varasi M. Targets Heterocycl. Syst. 1999, 3, 301–339. [Google Scholar]
- Algay V.; Singh I.; Heaney F. Org. Biomol. Chem. 2010, 8, 391–397. [DOI] [PubMed] [Google Scholar]
- van der Peet P. L.; Connell T. U.; Gunawan C.; White J. M.; Donnelly P. S.; Williams S. J. J. Org. Chem. 2013, 78, 7298–7304. [DOI] [PubMed] [Google Scholar]
- Gutsmiedl K.; Fazio D.; Carell T. Chem.—Eur. J. 2010, 16, 6877–6883. [DOI] [PubMed] [Google Scholar]
- Singh I.; Zarafshani Z.; Lutz J.-F.; Heaney F. Macromolecules 2009, 42, 5411–5413. [Google Scholar]
- Liu K.-C.; Shelton B. R.; Howe R. K. J. Org. Chem. 1980, 45, 3916–3918. [Google Scholar]
- Crystal data for 9, C21H16N3O6Re,CH4O, Mr = 624.61 g mol–1, triclinic, space group P1̅, a = 8.1342(8) Å, b = 12.5836(13) Å, c = 12.8618(13) Å, α = 117.169(5)°, β = 101.227(5)°, γ = 97.439(5)°, V = 1112.28 Å3, T = 293(2) K, Z = 2, μ(Mo Kα) = 5.512 mm–1, 9749 reflections measured, 5274 independent reflections (Rint = 0.029). The final R(F) values were 4562 [I > 2σ(I)]. The final Rw(F2) values were 0.066 [I > 2σ(I)]. The final R(F) values were 0.044 (all data). The final Rw(F2) values were 0.071 (all data). The goodness of fit on F2 was 1.051. CCDC deposition number: 962881.
- Kelly D. R.; Baker S. C.; King D. S.; de Silva D. S.; Lord G.; Taylor J. P. Org. Biomol. Chem. 2008, 6, 787–796. [DOI] [PubMed] [Google Scholar]
- Nitta M.; Kobayashi T. J. Chem. Soc., Chem. Commun. 1982, 877–878. [Google Scholar]
- Grundmann C.; Mini V.; Dean J. M.; Frommeld H.-D. Justus Liebigs Ann. Chem. 1965, 687, 191–214. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.