

Abstract
Peptide mass spectrometry relies crucially on algorithms that match peptides to spectra. We describe a method to evaluate the accuracy of these algorithms based on the masses of parent proteins before trypsin endoprotease digestion. Measurement of conformance to parent proteins provides a score for comparison of the performances of different algorithms as well as alternative parameter settings for a given algorithm. Tracking of conformance scores for spectrum matches to proteins with progressively lower expression levels revealed that conformance scores are not uniform within data sets but are significantly lower for less abundant proteins. Similarly peptides with lower algorithm peptide-spectrum match scores have lower conformance. Although peptide mass spectrometry data is typically filtered through decoy analysis to ensure a low false discovery rate, this analysis confirms that the filtered data should not be considered as having a uniform confidence. The analysis suggests that use of different algorithms and multiple standardized parameter settings of these algorithms can increase significantly the numbers of peptides identified. This data set can be used as a resource for future algorithm assessment.
Keywords: peptide mass spectrometry, trypsin, OMSSA, SEQUEST, Mascot, algorithm parameter sets, parent-protein conformance, decoy analysis
1. Introduction
Peptide mass spectrometry provides a powerful method to analyze proteome expression in cell lysates. At the core of this method, experimental mass spectra of fragmented peptides are matched with theoretical mass ladders based on peptide sequences. The accuracy of peptide matching depends critically on the effectiveness of algorithms that match the theoretical and experimental spectra. Accurate matching is a major challenge for these algorithms. We present here a method to evaluate algorithms to obtain high-confidence interrogation of proteomes.
In typical experiments, proteins in cell lysates are digested with an endoprotease, typically trypsin, to obtain peptides in size ranges that can be successfully analyzed by tandem mass spectrometry (MS/MS). Trypsin fragments with pronounced peaks in the first MS are selected for collision induced dissociation (CID) in the second MS. CID causes fragmentation of the trypsin peptides, typically at amide bonds, producing N-terminal b ions and C-terminal y ions that give rise to a set of detected mass to charge (m/z) peaks. Peptide-searching algorithms compare experimental m/z spectra with theoretical ion ladders derived from tryptic fragments of an input sequence ″database″ of all proteins in the proteome. Each spectrum-matching algorithm, however, is different in its design and structure. The best peptide-spectrum matches are determined by techniques such as cross-correlation (e.g., SEQUEST1) or by model-based approaches using statistical significance (OMSSA,2 Mascot3).
Each algorithm has multiple parameter settings, including mass tolerances between theoretical and observed trypsin fragments (precursor mass tolerance) or theoretical and CID fragments (fragment mass tolerance); which and how many optional mass modifications to allow per peptide; and which CID ion series (e.g., a, b, y) to assess. It is important to choose appropriate parameter settings of algorithms for accurate peptide identifications. Given the many combinations of choices, what are good strategies to determine parameter settings? Do different parameter settings of algorithms applied to a given data set provide different samplings of the proteome, and are these samplings of high quality?
The effectiveness of an algorithm is typically assessed through decoy analysis.4,5 For each forward protein sequence in the sequence “database”, the algorithm is also presented with the reverse of that sequence. The forward and reverse sequences are processed blindly together, including a theoretical trypsin digestion, and the frequency that decoy reverse peptides are chosen by the algorithm indicates the false discovery rate (FDR5) for that particular choice of parameter settings. Given that the algorithms output quality scores for each peptide-spectrum match (PSM), scoring thresholds can be computed and used to filter the output data to ensure an FDR below a desired level (e.g., 1% or 5%). Decoy analysis is an excellent strategy to assess algorithm parameter settings, especially since the approach is inherently independent of any particular algorithm.
We wished to develop additional approaches to assess algorithm performance: methods to be used alongside decoy analysis in order to build confidence in peptide matches by different standardized parameter settings of algorithms. Expanding on a protocol suggested by Park et al.,6 we present a strategy that evaluates peptide-spectrum matches by assessing the masses of parent proteins prior to trypsin digestion, an approach that can be applied to any algorithm using the spectra sets presented here (Supporting Information) or new data sets if desired. This parent-protein profiling approach uses a gel slice strategy to partition cell lysates according to parent-protein mass. Application of this approach suggests that different algorithms can provide different, yet valid, samplings of the proteome, and that it can also be extremely productive to run algorithms multiple times with different parameter settings, an approach that is becoming increasingly possible given the availability of increased computational power. We first focus our discussion on the SEQUEST and OMSSA algorithms and then present equivalent analysis of the Mascot algorithm, which revealed similar results.
2. Methods
2.1. Gel Slice Preparation
Conformance to parent proteins before digestion with trypsin was used to assess algorithm matches of spectra to tryptic peptides. Yeast cell lysates were partitioned into gel slices of known molecular weight size ranges (25–37, 37–50, and 50–75 kDa). Although the algorithms had no knowledge of the parent-protein sizes before trypsin digestion, peptide matches would be expected to conform to the correct parent-protein size ranges if the algorithm was matching successfully. This has been shown previously in MS/MS-based gel-band analysis of the proteome of Pseudomonas putida bacteria.6
Protein samples were prepared as follows: 100 mL of YSH474 cells were grown to mid-log phase in YPD and lysed with RIPA buffer (150 mM NaCl, 1% Igepal, 0.1% SDS, 50 mM Tris pH 8.0), and acid-washed glass beads. To prevent degradation, protease inhibitors (Roche) and PMSF were added, and samples were chilled on ice during lysis. The lysate was spun at 5,000 rpm, and samples of 500 or 1,000 μg were run alongside protein standard markers (Bio-Rad) on 4–20% SDS-PAGE gels (Bio-Rad). Protein standard bands served as a guide for the excision of gel slices of various molecular weight size ranges (25–37, 37–50, and 50–75 kDa; Figure 1). Samples were subjected to reduction and alkylation followed by overnight in-gel trypsin digestion.7 Extracted peptides were resuspended in 0.1% TFA, loaded onto a c18 packed (Michrom) nanospray column (Polymicro), and run with a 180-min gradient on a LCQ Deca XP (Thermo-Scientific) coupled to a high-performance liquid chromatography (HPLC) system (Agilent 1100 series) and a nanoelectrospray (nano-ESI) ion source. Preliminary tests indicated that gels with visible degradation had limited conformance to parent-protein masses. Hence, gels were discarded and not analyzed if their appearance suggested visible degradation.
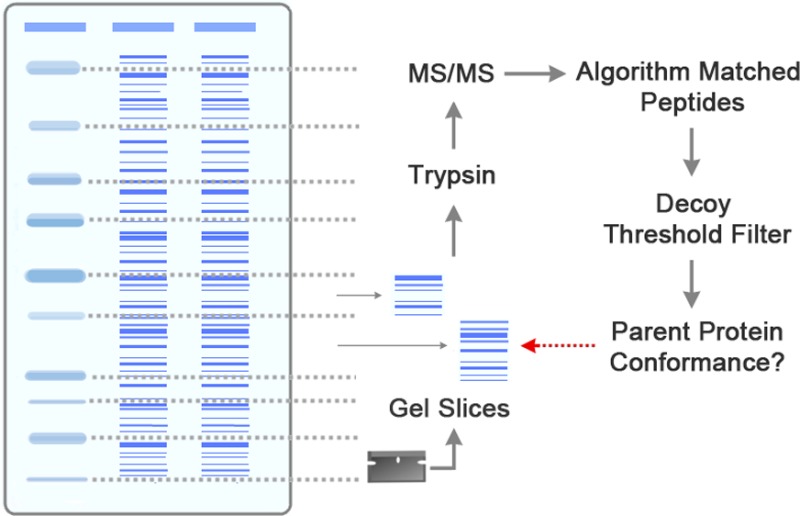
Figure 1.

Schematic summary of parent-protein profiling approach.
2.2. Peptide Spectrum Matching Algorithms
Peptide matches were identified using the SEQUEST algorithm (Proteome Discoverer v.1.2) run on a Dell Alienware Aurora R4 server, the open mass spectrometry search algorithm (OMSSA) run on a 90-node cluster, and the Mascot algorithm run on a Dell XPS 8300 server. Algorithm parameters were set up to search for either the standard b and y ions following CID or with the addition of a ions. Optional mass increases to peptides included dynamic modifications of +42 Da for acetylation of any N-terminal amino acid residue and +16 Da for oxidation of methionine residues, and static modifications included +57 Da for carbamidomethyl modification of cysteine residues. The SEQUEST algorithm parameter file included a precursor mass tolerance of 3.0 Da and a fragment mass tolerance of 1.0 Da, while the OMSSA algorithm was run using five different standard sets of parameters (Table 1) and Mascot was run using four parameter sets (Figure 6A); the SEQUEST parameter set is similar to that reported for PeptideAtlas yeast data (http://www.peptideatlas.org/); the OMSSA and Mascot parameter sets are similar to the algorithm default parameters. Precursor peptides for liquid chromatography MS/MS (LC–MS/MS) analysis were prepared by trypsin digestion, which cuts after arginine or lysine, except when flanked by proline. For the SEQUEST, OMSSA, and Mascot analysis, we required trypsin-cleavage sites at both ends of the precursor peptides (or one end if a terminal peptide). A sequence “database” file containing protein translations of annotated and downstream open reading frames (dnORFs) in FASTA format was constructed as described previously.8
Table 1. Comparison of SEQUEST and OMSSA Parameter Settings.
| OMSSA |
||||||
|---|---|---|---|---|---|---|
| SEQUEST | 0 | 1 | 2 | 3 | 4 | |
| max missed cleavage sites | 1 | 1 | 1 | 1 | 1 | 1 |
| precursor mass tolerance (Da) | 3.0 | 3.0 | 1.5 | 2.0 | 1.5 | 1.5 |
| fragment mass tolerance (Da) | 1.0 | 1.0 | 0.5 | 0.8 | 0.5 | 1.0 |
| precursor ion search type | mono | avg | avg | avg | mono | avg |
| fragment ion search type | mono | mono | mono | avg | mono | mono |
| mass tolerance charge scaling | N/A | none | none | none | none | linear |
Figure 6.

Assessment of the Mascot algorithm using parent-protein profiling. Spectra sets from the same 22 gel-slice LC–MS/MS experiments were analyzed with the Mascot algorithm using 5% FDR. The Mascot algorithm shows trends similar to those of SEQUEST and OMSSA. (A) The Mascot algorithm was run using similar parameter settings to those used with OMSSA. (B) Parent-protein conformance scores for b/y ion screen. (C) Parent protein conformance scores for a/b/y ion screen. (D) Few peptides were detected by the b/y ion screen alone, and these had relatively lower conformance. (E) Peptides detected by only one of the Mascot standardized parameter sets have lower conformance compared to those detected with multiple parameter sets. (F) Conformance scores are depressed for detected proteins with low expression (p < 1.05 × 10–18; chi-squared); for example, the set of proteins where protein molecules per cell <1000 (i.e., log(PE) < 3) have a conformance level of 53.9%. (G) Similarly, conformance scores are depressed for peptide matches that score close to the decoy FDR threshold. This is the case for the subsets of PSMs with scores below 77.6%, the 1% score threshold from bootstrap analysis. In this assessment, the scoring confidence, d = −log10(PSM_prob_score/FDR_threshold), is computed using PSM probabilities from the Mascot output: score = −10·log10(PSM_prob_score).
Output data from the three algorithms were uploaded to a relational database and analyzed with stored procedures written in MS-SQL to compute decoy false discovery rates (FDRs) and parent-protein conformance scores. Reverse-sequence decoy analysis was performed as described previously,8 and peptide matches were filtered to give a target FDR of ≤5%. Before computing decoy score thresholds, for each LC–MS/MS run, we excluded matches with internal trypsin sites and matches with an initial ranking (Rank) > 1 if a SEQUEST or Mascot matched peptide. The decoy score thresholds were then applied to the output data after first excluding OMSSA matches (which are not ranked) where multiple nondecoy peptides matched to the same spectrum. As discussed in Section 3.2 below, the peptide matching by OMSSA is stringent, and the false detection rate was typically below 5% after application of these filters (1.6–5.1% depending on parameter settings and CID ions assessed). For all three algorithms, we also excluded peptides that mapped to multiple parent proteins; although these were likely correct identifications, these peptides were excluded because they could not be assigned to unique parent proteins.
2.3. Conformance Score Computation
Parent-protein conformance scores were computed from forward peptide matches classified as conforming or nonconforming peptides according to the known molecular weight size range of the gel slice (25–37, 37–50, and 50–75 kDa). We analyzed data from 22 gel slices processed in 22 independent LC–MS/MS runs (Supplementary Figure 1). Peptides were counted once per LC–MS/MS run, even if detected by multiple spectra. To account for aberrant protein travel through the gel or possible post-translational modifications, mass tolerances of ±10% of the molecular weight size range were applied. For example, peptide matches from the 25–37 kDa gel slice range were categorized as conforming if the parent proteins were between 22.5 and 40.7 kDa.
Conformance scores for individual LC–MS/MS runs were computed as follows:
An overall conformance score (for a single set of parameter settings for the algorithm) was computed by summing the number of conforming peptides and nonconforming peptides across all 22 LC–MS/MS runs counting each matched peptide a maximum of once per run:
3. Results and Discussion
3.1. Parent-Protein Profiling Evaluates Algorithm Performance
Peptide-spectrum matching algorithms score individual peptide matches according to how well the masses of expected CID fragments of a tryptic peptide match the m/z peaks in a detected spectrum. We developed a parent-protein evaluation method to assess algorithm performance. The approach is based on the fact that algorithms have no knowledge of the masses of parent proteins prior to trypsin digestion. By partitioning parent proteins into known molecular weight size ranges (25–37, 37–50, and 50–75 kDa) using SDS-PAGE and processing individual gel slices for assessment through LC–MS/MS (Figure 1), we investigated whether peptide matches by algorithms conformed to the expected parent-protein size range. Allowing a mass tolerance of 10% to compensate for experimental variations inherent in the gel slice approach, we computed conformance scores indicative of the effectiveness of an algorithm parameter set (Table 2A.).
Table 2. Conformance Scoring Based on Distinct Peptide Matches per LC–MS/MS Run.
| algorithma | OMSSA parameter set |
distinct peptidesb |
conforming peptides |
nonconforming peptides |
overall conformance score |
overall decoy conformance score |
|
|---|---|---|---|---|---|---|---|
| (A) | b/y ion screen | ||||||
| SEQUEST | 4,480 | 3,781 | 699 | 84.4 | 14.6 | ||
| OMSSA | 0 | 3,060 | 2,717 | 343 | 88.8 | 23.6 | |
| OMSSA | 1 | 3,644 | 3,196 | 448 | 87.7 | 20.3 | |
| OMSSA | 2 | 2,134 | 1,893 | 241 | 88.7 | 23.5 | |
| OMSSA | 3 | 3,295 | 2,887 | 408 | 87.6 | 17.8 | |
| OMSSA | 4 | 3,035 | 2,696 | 339 | 88.8 | 23.9 | |
| (B) | a/b/y ion screen | ||||||
| SEQUEST | 4,757 | 4,021 | 736 | 84.5 | 17.0 | ||
| OMSSA | 0 | 2,583 | 2,282 | 301 | 88.3 | 18.0 | |
| OMSSA | 1 | 3,393 | 2,960 | 433 | 87.2 | 21.2 | |
| OMSSA | 2 | 1,702 | 1,482 | 220 | 87.1 | 19.8 | |
| OMSSA | 3 | 3,065 | 2,670 | 395 | 87.1 | 15.9 | |
| OMSSA | 4 | 2,556 | 2,250 | 306 | 88.0 | 16.7 | |
The same spectra from 22 LC–MS/MS runs were analyzed by the SEQUEST and OMSSA algorithms. FDR threshold values were (A) SEQUEST: 0.081; OMSSA parameter sets 0 to 4: 1.0. (B) SEQUEST: 0.164025; OMSSA parameter sets 0, 1, 3, 4: 1.0; OMSSA parameter set 2: 0.9.
Distinct peptides counted once per LC–MS/MS run. A total of 2,842 distinct peptides were detected when counted only once regardless of which algorithm, parameter set, ion screen, or gel-slice range. Of these, 10.5% (298 distinct peptides) were detected in more than one gel-slice range, and 5.9% (168 distinct peptides) were scored as both conforming and nonconforming depending on size range.
We ran individual LC–MS/MS runs for each of the 22 gel slices and assessed spectra from the runs using a standard parameter set for the SEQUEST algorithm and five different parameter sets for the OMSSA algorithm (Table 1). After employing decoy analysis to ensure false discovery rates (FDRs) below 5%,4,5 we used the convention that even if a peptide were detected with multiple spectra, each peptide was counted only once per LC–MS/MS run. (The same peptide was counted more than once if detected in multiple runs, which in some cases were from gel slices with different size ranges.) In a standard b/y ion screen, 84.4% of the 4,480 peptides detected by SEQUEST had parent proteins conforming to the expected size range. Using the same spectra, OMSSA detected between 2,134 and 3,644 peptides with conformance scores ranging from 87.6% to 88.8% depending on the particular parameter set (Table 2A).
The conformance scores provide a relative measure of the peptide matching accuracy of each set of parameter settings of an algorithm. For example, the standardized OMSSA parameter sets have somewhat higher conformance scores than SEQUEST. However, although correlated, the conformance score is not numerically equal to the matching efficiency of the algorithm due to several factors:
• Parent proteins may run aberrantly during gel electrophoresis.9
• Post-translational modifications of parent proteins, such as glycosylation10 or proteolytic cleavage,11 may substantially change their molecular weights.
• Parent proteins of peptides randomly matched by the algorithm may be randomly assigned to the correct size range; for example, 25% of annotated yeast proteins have masses between 22.5 and 40.7 kDA, so 25% of random matches would conform to this size range. Indeed, the overall conformance scores for the decoy reverse peptides are 20.6% (Table 2A).
Because these contributing factors likely apply equivalently across all parameter settings of algorithms analyzing the same spectrum data sets, differences in conformance scores nevertheless provide an excellent assessment of the relative accuracies of the different algorithms and can be used to assess new algorithms or new parameter settings of current algorithms.
3.2. Algorithm Detection of Decoys
The five parameter sets of OMSSA all showed higher parent-protein conformance scores compared with SEQUEST. Indeed, the conformance score distributions from bootstrap analysis indicated that these differences are significant (Supplementary Figure 2). Given that the data sets were filtered to give a 5% FDR, the difference in conformance scores indicates that the detection of reverse-sequence decoys by the two algorithms was not equivalent. Indeed, even before applying a 5% FDR scoring filter, the standard implementation of OMSSA returned decoys at rates of 1.6–3.9% depending on the parameter set, indicating that the OMSSA algorithm is quite stringent in its interpretation of acceptable PSMs. Moreover, unlike SEQUEST, OMSSA does not standardly output a ranking of PSMs; if only the best-scoring PSMs for each spectrum are considered, then the decoy rates are even lower for the OMSSA algorithm (0.6–1.7%). For comparison, we reassessed the SEQUEST output using a 0.6% FDR threshold instead of 5%. This resulted in fewer matches (3,398 instead of 4,480) and a significantly higher parent-protein conformance rate (88.2% instead of 84.4%) comparable to those of OMSSA (Supplementary Figure 2). However, for the analysis that follows we used standard implementations of both algorithms with 5% FDR thresholds and filters as described in Methods.
3.3. Using Multiple Standardized Parameter Settings of an Algorithm
Counting peptides once even if seen with multiple OMSSA parameter sets, we classified peptides according to the number of standardized parameter sets (i.e., 1–5) for which a peptide was detected. Although conformance scores for the individual parameter sets indicated high confidence in peptide matches, only 42.9% of the 3,922 peptide matches were detected by all five OMSSA standardized parameter sets (Figure 2A.). The different samplings of peptides from different parameter settings suggest that using multiple standardized settings of OMSSA can increase the yield of high-confidence detected peptides. Indeed, when probing the same spectrum data set, the union of the outputs from five OMSSA parameter sets gave 3,922 detected peptides at an overall conformance score of 86.2%. Furthermore, we found that peptides detected by only one of the five standardized parameter sets of OMSSA had a considerably lower conformance score (61.1%) and accounted for only 5.0% of the detected peptides (Figure 2A). This suggests that when using multiple settings of OMSSA it may be appropriate to exclude any peptides detected by only one of the standardized parameter sets given that this class of orphan peptides is found to be of lower confidence based on the parent-protein profiling approach.
Figure 2.

Union of outputs from multiple OMSSA parameter sets increases the yield of detected peptides. Detection by multiple parameter sets increases confidence in detected peptides. Conformance scores calculated from distinct peptides detected in the b/y (A) or a/b/y (B) ion screens in OMSSA; peptides were counted once even if seen with multiple standardized parameter sets.
3.4. Performing Different CID Ion Screens
Collision induced dissociation (CID) most commonly cleaves peptides at the amide bonds (between the C and N atoms) to give b and y ions. These are the two ion types typically assessed by the matching algorithms (b/y ion screen). However, a ions can also be produced if the cleavage position is shifted by one carbon, and algorithms can be configured to screen for a ions in addition to the b and y ions (a/b/y ion screen). Since assessment of a ions is sometimes included in specialized screens (e.g., of glutaraldehyde modified peptides12), we tested whether inclusion of the a ions might increase peptide detections. We performed an a/b/y ion screen on the same spectra data sets from the parent-protein profiling experiments above and examined the conformance scores (Table 2B).
Using SEQUEST, we detected 5,016 peptides, counting a peptide once per LC–MS/MS run whether seen in one or both of the b/y and a/b/y ion screens (Figure 3B). This corresponds to 2,261 unique peptides, of which 1,752 (77.5%) peptides were detected by both the b/y and a/b/y ion screens. Bootstrap analysis (Figure 3C, p < 0.001) indicated that peptides detected by SEQUEST in both the b/y and a/b/y ion screens had significantly higher conformance scores compared to peptides detected by either the b/y or a/b/y ion screens alone (Figure 3A). This suggests that confidence in SEQUEST peptide matches can be increased by performing both b/y and a/b/y ion screens and retaining only the peptide matches detected by both screens (the intersection of the outputs).
Figure 3.

Conformance scores for a/b/y and b/y ion screens. (A) Conformance scores were computed on the basis of distinct peptides, counted once per LC–MS/MS run, detected by SEQUEST in the a/b/y ion screen alone, the b/y ion screen alone, or both a/b/y and b/y ion screens. (B) SEQUEST detected 5,016 peptides, counting each peptide once per LC–MS/MS run. Of the 5,016 peptides, 4,221 peptides were detected by both ion screens, while 536 and 259 peptides were detected by a/b/y and b/y ion screens alone, respectively. (C) Bootstrap analysis shows significantly depressed conformance scores for b/y-alone and a/b/y-alone SEQUEST matches. Dotted lines and percentages represent the conformance score based on distinct peptides detected by b/y or a/b/y ion screens alone. Dashed lines and percentages represent the 1st and 99th percentiles. Left panel: Distribution of 1,000 conformance scores calculated after random sampling with replacement of 536 samples from a full set of 4,757 distinct peptides per LC–MS/MS run detected in the a/b/y ion screen. Right panel: Distribution of 1,000 conformance scores calculated after random sampling with replacement of 259 samples from a full set of 4,480 distinct peptides per LC–MS/MS run detected in the b/y ion screen. (D) Similar analysis with OMSSA. (E) Percentages of distinct peptides detected in either the a/b/y ion screen alone, the b/y ion screen alone, or both the a/b/y and b/y ion screens.
In contrast, the equivalent analysis with OMSSA did not give similar results. Higher percentages of the total number of distinct peptides (counted once per LC–MS/MS run) were detected by OMSSA in either the b/y or a/b/y ion screen alone as compared to the SEQUEST counterpart (Figure 3D, E). Additionally, the high conformance scores (Figure 3D) of peptides detected by OMSSA in either the b/y or a/b/y ion screen alone suggest that in order to increase significantly the numbers of detected peptides in OMSSA studies, it may be beneficial to perform both b/y and a/b/y ion screens and to take the union of the output results (rather than the intersection as with SEQUEST). We note, however, that this would approximately double the required computational time.
These results indicate that algorithms can have qualitatively different behaviors, emphasizing the importance of having evaluation tools to assess different standardized parameter settings of the algorithms.
3.5. Protein Expression
We investigated whether the striking differences in performance between SEQUEST and OMSSA in the b/y and a/b/y ion screens might be related to parent-protein expression levels of the detected peptides. Using data from genomic-scale Western analyses in yeast,13 we found that parent-protein expression values for peptides detected by SEQUEST in the b/y or a/b/y ion screens alone had significantly lower protein expression distributions compared to the distributions for peptides detected by both b/y and a/b/y ion screens (Figure 4A; p < 1.09 × 10–18 for b/y only, p < 1.12 × 10–5 for a/b/y only). However, this was not the case for the corresponding OMSSA analysis (Figure 4B, Supplementary Figure 3), potentially accounting for the high confidence in the b/y-only and a/b/y-only matches.
Figure 4.

Parent protein expression of distinct peptide matches (per LC–MS/MS run) detected in the a/b/y ion screen alone, the b/y ion screen alone, or both a/b/y and b/y ion screens. Protein expression values (PE; estimated molecules per cell) were obtained from genomic-scale Western analyses in yeast.13 Proteins of unknown PE values or values of zero (undetected) are not included. Distribution lines represent parent-protein PE values of peptides detected by the SEQUEST (A) and OMSSA parameter set 0 (B) screens. Also see Supplementary Figure 3.
The significantly lower parent-protein expression levels, in combination with the lower conformance scores of peptide matches detected by the b/y or a/b/y ion screens alone, suggest that, compared to OMSSA, the SEQUEST algorithm can detect peptides of lower abundance, but that confidence in these lower-abundance peptides is decreased. The qualitative difference in the results from SEQUEST and OMSSA raise the possibility that the two algorithms provide different samplings of the same cell lysate MS/MS data. Counting each peptide once regardless of how many MS/MS experiments, which standardized parameter sets of the algorithm, or which CID ion screens revealed the peptide, we find that 53.3% of the peptides are detected by both algorithms. SEQUEST detected 2,261 distinct peptides from the union of both the a/b/y and b/y ion screens, while OMSSA detected 2,097 distinct peptides from the union of the five standardized parameter settings of both the a/b/y and b/y ion screens. A total of 1,516 peptides were detected by both algorithms (Figure 5A) and had significantly higher parent conformance than peptides detected by either algorithm alone (89.0% of 3,382 peptides, counting peptides once per LC–MS/MS run; bootstrap p < 0.001; Figure 5B, Supplementary Figure 4). However, conformance rates are higher (yet still depressed) for SEQUEST-alone peptides if detected by both b/y and a/b/y screens (Figure 5C); similarly, OMSSA-alone peptides have higher (yet still depressed) conformance rates if detected by more than one parameter set (Figure 5C).
Figure 5.

Conformance of distinct peptides detected by OMSSA and SEQUEST alone or by both algorithms. (A) Counting each peptide once, regardless of which parameter set of the algorithm, how many MS/MS experiments, or which CID ion screens revealed the peptide, we find that 53.34% of the peptides are detected by both algorithms. Of the 2,261 distinct peptides detected by SEQUEST and the 2,097 distinct peptides detected by OMSSA, 1,516 peptides are found in both SEQUEST and OMSSA, while 745 peptides are unique to SEQUEST and 581 peptides are unique to OMSSA. (B) Conformance scores are computed by counting peptides once per LC–MS/MS run, even if seen in both ion screens or in multiple parameter sets, for peptides detected by both algorithms, SEQUEST alone, or OMSSA alone. (C) Conformance scores are computed after applying two filters limiting distinct peptides (i) from OMSSA to those that are detected in >1 parameter set and (ii) from SEQUEST to those that are detected in both b/y and a/b/y ion screens.
3.6. Spectrum Matching Performance Varies within Data Sets
The above results suggest that low protein expression is associated with poor conformance to parent proteins. Indeed, subsets of SEQUEST spectrum matches with progressively lower protein expression reveal that parent-protein conformance ranges from 59.4% (protein expression <103 molecules per cell) to 86.8% (protein expression >105 molecules per cell) (Table 3A). OMSSA matches show a similar progression. Interestingly, proteins in the “undetected” set (protein expression value = 0)13 have high conformance rates, suggesting that they were undetected for experimental reasons (e.g., inaccessible epitope tag) rather than low protein expression.
Table 3. Confidence in Algorithm Detected Peptides Depends on Parent Protein Expression and Distance from Decoy FDR Threshold.
| SEQUEST |
OMSSA |
||||||
|---|---|---|---|---|---|---|---|
| conforming peptidesb | nonconforming peptides | conformance scored | conforming peptides | nonconforming peptides | conformance scored | ||
| (A) | log(PE)a | ||||||
| undetected | 669 | 115 | 85.3 | 558 | 74 | 88.3 | |
| x ≤ 3 | 171 | 117 | 59.4 | 102 | 62 | 62.2 | |
| 3 < x ≤ 4 | 668 | 228 | 74.6 | 652 | 169 | 79.4 | |
| 4< x ≤ 5 | 1467 | 258 | 85.0 | 1446 | 215 | 87.1 | |
| x >5 | 1093 | 166 | 86.8 | 850 | 133 | 86.5 | |
| (B) | scoring confidencec (d) | ||||||
| d ≤ 1 | 724 | 420 | 63.3 | 431 | 270 | 61.5 | |
| 1 < d ≤ 3 | 1291 | 241 | 84.3 | 741 | 135 | 84.6 | |
| 3 < d ≤ 5 | 938 | 135 | 87.4 | 787 | 106 | 88.1 | |
| 5 < d ≤ 7 | 597 | 66 | 90.0 | 642 | 93 | 87.3 | |
| d >7 | 561 | 43 | 92.9 | 1123 | 101 | 91.7 | |
Protein expression (PE; estimated protein molecules per cell) based on large scale Western analysis.13
Peptides are counted once per LC–MS/MS run even if detected by both the b/y and a/b/y ion screens and, in the case of OMSSA, even if detected by multiple parameter sets.
Scoring confidence d = −log10(PSM_score/FDR_threshold). For OMSSA, the algorithm PSM score used was the e-value. For SEQUEST, implemented in Proteome Discoverer 1.2, we used the probability outputs to compute a PSM score = 10(probability/–10).
Conformance scores are significantly depressed for lower abundance proteins (chi-squared: SEQUEST, p < 6.18 × 10–36; OMSSA, p < 3.46 × 10–20) and for lower d scores (SEQUEST, p < 7.23 × 10–80; OMSSA, p < 6.25 × 10–72)
This result indicates that parent-protein conformance is not a uniform property within data sets but instead varies with protein expression levels: the algorithms are less effective at matching spectra for lower abundance proteins as might be expected.14 We examined the relationship between protein expression and the algorithm peptide-spectrum match (PSM) scores (SEQUEST probability score, OMSSA e-value score) and found that higher-confidence matching scores tend to be associated with higher expression proteins, whereas lower-confidence matching scores are common for both high and low expression proteins (Supplementary Figure 5). For this analysis we used a distance measure:
which measures the relative distance between an algorithm PSM score and the 95% confidence threshold for each experimental series. For example a d-score of 2 implies that the spectrum match is 102 fold better than the FDR threshold, which is the least stringent acceptable score for inclusion in the data set based on decoy analysis.
Not surprisingly, given this relationship between protein expression and algorithm PSM scores, we found that conformance to parent proteins tends to be higher for subsets of matches with higher-confidence algorithm PSM scores. We examined parent-protein conformance for subsets of spectrum matches with progressively better score ranges (Table 3B). This revealed that for both algorithms, matches with scores within 10-fold of the FDR threshold have parent conformance scores of only 61–64%, whereas score bins with greater distances from the FDR threshold have conformance scores that increase progressively up to 91–93%.
This analysis suggests that a given parent-protein conformance rate for a data set represents an average rate for the set of spectrum matches, and that the conformance rate is much lower for matches close to the FDR threshold and correspondingly higher for those further from the threshold. Similarly, as discussed previously,14 FDRs measure the average values for a data set and subsets of data with algorithm scores closer to the FDR threshold have elevated rates of false identification and hence are of lower confidence compared to matches with scores further from the FDR threshold.
We note that although decoy analysis4,5 can be employed to assess subsets of poor-scoring PSMs as discussed above, it would not be practical to use decoy analysis to assess algorithm performance with the other special subsets of detected peptides presented above, such as the outputs from different ion screens or different algorithms, due to difficulties in determining appropriate subsets of decoys for computing FDRs.
3.7. A Resource for Algorithm Assessment
The parent-protein profiling data set analyzed in this study provides a useful resource for assessing spectrum-peptide matching algorithms. Our 22 LC–MS/MS runs from the parent-protein profiling approach may be used as a benchmark data set for future yeast proteomic studies that are based on CID fragmentation and a mass spectrometer of similar resolution and sensitivity to the LCQ Deca XP (Supplementary Materials). For example, the spectra from the 22 gel slice experiments were used to evaluate several parameter settings of the Mascot algorithm,3 which is commonly used by many groups (Figure 6A). Peptide-spectrum matches identified by Mascot (Figure 6B, C) showed similar parent-protein conformance rates as SEQUEST and OMSSA and a similar graded dependence of conformance on protein expression (Figure 6F) and scoring confidence (Figure 6G). Like OMSSA, peptides detected with only one set of parameter settings of Mascot had poorer conformance compared to those detected by multiple parameter sets (Figure 6E). However, the behavior of the b/y and a/b/y ion screens was somewhat different for Mascot in that all matches detected by the a/b/y ion screen were also detected by the b/y screen, and the few matches detected by the b/y ion screen alone had poor parent-protein conformance (Figure 6D).
3.8. Conclusions
With the ongoing improvements in the design of currently available peptide-spectrum matching algorithms, as well as the development of new algorithms, our parent-protein profiling approach provides an unbiased and valid evaluation for assessing different algorithms and choosing effective parameter settings to obtain high confidence peptide matches. In particular, conformance rates provide a relative measure for comparing different algorithms and different standardized parameter settings. Assessment of peptide-spectrum matches from our 22 LC–MS/MS runs also calls for the use of multiple algorithms and parameter settings to increase the yield of identified peptides. In the case of SEQUEST, taking the intersection of the output from the b/y and a/b/y ion screens may increase confidence in peptide matches. In the case of OMSSA, taking the union of the outputs from multiple parameter sets and excluding PSMs detected by a single parameter set may increase confidence in peptide matches. In the case of Mascot, combining both of these filters (taking the intersection of b/y and a/b/y matches and excluding PSMs detected by only one parameter set) may increase confidence. As expected,14 assessment of PSMs in the context of known protein expression levels of the detected parent proteins indicates that confidence in spectrum matching by an algorithm varies within a data set and is lower for matches to low abundance proteins and matches with low-confidence algorithm PSM scores.
Acknowledgments
We thank Michael Rice, Scott Holmes, and Ruth Johnson for discussions and the anonymous reviewers for suggestions. This work was supported in part by funds from the Howard Hughes Medical Institute to support undergraduate initiatives in the life sciences, NSF grant CNS-0959856 and NIH grant 1R15GM096228.
Glossary
Abbreviations
- FDR
false discovery rate
- LC–MS/MS
liquid chromatography tandem mass spectrometry
- CID
collision induced dissociation
- OMSSA
open mass spectrometry search algorithm
- PSM
peptide spectrum match
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org. Raw data files can be found at http://www.peptideatlas.org/.
Author Contributions
§ These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Eng J.; McCormack A. L.; Yates J. R. 3rd An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Geer L. Y.; Markey S. P.; Kowalak J. A.; Wagner L.; Xu M.; Maynard D. M.; Yang X.; Shi W.; Bryant S. H. Open mass spectrometry search algorithm. J. Proteome Res. 2004, 35958–64. [DOI] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20183551–67. [DOI] [PubMed] [Google Scholar]
- Elias J. E.; Gygi S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 43207–14. [DOI] [PubMed] [Google Scholar]
- Fitzgibbon M.; Li Q.; McIntosh M. Modes of inference for evaluating the confidence of peptide identifications. J. Proteome Res. 2008, 7135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park G. W.; Kwon K. H.; Kim J. Y.; Lee J. H.; Yun S. H.; Kim S. I.; Park Y. M.; Cho S. Y.; Paik Y. K.; Yoo J. S. Human plasma proteome analysis by reversed sequence database search and molecular weight correlation based on a bacterial proteome analysis. Proteomics 2006, 641121–32. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Tomas H.; Havlis J.; Olsen J. V.; Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 162856–60. [DOI] [PubMed] [Google Scholar]
- Fournier C. T.; Cherny J. J.; Truncali K.; Robbins-Pianka A.; Lin M. S.; Krizanc D.; Weir M. P. Amino termini of many yeast proteins map to downstream start codons. J. Proteome Res. 2012, 11125712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakoucheva L. M.; Kimzey A. L.; Masselon C. D.; Smith R. D.; Dunker A. K.; Ackerman E. J. Aberrant mobility phenomena of the DNA repair protein XPA. Protein Sci. 2001, 1071353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung L. A.; Tao S. C.; Qian J.; Smith M. G.; Snyder M.; Zhu H. Global analysis of the glycoproteome in Saccharomyces cerevisiae reveals new roles for protein glycosylation in eukaryotes. Mol. Syst. Biol. 2009, 5, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox C. A.; Fuller R. S. Posttranslational processing of the prohormone-cleaving Kex2 protease in the Saccharomyces cerevisiae secretory pathway. J. Cell Biol. 1991, 1152297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A.; Chandramouli N.; Zhang L.; Deng H. Reductive glutaraldehydation of amine groups for identification of protein N-termini. J. Proteome Res. 2008, 794178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S.; Huh W. K.; Bower K.; Howson R. W.; Belle A.; Dephoure N.; O’Shea E. K.; Weissman J. S. Global analysis of protein expression in yeast. Nature 2003, 4256959737–41. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A. I. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics 2010, 73112092–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.