

Abstract
Exposure to high or repeated doses of methamphetamine can cause hyperthermia and neurotoxicity, which are thought to increase the risk of developing a variety of neurological conditions. Sigma receptor antagonism can prevent methamphetamine-induced hyperthermia and neurotoxicity, but the underlying cellular targets through which the neuroprotection is conveyed remain unknown. Differentiated NG108-15 cells were thus used as a model system to begin elucidating the neuroprotective mechanisms targeted by sigma receptor antagonists to mitigate the effects of methamphetamine. In differentiated NG108-15 cells, methamphetamine caused the generation of reactive oxygen/nitrogen species, an increase in PERK-mediated endoplasmic reticulum stress and the activation of caspase-3, -8 and -9, ultimately resulting in apoptosis at micromolar concentrations, and necrotic cell death at higher concentrations. The sigma receptor antagonist, 6-acetyl-3-(4-(4-(4-fluorophenyl)piperazin-1-yl)butyl)benzo[d]oxazol-2(3H)-one (SN79), attenuated methamphetamine-induced increases in reactive oxygen/nitrogen species, activation of caspase-3,-8 and-9 and accompanying cellular toxicity. In contrast, 1,3-di(2-tolyl)-guanidine (DTG), a sigma receptor agonist, shifted the dose response curve of methamphetamine-induced cell death towards the left. To probe the effect of temperature on neurotoxicity, NG108-15 cells maintained at an elevated temperature (40 °C) exhibited a significant and synergistic increase in cell death in response to methamphetamine, compared to cells maintained at a normal cell culture temperature (37 °C). SN79 attenuated the enhanced cell death observed in the methamphetamine-treated cells at 40 °C. Together, the data demonstrate that SN79 reduces methamphetamine-induced reactive oxygen/nitrogen species generation and caspase activation, thereby conveying neuroprotective effects against methamphetamine under regular and elevated temperature conditions.
Keywords: Apoptosis, Caspase, Methamphetamine, Necrosis, Reactive oxygen species, Sigma receptors
1. Introduction
Methamphetamine-induced neurotoxicity and hyperthermia may lead to motor and cognitive deficits and even death (Cruickshank and Dyer, 2009; Suchard, 2007). Methamphetamine interacts with sigma receptors in addition to monoamine transporters and causes cell death and damage in various brain regions and in vitro cell cultures (Krasnova and Cadet, 2009). Earlier studies have shown that pretreatment with sigma receptor antagonists, including AC927 (1-(2-phenethyl)piperidine oxalate), CM156 (3-(4-(4-cyclohexylpiperazin-1-yl)butyl)benzo[d]thiazole-2(3H)-thione) and SN79 (6-acetyl-3-(4-(4-(4-florophenyl)piperazin-1-yl)butyl)benzo[d]oxazol-2(3H)-one), attenuate the neurotoxic effects of methamphetamine in vivo (Kaushal et al., 2011b,d; Matsumoto et al., 2008; Seminerio et al., 2011). However, the mechanisms through which sigma[g80]receptor antagonists convey neuroprotection against methamphetamine remain unclear.
Recently, differentiated NG108-15 cells were confirmed to elicit the release of dopamine, the generation of reactive oxygen/nitrogen species, and cytotoxicity in response to micromolar concentrations of methamphetamine (Kaushal et al., 2012). Differentiated NG108-15 cells also exhibit neurite outgrowth, electrical excitability, and express neuronal and glial proteins (Ma et al., 1998). They have been used extensively for delineating the cellular actions of sigma receptors (Hayashi and Su, 2003). Since the sigma receptor antagonist AC927 can attenuate methamphetamine-induced cell death, dopamine release and reactive oxygen/nitrogen species generation in NG108-15 cells (Kaushal et al., 2012), we continue using this model to gain new insights into mechanisms that contribute to the neuroprotective actions of sigma ligands.
For the current studies, SN79 served as the prototypic sigma receptor antagonist because: i) it has significant and preferential affinity for sigma1 and sigma2 receptors (Ki 27 and 7 nM, respectively) compared to nearly 60 other non-sigma sites (Kaushal et al., 2011c), and ii) its long half life and low propensity for toxicity makes it a potential medication development lead (Kaushal et al., 2011c). The well established sigma receptor agonist, 1,3-di(2-tolyl)guanidine (DTG), was used for comparison in select studies.
Although the sigma receptor antagonist AC927 has been reported to mitigate methamphetamine-induced cell death in NG108-15 cells, agonist-antagonist relationships through sigma receptors have not yet been examined in this model system. Thus, we first tested SN79 and DTG alone and in combination with methamphetamine against apoptotic and necrotic cell damage in differentiated NG108-15 cells. Since elevated body temperature can exacerbate methamphetamine neurotoxicity in vivo, the influence of elevated cell culture temperatures on methamphetamine neurotoxicity and the ability of SN79 to mitigate cytotoxicity under these conditions were also examined to assess the temperature dependence of the effects. Finally, the involvement of PERK-mediated endoplasmic reticulum stress and caspase activation in the neuroprotective effects of SN79 against methamphetamine was assessed. Gene and protein analysis of PERK endoplasmic reticulum stress mediators and caspase-3 activation were probed, as well as the activation of intrinsic (caspase-9) and extrinsic apoptotic pathways (caspase-8) (Galluzzi et al., 2012). Together, the studies were aimed at identifying cellular processes targeted by sigma receptor antagonists to convey neuroprotective actions against methamphetamine.
2. Materials and methods
2.1. Cell growth and differentiation
The neuroblastoma × glioma, hybridoma cell line NG108-15 (Hamprecht et al., 1985) was obtained from ATCC (Rockville, MD). Cells were maintained in 25 cm2 (T25) or 75 cm2 (T75) culture flasks (Corning-Costar, Lowell, MA) in hi-glucose (4.5 g/l) Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin, and hypoxanthine-aminopterin-thymidine (HAT) at 37 C in a humidified incubator in 5% CO2. To differentiate the cells, the culture medium was replaced with differentiation medium (DMEM with 0.5% FBS, penicillin, streptomycin, HAT and 1% dimethylsulfoxide) and the cells allowed to grow for a further 3–4 days.
2.2. Hoechst and propidium iodide staining
NG108-15 cells were grown and differentiated in 96-well culture plates. Cells were exposed for 24 h at regular (37 °C) or elevated (40 °C) cell culture temperatures to one of the following drug treatments: methamphetamine (0–1 mM); SN79 (1–100 nM); DTG (0.1–10,000 nM); SN79 (1–100 nM) + methamphetamine (0–1 mM); DTG (0.1–10,000 nM) + methamphetamine (0–1 mM). Hoechst stain (20 μg/ml final concentration; Invitrogen, Carlsbad, CA) and propidium iodide stain (20 μg/ml final concentration; Invitrogen, Carlsbad, CA) were added to each well and incubated at room temperature for 10 min. After the incubation step, the plate was placed under the microscope with UV light and 2–3 pictures/well were taken using Leica Application Suite: Imaging software (Leica Microsystems Inc., Buffalo Grove, IL). The percentage of apoptotic and necrotic cells were determined using ImageJ software (NIH, Bethesda, MD). Cells with brightly stained and fragmented nuclei were counted as apoptotic; red cells were counted as necrotic.
2.3. Caspase-3, -9 and -8 assays
NG108-15 cells were grown and differentiated in 96-well culture plates for time course and inhibition studies. For the time course studies, cells were exposed to various concentrations of methamphetamine (0–1 mM) for 1.5, 3, 6, 9, 12, 18 and 24 h. For the inhibition studies, the cells were treated with either methamphetamine (0–1 mM), SN79 (1–100 nM), or SN79 (1–100 nM) + methamphetamine (0–1 mM) for the time point at which a peak response was observed for caspase-3, -9 or -8 activation in response to methamphetamine in the earlier time course studies. At appropriate time points for the time course or inhibition studies, reagents from the Caspase-Glo luminescence assay kit for caspase-3/7, -9, -8 (Invitrogen, Carlsbad, CA) were added to the wells and incubated for 2 h. Each plate was read using a luminescence plate reader (Chameleon V Microplate Reader, Hidex, Finland). The “no cell” blank reading was subtracted from each of the treatment readings. The results were expressed as a percentage of the untreated control cells at that time point.
2.4. Reactive oxygen/nitrogen species
Cells were grown and differentiated in 96 well plates (104 cells per well). The culture medium was replaced with phenol red free medium containing 0.5% FBS and 5 μM of the fluorigenic, oxidizable dye CM-H2DCFDA (5-(and-6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate) and returned to the incubator at 37 °C for 45–60 min. Cells were treated with methamphetamine (0.1–3000 μM) and/or SN79 (1–100 nM). Exogenously added 100 μM H2O2 served as a positive control in this assay. All compounds were added in replicates of 3–4 and the plates returned to the incubator. In the assay, SN79 (1–100 nM) was added 15 min prior to the addition of methamphetamine (0.1–3000 μM) or H2O2 (100 μM). After 20 min, plates were read using a fluorescence plate reader (Chameleon V Microplate Reader, Hidex, Finland) with excitation/emission filter wavelengths set at 485/535nm. Data were obtained as relative fluorescence units after background subtraction. Relative fluorescence units for untreated controls for each assay were averaged and the values for each treated sample normalized to this average followed by statistical analyses.
2.5. Quantitative real-time polymerase chain reaction (PCR)
Differentiated NG108-15 cells cultured as described above underwent their respective treatments. For the antagonist experiments, cells were treated with either vehicle (media) or SN79 (100 nM) 15 min prior to adding methamphetamine (1000 μM). The methamphetamine and SN79 concentrations were selected based on dose response characterizations described in earlier sections. A single time point of 6 h was selected for use in the antagonist experiments because methamphetamine alone significantly increased expression levels of PERK-mediated endoplasmic reticulum stress genes at this early time point in initial time course studies. Additionally, at the later 24 h time point, studies performed as part of the earlier sections of this manuscript showed that methamphetamine results in significant cellular toxicity, an effect mitigated by treatment with SN79.
RNA isolations were conducted by removing media and adding TRIzol cell lysis reagent to extract total RNA according to manufacturer's instructions (Invitrogen, Carlsbad, CA). The quality and quantity of RNA was assessed using a spectrophotometer (Biochrom, Cambridge, UK). To synthesize cDNA, 1 μg of RNA was used in conjunction with high capacity Superscript-H cDNA reverse transcription kits (Applied Biosystems, Foster City, CA). Respective cDNA was then used as a template for TaqMan quantitative real-time PCR with oligonucleotide primer sets specific for 18S rRNA (Hs99999901_s1) as an endogenous control and activating transcription factor 3 (ATF3, Mm00476032_m1), C/EBP homologous protein (CHOP, Mm00492097_m1) and protein phosphatase 1 regulatory subunit 15A (GADD34, Mm01205601_g1) (Applied Biosystems, Foster City, CA) as PERK-mediated endoplasmic reticulum stress gene targets. All samples were run using TaqMan real-time PCR universal master mixture (Applied Biosystems) for a total of 45 cycles using a StepOnePlus real-time PCR system (Applied Biosystems). The difference in cycles to threshold for each sample as compared to the respective 18S endogenous control for each sample was recorded. Threshold was set at 0.2 for all gene probes tested. The change in the difference in cycles to threshold method (ΔΔCT method) was used to calculate relative quantities of each gene in each respective sample obtained from NG108-15 cells.
2.6. Immunoblots
To evaluate potential regulation of CHOP by SN79 at the translational level, differentiated NG108-15 cells were cultured and treated as described above with either vehicle, methamphetamine (1000 μM), SN79 (100 nM), or SN79 + methamphetamine for a period of 24 h. Following the treatment period, the cells underwent protein isolations using TRIzol cell lysis reagent according to manufacturer's instructions (Invitrogen). The protein concentration of each sample was measured using a Coomassie Plus Bradford Assay Kit (Pierce, Rockford, IL) which is based upon a modified form of the Bradford assay (Bradford, 1976). Samples were run using 30 μg of protein/well using Tris-hydrochloride 12% pre-cast 15-well gels (Bio-Rad, Hercules, CA) in combination with 5X Lammeli sample buffer. Gels were run using a mini-PROTEAN system (Bio-Rad) and gels were equilibrated for 15 min in Towbin's buffer prior to transfer to polyvinylidene fluoride membranes (Bio-Rad) using semi-dry electrophoretic transfer cells (Bio-Rad). Membranes were blocked using 5% fat-free milk/tris buffered saline-T for 2 h at room temperature. Membranes were incubated with primary CHOP antibody (Cell Signaling, Danvers, MA) at a concentration of 1:1000 overnight at 4 °C. Anti-rabbit IgG horseradish peroxidase-linked antibody (Cell Signaling) was used at a concentration of 1:2000 with gentle shaking for 2 h. Imaging was conducted using LumiGLO chemiluminescent substrate (Cell Signaling) according to manufacturer's instructions. Horseradish peroxidase-conjugated β-actin rabbit mAB (Cell Signaling) was used as an endogenous control for all respective samples at a concentration of 1:10,000 and incubated for a period of 1 h. Molecular weight determination was conducted using a biotinylated protein ladder (Cell Signaling). Images were converted to 8-bit and analyzed using densitometry with background subtraction and normalized to β-actin using ImageJ software (NIH; http://rsbweb.nih.gov/ij/).
2.7. Statistical analysis
Statistical analyses of the data were performed using one or two way ANOVA with Dunnett's, Tukey's or Bonferroni's post-hoc tests employing GraphPad Prism (GraphPad Software, San Diego, CA).
3. Results
3.1. SN79 attenuates methamphetamine-induced apoptosis and necrosis
Exposure to methamphetamine significantly increased the percentage of apoptotic cells (F(7,192) = 22.14, P < 0.0001), with post-hoc Dunnett's tests revealing significant differences from control at the following concentrations: 10, 30, 100, 300 and 1000 μM (q = 3.79–9.77, P < 0.01–0.001). SN79 pretreatment significantly attenuated the apoptotic effects of methamphetamine (Fig. 1A). Two-way ANOVA revealed a significant effect of methamphetamine treatment (F(7,538) = 14.63, P < 0.0001), SN79 pretreatment (F(3,538) = 100.90, P < 0.0001), and SN79 pretreatment × methamphetamine treatment (F(21,538) = 6.47, P < 0.0001). Bonferroni's post-hoc tests showed that SN79 (1, 10 and/or 100 nM) pretreatment significantly attenuated the apoptotic effects of the following concentrations of methamphetamine: 3, 10, 30, 100, 300 and 1000 μM (t = 2.80–11.00, P < 0.05–0.001). On its own, SN79 did not affect apoptotic cell death in NG108-15 cells when compared to untreated controls (t = 0.01–1.29, not significant).
Fig. 1.

SN79 protects against methamphetamine (METH)-induced apoptosis (A) and necrosis (B). Differentiated NG108-15 cells were pretreated with SN79 (0–100 nM) prior to exposure to methamphetamine (0–1 mM) for 24 h. After 24 h, the wells were incubated with Hoechst 33342 and propidium iodide stains (20 μg/ml each) to obtain percentages of cells that were apoptotic (A) and necrotic (B). Data represent means from three separate experiments (n = 3/experiment) ± S.E.M. **P < 0.01 (control versus methamphetamine treated). #P < 0.05; ##P < 0.01; ###P < 0.001 (methamphetamine alone vs. methamphetamine with SN79).
Exposure to methamphetamine significantly increased the percentage of necrotic cells (F(7,192) = 8.28, P < 0.0001), with post-hoc Dunnett's tests confirming that 300 and 1000 μM methamphetamine differed significant from controls (q = 4.45–6.31, P < 0.01). SN79 pretreatment significantly attenuated the necrotic effects of methamphetamine (Fig. 1B). Twoway ANOVA showed a significant effect of SN79 pretreatment (F(3,538) = 14.71, P < 0.0001), methamphetamine treatment (F(7,538) = 6.98, P < 0.0001) and SN79 pretreatment × methamphetamine treatment interaction (F(21,538) = 1.82, P < 0.05). Post-hoc Bonferroni's tests confirmed that pretreatment with SN79 (1, 10 and 100 nM) attenuated the necrotic effects of 300 μM methamphetamine (t = 2.98–3.57, P < 0.05–0.01) and 1000 μM methamphetamine (t = 2.85–5.89, P < 0.05–0.001). On its own, SN79 did not elicit necrotic cell death in NG108-15 cells when compared with no treatment controls (t = 0.10–0.79, not significant).
3.2. DTG potentiates methamphetamine-induced apoptosis and necrosis
Two way ANOVA revealed a significant effect of methamphetamine treatment (F(6,889) = 37.83, P < 0.0001) and DTG pretreatment (F(6,889) = 7.49, P < 0.0001), but the methamphetamine treatment × DTG pretreatment interaction was not statistically significant (F(36,889) = 0.68, not significant). Fig. 2A shows that DTG pretreatment displayed a trend towards a shift in the dose response curve of methamphetamine-induced apoptosis towards the left. Bonferroni's post-hoc tests confirmed that DTG pretreatment at 10 nM and 1 μM with the methamphetamine 1 μM concentration significantly differed from the methamphetamine 1 μM treatment alone (t = 2.88–2.92, P < 0.05).
Fig. 2.

Effect of DTG pretreatment on methamphetamine (METH)-induced apoptosis (A) and necrosis (B). NG108-15 cells were exposed to DTG (0.1 nM-10 μM) and/or methamphetamine (0–1000 μM) for 24 h. After 24 h, the wells were incubated with Hoechst 33342 and propidium iodide stains (20 μg/ml each) to obtain percentages of cells that were apoptotic (A) and necrotic (B). Data represent means from two separate experiments (n = 3/experiment) ± S.E.M. *P < 0.05; **P < 0.01 (control vs. methamphetamine). ##P < 0.01; ###P < 0.001 (DTG+ methamphetamine vs. methamphetamine).
Fig. 2B shows that DTG pretreatment at intermediate concentrations shifted the dose response curve of methamphetamine towards the left, and at even higher concentrations, showed an upward and leftward shift in the dose response curve. Two way ANOVA confirmed a significant effect of methamphetamine treatment (F(6,948) = 55.75, P < 0.0001), DTG pretreatment (F(6,948) = 77.15, P < 0.0001), and methamphetamine treatment × DTG pretreatment interaction (F(36,948) = 1.80, P < 0.005). Bonferroni's post-hoc tests revealed that DTG (10, 100, 1000 and/or 10,000 nM) in combination with the following concentrations of methamphetamine significantly differed from methamphetamine treatment alone at those concentrations: 0.01 μM (t = 2.75–4.49, P < 0.05–0.001), 0.1 μM (t = 5.18, P < 0.001), 1 μM (t = 5.44–7.39, P < 0.001), 10 μM (t = 3.07–8.31, P < 0.05–0.001), 100 μM (t = 4.59–10.08, P < 0.001), and 1000 μM (t = 4.02–5.21, P < 0.001). In addition, the following concentrations of DTG alone differed significantly from no treatment controls: 1 and 10 μM (t = 2.85–6.87, P < 0.05–0.001).
3.3. Elevated temperature (40 °C) increases methamphetamine-induced apoptosis and necrosis
Methamphetamine caused concentration-dependent increases in apoptosis in NG108-15 cells at both 37 and 40 °C. At 37 °C, the methamphetamine effect was statistically significant (F(9,436) = 33.37, P < 0.0001), with Dunnett's post-hoc tests confirming significant differences from no treatment controls at the following concentrations of methamphetamine: 10, 30, 100, 300 and 1000 μM (q = 4.77–13.30, P < 0.01). At 40 °C, there was also a significant increase in methamphetamine-induced apoptosis (F(9,217) = 5.80, P < 0.0001), with Dunnett's post-hoc tests confirming significant differences from no treatment controls at the following concentrations of methamphetamine: 10, 30, 100, 300 and 1000 μM (q = 3.42–5.16, P < 0.01).
Upon comparing the methamphetamine-treated NG108-15 cells at 37 and 40 °C, cells maintained at 40 °C had a higher percentage of apoptotic cells in the no treatment control as well as methamphetamine treatment, compared to cells maintained at 37 °C (Fig. 3A). The dose response of methamphetamine at 40 °C showed a parallel and upward shift in apoptosis. Two way ANOVA confirmed a significant effect of methamphetamine treatment (F(9,635) = 21.52, P < 0.0001) and temperature (F(1,635) = 31.49, P < 0.0001). Bonferroni's post-hoc tests revealed a significant difference in apoptosis at 37 vs. 40 °C at the following concentrations of methamphetamine: 1, 10 and 100 μM (t = 2.90–4.47, P < 0.05–0.001).
Fig. 3.

Effect of temperature (37 vs. 40 °C) on methamphetamine (METH)-induced apoptosis (A) and necrosis (B). NG108-15 cells were exposed to methamphetamine (0–1000 μM) for 24 h at 37 or 40 °C. After 24 h, the wells were incubated with Hoechst 33342 and propidium iodide stains (20 μg/ml each) to obtain percentages of cells that were apoptotic (A) and necrotic (B). Data represent means from two separate experiments (n = 3/experiment) ± S.E.M. *P < 0.05; **P < 0.01 (control vs. methamphetamine treated at a particular temperature). #P < 0.05; ###P < 0.001 (treatment at 37 °C vs. 40 °C).
Methamphetamine significantly increased necrosis in NG108-15 cells maintained at 37 °C (F(9,428) = 32.93, P < 0.0001), with Dunnett's post-hoc tests confirming significant differences from no treatment control cells at the following concentrations of methamphetamine: 300 and 1000 μM (q = 6.75–14.02, P < 0.01). At an elevated temperature of 40 °C, methamphetamine significantly increased necrosis (F(9,205) = 9.63, P < 0.0001), when compared to no treatment control cells at the same temperature at the following concentrations: 1, 3, 10, 30, 100, 300 and 1000 μM (q = 2.75–7.33, P < 0.05–0.01).
Upon comparing the methamphetamine-treated NG108-15 cells at 37 and 40 °C, cells maintained at 40 °C exhibited a significantly higher percentage of necrosis in the no treatment control as well as methamphetamine treatment, when compared to cells maintained at 37 °C (Fig. 3B). The dose response of methamphetamine at 40 °C showed an upward and leftward shift in necrotic cell death. Two way ANOVA confirmed a significant effect of methamphetamine treatment (F(9,615) = 19.76, P < 0.0001), temperature (F(1,615) = 600, P < 0.0001), and methamphetamine treatment × temperature interaction (F(9,615) = 8.81, P < 0.0001). Bonferroni's post-hoc tests revealed that the following concentrations of methamphetamine caused a significant increase in necrosis when maintained at 40 versus 37 °C: 0, 0.01, 0.1, 1, 3, 10, 30, 100, 300 and 1000 μM (t = 3.24–17.32, P < 0.05–0.001).
3.4. SN79 attenuates apoptosis and necrosis in the absence and presence of methamphetamine at elevated temperature (40 °C)
Exposure to SN79 alone significantly reduced the elevated apoptotic cell death caused by increased temperature (40 °C). One way ANOVA showed a significant effect of SN79 treatment (F(3,120) = 8.21, P < 0.0001), with Dunnett's post-hoc tests confirming that apoptosis at elevated temperature in control untreated cells was significantly attenuated with the following concentrations of SN79: 1, 10 and 100 nM (q = 3.16–4.32, P < 0.01).
SN79 also attenuated the combined apoptotic effect of elevated temperature and all the concentrations of methamphetamine (Fig. 4A). One way ANOVA showed a significant effect of SN79 pretreatment (1, 10 and 100 nM) prior to the following concentrations of methamphetamine treatment: 0.01 μM (F(3,40) = 12.06, P < 0.0001), 0.1 μM (F(3,39) = 3.18, P < 0.05), 1 μM (F(3,114) = 20.05, P < 0.0001), 3 μM (F(3,65) = 7.63, P < 0.0005), 10 μM (F(3,108) = 30.55, P < 0.0001), 30 μM (F(3,74) = 23.44, P < 0.0001), 100 μM (F(3,115) = 31.41, P < 0.0001), 300 μM (F(3,73) = 15.99, P < 0.0001) and 1000 μM (F(3,110) = 13.29, P < 0.0001). Dunnett's post-hoc tests confirmed that SN79 pretreatment (1, 10 and/or 100 nM) attenuated the apoptotic effects of the following concentrations of methamphetamine: 0.01, 0.1, 1, 3, 10, 30, 100, 300 and 1000 μM (1 nM: q = 3.90–8.27, P < 0.01; 10 nM: q = 4.20–8.29, P < 0.01; 100 nM: q = 2.96–7.35, P < 0.05–0.01).
Fig. 4.

SN79 protects against methamphetamine (METH) and elevated temperature (40 °C)-induced apoptosis and necrosis. NG108-15 cells were pretreated with SN79 (1–100 nM) prior to exposure to methamphetamine (0–1000 μM) for 24 h at 40 °C. After 24 h, the wells were incubated with Hoechst 33342 and propidium iodide stains (20 μg/ml each) to obtain percentages of apoptotic and necrotic cells. (A) apoptotic cells; (B) necrotic cells; Data represent means from two separate experiments (n = 3/experiment) ± S.E.M. *P < 0.05; **P < 0.01 (control vs. methamphetamine treated). ##P < 0.01 (control/methamphetamine alone vs. control/METH with SN79).
In terms of necrosis, exposure to SN79 alone significantly reduced the elevated necrotic cell death caused by increased temperature (40 °C) in control untreated cells. One way ANOVA showed a significant effect of SN79 treatment (F(3,120) = 12.57, P < 0.0001), with Dunnett's post-hoc tests confirming significant attenuation of necrosis produced by elevated temperature at the following concentrations of SN79: 1, 10 and 10 nM (q = 4.82–5.04, P < 0.01).
SN79 also attenuated the combined necrotic effect of elevated temperature and all the concentrations of methamphetamine (Fig. 4B). One way ANOVA showed a significant effect of SN79 pretreatment (1, 10 and 100 nM) prior to the following concentrations of methamphetamine treatment: 0.01 μM (F(3,40) = 11.98, P < 0.0001), 0.1 μM (F(3,70) = 36.99, P < 0.0001), 1 μM (F(3,114) = 18.84, P < 0.0001), 3 μM (F(3,65) = 29.95, P < 0.0001), 10 μM (F(3,108) = 10.09, P < 0.0001), 30 μM (F(3,74) = 15.68, P < 0.0001), 100 μM (F(3,115) = 24.65, P < 0.0001), 300 μM (F(3,73) = 17.50, P < 0.0001) and 1000 μM (F(3,110) = 24.19, P < 0.0001). Dunnett's post-hoc tests confirmed that SN79 pretreatment (1, 10 and/or 100 nM) significantly attenuated the necrotic effects of the following concentrations of methamphetamine: 0.01, 0.1, 1, 3, 10, 30, 100, 300 and 1000 μM (1 nM: q = 4.30–9.43, P < 0.01; 10 nM: q = 4.90–9.61, P < 0.01; 100 nM: q = 3.82–9.15, P < 0.01).
3.5. SN79 attenuates methamphetamine-induced caspase-3 activation
Methamphetamine treatment significantly increased caspase-3 activation in differentiated NG108-15 cells (Fig. 5A). Two way ANOVA showed a significant effect of methamphetamine treatment (F(3,104) = 16.78, P < 0.0001), time (F(4,104) = 47.59, P < 0.0001), and methamphetamine treatment × time interaction (F(12,104) = 16.17, P < 0.0001). Bonferroni's post-hoc tests confirmed that the 10, 100 and 1000 μM concentrations of methamphetamine caused a significant increase in caspase-3 activation at the 24 h time point (t = 3.34–19.27, P < 0.05–0.0001).
Fig. 5.

Effects on caspase-3. (A) Dose response and time course of methamphetamine (METH) treatment on caspase-3 activation. Differentiated NG108-15 cells were treated with methamphetamine (0–1000 μM) and caspase-3 activation determined using Caspase-3/7 Glo Luminescence assays at 1.5, 3, 6, 12, and 24 h time points. At the 24 h time point, methamphetamine (10, 100 and 1000 μM) caused significant increases in caspase-3 activation. (B) Effect of SN79 pretreatment on methamphetamine-induced caspase-3 activation at 24 h. Differentiated NG108-15 cells were pretreated with SN79 (0–100 nM), 15 min prior to adding methamphetamine (1, 10, 100, 1000 μM). At the 24 h time point, SN79 attenuated the caspase-3 activation caused by methamphetamine at 100 and 1000 μM without significantly altering caspase-3 activation on its own. *P < 0.05; ****P < 0.0001, vs. no treatment control. #P < 0.05; ##P < 0.01; ####P < 0.0001, vs. methamphetamine.
SN79 pretreatment significantly attenuated methamphetamine-induced caspase-3 activation at the 24 h time point (Fig. 5B). Two way ANOVA showed a significant effect of methamphetamine treatment (F(4,128) = 109.50, P < 0.0001), SN79 pretreatment (F(3,128) = 14.82, P < 0.0001), and methamphetamine × SN79 pretreatment interaction (F(12,128) = 4.73, P < 0.0001). Bonferroni's post-hoc tests showed that SN79 (1, 10 and/or 100 nM) significantly attenuated caspase-3 activation caused by 100 μM methamphetamine (t = 3.20–4.02, P < 0.05–0.01) and 1000 μM methamphetamine (t = 5.32–7.52, P < 0.0001).
3.6. SN79 attenuates methamphetamine-induced caspase-8 activation
Methamphetamine treatment significantly increased caspase-8 activation in differentiated NG108-15 cells (Fig. 6A). Two way ANOVA showed a significant effect of methamphetamine treatment (F(4,319) = 41.49, P < 0.0001), time (F(5,319) = 10.95, P < 0.0001), and methamphetamine treatment × time interaction (F(20,319) = 3.13, P < 0.0001). Bonferroni's post-hoc tests confirmed that the 1, 10, 100 and 1000 μM concentrations of methamphetamine caused a significant increase in caspase-8 activation at the 9 h time point (t = 3.56–16.39, P < 0.05–0.0001). Methamphetamine (1000 μM) also caused significant caspase-8 activation at the 1.5, 3, 6, 12, and 24 h time points (t = 3.37–4.93, P < 0.05–0.0001).
Fig. 6.

Effects on caspase-8. (A) Dose response and time course of methamphetamine (METH) treatment on caspase-8 activation. Differentiated NG108-15 cells were treated with methamphetamine (0–1000 μM) and caspase-8 activation determined using Caspase-8 Glo Luminescence assays at 1.5, 3, 6, 9, 12, and 24 h time points. At the 9 h time point, methamphetamine (1, 10, 100 and 1000 μM) caused significant increases in caspase-8 activation. (B) Effect of SN79 pretreatment on methamphetamine-induced caspase-8 activation at 9 h. Differentiated NG108-15 cells were pretreated with SN79 (0–100 nM), 15 min prior to adding methamphetamine (1, 10, 100, 1000 μM). At the 9 h time point, SN79 attenuated caspase-8 activation caused by methamphetamine at 10, 100 and 1000 μM without significantly altering caspase-8 activation on its own. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, vs. no treatment control. #P < 0.05; ##P < 0.01; ###P < 0.001, vs. methamphetamine.
SN79 pretreatment significantly attenuated methamphetamine-induced caspase-8 activation at the 9 h time point (Fig. 6B). Two way ANOVA showed a significant effect of methamphetamine treatment (F(4,278) = 88.61, P < 0.0001), SN79 pretreatment (F(3,278) = 19.50, P < 0.0001), and methamphetamine × SN79 pretreatment interaction (F(12,278) = 3.60, P < 0.0001). Bonferroni's post-hoc tests showed that SN79 (1, 10 and/or 100 nM) attenuated caspase-8 activation caused by treatment with 10 μM methamphetamine (t = 3.26, P < 0.05), 100 μM methamphetamine (t = 3.36–4.22, P < 0.05–0.001) and 1000 μM methamphetamine (t = 3.05–3.92, P < 0.05–0.01).
3.7. SN79 attenuates methamphetamine-induced caspase-9 activation
Methamphetamine treatment significantly increased caspase-9 activation in differentiated NG108-15 cells (Fig. 7A). Two way ANOVA showed a significant effect of methamphetamine treatment (F(4,511) = 52.31, P < 0.0001), time (F(6,511) = 19.18, P < 0.0001), and methamphetamine treatment × time interaction (F(24,511) = 3.13, P < 0.0001). Bonferroni's post-hoc tests confirmed that the 1000 μM concentration of methamphetamine caused a significant increase in caspase-9 activation at the 3, 6, 9, 12, 18 and 24 h (t = 2.74–20.93, P < 0.05–0.001) time points. Methamphetamine (100 μM) also caused caspase-9 activation at the 9 and 24 h (t = 4.12–6.24, P < 0.001) time points.
Fig. 7.

Effects on caspase-9. (A) Dose response and time course of methamphetamine (METH) treatment on caspase-9 activation. Differentiated NG108-15 cells were treated with methamphetamine (0–1000 μM) and caspase-9 activation determined using Caspase-9 Glo Luminescence assays at 1.5, 3, 6, 9, 12, 18, and 24 h time points. At the 9 h time point, methamphetamine (1, 10, 100 and 1000 μM) caused significant increases in caspase-9 activation. (B) Effect of SN79 pretreatment on methamphetamine-induced caspase-9 activation at 24 h. Differentiated NG108-15 cells were pretreated with SN79 (0–100 nM), 15 min prior to adding methamphetamine (1, 10, 100, 1000 μM). At the 24 h time point, SN79 attenuated caspase-9 activation caused by methamphetamine at 10, 100 and 1000 μM without significantly altering caspase-9 activation on its own. *P < 0.05; **P < 0.01; ***P < 0.001, vs. no treatment control. #P < 0.05; ###P < 0.001, vs. methamphetamine.
SN79 pretreatment significantly attenuated methamphetamine-induced caspase-9 activation at the 24 h time point (Fig. 7B). Two way ANOVA showed a significant effect of methamphetamine treatment (F(4,269) = 82.60, P < 0.0001), SN79 pretreatment (F(3,269) = 13.00, P < 0.0001), and methamphetamine × SN79 pretreatment interaction (F(12,269) = 3.89, P < 0.0001). Bonferroni's post-hoc tests showed that the 10 nM SN79 concentration significantly attenuated caspase-9 activation caused by 1000 μM methamphetamine (t = 5.24, P < 0.001), and that the 100 nM SN79 concentration significantly attenuated caspase-9 activation caused by the 100 and 1000 μM concentrations of methamphetamine (t = 2.91–7.69, P < 0.05–0.001).
3.8. SN79 attenuates methamphetamine-induced generation of reactive oxygen/nitrogen species
SN79 pretreatment significantly attenuated reactive oxygen/nitrogen species generated by methamphetamine (Fig. 8). Two-way ANOVA showed a significant effect of SN79 pretreatment (F(4,824) = 35.27, P < 0.0001), methamphetamine or H2O2 treatment (F(9,824) = 75.77, P < 0.0001), and SN79 pretreatment × methamphetamine or H2O2 treatment interaction (F(36,824) = 2.24, P < 0.001). Dunnett's post-hoc tests confirmed that the following concentrations of methamphetamine generated reactive oxygen/nitrogen species: 1, 3, 10, 30, 100, 300, 1000 and 3000 μM (q = 4.46–7.15, P < 0.01); in addition, 100 μM H2O2 generated reactive oxygen/nitrogen species (q = 14.86, P < 0.01). Bonferroni's post-hoc tests confirmed that SN79 (1, 10, 100 and/or 1000 nM) significantly inhibited the reactive oxygen/nitrogen species generated by the following concentrations of methamphetamine: 1, 3, 30, 100, 300, 1000 and 3000 μM (t = 2.88–5.29, P < 0.05–0.001). SN79 (1000 nM) also significantly attenuated the reactive oxygen/nitrogen species generated by H2O2 (t = 4.26, P < 0.001).
Fig. 8.
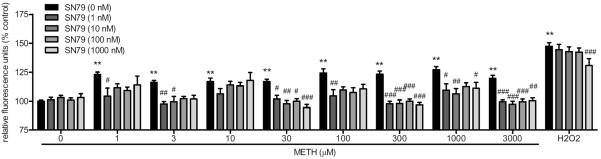
Effect of SN79 pretreatment on methamphetamine (METH)-induced reactive oxygen/nitrogen species generation. The production of reactive oxygen/nitrogen species capable of oxidizing CM-H2DCFDA was assessed in differentiated NG108-15 cells in the presence of 0 to 300 μM methamphetamine or 100 μM H2O2 in the absence (solid bars) or presence (cross hatched bars) of SN79 (1–1000 nM) as described in the methods. Data represent averages of relative fluorescence units (rfu) from 3–6 different experiments (n = 3–4/experiment) obtained 20 min after addition of drug ± S.E.M. **, P < 0.01 (control vs. methamphetamine or H2O2 treated). #P < 0.05; ##P < 0.01; ###P < 0.001 (methamphetamine alone vs. methamphetamine with SN79).
3.9. Methamphetamine increases PERK-mediated endoplasmic reticulum stress genes
Methamphetamine treatment dose-dependently increased the expression of PERK-mediated endoplasmic reticulum stress genes at all three time points tested (6, 12, 24 h) (Fig. 9A). Two-way ANOVA confirmed that methamphetamine treatment resulted in a dose dependent difference in atf3 mRNA expression (P < 0.0001). Bonferroni's post-hoc analysis revealed that the 300 and 1000 M concentrations of methamphetamine increased atf3 expression at all three time points (t = 3.18–7.58, P < 0.001).
Fig. 9.
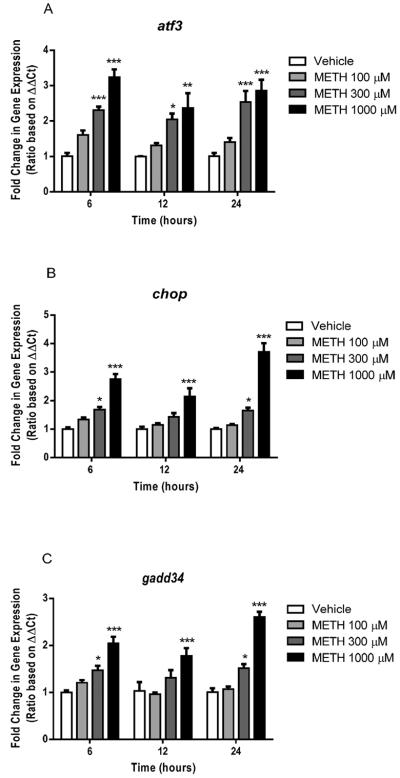
Effects of methamphetamine (METH) on genes involved in PERK-mediated endoplasmic reticulum stress in NG108-15 cells. (A) METH increased atf3 mRNA expression after 6, 12 and 24 h of treatment (*P < 0.05, **P < 0.01, ***P < 0.001; vs. vehicle control). (B) METH significantly upregulated chop at all three time points (*P < 0.05, ***P < 0.001; vs. vehicle control). (C) gadd34 expression was upregulated by METH after 6, 12 and 24 h of treatment (*P < 0.05, ***P < 0.001; vs. vehicle control).
Methamphetamine also increased the expression of chop, a downstream mediator of the PERK endplasmic reticulum stress pathway (Fig. 9B). Two-way ANOVA revealed a significant dose and time effect of methamphetamine treatment on chop expression, as well as their interaction (P < 0.01–0.0001). Bonferroni's post-hoc analysis revealed that methamphetamine treatment at concentrations of 300 and 1000 M increased chop mRNA expression 6 and 24 h post-treatment as compared to vehicle controls (t = 3.06–12.69, P < 0.05–0.001). Twelve hours post-treatment with methamphetamine (1000 M) significantly increased chop mRNA expression (t = 4.80, P < 0.001).
Additionally, two-way ANOVA revealed a significant dose, time and interaction effect of methamphetamine treatment on gadd34 expression (P < 0.05–0.0001) (Fig. 9C). Similar to methamphetamine-induced increases in chop expression, methamphetamine increased gadd34 mRNA expression 6 and 24 h post-treatment, at concentrations of 300 and 1000 M (Bonferroni's post-hoc tests, t = 2.94–10.00, P < 0.05–0.001). The 1000 M concentration of methamphetamine also increased gadd34 mRNA expression 12 h post-treatment (t = 4.68, P < 0.001).
3.10. SN79 fails to mitigate methamphetamine-induced increases in PERK-mediated endoplasmic reticulum stress genes
SN79 treatment had little effect on methamphetamine-induced PERK-mediated endoplasmic reticulum stress. One-way ANOVA revealed significant differences between treatment groups in atf3 expression (F(3,23) = 8.98, P < 0.001). Tukey's post-hoc test confirmed that SN79 pretreatment had no effect on methamphetamine-induced increases in atf3 expression (q = 1.56, n.s.), although cells treated with SN79 in combination with methamphetamine did significantly differ from vehicle treated controls (q = 5.96, P < 0.01). Additionally, SN79 displayed no effects on its own (q = 0.21, n.s.) (Fig. 10A).
Fig. 10.

Effects of SN79 on methamphetamine (METH)-induced upregulation of PERK-mediated endoplasmic reticulum stress genes after 6 h of treatment. (A) SN79 failed to alter methamphetamine-induced increases in atf3 mRNA expression (*P < 0.05, **P < 0.01; vs. vehicle control). (B) METH-induced increases in chop expression were not altered by SN79 treatment (*P < 0.05, **P < 0.01; vs. vehicle control). (C) METH-induced increases in gadd34 expression were unaltered by SN79 treatment (**P < 0.01, ***P < 0.001; vs. vehicle control).
SN79 treatment also had no effect on methamphetamine-induced chop mRNA expression (Fig. 10B). One-way ANOVA revealed significant differences between treatment groups (F(3,23) = 14.22, P < 0.0001). Methamphetamine increased chop mRNA expression, as compared to vehicle-treated controls (Tukey's test, q = 4.95, P < 0.05). SN79 treatment had no effect on methamphetamine-induced increases in chop mRNA expression (q = 3.40, n.s.). Treatment with SN79 alone had no significant effects on chop mRNA expression as compared to vehicle-treated controls (q = 1.30, n.s.).
One-way ANOVA revealed significant differences between treatment groups in gadd34 mRNA expression (F(3,23) = 13.89, P < 0.0001). Tukey's post-hoc analysis found that methamphetamine treatment increased gadd34 expression as compared to vehicle-treated controls (q = 5.19, P < 0.01). SN79 in combination with methamphetamine increased expression of gadd34 mRNA, as compared to vehicle-treated controls (q = 8.10, P < 0.001); however, SN79 treatment had no effect on methamphetamine-induced increases in gadd34 expression (q = 2.90, n.s.) (Fig. 10C).
3.11. SN79 fails to mitigate methamphetamine-induced increases in CHOP protein expression
One-way ANOVA found significant differences between treatment groups in CHOP protein expression (F(3,23) = 5.98, P < 0.005). As shown in Fig. 11A, methamphetamine dose dependently increased CHOP expression as compared to vehicle controls at 24 h (Dunnett's test, q = 3.94, P < 0.001). SN79, however, was unable to mitigate methamphetamine-induced increases in CHOP expression at 24 h (One-way ANOVA (F(3,23) = 47.34, P < 0.0001), Tukey's test, q = 2.15, n.s.) (Fig. 11B).
Fig. 11.

Effects of methamphetamine (METH) on CHOP protein expression. (A) Methamphetamine dose dependently increased CHOP protein expression after 24 h of treatment (**P < 0.01; vs. vehicle control). (B) The sigma receptor antagonist SN79 did not alter METH-induced increases in CHOP expression. After 24 h of treatment, METH(1000 μM) significantly upregulated CHOP (***P < 0.001; vs. vehicle control), but SN79 was unable to significantly alter the METH-induced increases in CHOP expression (not significant, SN79 + METH vs. METH).
4. Discussion
This study showed that methamphetamine caused apoptosis in differentiated NG108-15 cells at physiologically relevant micromolar concentrations and necrosis at millimolar concentrations. Methamphetamine also elicited reactive oxygen/nitrogen species generation, PERK-mediated endoplasmic reticulum stress, and caspase-3, -8 and -9 activation at earlier time points, effects hypothesized to contribute to methamphetamine-induced cellular toxicity. Notably, SN79 attenuated methamphetamine-induced cell death, reactive oxygen/nitrogen species generation and caspase activation at normal and elevated cell culture temperatures.
The attenuation of methamphetamine-induced apoptosis and necrosis by SN79 is consistent with the role of sigma receptors in apoptosis in tumor models (Bowen, 2000; Kaushal et al., 2012; Marrazzo et al., 2011), and the ability to AC927, another putative antagonist, to attenuate necrosis in these cells (Kaushal et al., 2012). Although there are only a few reports linking sigma receptors to necrosis, it is not surprising that SN79 could protect against both necrotic and apoptotic cell death. Earlier studies suggest considerable overlap between the two forms of cell death as evidenced by coining of terms like `necroptosis' and `aponecrosis' (Berghe et al., 2010; Formigli et al., 2000; Kroemer et al., 2009; Jiang et al., 2011) and suggest that the terminal degeneration caused by methamphetamine in vivo involve similar processes. Additionally, the apoptotic cells may appear necrotic at later time points since phagocyte cells are absent in the in vitro system.
Confirmation that methamphetamine-induced cell death is sigma receptor sensitive stems from the ability of DTG to shift the dose response curve of methamphetamine-induced cell death towards the left. At higher concentrations, DTG alone displays toxic effects, consistent with the ability of sigma receptor agonists (especially the sigma2 subtype) to cause cytotoxicity (Bowen, 2000; Crawford and Bowen, 2002; Vilner et al., 1995). In this study, DTG had a more pronounced effect on methamphetamine-induced necrotic (vs. apoptotic) cell death. This may result because NG108-15 is a sensitive cell line and induction of stressors/pro-death mediators beyond a certain threshold can shift cell death from apoptosis to necrosis (Nicotera and Melino, 2004; Soti et al., 2003).
The second part of the study evaluated the influence of temperature on methamphetamine neurotoxicity and its interaction with the neuroprotective effects of SN79. In animals, sigma receptor antagonists, including SN79, can attenuate both the hyperthermia and neurotoxicity of methamphetamine (Kaushal et al., 2011b; Matsumoto et al., 2008; Seminerio et al., 2011). In fact, there is a strong correlation between the body temperatures of the animals and the level of neurotoxicity subsequently observed across the treatment groups (Kaushal et al., 2011b; Robson et al., 2013; Seminerio et al., 2011).
To assess the contribution of methamphetamine, hyperthermia and their interaction, NG108-15 cells were treated with methamphetamine at 37 or 40 °C, corresponding to normal or elevated cell culture temperatures, respectively. The results demonstrated that by maintaining the cells at an elevated temperature, an increase in cell death (predominantly necrosis) was observed. The ability of SN79 to attenuate both the elevated temperature-induced toxicity and the enhanced toxicity caused by the interaction of methamphetamine plus elevated temperature is consistent with the potential role of sigma receptors in various cell stress and death mechanisms (Kaushal and Matsumoto, 2011a). These in vitro results clearly demonstrate that sigma receptor-mediated neuroprotection can occur independently of changes in temperature.
In the current study, caspase-3 activation was observed 24 h following methamphetamine exposure, suggesting that caspase-dependent mechanisms are involved in this cell death. SN79 attenuated methamphetamine-induced caspase-3 activation, consistent with the role of sigma receptors in caspase-dependent cell death mechanisms (Crawford and Bowen, 2002; Hornick et al., 2010; Jonhede et al., 2010). Since caspase-3 can be activated via extrinsic death receptors (caspase-8 activation) or intrinsic mitochondrial death pathways (caspase-9 activation), other caspases associated with each of these pathways were also examined.
The activation of caspase-8 by methamphetamine at the 9 h time point suggests the involvement of an extrinsic cell death pathway. Methamphetamine-induced Fas/FasL pathway and caspase-8 activation has also been previously reported in animal models (Jayanthi et al., 2005). One of the most likely mechanisms of methamphetamine-induced caspase-8 activation in NG108-15 cells is reactive oxygen/nitrogen species production which can increase cellular death receptor expression (Bauer et al., 1998), and also reactive oxygen species-induced death receptor independent caspase-8 activation via the p38 MAPK pathway (El Mchichi et al., 2007). Once activated, caspase-8 has been reported to cleave Bid which causes cytochrome c release from the mitochondria, and subsequent caspase-9 activation (Li et al., 1998). In this study, caspase-8 activation was followed by caspase-9 activation at 9 h which peaked at the 24 h time point. This indicates that NG108-15 cells may be type II cells that involve the mitochondrial death loop for the processing of apoptotic cell death.
The ability of SN79 to attenuate caspase-8 activation is most likely due to the role of sigma receptors in the modulation of cellular reactive oxygen/nitrogen species levels (Kaushal et al., 2012; Ostenfeld et al., 2005; Pal et al., 2012). In addition, the localization of sigma receptors in lipid rafts (Gebreselassie and Bowen, 2004; Hayashi and Su, 2003) may allow them to modulate death receptor signaling (Cahuzac et al., 2006; Gajate and Mollinedo, 2011). Sigma2 receptor agonists have also been reported to elicit caspase-dependent Bid cleavage and the release of apoptosis-promoting factors from the mitochondria (Wang and Bowen, 2006), providing another mechanism through which SN79 may mitigate the cytotoxic effects of methamphetamine. The decrease in methamphetamine-induced caspase-9 activation at the 24 h time point may be due to the ability of SN79 to attenuate the caspase-8 signal. Alternately, sigma receptors are located in the endoplasmic reticulum and mitochondria and can regulate calcium signaling between these organelles (Cassano et al., 2009; Vilner and Bowen, 2000; Wei et al., 2006). Previous studies in tumor models have indicated a role of sigma receptors in the modulation of mitochondrial death cascades (Wei et al., 2006). Caspase-9 can also be activated by other methamphetamine-induced cellular stress signals like reactive oxygen/nitrogen species generation (Kadenbach et al., 2004), which can be regulated by sigma receptors.
Reactive oxygen/nitrogen species generation is one of the main mediators of methamphetamine neurotoxicity, which occurs through various sources including excessive dopamine or glutamate release, microglial activation, and mitochondrial dysfunction (Krasnova and Cadet, 2009). These reactive oxygen/nitrogen species generated can injure neurons and surrounding cells via oxidative damage to cellular components such as lipids, proteins, and DNA, or cause endoplasmic reticulum stress and activation of mitochondrial death cascades, ultimately leading to nerve terminal degeneration or cell death (Krasnova and Cadet, 2009). In this study, methamphetamine caused reactive oxygen/nitrogen species generation in differentiated NG108-15 cells. SN79 pretreatment attenuated the reactive oxygen/nitrogen species generated by methamphetamine. This effect of SN79 at lower concentrations (1–100 nM) does not seem to be a general antioxidant effect as it cannot prevent the H2O2-mediated reactive oxygen/nitrogen species signal. However, at a high concentration of 1000 nM, SN79 decreases H2O2-mediated reactive oxygen/nitrogen species, which may be due to some intrinsic antioxidant activity at this concentration or to potential up regulation of cellular antioxidant defenses. Mechanistically, SN79 can act via sigma receptors to modulate cellular reactive oxygen/nitrogen species generation and antioxidant systems. Previous studies in tumor cells have shown that sigma2 receptor activation causes reactive oxygen species generation (Ostenfeld et al., 2005). Sigma1 receptors are involved in oxidative stress-mediated toxicities and have been shown to provide protective effects (Pal et al., 2012). Sigma receptors also play a regulatory role in the redox state of the cell, and can further modulate nNOS and iNOS enzyme activity (Tsai et al., 2009; Vagnerova et al., 2006; Yang et al., 2010).
We hypothesized that the neuroprotective effects of sigma receptor antagonists against methamphetamine may also result from the modulation of endoplasmic reticulum stress signaling. The inability of a sigma receptor antagonist to modulate PERK-mediated endoplasmic reticulum stress elicited by methamphetamine is surprising. Overexpression of sigma1 receptors has previously been shown to attenuate PERK phosphorylation, indicative of an upstream blockade of PERK-mediated endoplasmic reticulum stress, in response to the endoplasmic reticulum stressor thapsigargin in CHO cells (Hayashi and Su, 2007). Thapsigargin functions, however, by depleting endoplasmic reticulum calcium stores and it is possible that methamphetamine results in endoplasmic reticulum stress solely by altering the level of unfolded proteins within the endoplasmic reticulum irrespective of endoplasmic reticulum calcium levels. This is one potential explanation for the disparities between data presented here utilizing methamphetamine and reports using thapsigargin to induce PERK-mediated endoplasmic reticulum stress.
In summary, this study demonstrates that physiologically significant concentrations of methamphetamine can cause apoptosis in an in vitro system representative of a differentiated neuronal cell, which mechanistically involves induction of reactive oxygen/nitrogen species and activation of caspases. Importantly, the sigma receptor antagonist, SN79, can attenuate methamphetamine-induced cell death and neurotoxic mediators such as caspases and reactive oxygen/nitrogen species. SN79 can also attenuate the enhanced toxicity caused by elevated temperature, and the interaction of methamphetamine plus elevated temperature. The ability of SN79 and other sigma receptor antagonists to intervene at multiple points and mechanisms in the neurotoxic cascade is consistent with the location of sigma receptors in various subcellular organelles (Kaushal and Matsumoto, 2011a; McCann et al., 1994; Zeng et al., 2007). Future studies are needed to delineate the individual contributions of each of the sigma receptor subtypes in the effects observed herein.
Acknowledgements
This study was supported by grants from the National Institute on Drug Abuse at the National Institutes of Health (DA013978 and DA023205). Abagail Rosen was supported by an ARRA supplement for a summer undergraduate research experience. The funding source had no other involvement in the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Abbreviations
- AC927
1-(2-phenethyl)piperidine oxalate
- ATF3
activating transcription factor 3
- CHOP
C/EBP homologous protein
- CM156
3-(4-(4-cyclohexylpiperazin-1-yl)butyl)benzo[d]thiazole-2(3H)-thione
- CM-H2DCFDA
5-(and-6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate
- DMEM
Dulbecco's modified Eagle's medium
- DTG
1,3-di(2-tolyl)guanidine
- EDTA
ethylenediaminetetraacetic acid
- FBS
fetal bovine serum
- GADD34
protein phosphatase 1 regulatory subunit 15A
- HAT
hypoxanthine-aminopterin-thymidine
- PERK
PKR-like ER kinase
- SN79
6-acetyl-3-(4-(4-(4-fluorophenyl)piperazin-1-yl)butyl)benzo[d]oxazol-2(3H)-one
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bauer MK, Vogt M, Los M, Siegel J, Wesselborg S, Schulze-Osthoff K. Role of reactive oxygen intermediates in activation-induced CD95 (APO-1/Fas) ligand expression. J. Biol. Chem. 1998;273:8048–8055. doi: 10.1074/jbc.273.14.8048. [DOI] [PubMed] [Google Scholar]
- Berghe TV, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell. Death Differ. 2010;17:922–930. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- Bowen WD. Sigma receptors: recent advances and new clinical potentials. Pharm. Acta Hel. 2000;74:211–218. doi: 10.1016/s0031-6865(99)00034-5. [DOI] [PubMed] [Google Scholar]
- Cahuzac N, Baum W, Kirkin V, Conchonaud F, Wawrezinieck L, Marguet D, Janssen O, Zornig M, Hueber AO. Fas ligand is localized to membrane rafts, where it displays increased cell death-inducing activity. Blood. 2006;107:2384–2391. doi: 10.1182/blood-2005-07-2883. [DOI] [PubMed] [Google Scholar]
- Canete E, Diogene J. Comparative study of the use of neuroblastoma cells (Neuro-2a) and neuroblastoma × glioma hybrid cells (NG108-15) for the toxic effect quantitation of marine toxins. Toxicon. 2008;52:541–550. doi: 10.1016/j.toxicon.2008.06.028. [DOI] [PubMed] [Google Scholar]
- Cassano G, Gasparre G, Niso M, Contino M, Scalera V, Colabufo NA. F281, synthetic agonist of the sigma-2 receptor, induces Ca2+ efflux from the endoplasmic reticulum and mitochondria in SK-N-SH cells. Cell. Calcium. 2009;45:340–345. doi: 10.1016/j.ceca.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Crawford KW, Bowen WD. Sigma-2 receptor agonists activate a novel apoptotic pathway and potentiate antineoplastic drugs in breast tumor cell lines. Cancer Res. 2002;62:313–322. [PubMed] [Google Scholar]
- Cruickshank CC, Dyer KR. A review of the clinical pharmacology of methamphetamine. Addiction. 2009;104:1085–1099. doi: 10.1111/j.1360-0443.2009.02564.x. [DOI] [PubMed] [Google Scholar]
- El Mchichi B, Hadji A, Vazquez A, Leca G. p38 MAPK and MSK1 mediate caspase-8 activation in manganese-induced mitochondria-dependent cell death. Cell Death Differ. 2007;14:1826–1836. doi: 10.1038/sj.cdd.4402187. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Wilkins DG, Gibb JW, Hanson GR. Interaction between hyperthermia and oxygen radical formation in the 5-hydroxytryptaminergic response to a single methamphetamine administration. J. Pharmacol. Exp .Ther. 1997;283:281–285. [PubMed] [Google Scholar]
- Formigli L, Papucci L, Tani A, Schiavone N, Tempestini A, Orlandini GE, Capaccioli S, Orlandini SZ. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell. Physiol. 2000;182:41–49. doi: 10.1002/(SICI)1097-4652(200001)182:1<41::AID-JCP5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Gajate C, Mollinedo F. Lipid rafts and Fas/CD95 signaling in cancer chemotherapy. Recent Pat. Anticancer Drug Discov. 2011;6:274–283. doi: 10.2174/157489211796957766. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Barhrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottleib E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon H-U, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Diff. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebreselassie D, Bowen WD. Sigma-2 receptors are specifically localized to lipid rafts in rat liver membranes. Eur. J. Pharmacol. 2004;493:19–28. doi: 10.1016/j.ejphar.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Hamprecht B, Glaser T, Reiser G, Bayer E, Propst F. Culture and characteristics of hormone-responsive neuroblastoma × glioma hybrid cells. Methods Enzymol. 1985;109:316–341. doi: 10.1016/0076-6879(85)09096-6. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 receptors (σ1 binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 2003;306:718–725. doi: 10.1124/jpet.103.051284. [DOI] [PubMed] [Google Scholar]
- Hazelwood S, Bowen WD. Sigma-2 receptor-mediated apoptosis in human SK-N-SH neuroblastoma cells: role of lipid rafts, caspases, and mitochondrial depolarization. Proc. Am. Assoc. Cancer Res. 2006;47:4932. [Google Scholar]
- Hornick JR, Xu J, Vangveravong S, Tu Z, Mitchem JB, Spitzer D, Goedegebuure P, Mach RH, Hawkins WG. The novel sigma-2 receptor ligand SW43 stabilizes pancreas cancer progression in combination with gemcitabine. Mol .Cancer. 2010;9:298. doi: 10.1186/1476-4598-9-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S, Deng X, Noailles PA, Ladenheim B, Cadet JL. Methamphetamine induces neuronal apoptosis via cross-talks between endoplasmic reticulum and mitochondria-dependent death cascades. FASEB J. 2004;18:238–251. doi: 10.1096/fj.03-0295com. [DOI] [PubMed] [Google Scholar]
- Jayanthi S, Deng X, Ladenheim B, McCoy MT, Cluster A, Cai NS, Cadet JL. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA. 2005;102:868–873. doi: 10.1073/pnas.0404990102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YG, Peng Y, Koussougbo KS. Necroptosis: a novel therapeutic target for glioblastoma. Med. Hypotheses. 2011;76:350–352. doi: 10.1016/j.mehy.2010.10.037. [DOI] [PubMed] [Google Scholar]
- Jonhede S, Petersen A, Zetterberg M, Karlsson JO. Acute effects of the sigma-2 receptor agonist siramesine on lysosomal and extra-lysosomal proteolytic systems in lens epithelial cells. Mol. Vis. 2010;16:819–827. [PMC free article] [PubMed] [Google Scholar]
- Kadenbach B, Arnold S, Lee I, Huttemann M. The possible role of cytochrome c oxidase in stress-induced apoptosis and degenerative diseases. Biochim. Biophys. Acta. 2004;1655:400–408. doi: 10.1016/j.bbabio.2003.06.005. [DOI] [PubMed] [Google Scholar]
- Kaushal N, Matsumoto RR. Role of sigma receptors in methamphetamine-induced neurotoxicity. Curr. Neuropharmacol. 2011a;9:54–57. doi: 10.2174/157015911795016930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal N, McCurdy CR, Matsumoto RR. Society for Neuroscience meeting 2011 Program #367.03. 2011b. SN79 attenuates the neurotoxic effect of methamphetamine: In vivo and in vitro studies. [Google Scholar]
- Kaushal N, Robson MJ, Vinnakota H, Narayanan S, Avery BA, McCurdy CR, Matsumoto RR. Synthesis and pharmacological evaluation of 6-acetyl-3-(4-(4-(4-fluorophenyl)piperazin-1-yl)butyl)benzo[d]oxazol-2(3H)-one (SN79), a cocaine antagonist, in rodents. AAPS J. 2011c;13:336–346. doi: 10.1208/s12248-011-9274-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal N, Seminerio MJ, Shaikh J, Medina MA, Mesangeau C, Wilson LL, McCurdy CR, Matsumoto RR. CM156, a high affinity sigma ligand, attenuates the stimulant and neurotoxic effects of methamphetamine in mice. Neuropharmacology. 2011d;61:992–1000. doi: 10.1016/j.neuropharm.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal N, Elliott M, Robson MJ, Iyer AK, Rojanasakul Y, Coop A, Matsumoto RR. AC927, a σ receptor ligand, blocks methamphetamine-induced release of dopamine and generation of reactive oxygen species in NG108-15 cells. Mol. Pharmacol. 2012;81:299–308. doi: 10.1124/mol.111.074120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin EA, Brown PL, Sharma HS. Brain edema and breakdown of the blood-brain barrier during methamphetamine intoxication: critical role of brain hyperthermia. Eur. J. Neurosci. 2007;26:1242–1253. doi: 10.1111/j.1460-9568.2007.05741.x. [DOI] [PubMed] [Google Scholar]
- Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res. Rev. 2009;60:379–407. doi: 10.1016/j.brainresrev.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell. Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Ma W, Prancrazio JJ, Coulombe M, Dumm J, Sathanoori R, Barker JL, Kowtha VC, Stenger DA, Hickman JJ. Neuronal and glial epitopes and transmitter-synthesizing enzymes appear in parallel with membrane excitability during neuroblastoma × glioma hybrid differentiation. Brain Res. Dev. Brain Res. 1998;106:155–163. doi: 10.1016/s0165-3806(97)00208-3. [DOI] [PubMed] [Google Scholar]
- Marrazzo A, Fiorito J, Zappala L, Prezzavento O, Ronsisvalle S, Pasquinucci L, Scoto GM, Bernardini R, Ronsisvalle G. Antiproliferative activity of phenylbutyrate ester of haloperidol metabolite II [(+/−)-MRJF4] in prostate cancer cells. Eur. J. Med. Chem. 2011;46:433–438. doi: 10.1016/j.ejmech.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Matsumoto RR, Shaikh J, Wilson LL, Vedam S, Coop A. Attenuation of methamphetamine-induced effects through the antagonism of sigma (σ) receptors: Evidence from in vivo and in vitro studies. Eur. Neuropsychopharmacol. 2008;18:871–881. doi: 10.1016/j.euroneuro.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann DJ, Weissman AD, Su TP. Sigma-1 and sigma-2 sites in rat brain: comparison of regional, ontogenetic, and subcellular patterns. Synapse. 1994;17:182–189. doi: 10.1002/syn.890170307. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Melino G. Regulation of the apoptosis-necrosis switch. Oncogene. 2004;23:2757–2765. doi: 10.1038/sj.onc.1207559. [DOI] [PubMed] [Google Scholar]
- Numachi Y, Ohara A, Yamashita M, Fukushima S, Kobayashi H, Hata H, Watanabe H, Hall FS, Lesch KP, Murphy DL, Uhl GR, Sora I. Methamphetamine-induced hyperthermia and lethal toxicity: role of the dopamine and serotonin transporters. Eur. J. Pharmacol. 2007;572:120–128. doi: 10.1016/j.ejphar.2007.06.022. [DOI] [PubMed] [Google Scholar]
- Ostenfeld MS, Fehrenbacher N, Hoyer-Hansen M, Thomsen C, Farkas T, Jaattela M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005;65:8975–8983. doi: 10.1158/0008-5472.CAN-05-0269. [DOI] [PubMed] [Google Scholar]
- Pal A, Fontanilla D, Gopalakrishnan A, Chae Y-K, Markley JL, Ruoho AE. The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur. J. Pharmacol. 2012;682:12–20. doi: 10.1016/j.ejphar.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminerio MJ, Kaushal N, Shaikh J, Huber JD, Coop A, Matsumoto RR. Sigma (σ) receptor ligand, AC927 (N-phenethylpiperidine oxalate), attenuates methamphetamine-induced hyperthermia and serotonin damage in mice. Pharmacol. Biochem. Behav. 2011;98:12–20. doi: 10.1016/j.pbb.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma HS, Sjoquist PO, Ali SF. Drugs of abuse-induced hyperthermia, blood-brain barrier dysfunction and neurotoxicity: neuroprotective effects of a new antioxidant compound H-290/51. Curr. Pharm. Des. 2007;13:1903–1923. doi: 10.2174/138161207780858375. [DOI] [PubMed] [Google Scholar]
- Spruce BA, Campbell LA, McTavish N, Cooper MA, Appleyard MV, O'Neill M, Howie J, Samson J, Watt S, Murray K, McLean D, Leslie NR, Safrany ST, Ferguson MJ, Peters JA, Prescott AR, Box G, Hayes A, Nutley B, Raynaud F, Downes CP, Lambert JJ, Thompson AM, Eccles S. Small molecule antagonists of the sigma-1 receptor cause selective release of the death program in tumor and self-reliant cells and inhibit tumor growth in vitro and in vivo. Cancer Res. 2004;64:4875–4886. doi: 10.1158/0008-5472.CAN-03-3180. [DOI] [PubMed] [Google Scholar]
- Suchard JR. Recovery from severe hyperthermia (45 degrees C) and rhabdomyolysis induced by methamphetamine body-stuffing. West J Emerg Med. 2007;8:93–95. [PMC free article] [PubMed] [Google Scholar]
- Soti C, Sreedhar AS, Csermely P. Apoptosis, necrosis and cellular senescence: chaperone occupancy as a potential switch. Aging Cell. 2003;2:39–45. doi: 10.1046/j.1474-9728.2003.00031.x. [DOI] [PubMed] [Google Scholar]
- Tsai SY, Hayashi T, Mori T, Su TP. Sigma-1 receptor chaperones and diseases. Cent. Nerv. Syst. Agents Med. Chem. 2009;9:184–189. doi: 10.2174/1871524910909030184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnerova K, Hurn PD, Bhardwaj A, Kirsch JR. Sigma 1 receptor agonists act as neuroprotective drugs through inhibition of inducible nitric oxide synthase. Anesth. Analg. 2006;103:430–434. doi: 10.1213/01.ane.0000226133.85114.91. [DOI] [PubMed] [Google Scholar]
- Vilner BJ, de Costa BR, Bowen WD. Cytotoxic effects of sigma ligands: sigma receptor-mediated alterations in cellular morphology and viability. J. Neurosci. 1995;15:117–134. doi: 10.1523/JNEUROSCI.15-01-00117.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilner BJ, Bowen WD. Modulation of cellular calcium by sigma-2 receptors: release from intracellular stores in human SK-N-SH neuroblastoma cells. J. Pharmacol. Exp. Ther. 2000;292:900–911. [PubMed] [Google Scholar]
- Wang X, Bowen WD. Sigma-2 receptors mediate apoptosis in SK-N-SH neuroblastoma cells via caspase-10-dependent Bid cleavage and mitochondrial release of endonuclease G and apoptosis-inducing factor. Soc. Neurosc. Abst. 2006 #90.1. [Google Scholar]
- Wei Z, Mousseau DD, Dai Y, Cao X, Li XM. Haloperidol induces apoptosis via the sigma2 receptor system and Bcl-XS. Pharmacogenomics J. 2006;6:279–288. doi: 10.1038/sj.tpj.6500373. [DOI] [PubMed] [Google Scholar]
- Xie T, McCann UD, Kim S, Yuan J, Ricaurte GA. Effect of temperature on dopamine transporter function and intracellular accumulation of methamphetamine: implications for methamphetamine-induced dopaminergic neurotoxicity. J. Neurosci. 2000;20:7838–7845. doi: 10.1523/JNEUROSCI.20-20-07838.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZJ, Carter EL, Torbey MT, Martin LJ, Koehler RC. Sigma receptor ligand 4-phenyl-1-(4-phenylbutyl)-piperidine modulates neuronal nitric oxide synthase/postsynaptic density-95 coupling mechanisms and protects against neonatal ischemic degeneration of striatal neurons. Exp. Neurol. 2010;221:166–174. doi: 10.1016/j.expneurol.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Vangveravong S, Xu J, Chang KC, Hotchkiss RS, Wheeler KT, Shen D, Zhuang ZP, Kung HF, Mach RH. Subcellular localization of sigma-2 receptors in breast cancer cells using two-photon and confocal microscopy. Cancer Res. 2007;67:6708–6716. doi: 10.1158/0008-5472.CAN-06-3803. [DOI] [PubMed] [Google Scholar]