

Abstract
This study investigates the incorporation of hyaluronan (HA) binding peptides into poly(ethylene glycol) (PEG) hydrogels as a mechanism to bind and retain hyaluronan for applications in tissue engineering. The specificity of the peptide sequence (native RYPISRPRKRC vs non-native RPSRPRIRYKC), the role of basic amino acids, and specificity to hyaluronan over other GAGs in contributing to the peptide–hyaluronan interaction were probed through experiments and simulations. Hydrogels containing the native or non-native peptide retained hyaluronan in a dose-dependent manner. Ionic interactions were the dominating mechanism. In diH2O the peptides interacted strongly with HA and chondroitin sulfate, but in phosphate buffered saline the peptides interacted more strongly with HA. For cartilage tissue engineering, chondrocyte-laden PEG hydrogels containing increasing amounts of HA binding peptide and exogenous HA had increased retention and decreased loss of cell-secreted proteoglycans in and from the hydrogel at 28 days. This new matrix-interactive hydrogel platform holds promise for tissue regeneration.
Introduction
Hyaluronic acid (HA) is a naturally occurring glycosaminoglycan (GAG) that offers many advantages to the design of biomaterials.1 For example, HA can interact with cells via cell surface receptors, can be degraded by cell-secreted enzymes, and is considered nonimmunogenic.2,3 Furthermore, HA plays a role in several key processes in the body including angiogenesis, wound healing, mediation of long-term inflammation, and extracellular matrix (ECM) homeostasis.2 HA is a major component of the ECM. For example, aggrecan, the major proteoglycan in cartilage, is retained through its interaction with HA, forming large aggregates of aggrecan along a HA backbone and enabling cartilage to resist mechanical loads.
The many diverse biological functions of HA have led to its use in a wide range of biomaterial applications. For example, HA hydrogel films have been applied to full-thickness wounds leading to accelerated healing.4 Scaffolds formed from Hyaff, a benzyl ester derivatized HA, have been used in numerous applications ranging from for example skin, cartilage, nerve and vascular tissue engineering.5 HA has also been modified with (meth)acrylates to enable cross-linking by radical mediated polymerization offering a platform to encapsulate cells.6 This hydrogel platform has shown promise in cartilage tissue engineering, whereby tuning cross-link density7 or incorporating hydrolytically cleavable segments of caprolactone8 created environments supportive for cartilage cells and for chondrogenesis of mesenchymal stem cells, respectively, resulting in deposition of cartilage ECM molecules, aggrecan and collagen II.
HA is often chemically modified with functional groups, such as those described above, enabling it to be fabricated into a biomaterial.9 This enables modified HA to be reacted with other chemistries8,10,11 offering control over the amount of HA, and therefore its bioactivity, in a biomaterial. Chemical modification of HA, however, may affect its ability to be degraded by enzymes (i.e., hyaluronidases) as well as its biological function.12,13 Upon degradation, the size of the HA fragments can have significant biological effects.11 For example, the size of HA fragments has been shown to influence tissue synthesis11 and as well low molecular weight HA oligomers can shift HA from being noninflammatory to pro-inflammatory.11,14,15 Nonetheless, chemical modification of HA has many benefits for creating bioactive biomaterials and has been used in a wide range of tissue engineering applications.
This study investigates an alternative strategy to incorporating HA into a hydrogel biomaterial. Rather than using HA as a building block of the biomaterial, HA is noncovalently tethered into a bioinert hydrogel thereby introducing bioactivity without contributing to the overall structure. This strategy leverages the native interaction that HA has with many proteins. In vivo there are a large number of HA binding proteins, some of which bind to HA via a linear 8–11 amino acid peptide motif containing multiple basic amino acids.16 Therefore, the objective of this study was to develop a poly(ethylene glycol) (PEG) hydrogel platform containing a peptide motif with HA binding affinity. The basic amino acid sequence, RYPISRPRKRC found in link protein, has been implicated as a HA binding motif16−18 and therefore was chosen for this study. A series of experiments were designed to investigate (a) the role of the basic amino acids in the binding of the peptide to HA and (b) the specificity of the peptide to HA when the peptide is tethered into a hydrogel. A combination of experimental approaches and atomistic molecular dynamics simulations were employed to investigate these interactions. The biological functionality of this hydrogel platform was evaluated for cartilage tissue engineering.
Experimental Section
Macromolecular Monomer Synthesis
PEG-tetranorbornene (PEGTNB) was synthesized by combining 4arm-PEG-NH2 (5000 Da, JenKemUSA) with 4 molar excess 5-norbornene-2-carboxylic acid (Sigma) in dimethylformamide in the presence of 2 molar excess 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium (HATU, AKSci) and 2 molar excess N,N-diisopropylethylamine (DIEA, Sigma). PEGTNB was precipitated in ice-cold diethyl ether, dialyzed against diH2O (SpectraPor7, MWCO1000), sterile filtered, and the final product was collected after lyophilization. The functionalization of PEG with norbornene was determined using 1H NMR imaging. The alkenes associated with the norbornene (∼6 ppm) were compared to the methylene groups associated with the PEG molecule (∼3.6 ppm) to determine percent substitution (>95%). PEG-dithiol (PEGDSH) was purchased (3400 Da, LaysanBio). RYPISRPRKRC (HA binding peptide), RPSRPRIRYKC (non-native, scrambled HA binding peptide sequence), and GYPISGPGGGC (charge control peptide) were either purchased (GenScript or University of Colorado Peptide and Protein Chemistry Core Facility) or synthesized using solid-phase peptide synthesis (SPPS) on an Applied Biosystems model 433A peptide synthesizer, followed by HPLC purification, and confirmation by matrix-assisted laser desorption ionization based on the molecular weight. All peptide purity was >95%.
Fluorescent Hyaluronan (f-HA)
Fluorescently labeled hyaluronan was synthesized as described by Nagata et al.19,20 Briefly, hyaluronan (Mn ∼ 37 kDa) dissolved in 75% (v/v) 1 M HCl and 25% (v/v) pyridine solution and combined with 5 aminofluorescein (1.6 mol equivalent per disaccharide unit) dissolved in a 50% (v/v) 1 M HCl and 50% (v/v) pyridine solution and the pH adjusted to 4.75. 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide HCl (EDC; 49 mol equiv per disaccharide unit) was added to the solution and agitated for 2 h at room temperature. The product was purified by dialysis and recovered by precipitation in chilled ethanol with 1.25% sodium acetate followed by centrifugation. The resulting pellet was dissolved in NaOH (0.1 M) for 20 h at 37 °C, and neutralized prior to recovering by a second precipitation and centrifugation step. The pellet was dissolved in diH2O and dialyzed overnight. The final product was recovered by lyophilization and stored at −20 °C protected from light.
Hydrogel Fabrication
PEGTNB and PEGDSH (1 ene: 0.8 thiol) were dissolved in diH2O or PBS to yield a final monomer solution of 10 wt % with 2.2 mM photoinitiator (I2959). Peptide (RYPISRPRKRC, RPSRPRIRYKC, or GYPISGPGGGC) was added to final concentrations between 0 and 5 mM. The macromer solution was photopolymerized using 352 nm light (Sankyo Denki) at an intensity of 6 mW/cm2 for 10 min to form hydrogels (∼5 mm in diameter by ∼1 mm thick). The resulting gels were allowed to swell 48 h in diH2O (pH = 6–9) or PBS (pH = 7.4) to remove any unreacted monomer and peptide.
Glycosaminoglycan Loading and Release
PEG hydrogels (n = 5) with and without peptide (0–5 mM) were removed from diH2O or PBS and placed in 200 μL containing 0.5 mg f-HA or chondroitin sulfate (ChS; primarily chondroitin-4-sulfate sodium salt; Sigma) per mL in diH2O or PBS on a figure-of-eight shaker (60 rpm) for 48 h at 37 °C. The liquid was collected and hydrogels were subsequently placed in diH2O or PBS for 48 h on a figure-of-eight shaker (60 rpm) at 37 °C to release any unbound GAGs. All liquid fractions were probed for GAGs by absorbance analysis using a spectrophotometer (FLUOstar Optima plate reader (BMG Labtech)) with 485 nm bandpass filter for f-HA and by dimethylmethylene blue dye method (DMMB)21 for ChS. Preliminary studies confirmed similar loading and release of f-HA and unmodified HA using a uronic acic carbazole reaction assay and therefore f-HA was used for this study.
F-HA and ChS Temporal Loading
PEG hydrogels (n = 3) with and without peptide (5 mM) were removed from diH2O and placed in 200 μL of diH2O containing 0.5 mg f-HA or ChS per mL on a figure-of-eight shaker (60 rpm) for 144 h at 37 °C. Liquid fractions were collected over time and assessed for f-HA by spectrophotometric analysis for absorbance using a NanoDrop1000 (Thermo Scientific) at 492 nm and for ChS by DMMB dye method.21
F-HA and ChS Release in Varying NaCl Concentrations
PEG hydrogels (n = 5) with the native peptide (RYPISRPRKRC, 5 mM) were removed from diH2O and placed in 200 μL diH2O containing 0.5 mg f-HA or ChS per ml on a figure-of-eight shaker (60 rpm) for 48 h at 37 °C. The liquid was collected. In order to release any unbound GAGs, the hydrogels were subsequently placed in diH2O for 48 h at 37 °C on a figure-of-eight shaker (60 rpm). The liquid was collected and the hydrogels were transferred to diH2O containing NaCl in varying concentrations (0.15, 0.5, 1, or 2 M) for 48 h at 37 °C on a figure-of-eight shaker (60 rpm). The liquid was collected and ran through a desalting column using Amicon Ultra-4, PLGC Ultracel-PL Membrane, 10 kDa columns (Millipore) according to the manufacturer’s protocol. All liquid fractions were assayed for f-HA by absorbance (485 nm) and for ChS by DMMB dye method.21
Chondrocyte Isolation and Encapsulation
Chondrocytes were isolated from the femoral-patellar groove of a 1–3 week old calf (Research 87, Marlboro, MA), as described elsewhere.22 Freshly isolated chondrocytes (50 million cells per mL) were combined with the sterile monomer/peptide/photoinitiator solution described above in phosphate buffered saline (PBS) with 1 mg/g of HA (37 kDa, Lifecore Biomedical). The solution was exposed to 352 nm light (Sankyo Denki) at an intensity of 6 mW/cm2 for 10 min producing hydrogel disks that were 5 mm diameter and 2.5 mm in height. The cell–hydrogel constructs were cultured on figure-of-eight shaker (60 rpm) at 37 °C in a humid environment with 5% CO2 in chondrocyte medium (Dulbecco’s Modified Eagle Medium supplemented (DMEM) with 10% fetal bovine serum (v/v), 0.04 mM l-proline, 50 mg/L l-ascorbic acid,10 mM HEPES, 0.1 M MEM-nonessential amino acids, 1% penicillin-streptomycin, 0.5 μg/mL fungizone, and 20 μg/mL gentamicin). Medium was replaced every 2–3 days. Removed medium was frozen and stored at −80 °C. Viability of the encapsulated cells was determined by the Live/Dead Cell Viability Assay (Invitrogen), which stains live cells green and dead cells red, and imaged on a Zeiss LSM 5 Pascal confocal microscope.
Biochemical Analysis
Constructs were lyophilized, homogenized, and digested in a papain solution [100 mM sodium phosphate buffer, 10 mM Na2EDTA, 10 mM l-cysteine, 0.125 mg/mL papain (Worthington)] for 16 h at 60 °C. Sulfated glycosaminoglycan content in the constructs and medium was determined using the DMMB method.21
Statistical Analysis
Data are expressed as mean with standard deviation as error bars (mean(SD)). Data were compared using analyses of variance (ANOVA) with subsequent analysis using unpaired t tests. Post-hoc analyses for the temporal profile of f-HA and ChS loading (Figure 4) were conducted using the Bonferroni test.
Figure 4.

Loading of (a) f-HA and (b) ChS over 144 h (6 days) for the native peptide (square, solid line), non-native peptide (×, dashed line), control peptide (circle, solid line), and no peptide (diamond, dashed line).
Simulations
Simulation Set-Up
Atomistic molecular dynamics simulations of peptide-glycosaminoglycan binding were conducted with the pmemd program in the Amber suite software suite (http://ambermd.org). Three peptides were simulated: HA binding peptide (peptide sequence RYPISRPRKRC); non-native, scrambled HA binding peptide sequence (RPSRPRIRYKC); and the charge control peptide (GYPISGPGGGC). The GAGs simulated were pentamers of hyaluronic acid (HA; i.e., five repeat units of N-acetyl-glucosamine and glucuronate linked by β 1–3 and β 1–4 glycosidic bonds) and chondroitin-4-sulfate (i.e., five repeat units of N-acetyl-galactosamine-4-sulfate and glucuronate linked by by β 1–3 and β 1–4 glycosidic bonds), the latter of which was the primary component of the chondroitin sulfate used in the experiments. Each simulated system contained one of the GAGs (HA or ChS) and one of the three peptides (native HA binding peptide, non-native HA binding peptide, or charge control peptide) for a total of six systems. Each of these systems contained only neutralizing counterions (i.e., the number of Na+ ions is equal to the number of negatively charged groups, and the number of Cl– ions is equal to the number of positively charged groups), which is equivalent to deionized water (i.e., an added salt concentration of zero).
All systems were solvated with explicit TIP3P water molecules, and TIP3P-optimized Na+ and Cl– counterions were used.23 The Amber force field was used to parametrize the peptides, water molecules, and counterions, while the GLYCAM06 force field (revision h-1) was used to parametrize the glycosaminoglycans.23−26 All systems were constructed with the tleap program in the Amber suite.
To examine peptide–GAG binding, we conducted unbiased atomistic molecular dynamics simulations of a peptide and a GAG initially separated by approximately 25 Å, in a water box of approximately (70 Å)3 with Na+ and Cl– counterions, and observed the behavior of the two biomolecules over the course of 30 ns simulations. To equilibrate the systems, we first minimized the energy with 1000 steps of steepest descent minimization followed by 1000 steps of conjugate gradient minimization, followed by slow heating at constant volume from 0 to 300 K over 20 ps, followed by constant pressure equilibration to 1 atm at 300 K over 20 ps. After these equilibration steps, production simulations were conducted at 1 atm and 300 K at constant pressure and constant temperature (NPT ensemble). Temperature was controlled with a Langevin thermostat with a collision frequency of 5 ps–1, and pressure was controlled by isotropic position rescaling with a weak-coupling algorithm with a relaxation time constant of 2 ps. All bonds with hydrogen atoms were constrained to their equilibrium lengths using the SHAKE algorithm with a tolerance of 10–5 Å, and a 2 fs time step was used.27,28 Snapshots were recorded every 2 ps. Short-range nonbonded interactions were cut off at 9 Å, and the particle mesh Ewald (PME) method was used to calculate long-range electrostatic interactions (PME parameters: interpolation order of 4, tolerance of 10–5, and maximum grid spacing of 1 Å). The simulation box was periodic in all three dimensions. As required by the Amber and GLYCAM force fields, differing scaling factors for 1–4 interactions were used for the peptide (scaling factor 0.5 for van der Waals and 0.833 for electrostatics) and carbohydrate (scaling factor 1.0 for both van der Waals and electrostatics) molecules.
Simulation Analysis Methods
The peptide–GAG complexes formed during the 30 ns production simulations were characterized by calculating the peptide–GAG total nonbonded interaction energy (decomposed into van der Waals and electrostatic contributions) and by calculating the number of electrostatic contacts and hydrogen bonds between the peptide and the GAG. An electrostatic contact was defined to be formed when two oppositely charged chemical groups (i.e., primary amine group of lysine, guanidium group of arginine, carboxyl group, or sulfate group) were within the Bjerrum length of each other. The Bjerrum length is the ratio of the electrostatic attraction of two point charges to the thermal energy and is approximately 7 Å in water at 300 K. A hydrogen bond was defined to be formed when the donor and acceptor atoms were separated by less than 3.5 Å and the angle formed by the donor, hydrogen, and acceptor atoms is greater than 120°. The peptide–GAG complexes were considered to be equilibrated after 15 ns of simulation time because the total peptide–GAG interaction energy had reached a stable value, and only the last 15 ns of each simulation were used for analysis. All visualization and the analysis of electrostatic contacts and hydrogen bonds was conducted with the VMD program,29 while nonbonded energy decomposition was conducted with the NAMD program.30
Results and Discussion
HA Binding to PEG Hydrogels Modified with HA Binding Peptides
Three peptides based on the B-X-B HA binding motif,16 where B is a basic amino acid and X is any nonacidic amino acid, were investigated for their ability to bind HA within PEG hydrogels: a native HA binding peptide (RYPISRPRKRC),18 a non-native peptide, but with the same charge density where the location of the charged amino acids are scrambled (RPSRPRIRYKC), and a peptide where the basic amino acids are replaced with glycine, yielding a net electrically neutral peptide at physiological pH (GYPISGPGGGC). The peptides were tethered into a PEG hydrogel via thiol–ene covalent bonds (Figure 1).
Figure 1.
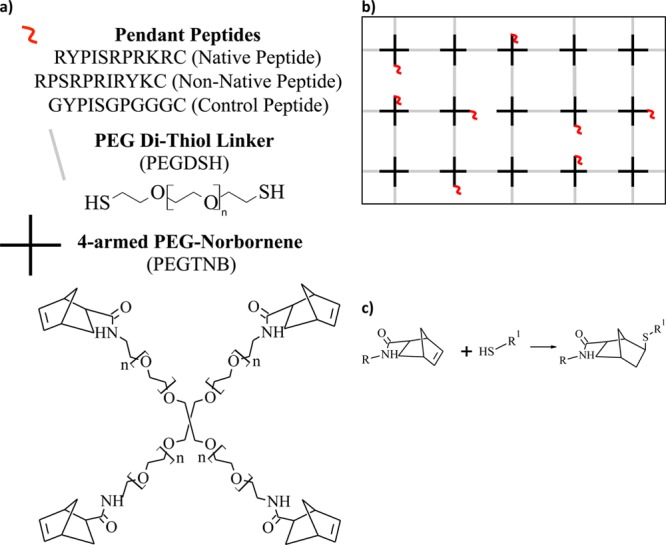
(a) Macromolecular monomers used for the fabrication of PEG-based gels. The precursors included PEGTNB (n ∼ 30), the dithiol linker PEG (n ∼ 80), and cysteine-terminated peptide. (b) Depiction of an idealized cross-linked network formed these macromolecular monomers. (c) Reaction scheme between the thiol containing monomer with the norbornene containing monomer. R denotes PEG and R1 denotes PEG or peptide.
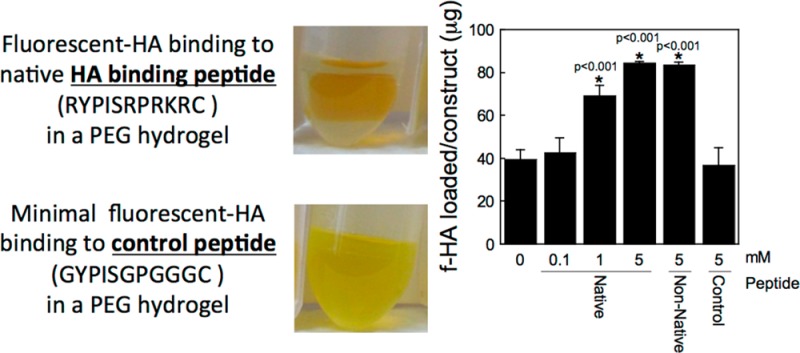
To characterize the interaction of HA with the peptides when tethered into a PEG hydrogel, preswollen hydrogels were placed in a bath of fluorescently labeled HA (f-HA) in distilled water (diH2O) as depicted in the schematic and photographs in Figure 2a,b. The amount of f-HA loaded into the hydrogel increased with increasing concentration of HA binding peptide (p < 0.0001; Figure 2c). Hydrogels containing no peptide loaded 39(4) μg of the f-HA from the bath, which was similar to the charge control peptide (37(8) μg). PEG hydrogels with 5 mM native HA binding peptide or non-native HA binding peptide loaded 85(1) and 84(2) μg of f-HA, respectively, of the f-HA from the bath. The higher amount of f-HA loaded into the gels containing the HA binding peptide or the non-native HA binding peptide compared to that which was loaded in PEG-only hydrogels suggests an interaction between the peptide and hyaluronan.
Figure 2.

Hyaluronan (HA) loading and retention capabilities of a HA binding peptide (RYPISRPRKRC), non-native HA binding peptide (RPSRPRIRYKC), and charge control peptide (GYPISGPGGGC). (a) Schematic of the experimental setup (f-HA loading for 48 h, f-HA release in diH2O for 48 h). (b) Representative images of hydrogels (containing the native HA binding peptide or charge control peptide) in solution corresponding to each 48 h step (i.e., the latter two images of the experimental setup). The fluorescein can be visualized by the yellow color showing retention of the f-HA in hydrogels with the native HA binding peptide in diH2O and with the charge control peptide showing continued release of f-HA in diH2O. (c) Total amount of f-HA loading into hydrogels after 48 h of immersion in a solution of 100 μg of f-HA in diH2O. (d) Release of loaded f-HA after 48 h of immersion in diH2O. (e) Percent-retained of loaded f-HA after immersion in diH2O. Data represent mean(SD) with a sample size of 5; *indicates samples were compared to no peptide control (0 mM).
To determine how much f-HA was simply absorbed into each of the hydrogels, hydrogels were transferred to fresh diH2O and the amount of f-HA release was determined as depicted in Figure 2a,b. In diH2O, the amount of the f-HA released decreased with increasing HA binding peptide concentration (p < 0.001, Figure 2d). PEG hydrogels containing 5 mM HA binding peptide or 5 mM non-native, scrambled HA binding peptide released 15(1) μg and 14(1) μg, respectively. Based on these data, the % f-HA retained in the hydrogel was calculated and presented in Figure 2e showing significantly higher retention of f-HA with increasing native peptide concentration. PEG hydrogels with 5 mM native HA binding peptide or non-native HA binding peptide retained 82(1) and 83(1)% of the originally loaded f-HA. A small amount of f-HA was retained in the no peptide and control peptide, which was confirmed visually by a slight yellow appearance in the hydrogels. This observation suggests that nonionic interactions between f-HA and PEG and between f-HA and peptide may exist, such as hydrogen bonding and van der Waals, which have been implicated in the binding affinity of HA to some proteins31 and to other HA molecules.32
Nonspecific Binding of GAGs to HA Binding Peptides in PEG Hydrogels
To probe the specificity of the interaction of the HA binding peptide to HA, chondroitin-sulfate, a negatively charged GAG that is one of the primary GAGs in aggrecan, was investigated. Similar experiments were performed where loading and subsequent release of ChS in diH2O was probed. There was no obvious trend in ChS loading with HA binding peptide concentration (Figure 3a), a contrast to the HA studies (Figure 2c). However, the native and non-native HA binding peptide resulted in high loading of ChS loading with 80(8) and 65(13) μg of ChS, respectively. The amount of ChS released in diH2O decreased with increasing HA binding peptide concentration (p < 0.001, Figure 3b), similar to that observed with f-HA. Based on these data, the % ChS retained in the hydrogel was calculated and is presented in Figure 3c showing high retention of ChS with 5 mM native or non-native HA binding peptide. A small fraction of ChS was retained in the hydrogel with no peptide and control peptide, suggesting that some nonionic interactions may exist with PEG and the peptide in the hydrogels. Overall, these results confirm that the native and non-native HA binding peptide interact with ChS.
Figure 3.

Chondroitin Sulfate (ChS) loading and retention capabilities of a HA binding peptide (RYPISRPRKRC), non-native HA binding peptide (RPSRPRIRYKC), and charge control peptide (GYPISGPGGGC). (a) Total amount of ChS loading into hydrogels after 48 h of immersion in a solution of 100 μg of ChS in diH2O. (b) Release of loaded ChS after 48 h of immersion in diH2O. (c) Percent-retained of loaded ChS after immersion in diH2O. Data represent mean(SD) with a sample size of 5; *indicates samples were compared to no peptide control (0 mM).
Temporal Loading of GAGs into PEG Hydrogels Containing HA Binding Peptides
GAG loading into the PEG hydrogels will be influenced by a combination of diffusion and peptide chemistry. To assess the kinetics of GAG loading, temporal loading experiments were performed in diH2O. The loading profiles for both f-HA (Figure 4a) and ChS (Figure 4b) in hydrogels with the native or non-native HA binding peptide (5 mM) were statistically similar for both GAGs. For hydrogels with the control peptide (5 mM) or no peptide, GAG loading was significantly reduced (p < 0.0001) compared to the native and non-native peptides. Equilibrium was reached within 3 h and was similar for all conditions suggesting that diffusion into the hydrogels for both GAGs, which are similar in molecular weight, was comparable.
Ionic Interactions in the Binding of GAGs to HA Binding Peptides in PEG Hydrogels
To assess if the f-HA and ChS retained in the hydrogels containing the native HA binding peptide was due to ionic interactions a series of experiments were performed in water with an increasing concentration of ions (NaCl). With 0.15 M NaCl, partial GAG release was observed, demonstrating that ions can interfere with the binding between GAGs and the native HA binding peptide. Interestingly in 0.15 M NaCl a higher fraction of the retained f-HA was released (45(5)%, Figure 5a) compared to ChS (25(4)%, Figure 5b) suggesting that ChS interacts more strongly with the native HA binding peptide than f-HA. With 0.5 M NaCl, majority of GAGs was released (86(18)% for f-HA and 90(6)% for ChS) and higher salt concentrations did not lead to any additional release suggesting that all ionic interactions were disrupted.
Figure 5.

Amount of GAG retained within constructs having 5 mM native peptide after GAG-loaded constructs were immersed for 48 h in diH2O, similar to Figures 2c and 3c, is given to the left of the dashed line. To the right of the dashed line, the % of the total loaded GAG that was released from hydrogels containing 5 mM native peptide in varying concentrations of NaCl ranging from the physiological range (0.15 M) to supraphysiological (0.5–2 M NaCl). This was evaluated with both (a) f-HA and (b) ChS.
Overall, these results confirm that the HA binding peptide interacts largely through ionic interactions with HA and ChS leading to greater retention of GAGs in the hydrogel. Our findings agree with other reports of HA describing that ionic interactions are critical to how proteins interact with and bind HA.16−18 In particular, basic amino acids are proposed to be the major determinant in the binding of aggrecan and link protein to HA. These amino acids, namely arginine and lysine, form ionic bonds with the carboxylic acid group of glucuronic acid in HA.33 It has been suggested that a charged amino acid motif having a B-X-B motif (where B is basic amino acid and X is any nonacidic amino acid) is the primary amino acid sequence that binds HA and that there is not a specific conserved primary amino acid sequence for HA binding16 nor is a generalized basic charge density sufficient. The latter was confirmed using polylysine, which was not able to block the binding of link protein to HA.18 Substitution of the arginine or lysine residues in a peptide containing the B-X-B motif with histidine, also a basic amino acid, abolished the peptides ability to bind to HA17 suggesting that arginine and lysine are the key B amino acids. However, the exact location and degree of the basic charge may16 or may not31 be important. Our data suggest that this interaction, at least in diH2O, is nonspecific, and the B-X-B motif can interact with both HA and ChS.
Simulation of Binding of GAGs to HA Binding Peptides
To further characterize the nature of the interactions between the peptides and GAGs, atomistic molecular dynamics simulations were performed for the three peptides (native HA binding peptide, non-native HA binding peptide, and charge control peptide) interacting with, or binding to, the two GAGs (HA and chondroitin-4-sulfate) under simulation conditions corresponding to diH2O. These simulations were performed with the GAGs and peptides free in bulk solution (i.e., not tethered into hydrogels). Representative snapshots of several peptide-GAG complexes are shown in Figure 6a.
Figure 6.

(a) Representative simulation snapshots of peptide–GAG complexes for the positively charged native HA binding peptide (RYPISRPRKRC) and the electrically neutral charge control peptide (GYPISGPGGGC). The simulation snapshots for non-native peptide–GAG systems are similar visually to that of the native peptides, so it is not shown for brevity. Glycosaminoglycans are shown with a “ball-and-stick” representation, while peptides are shown with a “licorice” representation. Atoms are colored in the following manner: carbon, cyan; hydrogen, white; oxygen, red; nitrogen, blue; sulfur, yellow. Hydrogen bonds are indicated with pink dashed lines. Panels (b) and (c) quantify the energetics of the interactions of the native HA binding peptide (RYPISRPRKRC), non-native HA binding peptide (RPSRPRIRYKC), or charge control peptide (GYPISGPGGGC) with either hyaluronan (HA) or chondroitin-4-sulfate (ChS). (b) Electrostatic and total interaction energy (sum of van der Waals and electrostatic energy) between peptide and GAG. The interaction is primarily electrostatic, and the interaction of the GAG with the charged peptides (native and non-native HA binding peptides) is therefore much stronger than with the electrically neutral charge control peptide. Interactions with ChS are much stronger than those with HA because of the higher charge density of ChS. (c) Total peptide–GAG interaction energy of each residue along the peptides. All of the residues in the charge control peptide generally show weak interactions with the GAG. The charged residues in the positively charged peptides are the primary source of the peptide-GAG interaction energy. Data represent mean(SD) of three independent simulation trials.
In agreement with the experiments, it was found that there was a much greater total energetic attraction between the positively charged peptides (i.e., native HA binding peptide and non-native, scrambled HA binding peptide) and the GAGs than between the electrically neutral charge control peptide and the GAGs. Also, in accord with the experiments, it was found that both the native and non-native HA binding peptides bound to the GAGs with comparable strength (Figure 6b). Simulations indicated that the peptide–GAG interaction energy was almost entirely electrostatic in origin (Figure 6b). The positively charged residues (i.e., lysine and arginine) were the primary source of peptide-GAG attraction, and replacing the positively charged residues in the HA binding peptide with neutral glycine residues (i.e., the charge control peptide) largely eliminated peptide-GAG attraction (Figure 6c). Furthermore, for the native and non-native HA binding peptides there was a much greater electrostatic attraction to ChS than to HA, likely due to the higher charge density of ChS. Lastly, it should be noted that the terminal residues (#1 and #11) can also induce an attractive or repulsive peptide–GAG interaction because they contain the positively charged N-terminus and the negatively charged C-terminus, respectively (Figure 6c).
The molecular-level interactions mediating the electrostatic attraction of the positively charged residues to the GAG included electrostatic contacts and hydrogen bonds (Supporting Information, Figure 1). As expected, no electrostatic contacts and few hydrogen bonds formed between the charge control peptide and the GAGs (see also Figure 6a, where a small number of hydrogen bonds between the charge control peptide and GAGs are visible, as indicated by dashed pink lines). Conversely, numerous electrostatic contacts and hydrogen bonds formed between the positively charged peptides and the GAGs (see Supporting Information, Figure 1 for quantitative results and Figure 6a for visualization). The charged residues were responsible for approximately 80% of the total number of peptide–GAG hydrogen bonds (and, by definition, all of the electrostatic contacts), which further confirms that basic residues play an important role in peptide–GAG binding.
Binding of GAGs to HA Binding Peptide in PEG Hydrogels under Physiologically Relevant Ionic Strength
Both simulation and experimental results confirm that HA binding peptides bind negatively charged GAGs largely through electrostatic interactions. However, peptide–GAG interactions typically occur in a physiological environment where the ionic strength is ∼0.15 M. Therefore, the GAG–peptide interactions were also examined in a physiologically relevant ionic strength solution. In this set of experiments, only the highest peptide concentration (5 mM) was investigated and PBS was used to emulate the physiological environment. Contrary to the previous experiments, GAGs were loaded into the hydrogels in the presence of PBS, which contains NaCl, KCl, Na2HPO4, and KH2PO4 and therefore ions than can shield the charges on the peptides and GAGs. There was greater f-HA loading in the presence of native HA binding peptide (56(8) μg) or non-native HA binding peptide (46(5) μg), when compared to no peptide (28(3) μg) or the charge control peptide (30(4) μg; Figure 7a). The amount of f-HA loaded into the HA binding peptide hydrogels was substantially lower in PBS compared to diH2O. Subsequent release of the loaded f-HA was lower with the HA binding peptides compared to the control peptide (p = 0.001, Figure 7b), supporting the existence of electrostatic interactions between the peptide and HA in PBS. There was also greater loading of ChS with the native HA binding peptide (23(5) μg) and to a lesser extent with the non-native HA binding peptide (9(4) μg), when compared to no peptide (3(3) μg) or the charge control peptide (3(3) μg). Similar to HA, ChS loading was lower for all hydrogel formulations in PBS (Figure 7c) when compared to diH2O (Figure 3a). Moreover, the ChS was entirely released in PBS in all formulations, with the exception of the non-native HA binding peptide (Figure 7d).
Figure 7.

(a) Total amount of f-HA loading into hydrogels after 48 h of immersion in a solution of 100 μg of f-HA in PBS. (b) Percent release of loaded f-HA after 48 h of immersion in PBS. (c) Total amount of ChS loading into hydrogels after 48 h of immersion in a solution of 100 μg of ChS in PBS. (d) Percent release of loaded ChS after 48 h of immersion in PBS. Data represent mean(SD) with a sample size of 5; *indicates samples were compared to no peptide control (0 mM).
Proposed Mechanisms Driving GAG Interactions with HA Binding Peptides
Ionic interactions have been suggested as one of the primary mechanisms by which proteins bind to and interact with many GAGs.16,34 ChS and HA are both GAGs, with the main difference between the two being a sulfate group in place of a hydroxyl group in the ChS.35 As such, ChS has two negative charges associated with the carboxyl and sulfate groups in each repeat unit, whereas HA has only one negative charge associated with the carboxyl group in each repeat unit. Moreover, the sulfate group has a lower pKa than the carboxyl group.36,37 Therefore, one would expect the electrostatic interactions between the HA binding peptides and ChS to be greater than between the HA binding peptides and HA, which is observed in the experimental and simulation results. Many studies report a certain degree of cross-reactivity between HA binding proteins and other GAGs, but interestingly show a stronger interaction with HA than with other GAGs. For example, it has been shown that the G1 domain of aggrecan (which is structurally similar to link protein38) binds to ChS with lower affinity that it does with HA.35 CD44, a cell surface receptor that binds to HA, has been shown to bind chondroitin-4-sulfate ∼100× weaker than with HA and even weaker to chondroitin-6-sulfate.33,39 However, these studies examine full proteins where it has been suggested that the presence of neighboring amino acids and protein confirmation may help to further regulate how a protein interacts with different GAGs. The simulations confirm electrostatic interactions between the HA binding peptide and HA, but also provide insight into the existence of nonionic HA–peptide interactions (e.g., hydrogen bonding). Hydrogen bonding and van der Waals have been suggested to play a role in the binding affinity of HA to some proteins31 and to other HA molecules.32
In the experiments, in a physiologically relevant salt buffer (i.e., PBS), the ability of the HA binding peptides to bind and retain either HA or ChS decreased. This finding is not surprising given that ions present in PBS can shield the negative charges in the GAGs and the positive charges in the peptide reducing electrostatic interactions. Interestingly, in PBS, the HA binding peptide interacted more strongly with HA and only weakly with ChS. Others have reported that link protein interacts strongly with HA in a buffered solution, while sulfated GAGs including chondroitin-4-sulfate do not; a finding that was in part attributed to conformational changes in the HA around the protein.40 Unbiased molecular dynamics simulations of peptide–GAG interactions in 2 M NaCl solution, mimicking the experimental high salt solutions, showed that the peptide and GAG did not interact strongly during the 30 ns simulation time (data not shown). However, due to the limited sampling of these unbiased simulations, no definitive conclusions could be drawn from the simulations about the role of salt in screening electrostatic interactions and reducing the total interaction energy. Overall, the experimental findings indicate that when electrostatics are partially screened the HA binding peptides interact more strongly with HA than ChS. These observations point to the involvement of nonionic interactions (e.g., van der Waals, hydrogel bonding, etc.) in the case of hyaluronan, which may be important to the selectivity of the B-X-B motif to HA over other GAGs in physiological environments.
Incorporating HA in PEG Hydrogel via HA Binding Peptides for Cartilage Tissue Engineering
Because HA has numerous benefits for cartilage tissue engineering including improved tissue synthesis7,41 and enhanced retention of chondrocyte-secreted sulfated GAGs,22 this study explored whether immobilizing HA into a PEG hydrogel via the HA binding peptides would further improve neocartilage formation. Demonstrating the utility of the peptides in a biologically relevant cell culture system over time is important, because it demonstrates the tissue engineering potential for the HA binding peptides despite potential confounding factors, such as peptide proteolysis. In a pilot study, chondrocytes were encapsulated in PEG hydrogels along with exogenous HA (at the same concentration), but with increasing concentrations of the native HA binding peptide tethered into the PEG hydrogel (Figure 6a). Cell viability was similar among all conditions (Supporting Information, Figure 2). Tissue synthesis was assessed by measuring sGAGs, which can be easily measured using biochemical assays, where sGAGs synthesized and secreted by chondrocytes are in the form of proteoglycans, with the majority being aggrecan. After 28 days of culture, a dose dependent response of the HA binding peptide was observed leading to increased accumulation of cell-secreted sGAG in the PEG hydrogel concomitant with a reduced release of sGAG with increasing peptide concentration. For example, PEG hydrogels containing the native or non-native HA binding peptide contained 148.8(6.6) μg and 164.6(3.1) μg of sGAG per construct, respectively, whereas the PEG-only constructs contained 121.8(16.0) μg sGAG (native peptide: p = 0.108 and non-native peptide: p = 0.038, Figure 8a). Sulfated GAG released into the culture media were not significantly different for any conditions between days 0 to 14, however between days 14–28, a reduction in sGAG release by 3.2-fold (p = 0.005) and 4.2-fold (p < 0.001) was observed for the native and non-native HA binding peptide, respectively, compared to PEG-only controls (Figure 8b). Taken together, these results demonstrate that the incorporation of HA, when either the native or non-native HA binding peptide is present, improves sGAG content in the hydrogel while minimizing its loss from the hydrogel, thus, overall improving neocartilage deposition.
Figure 8.

(a) Schematic of hydrogel formation from a solution of PEG macromers, peptide, HA and chondrocytes. Effect of peptide concentration and peptide chemistry on tissue synthesis showing total amount accumulated in the hydrogel after 28 days (b) and cumulative amount that was released to the culture medium between 14 and 28 days (c). Data represent mean(SD) with a sample size of 3–4; *indicates that samples were compared to nonpeptide control (0 mM).
The improved tissue engineering outcome is attributed to the ability of the peptides to retain the encapsulated HA. The biological effects of HA are then attributed to several possible factors. We have shown previously that the incorporation of HA can retain cell-secreted sGAGs.22 If the cell-secreted sGAGs are associated with aggrecan monomers having the globular 1 (G1) domain, which contains two HA binding regions, the exogenous HA could bind and thus retain any aggrecan monomers; a process similar to the native assembly of aggrecan aggregates. It is also possible that any peptide not already bound with HA could interact with endogenous, cell-secreted HA or HA already associated with aggrecan aggregates. However, the latter mechanism is unlikely given the large size of endogenous HA and aggrecan aggregates, which will limit their diffusion through the hydrogel. Moreover, it should be noted that some loss of sGAGs may be expected from the HA binding hydrogels. The DMMB assay used to measure sGAG will include aggrecan, any processed (i.e., catabolically cleaved) aggrecan that still have sGAGs attached to it, and any other sGAG containing molecules. Therefore, it is not expected that all of the measured sGAG interacts with the HA. This observation is further supported by our findings that sGAGs alone do not interact strongly with the HA binding peptides under physiological ionic strengths. However, the increased sGAG released from hydrogels without the HA binding peptide may represent increased degraded segments of aggrecan. The presence of HA in hydrogels containing the HA binding peptide may have led to a reduction in catabolic activity in the chondrocytes, as shown by others.42−44 The exact mechanism remains to be elucidated and is currently limited by the available antibodies for detecting degraded aggregan products. Despite lower total amount of sGAG produced by chondrocytes in hydrogels containing HA binding peptides, the HA binding peptides with HA led to higher amounts of sGAGs within the construct, which is critical for engineering cartilage.
Conclusions
Short, synthetic matrix binding peptides can be easily tethered into hydrogels to provide a facile means for retention of ECM molecules, such as hyaluronan. These ECM-interactive materials are promising candidates as biomaterials for tissue regeneration applications, due to the combined benefits of a tightly controlled, synthetic hydrogel, with the natural presentation of ECM matrix analogs that can actively retain bioactive molecules.
Acknowledgments
The authors are grateful to a Department of Education Graduate Assistantships in Areas of National Need (GAANN) Fellowship and University of Colorado-National Institute of Standards and Technology (CU-NIST) Material Science and Engineering Fellowship to J.J.R. and a State of Colorado Bioscience Discovery and Evaluation Grant. R.M.E. and A.J. acknowledge use of the Janus supercomputer, which is supported by the National Science Foundation (Award No. CNS-0821794) and the University of Colorado Boulder. The Janus supercomputer is a joint effort of the University of Colorado Boulder, the University of Colorado Denver and the National Center for Atmospheric Research. Research reported in this publication was also supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award No. R01 AR065441. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
The molecular-level interactions between peptide (native HA binding peptide, non-native HA binding peptide, and control peptide) and GAG decoupled into electrostatic contacts and hydrogen bonds. Cell viability based on a membrane integrity assay for chondrocytes cultured in PEG hydrogels containing the native or non-native HA binding peptide for 28 days. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Leach J. B.; Schmidt C. E. In Encyclopedia of Biomaterials and Biomedical Engineering; Wnek G. E., Bowlin G. L., Eds.; Informa Healthcare USA: New York, 2008; pp 1421–1431. [Google Scholar]
- Chen W. Y.; Abatangelo G. Wound Repair Regen. 1999, 7, 79–89. [DOI] [PubMed] [Google Scholar]
- Lapcik L. Jr.; Lapcik L.; De Smedt S.; Demeester J.; Chabrecek P. Chem. Rev. 1998, 98, 2663–2684. [DOI] [PubMed] [Google Scholar]
- Kirker K. R.; Luo Y.; Nielson J. H.; Shelby J.; Prestwich G. D. Biomaterials 2002, 23, 3661–3671. [DOI] [PubMed] [Google Scholar]
- Vindigni V.; Cortivo R.; Iacobellis L.; Abatangelo G.; Zavan B. Int. J. Mol. Sci. 2009, 10, 2972–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeds K. A.; Grinstaff M. W. J. Biomed. Mater. Res. 2001, 54, 115–121. [DOI] [PubMed] [Google Scholar]
- Chung C.; Mesa J.; Randolph M. A.; Yaremchuk M.; Burdick J. A. J. Biomed. Mater. Res., Part A 2006, 77, 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.; Beecham M.; Mauck R. L.; Burdick J. A. Biomaterials 2009, 30, 4287–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick J. A.; Prestwich G. D. Adv. Mater. 2011, 23, H41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuttelman C. R.; Rice M. A.; Rydholm A. E.; Salinas C. N.; Shah D. N.; Anseth K. S. Prog. Polym. Sci. 2008, 33, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters K. S.; Shah D. N.; Leinwand L. A.; Anseth K. S. Biomaterials 2005, 26, 2517–2525. [DOI] [PubMed] [Google Scholar]
- Segura T.; Anderson B. C.; Chung P. H.; Webber R. E.; Shull K. R.; Shea L. D. Biomaterials 2005, 26, 359–371. [DOI] [PubMed] [Google Scholar]
- Mano J. F.; Silva G. A.; Azevedo H. S.; Malafaya P. B.; Sousa R. A.; Silva S. S.; Boesel L. F.; Oliveira J. M.; Santos T. C.; Marques A. P.; Neves N. M.; Reis R. L. J. R. Soc., Interface 2007, 4, 999–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardue E. L.; Ibrahim S.; Ramamurthi A. Organogenesis 2008, 4, 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble P. W. Matrix Biol. 2002, 21, 25–29. [DOI] [PubMed] [Google Scholar]
- Amemiya K.; Nakatani T.; Saito A.; Suzuki A.; Munakata H. Biochim. Biophys. Acta 2005, 1724, 94–99. [DOI] [PubMed] [Google Scholar]
- Yang B.; Yang B. L.; Savani R. C.; Turley E. A. EMBO J. 1994, 13, 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetinck P. F.; Stirpe N. S.; Tsonis P. A.; Carlone D. J. Cell Biol. 1987, 105, 2403–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata H.; Kojima R.; Sakurai K.; Sakai S.; Kodera Y.; Nishimura H.; Inada Y.; Matsushima A. Anal. Biochem. 2004, 330, 356–358. [DOI] [PubMed] [Google Scholar]
- Ogamo A.; Matsuzaki K.; Uchiyama H.; Nagasawa K. Carbohydr. Res. 1982, 105, 69–85. [DOI] [PubMed] [Google Scholar]
- Farndale R. W.; Buttle D. J.; Barrett A. J. Biochim. Biophys. Acta 1986, 883, 173–177. [DOI] [PubMed] [Google Scholar]
- Roberts J. J.; Nicodemus G. D.; Giunta S.; Bryant S. J. J. Biomed. Mater. Res., Part A 2011, 97, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung I. S.; Cheatham T. E. J. Phys. Chem. B 2008, 112, 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar]
- Kirschner K. N.; Yongye A. B.; Tschampel S. M.; González-Outeiriño J.; Daniels C. R.; Foley B. L.; Woods R. J. J. Comput. Chem. 2008, 29, 622–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Miyamoto S.; Kollman P. A. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics Modell. 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day A. J.; Prestwich G. D. J. Biol. Chem. 2002, 277, 4585–4588. [DOI] [PubMed] [Google Scholar]
- Horkay F.; Basser P. J.; Londono D. J.; Hecht A. M.; Geissler E. J. Chem. Phys. 2009, 131, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahmann J. D.; O’Brien R.; Werner J. M.; Heinegard D.; Ladbury J. E.; Campbell I. D.; Day A. J. Structure 2000, 8, 763–774. [DOI] [PubMed] [Google Scholar]
- Sobel M.; Soler D. F.; Kermode J. C.; Harris R. B. J. Biol. Chem. 1992, 267, 8857–8862. [PubMed] [Google Scholar]
- Parkar A. A.; Day A. J. FEBS Lett. 1997, 410, 413–417. [DOI] [PubMed] [Google Scholar]
- Ng L. J.; Plaas A.; Grodzinsky A. J.; Ortiz C. Abstr. Pap. Am. Chem. Soc. 2002, 224, U408–U408. [Google Scholar]
- Hascall V.; Esko J. D. In Essentials of Glycobiology; Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, 2009; pp 219–228. [PubMed] [Google Scholar]
- Hardingham T. E. In Dynamics of Bone and Cartilage Metabolism; Seibel M. J., Robins S. P., Bilezikian J. P., Eds.; Elsevier: Burlington, 2006; Vol. 1, pp 85–98. [Google Scholar]
- Peach R. J.; Hollenbaugh D.; Stamenkovic I.; Aruffo A. J. Cell Biol. 1993, 122, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg L.; Tang L. H.; Pal S.; Johnson T. L.; Choi H. U. J. Biol. Chem. 1988, 263, 18071–18077. [PubMed] [Google Scholar]
- Kavalkovich K. W.; Boynton R. E.; Murphy J. M.; Barry F. In Vitro Cell. Dev. Biol.: Anim. 2002, 38, 457–466. [DOI] [PubMed] [Google Scholar]
- Manicourt D. H.; Devogelaer J. P.; Thonar E. J. In Dynamics of Bone and Cartilage Metabolism; Seibel M. J., Robins S. P., Bilezikian J. P., Eds.; Elsevier: Burlington, 2006; Vol. 1, pp 421–449. [Google Scholar]
- Fukuda K.; Dan H.; Takayama M.; Kumano F.; Saitoh M.; Tanaka S. J. Pharmacol. Exp. Ther. 1996, 277, 1672–1675. [PubMed] [Google Scholar]
- Homandberg G. A.; Hui F.; Wen C.; Kuettner K. E.; Williams J. M. Osteoarthr. Cartil. 1997, 5, 309–319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.