

Abstract
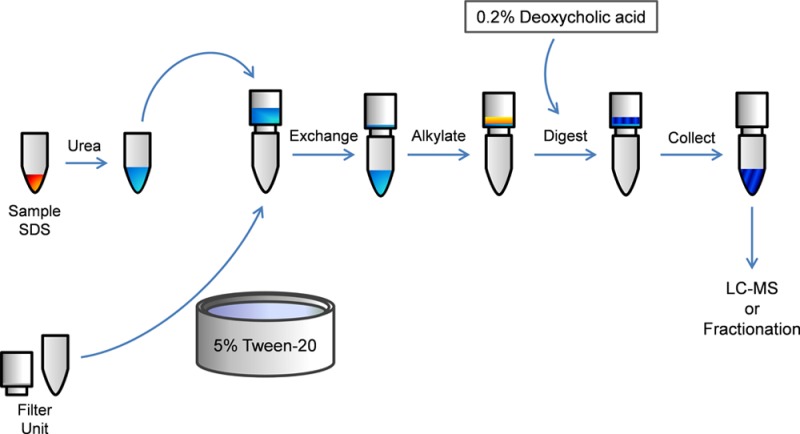
The integrity of quantitative proteomic experiments depends on the reliability and the robustness of the protein extraction, solubilization, and digestion methods utilized. Combinations of detergents, chaotropes, and mechanical disruption can yield successful protein preparations; however, the methods subsequently required to eliminate these added contaminants, in addition to the salts, nucleic acids, and lipids already in the sample, can result in significant sample losses and incomplete contaminant removal. A recently introduced method for proteomic sample preparation, filter-aided sample preparation (FASP), cleverly circumvents many of the challenges associated with traditional protein purification methods but is associated with significant sample loss. Presented here is an enhanced FASP (eFASP) approach that incorporates alternative reagents to those of traditional FASP, improving sensitivity, recovery, and proteomic coverage for processed samples. The substitution of 0.2% deoxycholic acid for urea during eFASP digestion increases tryptic digestion efficiency for both cytosolic and membrane proteins yet obviates needed cleanup steps associated with use of the deoxycholate sodium salt. For classic FASP, prepassivating Microcon filter surfaces with 5% TWEEN-20 reduces peptide loss by 300%. An express eFASP method uses tris(2-carboxyethyl)phosphine and 4-vinylpyridine to alkylate proteins prior to deposition on the Microcon filter, increasing alkylation specificity and speeding processing.
Keywords: filter-aided sample preparation, quantitative proteomics, detergent, ammonium deoxycholate, MSE
Introduction
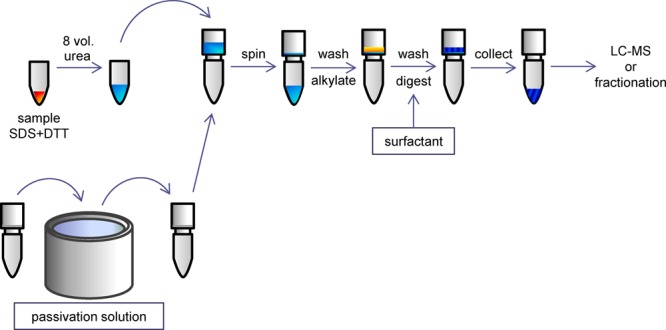
Extraction and solubilization of proteins, at the heart of any proteomic experiment, generally combine detergent and chaotrope treatment with mechanical disruption of cells. These components, as well as salts, nucleic acids, and lipids, must then be removed downstream, generally by organic solvent precipitation, affinity chromatography, or electrophoresis. Removal methods are generally subject to resolubilization problems, poor recoveries, or protein-to-protein variation. A recently introduced method for proteomic sample preparation, filter-aided sample preparation (FASP),1,2 cleverly circumvents many of the challenges associated with protein purification; however, it is associated with significant sample loss. Sodium dodecyl sulfate (SDS) prepared samples are diluted with a large volume of 8 M urea and then subjected to sequential buffer exchange steps in spin filter units to fully remove SDS. Proteins in the filter unit are alkylated, washed, and then digested. The eluting peptides are free of detergents, reductants, and alkylators as well as salts, lipids, and nucleic acids. We present modifications to the FASP workflow that quantitatively improve sensitivity, recovery, and proteomic coverage.
Filter-Aided Sample Preparation for Proteomics
In preparing proteomic samples, denaturant use is imperative to efficiently extract and solubilize matrix proteins. The common components of lysis buffers, anionic detergents (lauryl sulfate and lauroyl sarcosine) and chaotropes (urea and thiourea), function differently yet achieve the same goal. Although they are essential to protein solubilization, these denaturants can deleteriously affect downstream processing and analysis. Hence, they must be removed later.
Denaturants can be separated from proteinaceous components in many ways. The most common method is selectively precipitating proteins in large volumes of an organic solvent (usually acetone), with reductants, acids, and/or carrier compounds. Anionic detergents are removed by low pH precipitation or by selective binding to detergent-specific media. Dialysis may remove (or reduce) detergents and salts in an extract. Each purification method has unique problems associated with sample loss and residual interferences.
Manza et al. presented a novel method for sample purification and preparation in a spin filter unit, enabling SDS removal prior to enzymolysis.1 SDS, salts, nucleic acids, and lipids were removed by buffer exchange, after which the proteins remaining on the spin filter were reduced, alkylated, and digested. The resultant peptides were liberated by centrifugation and analyzed directly by LC–MS. While conceptually promising, the method yielded high sample losses and removed SDS incompletely.2 In 2009, Wisniewski et al. described a modified method, FASP, which incorporated urea to completely remove SDS.3
Urea usage enables two important advances. First, urea dissociates SDS from proteins and reduces the detergent concentration below its critical micelle concentration, enabling a one-step buffer exchange to remove it completely. Second, proteins remain denatured in urea, permitting Microcon ultrafiltration (UF) membranes with pore sizes up to 30 kDa to be employed, significantly reducing the time required for buffer exchange.3,4
One concern for the future use of FASP was the discontinuation of the Millipore Microcon centrifugal filters. Wisniewski et al. compared the retention of denatured proteins and FASP performance of alternative UF units.4 Vivapore and Millipore UF devices performed similarly for cell lysates, but the Millipore units outperformed Vivapore in FASP methods tailored for glycoproteins.1,2,4 By UV absorbance, that study estimated sample losses near 50%. Nevertheless, the authors were able to identify more than 6500 peptides and 2500 proteins.
Additions to the FASP workflow have aimed at performance and selectivity enhancements. Coupling FASP to strong anion exchange (SAX) performed on StageTips (Thermo) desalts samples, removes urea used during digestion, and enables small-scale fractionation. This FASP-SAX method was used to analyze membrane protein-enriched samples and to map C. elegans phosphorylation sites globally.1,5−7
Glycopeptide enrichment often relies on binding peptides to lectin-immobilized solid supports. FASP can be modified to circumvent immobilization steps by employing lectins larger than the molecular weight (MW) cutoff of the spin filter. Lectin-bound glycopeptides are prevented from passing through the filter, while nonglycopeptides accumulate in the filtrate. This enrichment strategy was applied successfully to map the mammalian N-glycoproteome.2,4,8
The unique ability of filter units to separate molecular species by size is the basis for MED-FASP, which uses multiple enzymes consecutively.3,9 This method responds to findings that sequence coverage increases when a second enzyme degrades large proteolytic peptides.3,4,10 The orthogonal peptide populations from each digestion may be analyzed separately. Trypsinolysis of materials retained after Lys-C digestion and peptide elution increased by 40% the proteins and phosphorylation sites identified versus using trypsin alone.9
Data-independent acquisition (DIA), in the form of LC–MSE, represents a strategy to increase the depth of precursor and fragment ion data acquired while simultaneously quantifying proteins within complex mixtures.11 MSE is a nonselective method that collects precursor and product ion data by continually alternating between low- and high-collision energies.12 Complete peptide elution profiles are collected, supporting both relative and absolute protein quantification.11,13 From each ascribed protein, the average MS signal from the three most intense peptide ions (Hi3) is compared with the Hi3 of a predigested internal standard protein, accurately dispensed to the sample to determine absolute quantity. The Hi3 for a protein may also be used to quantify relatively across samples. Free from isotope labeling, this quantitation method affords a near-comprehensive view of a cellular proteome. Isotope-labeled samples may be analyzed by LC–MSE to enable sample multiplexing, intra-run quantitative comparisons, and deeper proteome coverage than can be attained by data-dependent acquisition.
Increasing Proteome Coverage with Surfactants
Sodium deoxycholate (NaDOC) is a bile salt surfactant that aids lipid solubilization in the alimentary canal. Deoxycholate is secreted along with cholate and pancreatic enzymes, for example, chymotrypsin and trypsin. In 2006 and 2008, two groups proposed NaDOC inclusion in proteomic workflows to enhance trypsin digestion and increase membrane protein representation.14−18 Concentrations up to 10% (w/v) NaDOC are tolerated by trypsin, and concentrations as low as 0.01% were shown to improve digestion efficiency and sequence coverage.14,17,19 NaDOC can be removed quantitatively by acidification and phase transfer, a boon to its use in digestion.
Acid-insoluble NaDOC is removed from digests by phase transfer (PT) to a peptide-immiscible solvent phase, ethyl acetate (EA), following thorough phase mixing and centrifugation,14 affording a peptide-rich, surfactant-free aqueous layer. PT avoids peptide losses associated with precipitation.20
An additional benefit of PT is that many contaminants are soluble in EA, including SDS, n-octylglucoside, NP-40, and Triton X-100.21 Although NaDOC increases trypsin activity, the nonvolatile sodium counterions interfere with downstream processing, fractionation, and analysis without additional desalting.
By increasing peptide recovery from FASP, this study endeavors to provide quantitatively reproducible sample digests that recover hydrophobic species well (Figure 1). The goals are three-fold: (i) to enhance trypsin digestion by introducing removable surfactants to the FASP workflow, including the NaDOC-alternative, deoxycholic acid (DCA), that eliminates downstream desalting, (ii) to assess the utility of passivated UF units for decreasing sample losses, and (iii) to assess the impact on sample handling and processing time of employing the alternative reduction and alkylation reagents tris(2-carboxyethyl)phosphine (TCEP) and 4-vinylpyridine (4-VP).
Figure 1.
Enhancements to the FASP workflow. Samples are prepared in 4% SDS and diluted with 8 M urea to dissociate SDS from proteins. Microcon filter units and collection tubes are passivated by overnight incubation with 5% TWEEN-20, followed by thorough washing in MS-grade water. Diluted samples are then applied to passivated Microcon filters for buffer exchanges, eliminating SDS and contaminants. Proteins are alkylated with urea present, followed by successive buffer exchanges. Proteins are digested in the presence of surfactants, with the product peptides liberated by centrifugation. Extracting surfactant into an organic layer leaves behind pure peptides for fractionation or directly analysis by LC–MS.
Experimental Section
E. coli Extraction and SDS-PAGE with Deoxycholic Acid for Qualitative Studies
Lyophilized Escherichia coli strain K12 cells (EC1, Sigma-Aldrich) thrice washed with MS-grade water (J.T. Baker, Phillipsburg, NJ), were dispensed to four microfuge tubes and pelleted at 1000 g for 2 min. Each E. coli pellet (estimated ∼2 mg protein) was resuspended in 300 μL of extraction buffer (A, B, or C, Table 1A), sonicated on ice for three 10 s intervals (Sonic Dismembrator model 100, Thermo Fisher Scientific, Rockford, IL), and centrifuged at 16 000 g for 5 min. Sonication and centrifugation were repeated five times, after which debris was pelleted for 5 min at 2500 g. The four supernates were transferred to new tubes. Extraction buffers A–C (Table 1A) contained detergent, reductant, and 200 mM ammonium bicarbonate (ABC). As detergent, buffers A and B employed 12 mM DCA/12 mM N-lauroyl sarcosine, while C used 4% SDS (Sigma-Aldrich). Extraction buffer A included 10 mM TCEP as reductant, while B and C used 10 mM dithiothreitol (DTT). One of two E. coli extracts in buffer C was acetone-precipitated and resuspended in 300 μL of extraction buffer C to evaluate precipitation losses from SDS solutions. Unless specified otherwise, all reagents were obtained from Sigma-Aldrich, St. Louis, MO.
Table 1. (A) Compositions of E. coli Extraction Buffers, (B) Processing Conditions for the Eight 400 μg Aliquots of Standard Protein Mixture, (C) Filter-Aided Sample Preparation Buffer Components, and (D) E. coli Lysis Buffers Used to Evaluate eFASP with Surface Passivation.
| (A) E. coli Extraction Buffers | ||
|---|---|---|
| extraction buffer | chaotrope | reductant/buffer |
| A | 12 mM DCA, 12 mM N-lauroyl sarcosine | 10 mM TCEP, 200 mM ABC, pH 8 |
| B | 12 mM DCA, 12 mM N-lauroyl sarcosine | 10 mM DTT, 200 mM ABC, pH 8 |
| C | 4% SDS | 10 mM DTT, 200 mM ABC, pH 8 |
| (B) Standard Protein Mixture Processing Conditions | ||||||
|---|---|---|---|---|---|---|
| |
buffer composition |
|||||
| aliquot | title | suspensiona | reductionb | alkylationc | quenchingd | digestione |
| 1 | DTT/IAN | ABC | DTT | IAN | DTT | ABC |
| 2 | DCA/DTT/IAN | DCA | DTT | IAN | DTT | DCA |
| 3 | DCA/DTT/4VP | DCA | DTT | 4-VP | DTT | DCA |
| 4 | TCEP/IAN | ABC | TCEP | IAN | DTT | ABC |
| 5 | DCA/TCEP/IAN | DCA | TCEP | IAN | DTT | DCA |
| 6 | TCEP/4-VP | ABC | TCEP | 4-VP | DTT | ABC |
| 7 | DCA/TCEP/4-VP | DCA | TCEP | 4-VP | DTT | DCA |
| 8 | DCA/TCEP+DTT/4-VP | DCA | DTT/TCEP | 4-VP | DTT | DCA |
| (C) Filter-Aided Sample Preparation Solubilization, Exchange and Digestion Buffers | ||
|---|---|---|
| chaotrope/buffer | surfactant | |
| solubilization buffer A | 4% SDS, 200 mM ABC, pH 8, 7.5 mM TCEP | 0.2% DCA |
| exchange buffer A | 8 M urea, 100 mM ABC, pH 8 | none |
| exchange buffer B | 8 M urea, 100 mM Tris-HCl, pH 8 | 0.1% nOG |
| 0.1% DCA | ||
| 0.2% DCA | ||
| 0.1%DCA, 0.1% nOG | ||
| alkylation buffer A | 8 M urea, 100 mM ABC, pH 8, 50 mM IAN | |
| alkylation buffer B | 100 mM ABC, pH 8, 30 mM IAN | |
| FASP digestion buffer | 50 mM ABC | none |
| eFASP digestion buffer | 50 mM ABC | 0.1%nOG |
| 0.1% DCA | ||
| 0.2% DCA | ||
| 0.1% DCA, 0.1% nOG | ||
| 2D clean up digest buffer | 100 mM ABC, pH 8 | 0.1% DCA |
| (D) Lysis Buffer Compositions for E. coli Evaluation of eFASP on Passivated Surfaces | ||
|---|---|---|
| lysis buffer | chaotrope | reductant/buffer |
| A | 4% SDS, 0.2% nOG | 7.5 mM TCEP, 200 mM ABC, pH 8 |
| B | 4% SDS, 0.1% DCA, 0.1% nOG | 50 mM TCEP, 200 mM ABC, pH 8 |
ABC = 25 mM ABC. DCA = 0.1% DCA, 25 mM ABC.
DTT = 22 mM DTT. TCEP = 5.5 mM TCEP. DTT/TCEP = 11 mM DTT/2.8 mM TCEP.
IAN = 30 mM IAN. 4-VP = 25 mM 4-VP.
DTT = 22 mM DTT.
ABC = 25 mM ABC. DCA = 0.1% DCA, 25 mM ABC.
A 10 μg portion of each extract was devoted to SDS-PAGE. Aliquots were boiled for 5 min in NuPAGE LDS sample buffer (4×) and NuPAGE Sample Reducing Agent (10×) from Invitrogen (Carlsbad, CA) and subsequently centrifuged. Proteins were resolved on a NuPAGE Novex 4–12% Bis-Tris Gel, electrophoresed at 200 V, current-limited to 160 mA, for 90 min. The gel was stained with GelCode Blue Reagent (Thermo Fisher Scientific) and imaged on a flatbed scanner. After imaging, the gel was rinsed with water and silver-stained with the SilverQuest Staining Kit (Invitrogen) following the manufacturer’s instructions.
Processing Standard Protein Mixtures with DTT, TCEP, Iodoacetamide, and 4-VP
A standard mixture was prepared by suspending in 200 mM ABC the proteins α-casein (bovine), β-casein (bovine), enolase (yeast), apo-transferrin (human), carbonic anhydrase (bovine), and ribonuclease B (bovine) to concentrations of 6, 2, 4, 2.3, 2.5, and 2 μg/μL. Eight 400 μg aliquots of this mixture were alkylated and digested as described later (Table 1B).
Aliquots suspended in the pH 8 reducing buffers specified in Table 1B were incubated at 50 °C with shaking for 60 min, after which they were alkylated with 30 mM iodoacetamide (IAN) or 25 mM 4-VP, shaking for 30 min at 37 °C.
All alkylated samples were quenched by the addition of 200 mM DTT to a final concentration of 22 mM and then diluted 1:1 with either 25 mM ABC or 0.1% DCA in 25 mM ABC (Table 1B). Modified, sequencing-grade trypsin (Promega) was added to each sample (1:30 w/w). Digestion proceeded for 12 h on a 37 °C shaker. Aliquots (10 μg) were removed for SDS-PAGE analysis.
eFASP-Enhanced Filter-Aided Sample Preparation
eFASP without Microcon Filter Unit Passivation
Lyophilized E. coli cells (6.5 mg total protein) were washed in H2O as previously described and pelleted. The cell pellet was resuspended and incubated in solubilization buffer A (4% SDS, 0.2% DCA, 7.5 mM TCEP, 200 mM ABC, Table 1C) for 10 min at 90 °C followed by three 10 s intervals of sonication and 10 min of centrifugation at 16 000 g. Sonication and centrifugation were repeated once. Supernate aliquots (163 μg, Table 1C) were transferred to six microcentrifuge tubes. One aliquot was reserved for processing with the 2D clean up kit (GE Healthcare Biosciences, Pittsburgh, PA) and subsequent in-solution digestion (described later).
SDS buffer exchange was initiated on the other five aliquots by first diluting each with eight volumes of exchange buffer A (8 M urea, 100 mM ABC, pH 8) and depositing them onto five Microcon UF units (YM-30 30 kDa cutoff limit; Millipore, Billerica, MA). After 10 min of 14 000 g centrifugation, the filtrate was discarded, an additional 200 μL of exchange buffer A was deposited in each unit, and centrifugation resumed for 10 more minutes. This buffer addition/centrifugation step was repeated twice more.
The TCEP-reduced proteins were alkylated within the filter unit by adding alkylation buffer A (8 M urea, 50 mM IAN, and 100 mM ABC, pH 8) and incubating at 37 °C for 1 h with shaking. DTT was added to a final concentration of 50 mM to deactivate residual IAN, after which, one buffer exchange was performed with exchange buffer A, followed by three exchanges with FASP digestion buffer (50 mM ABC) or with an eFASP detergent-containing digestion buffer, that is, 50 mM ABC with 0.1% DCA, 0.2% DCA, 0.1% n-octyl glucoside (nOG), or 0.1% DCA and 0.1% nOG combined (Table 1C).
FASP or eFASP digestion buffer (100 μL) was added to each UF device, followed by 3.25 μg of trypsin (1:50 w/w). Digestion proceeded with shaking for 12 h at 37 °C. Peptides were recovered by transferring the UF filter to a new collection tube and spinning at 14 000 g for 10 min. To complete peptide recovery, we rinsed filters twice with 50 μL of 50 mM ABC that was collected by centrifugation.
Sample Preparation with the 2D Clean Up Kit
The reserved aliquot of TCEP-reduced E. coli lysate in solubilization buffer A (Table 1C) was precipitated with the 2D clean up kit according to the manufacturer’s directions. Next, the precipitate was resuspended and incubated in 22 mM DTT, 100 mM ABC at 50 °C for 60 min, followed by alkylation at 37 °C for 30 min with 50 mM IAN (alkylation buffer B). The alkylation reaction was quenched with 200 mM DTT, and the sample was diluted to a final concentration of 0.95 μg/ μL in 0.1% DCA/100 mM ABC (Table 1C). Trypsin (1:50 w/w) was added, and digestion proceeded for 12 h on a 37 °C shaker.
eFASP with Passivated Ultrafiltration Unit
UF filters and collection tubes were incubated overnight in MS-grade water, 5% v/v TWEEN-20 (T20, Sigma-Aldrich P7949), or 5% w/v ammonium lauryl sulfate (ALS, Sigma-Aldrich). Following incubation, the filter units and collection tubes were rinsed thoroughly by two immersions in 500 mL of MS-grade water.
For quantitative studies, two lysis buffers (Table 1D) were employed to prepare lysates from H2O-washed, lyophilized E. coli cells: lysis buffer A (4% SDS, 7.5 mM TCEP, 0.2% nOG, 200 mM ABC) or lysis buffer B (4% SDS, 50 mM TCEP 0.1% DCA, 0.1% nOG, 200 mM ABC). Pelleted cells were suspended in lysis buffer A (1.3 mg total protein, with 20 μg phosphorylase B (rabbit muscle; Protea Biosciences, Morgantown, WV)) or B (3 mg total protein, with 20 μg phosphorylase B) and incubated for 10 min at 90 °C. Suspensions were sonicated in three 10 s intervals followed by 10 min of centrifugation. Sonication and centrifugation were repeated once. Supernates and residual pellets were sonicated and allowed to cool to 37 °C. The reduced proteins were alkylated with 4-VP (25 mM final concentration) over 1 h at 37 °C. DTT was added to 40 mM to deactivate residual 4-VP. Aliquots from each lysate were prepared: six aliquots (163 μg) from lysis buffer A material and five aliquots (50 μg) from lysis buffer B material.
Buffer exchange to eliminate SDS from the lysates was initiated by adding eight volumes of exchange buffer B (8 M urea, 100 mM Tris-HCL (pH 8)) without or with one of four surfactants (0.1% nOG, 0.1% DCA, 0.2% DCA, or 0.1% DCA and 0.1% nOG) (Table 1C). Diluted lysates were dispensed to passivated UF filters (described later) and spun at 14 000 g for 10 min. The filtrates were discarded, and 200 μL of the appropriate version of exchange buffer B was added to each filter unit and then spun at 14 000 g for 10 min. This step was repeated twice. To remove urea, three buffer exchange steps were performed with FASP digestion buffer (50 mM ABC) or eFASP digestion buffer bearing appropriate surfactant (0.1% nOG, 0.1% DCA, 0.2% DCA, or 0.1%DCA and 0.1% nOG in 50 mM ABC). In some of our studies, we included Tris-HCl in the first steps of buffer exchange but ABC for later exchanges to minimize Tris contamination of the final peptide solution while reducing desalting steps. Certainly, the first buffer exchange does not need to be Tris-HCl, although it has been employed in some FASP protocols, and some investigators prefer employing nonvolatile buffers to control pH during alkylation.
FASP digestion buffer or eFASP digestion buffer (100 μL) was added to each Microcon filter unit, followed by trypsin (1:50 w/w). Digestion proceeded for 12 h on a 37 °C shaker. The UF filters were transferred to different collection tubes, passivated with or without their respective passivation reagents, for centrifugation to recover peptide retentate. Two wash and centrifugation steps, each with 50 μL of 50 mM ABC buffer, followed to complete the peptide recovery.
Sample Preparation for LC–MSE Analysis
DCA and nOG were removed postcleavage by acidification and PT to EA (J.T. Baker),14 but instead of simply withdrawing the lower aqueous layer,14 the upper organic layer was withdrawn and discarded, followed by two rounds adding and discarding EA to remove more surfactant, similar to the strategy Yeung et al. employed for nOG.21 Recovered peptides were dried in a SpeedVac, then resuspended in 50% methanol and vacuum-dried three more times to remove volatile salts completely. Dried peptides were suspended in 96.8% H2O/3% ACN/0.2% formic acid.
Analysis by nanoLC–MSE
nanoUPLC of peptides was performed on a Waters nanoACQUITY UPLC system equipped with a nanoACQUITY symmetry C18 180 μm × 20 mm trap column (Waters, Milford, MA) and a nanoACQUITY UPLC BEH C18 75 μm × 150 mm reversed-phase analytical column (Waters). Mobile phase A contained 0.1% formic acid in water, and mobile phase B contained 0.1% formic acid in acetonitrile. Five μL of the sample was loaded onto the trap and washed with 99% A and 1% B at 5 μL/min for 5 min and then injected onto the column in 3% mobile phase B. Peptides were eluted with a 3–40% B gradient over 60 or 90 min at 300 nL/min, followed by a 5 min rinse with 95% B. The column was re-equilibrated in 3% B for 20 min.
The lock mass compound, [Glu1]fibrinopeptide (0.5 pmol/μL, 25% ACN, 0.1% formic acid), and the analyte were each delivered at 300 nL/min to the NanoLockSpray dual electrospray ion source (Waters), which was equipped with a microcapillary for lockmass delivery and a PicoTip Emitter (New Objective, Woburn, MA) for the analyte. Two electrosprays are produced that may be selectively sampled by positioning the baffle.
The ion source was interfaced with a Waters Synapt qTOF HDMS or a Waters Xevo qTOF MS for mass spectrometric analysis. Spectra were acquired by positive nanoESI on instruments operated in V-mode (average resolution 9500). Accurate precursor and fragment ion mass data were collected over 100–2000 m/z, with acquisition alternating in 1 s intervals between low and elevated energy. MS and MSE data were continually calibrated by sampling the lock mass every 30 s.
Data Processing and Protein Identification
Raw data files processed in ProteinLynx Global Server 2.5.2 (PLGS, Waters) provided an inventory of precursor and respective product ions for searches against the HAMAP Escherichia coli database (March 2011), supplemented with entries for contaminants and protein standards.22 Database search settings employed a minimum of three peptides per protein, two missed cleavages, 4% protein false-positive rate, fixed carbamidomethyl or S-pyridylethyl modification of cysteine residues, and variable oxidation of methionine residues. Data were quantified employing the Waters Expression Informatics component of PLGS.11,13,23 (See separate Supporting Information for more details and listing of protein identifications.)
Custom software extracted protein and peptide data for comparative analysis, calculated peptide, and protein isoelectric points and hydrophobicities based on Kyte and Doolittle24 amino-acid-derived hydropathicities.
Results and Discussion
DCA Is Effective in Protein Extraction and Solubilization
A variety of surfactants and detergents at various concentrations have been tested for their impact on trypsin and Lys-C digestions. NaDOC is commonly used in lysis buffers, for example, RIPA buffer, and more recently has been found to improve protein digestion by trypsin and Lys-C.14,17 NaDOC can be quantitatively removed from a digest by acidification and transfer to EA.
Masuda et al. proposed to replace standard SDS lysis buffer with one combining a high concentration of NaDOC with sodium lauroyl sarcosine (NaLS). Less common NaLS is a strong anionic detergent shown to be removed readily by acidification and PT.16 NaDOC/NaLS lysis buffer is effective in extracting and solubilizing proteins from membranes, and, with dilution, improved trypsin digestion of those proteins. (16) After NaDOC and NaLS are removed by PT, samples must be desalted before postprocessing and MS analysis.
To evaluate the impact, when preparing buffers, of replacing the sodium salts of DCA and N-lauroyl sarcosine by the free acid and ammonium salt, respectively, we incorporated DCA into extraction buffers similar to those used by Masuda et al.16 and processed E. coli samples with each of three extraction buffers (Table 1A). Two buffers were composed of 12 mM DCA with 12 mM ammonium lauroyl sarcosine (AmLS) and either 10 mM TCEP or 10 mM DTT in 200 mM ABC. The third buffer contained 4% SDS with 10 mM DTT in 200 mM ABC. An additional sample was processed with 4% SDS/10 mM DTT/200 mM ABC, precipitated with acetone, and then resuspended in the same buffer. Each sample was run on an SDS-PAGE gel and silver-stained (Figure S1 in the Supporting Information).
Samples extracted with DCA and AmLS showed high extraction and solubilization efficiency across a broad MW range and appeared particularly good at solubilizing proteins beyond 115 kDa in size (Figure S1 in the Supporting Information). 4% SDS samples fared well, and, surprisingly, acetone precipitation affected the protein yield little, at least as assessed by 1D SDS-PAGE and silver staining, albeit a method limited in dynamic range. Precipitation losses are also less likely to be disabling from the milligram samples investigated here. Thus, DCA and AmLS are as powerful for protein extraction and solubilization as their sodium salts or as 4% SDS but without contributing involatile salts. Nevertheless, the 12.5 mM concentrations of DCA and AmLS employed here (0.49 and 0.34% w/v, respectively) were challenging to formulate due to slow dissolution at these concentrations. Future evaluations with reduced DCA/AmLS concentrations are suggested.
DCA Enhances Trypsin Digestion without Introducing Involatile Na+
Digestion of proteins with trypsin is a critical step in proteomic workflows, especially for quantitative experiments. After harsh denaturing agents (SDS, urea, and thiourea) are removed by precipitation, dialysis, or affinity chromatography, proteins must be reduced and alkylated. DTT and TCEP are commonly used to cleave disulfide bonds in preparation for alkylation. IAN is the most widely used alkylator of cysteine residues in proteomic applications, but a minority of studies have used vinylpyridine (4-VP or 2-VP). Because these Michael addition reagents may react with enzymes, they should be removed or diluted prior to enzyme addition.
While NaDOC has been shown to enhance trypsin digestion, there are no data assessing efficacy of DCA. To test the effect of DCA on trypsin activity, we tested a standard mixture of six proteins varying in molecular weight and cysteine content, processed with different combinations of reducing and alkylating agents (Table 1B). All samples were diluted 1:1 with ABC before adding trypsin. After digestion, aliquots were analyzed by SDS-PAGE (Figure S2 in the Supporting Information).
Excess residual reducing and alkylating agents, for example, the high concentrations employed here, impair trypsin activity, necessitating adequate dilution, or complete removal, prior to enzyme addition. Under the inhibited conditions in our 1:1 dilutions, enough proteolytic activity remained to digest a portion of the proteins, but significant quantities remained undigested (Figure S2 in the Supporting Information). Interestingly, the reduction/alkylation-agent-caused inhibition was compensated by the increase in trypsin activity in 0.1% DCA-containing solutions. Combining 2.8 mM TCEP with 11 mM DTT resulted in the poorest digestion efficiency, but the carbonic anhydrase, apo-transferrin, α-casein, and β-casein TCEP/DCA-protocol digestions still returned better results than DTT-protocol samples digested absent 0.1% DCA. Clearly, DCA enhances trypsin activity without contributing nonvolatile salts to the sample.
Comparing Solubilization and Digestion in FASP with or without Surfactants
One of the drawbacks of digesting in solution with facilitating additives present can be any cleanup procedures required for LC–MS compatibility. FASP benefits workflows by freeing protein samples of many pre- and post-alkylation-introduced contaminants.
Urea, present in FASP protocols through the alkylation step, is removed prior to enzymolysis. Although extending the urea presence through digestion (in 2 M urea) increases peptide IDs (∼20%), it does not greatly change the number of proteins or protein classes, for example, membrane and nuclear proteins, identified.3 Moreover, urea exposure increases the potential to artifactually carbamylate proteins. Noting sensitivity losses from exchanging urea out of the digestion buffer, we hypothesized that incorporating uniquely removable, trypsin activity-enhancing surfactants through digestion within the FASP workflow should increase the range of peptides and the overall sensitivity at which they are detected while avoiding chromatographic cleanup steps (Figure 1). DCA and nOG greatly enhance trypsin digestion in-solution and in-gel.14,25 They permit higher temperature digestions without risking cysteine, lysine, or arginine carbamylation.
Protein Identification Comparison with E. coli Lysates
Aliquots of an E. coli lysate (163 μg, Table 1C) were processed by precipitation (2D clean up kit, GE Life Sciences)/in-solution digestion, by FASP, or by eFASP. FASP samples were trypsin-digested in ABC, while eFASP-processed samples were digested in ABC with 0.1% nOG or 0.1% DCA. The precipitated sample was digested in ABC with 0.1% DCA. All digests were examined by LC–MSE.
Processing2 small quantities of sample by the standard FASP approach led to proportionally larger losses than processing by 2DCleanUpKit-0.1DCA, eFASP-0.1nOG, and eFASP-0.1DCA (Table 2). eFASP-0.1DCA delivered the best results, with 13 and 27% more protein identifications than eFASP-0.1nOG and 2DCleanUpKit-0.1DCA, respectively. While eFASP-0.1nOG did not identify as many peptides or proteins as eFASP-0.1DCA, both were significantly better than FASP and 2DCleanUpKit-0.1DCA, despite injecting twice as much product for the latter two. eFASP-0.1DCA allows for a 30% increase in Pass 1 peptide IDs (the highest quality peptides identified by PLGS that contain no variable modifications) compared with the 2DCleanUpKit-0.1DCA. These results suggest that FASP digestion or peptide recovery are greatly hindered in the absence of detergents or chaotropes.
Table 2. Analysis of an E. coli Lysate (163 μg) Processed and Digested by Traditional and Experimental Methodsa.
| peptides |
||||||
|---|---|---|---|---|---|---|
| prep method | digest buffer | quantity (μg) | proteins | pass 1 | pass 2 | masses |
| 2DCleanupKit-0.1DCA | 0.1% DCA, ABC | 2 | 344 | 1965 | 4576 | 29 972 |
| FASP | ABC | 2 | 14 | 26 | 77 | 3736 |
| eFASP-0.1nOG | 0.1% nOG, ABC | 1 | 408 | 2141 | 5568 | 31 472 |
| eFASP-0.1DCA-0.1nOG | 0.1% DCA, ABC | 1 | 471 | 2678 | 6145 | 31 744 |
| proteins identified
with specified number of TMHs |
||||
|---|---|---|---|---|
| no. TMHs | 2DCleanupKit-0.1DCA | FASP-ABC | eFASP-0.1nOG | eFASP-0.1DCA |
| 12 | 1 | 0 | 1 | 1 |
| 11 | 0 | 0 | 0 | 1 |
| 9 | 1 | 0 | 1 | 1 |
| 6 | 0 | 0 | 1 | 2 |
| 5 | 2 | 0 | 2 | 3 |
| 4 | 1 | 0 | 0 | 2 |
| 3 | 2 | 0 | 3 | 3 |
| 2 | 2 | 0 | 6 | 7 |
| 1 | 15 | 0 | 20 | 23 |
| total | 24 | 0 | 34 | 43 |
| % of all IDs | 7.0 | 0 | 8.3 | 9.1 |
Lysate was either precipitated and digested in-solution in the presence of 0.1% DCA or cleaned in a Microcon filter unit and digested in the presence of ABC (FASP), 0.1% nOG (eFASP-0.1nOG), or 0.1% DCA (eFASP-0.1DCA). Each injection (1 or 2 μg, assuming complete recovery) was analyzed by LC–MSE and searched for (a) protein and peptide content and (b) predicted transmembrane helices (TMHs) in the identified protein sequences.
Qualitative Comparison
Samples processed by eFASP-0.1DCA displayed better sensitivity and sample purity in base peak intensity (BPI) chromatograms compared with samples processed by 2DCleanUpKit-0.1DCA, despite the fact that twice as much material was analyzed from the latter. Large interference peaks in the first half of the BPI chromatogram from 2DCleanUpKit-0.1DCA suggested that contaminants were removed incompletely. Moreover, the eFASP-0.1DCA sample was superior (more complex) in the later, more hydrophobic, region of the gradient.
Comparing 0.1% DCA to 0.1% nOG eFASP revealed that both digests were exceptionally clean, but eFASP-0.1DCA showed more BPI features in the middle and later portions of the chromatographic gradient and overall larger peak intensities than eFASP-0.1nOG. This intensity disparity widens with increasing organic content in the mobile phase. eFASP-0.1DCA better reveals hydrophobic species and, perhaps, functions on a wider range of proteins than nOG.
Both the number of proteins identified and the fraction of those containing trans-membrane helices (TMHs) are affected by the choice of protocol (Table 2). Of protein identifications by eFASP-0.1DCA, 9.1% have at least 1 TMH, compared with 8.3 and 7.0% for eFASP-0.1nOG and 2DCleanUpKit-0.1DCA, respectively. The 11 TMH drug efflux protein, acriflavine resistance protein B (Uniprot AC P31224), was identified only by eFASP-0.1DCA. The loss of membrane proteins during precipitation is also highlighted by these data.
Quantitative Comparison
LC–MSE quantification compares precursor ion volumes. Our software tool, Data Extraction and Collation (DECO), assembled ion volume information for Pass 1 peptides identified in the samples. Ion volumes for peptides common to all processed samples were plotted against each other (Figure 2). Ion volumes from eFASP-0.1DCA versus those from eFASP-0.1nOG (Figure 2A) reveal groups of peptides that are more abundant under each condition; that is, the surfactants’ solubilizing abilities are not completely correlated (R2 = 0.733). Figure 2B, plotting peptide ion volumes from 2DCleanUpKit-0.1DCA against those obtained from eFASP-0.1nOG and eFASP-01DCA, reveals that the various peptide quantities recovered from the two eFASP protocols are much more similar to each other than to recoveries from the 2D Clean Up Kit. Compared with the 2DCleanUpKit-0.1DCA, eFASP protocols employing 0.1% DCA or nOG not only yield large gains in ion volumes but also increase the number of protein and peptide IDs. The reduced peptide ion volumes from the 2DCleanUpKit-0.1DCA as compared with eFASP-0.1DCA (R2 = 0.286) suggest that eFASP benefits by circumventing precipitation losses and persistent contaminants.
Figure 2.
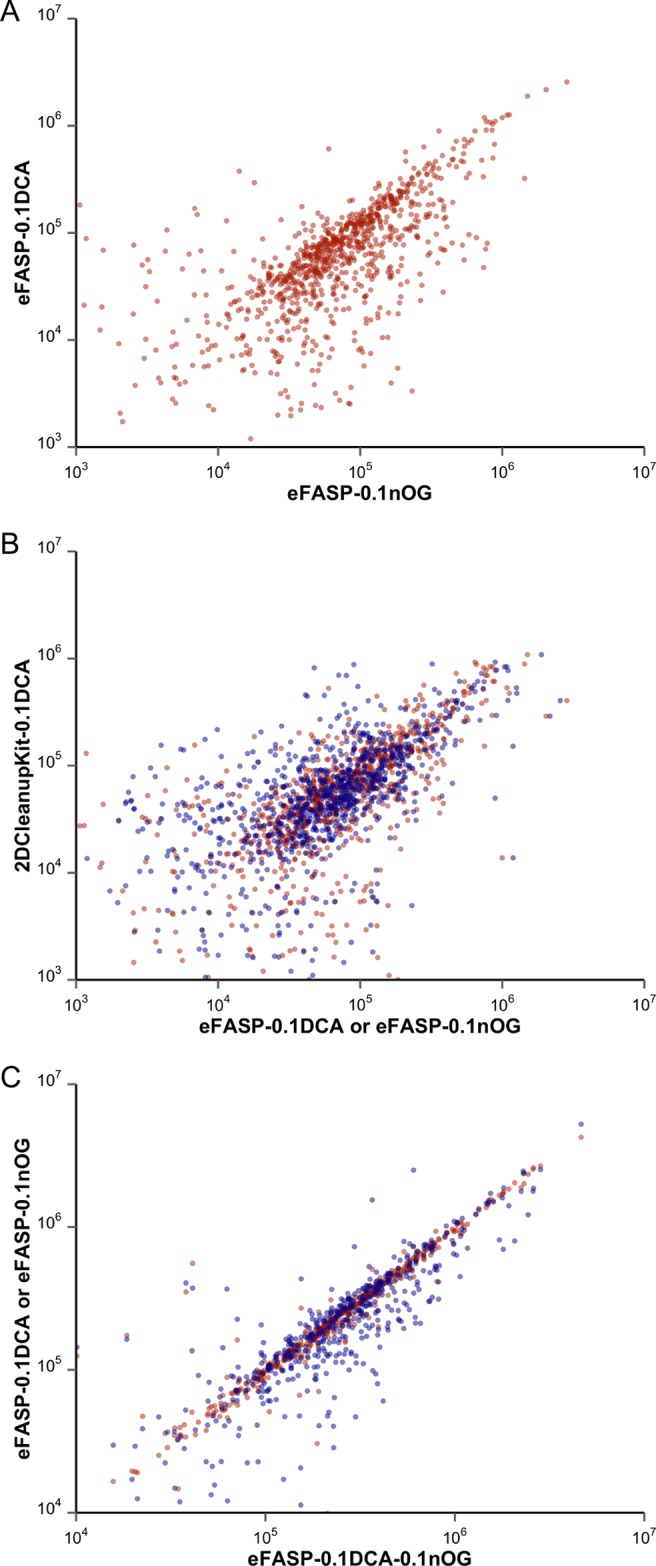
Ion volumes for peptides quantified from the protocols (A) eFASP with 0.1% DCA versus eFASP with 0.1% nOG and (B) 2DCleanUpKit-0.1DCA versus eFASP with 0.1% nOG or 0.1% DCA and (C) eFASP with 0.1% nOG (blue) or 0.1% DCA (red) versus eFASP employing 0.1% of both nOG and DCA.
Optimal DCA Concentration and Combination with nOG
Benefits from NaDOC inclusion in tryptic digests scale with detergent concentration from 0.01 to 1.0%, but the free acid is less soluble, with 0.2% being the upper limit for DCA in ABC buffer. Workflows digesting E. coli proteins with 0.1 and 0.2% DCA (eFASP-0.1DCA vs eFASP-0.2DCA) identified similar numbers of proteins and Pass 1 peptides (Table 3), and the peptides revealed at the two concentrations were highly correlated (R2 = 0.992, Figure S3 in the Supporting Information). eFASP-0.2DCA afforded a 21% increase in identified TMH proteins, as annotated by the TMHMM algorithm (Table 3), without sacrificing hydrophilic protein recovery or quantitative peptide yield. Uniprot AC P69786, containing 10 theoretical TMHs, was identified only by eFASP-0.2DCA.
Table 3. Analysis of an E. coli Lysate (50 μg) Processed by eFASP with Digestion Occurring in the Presence of 0.1% nOG, 0.1% DCA, 0.2% DCA, or the Combination of 0.1% nOG and 0.1% DCAa.
| peptides |
||||
|---|---|---|---|---|
| prep method | digest buffer | proteins | pass 1 | pass 2 |
| eFASP-0.1nOG | 0.1% nOG, ABC | 202 | 989 | 3766 |
| eFASP-0.1DCA | 0.1% DCA, ABC | 253 | 1304 | 4947 |
| eFASP-0.2DCA | 0.2% DCA, ABC | 256 | 1392 | 4842 |
| eFASP-0.1DCA-0.1 nOG | 0.1% DCA, 0.1% nOG, ABC | 249 | 1394 | 4689 |
| proteins identified
with specified number of TMHs |
|||
|---|---|---|---|
| no. TMHs | eFASP-0.1DCA | eFASP-0.2DCA | eFASP-0.1DCA-0.1nOG |
| 12 | 1 | 1 | 1 |
| 10 | 0 | 1 | 0 |
| 9 | 1 | 1 | 0 |
| 5 | 2 | 1 | 1 |
| 4 | 1 | 1 | 0 |
| 3 | 1 | 1 | 1 |
| 2 | 2 | 2 | 3 |
| 1 | 10 | 15 | 12 |
| total | 18 | 23 | 18 |
| % of all IDs | 7.1 | 9.0 | 7.2 |
Each injection (400 ng, assuming complete recovery) was analyzed by LC–MSE and searched for (a) protein and peptide content and (b) predicted TMHs in the identified protein sequences.
LC–MSE analyses of E. coli samples processed with 0.1% DCA, 0.2% DCA, and 0.1% DCA + 0.1% nOG reveal close similarities between eFASP-0.1DCA and eFASP-0.1DCA-0.1nOG, and combining them during digestion rescues deficiencies associated with 0.1% nOG alone. eFASP-0.1DCA-0.1nOG yields slightly more peptide identifications than eFASP-0.1DCA and a number equivalent to eFASP-0.2DCA (Table 3). The eFASP-0.1DCA-0.1nOG ion volume profile (Figure 2C) correlates closely to that of eFASP-0.1DCA (R2 = 0.986), and the latter method has already been shown to correlate with 0.2% DCA digestion. Proteins identified across the three conditions overlap by ∼50% (Figure 3). Although eFASP-0.1DCA-0.1nOG delivers a similar number of protein and Pass 1 peptide identifications to eFASP-0.2DCA, several proteins are only identified by eFASP-0.2DCA. Protein identification results from eFASP-0.2DCA are the most dissimilar to those from the other conditions. DCA/ABC solutions should be prepared immediately before use because DCA settles over time.
Figure 3.
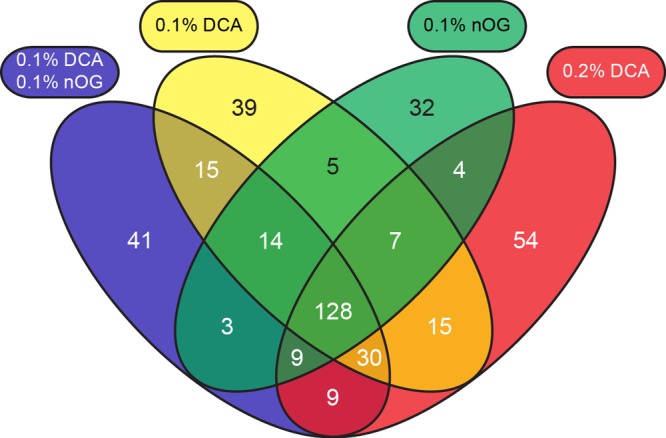
Protein identification overlap from eFASP processing of E. coli lysate with digestion buffers containing 0.1% nOG (green), 0.1% DCA (yellow), 0.2% DCA (red), and the combination of 0.1% DCA and 0.1% nOG (blue).
Grand average of hydropathy (GRAVY) values were calculated for all proteins identified from samples produced by the four eFASP variants (Figure 4). GRAVY values sum hydropathies based on the Kyte–Doolittle scale for all amino acids in a protein, divided by the amino acid length.24 Comparison of protein GRAVY values between eFASP-0.1DCA, eFASP-0.2DCA, and eFASP-0.1DCA-0.1nOG reveals that a greater number of unique proteins are identified by eFASP-0.2DCA, in concert with a larger number of challenging hydrophobic proteins.
Figure 4.
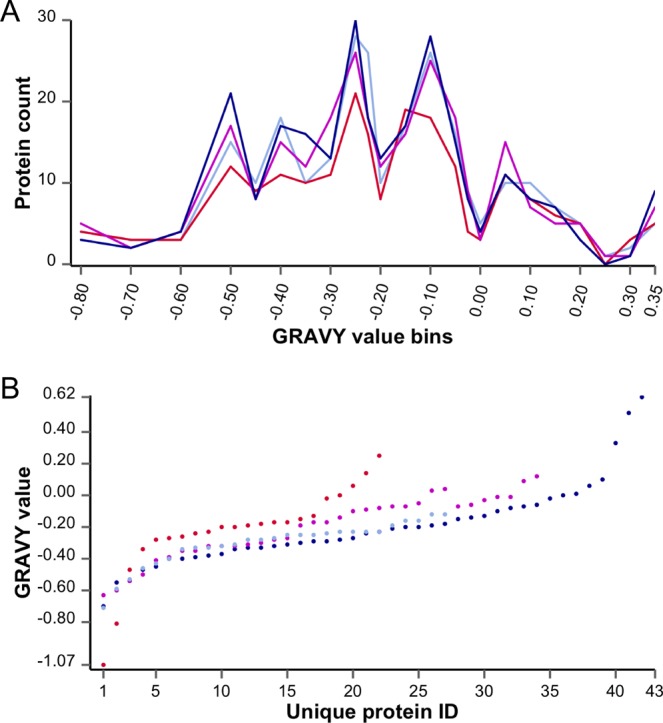
GRAVY values for proteins identified in E. coli lysates processed by eFASP using different digestion buffers. GRAVY values were calculated for (A) all proteins identified in each digest or (B) proteins unique to one digest condition. (A) GRAVY values for all proteins are binned and plotted for eFASP-0.1DCA (light blue), eFASP-0.2DCA (dark blue), eFASP-0.1DCA-0.1nOG (magenta), and eFASP-0.1nOG (red). (B) GRAVY values are plotted for proteins unique to eFASP-0.1DCA (light blue), eFASP-0.2DCA (dark blue), eFASP-0.1DCA-nOG (magenta), and eFASP-0.1nOG (red).
Passivating Plastic Surfaces
Protein and peptide binding to surfaces during sample preparation can remove proteins nonspecifically, a particular detriment to analyses employing small sample quantities. We hypothesized that nonspecific binding to the plastic surfaces and UF membrane of the Microcon filter unit were major factors in FASP’s estimated 50% sample loss.
Passivation of surfaces is not a novel concept and has been utilized for various surfaces that can bind analytes, for example, metal and plastic. Often manufacturers produce consumables optimized for minimal protein binding, for example, Eppendorf LoBind tubes, or they recommend reagents for surface pretreatment, for example, EDTA, to passivate metal surfaces.
Polyethylene glycol (PEG 20 000) and dextran T-70 have been utilized (0.5%) as carriers in FASP to increase peptide recoveries from high nanogram and low microgram range samples without compromising LC–MS analyses.26 Peptide yield improved by ∼30% and enabled analyses from <1000 cells acquired by laser capture microdissection of formalin-fixed paraffin-embedded tissues. Carriers are effective when <10 μg of starting material is available; no benefit was realized with larger amounts of starting material.26
Even when carriers are added to samples, many surfaces of the Microcon filter unit that contact peptides exclude polymeric carriers, including areas of the UF membrane and the walls of the collection tube. Additionally, spontaneous decomposition of PEG carriers in FASP contaminates downstream LC–MS analyses.26 We propose a surface passivation approach for the Microcon filter unit that avoids polymer decomposition-related contamination and reduces binding to all surfaces contacting proteins and peptides.
The Amicon Centricon manufacturer recommends that various reagents be included for use with very dilute protein samples, that is, 1% BSA, 1% IgG, 1% powdered milk, 5% T20, 5% SDS, 5% PEG, and 5% Triton-X.27 T20 and SDS are recognized choices for minimizing protein binding.
We tested high-purity T20 and ALS for passivating all surfaces of the Microcon filter units and the collection tubes, as described in the Experimental Section. Nonpassivated (H2O-incubated) or passivated (T20 or ALS) Microcon units and collection tubes were utilized to process 50 μg of E. coli by FASP, eFASP-0.1DCA, or eFASP-0.2DCA. Samples were analyzed by LC–MS in profile mode for qualitative evaluations and LC–MSE for identification and quantification.
Enhanced signal throughout the gradient was observed from samples prepared in passivated materials. By a small margin, eFASP-0.1DCA-T20 outperformed eFASP-0.1DCA-ALS, and both increased sensitivity about two-fold compared with eFASP-0.1DCA-H2O. More hydrophobic proteins were recovered from T20-passivated materials than from ALS- or nonpassivated surfaces.
In the absence of 0.2% DCA, passivating FASP surfaces with T20 increased protein identifications by 250% and peptide identifications by 300% (Table 4). In contrast, eFASP-0.2DCA processing with T20-passivated materials increased protein and peptide identification by only 12 and 7%, respectively, compared with nonpassivated materials. Peptide ion volumes correlated well for eFASP-0.2DCA processing with and without passivated materials (R2 = 0.954).
Table 4. Microcon Filter Units and Collection Tubes Were Incubated with Water or 5% T20 Overnighta.
| peptides |
|||||
|---|---|---|---|---|---|
| prep method | passivation | digest buffer | proteins | pass 1 | pass 2 |
| FASP | no | ABC | 21 | 85 | 229 |
| FASP-T20 | 5% T20 | ABC | 52 | 246 | 604 |
| eFASP-0.2DCA | no | 0.2% DCA, ABC | 252 | 1436 | 3645 |
| eFASP-0.2DCA-T20 | 5% T20 | 0.2% DCA, ABC | 284 | 1531 | 3671 |
Analysis of an E. coli lysate (50 μg) processed by eFASP with digestion occurring in the presence of ABC or 0.2% DCA. Each injection (100 ng, assuming complete recovery) was analyzed by LC–MSE and searched for (a) protein and peptide content and (b) predicted TMHs in the identified protein sequences.
Effective passivation agents, such as T20 and ALS, bind plastic surfaces to block protein and peptide binding. The minimal gains observed from prepassivation when eFASP procedures include DCA may reflect the surfactants’ ability to passivate surfaces.
Express eFASP
FASP protocols include two buffer-exchange steps before digestion. The first buffer exchange displaces SDS with urea and removes DTT prior to alkylation. The second buffer exchange removes residual IAN and urea prior to digestion. With YM-30 units, each step takes ∼45 min. We propose substituting reduction and alkylation reagents in the FASP workflow with TCEP and 4-vinylpyridine to enable one-step reduction/alkylation and increase specificity (Figure S2 in the Supporting Information, lane 9).
The alkylating agent 4-VP is highly specific for cysteine residues,28 reacting quickly and at neutral pH.29 Prolonged incubation with 4-VP rarely alkylates noncysteine residues, and the lower reaction pH assures that lysine ε-amino groups are unreactive. 4-VP is also ideal because it is reported to not be inactivated by reducing agent TCEP, allowing for one-step reduction and alkylation. TCEP and 4-VP have been used to enhance the resolution of basic proteins in 2DE experiments.30,31 Alkylating samples before applying them to the spin filter would eliminate the first series of buffer exchanges, saving time and effort. To examine the applicability of TCEP and 4-VP as replacements for DTT and IAN, we prepared two E. coli lysates with 4% SDS and either 100 mM DTT or 7.5 mM TCEP.
LC–MSE analysis of the sample processed by eFASP-0.2DCA-DTT/IAN yielded 284 protein identifications and 1531 first-pass peptides, which is ∼15% greater than those obtained for the eFASP-0.2DCA-TCEP/4VP sample (Table S1 in the Supporting Information). Further comparisons were done to address this disparity because chromatograms for the samples were similar in elution pattern and intensity.
Peptides identified in both eFASP-0.2DCA-DTT/IAN and eFASP-0.2DCA-TCEP/4VP were quantitatively compared (Figure S4 in the Supporting Information). While there are some deviations in the more abundant species, the overall correlation of peptide ion volume profiles is very good (R2 = 0.972). To assess sequence coverage of proteins in each sample, DECO was used to collect, bin, and tabulate information for all proteins identified in each sample (Figure 5). Samples processed with IAN/DTT or TCEP/4VP delivered similar sequence coverage.
Figure 5.
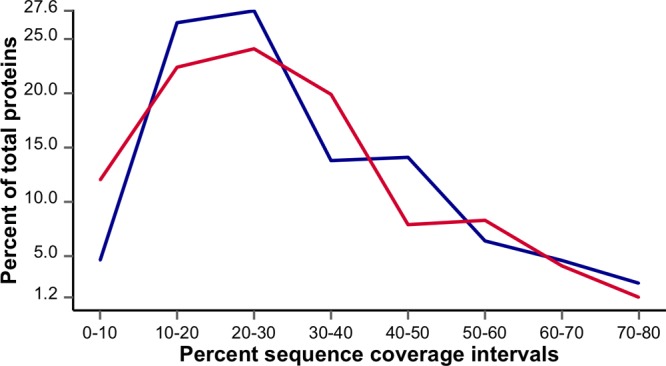
Sequence coverage for E. coli samples processed by eFASP with reduction and alkylation accomplished with either DTT/IAN (eFASP-0.2DCA-DTT/IAN, blue) or TCEP/4VP (eFASP-0.2DCA-TCEP/4VP, red).
The 104 S-pyridylethyl modified first-pass peptides identified from TCEP/4VP treated samples are far fewer than the 203 first-pass peptides identified with carbamidomethyl modifications from DTT/IAN treatment. Workflow parameters were modified to check for incomplete alkylation, but changing the S-pyridylethyl modification from fixed to variable, or removing it completely, did not improve results. These observations, in light of the high similarity between the DTT/IAN and the TCEP/4VP samples (chromatograms, sequence coverage, and identified masses), are consistent with complications in properly identifying or scoring S-pyridylethyl cysteine-containing peptides (e.g., due to the facile loss of the pyridylethyl ion, known for its characteristic ion signal at m/z 106).
Although a discrepancy remains between eFASP-0.2DCA performed with DTT/IAN and with TCEP/4VP, the presented workflow is effective in avoiding side reactions of IAN or urea32 and greatly reduces sample processing.
Conclusions
The eFASP approach incorporates alternative reagents to those of traditional FASP that quantitatively improve sensitivity, recovery, and proteomic coverage for processed samples. First, substituting 0.2% DCA for urea during eFASP digestion increases tryptic digestion efficiency for both cytosolic and membrane proteins, yet it obviates necessary cleanup steps associated with the usage of urea or the deoxycholate sodium salt. Second, prepassivating Microcon filter and collection tube surfaces with 5% T20 reduces peptide loss by 300% through precluding peptide binding. Incorporating 0.2% DCA and 5% T20 passivation into the FASP workflow produces peptides free of detergents, reductants, and alkylators as well as salts, lipids, and nucleic acids (Supplementary Protocols, eFASP: Standard in the Supporting Information). An express eFASP method variant is proposed that utilizes one-step protein reduction and alkylation with TCEP and 4-VP prior to deposition on the Microcon filter, increasing alkylation specificity and speeding processing (Supplementary Protocols, eFASP: Express in the Supporting Information). These optimized eFASP methods maintain significant advantages compared with traditional FASP and standard proteomics sample preparation methods.
Acknowledgments
This work was supported by the Ruth L. Kirschstein National Research Service Award (Grant GM007185, UCLA Cellular and Molecular Biology Training Grant, for J.E.) and the National Institutes of Health (U19 AI067769).
Supporting Information Available
Figure S1. Deoxycholic acid (DCA) and ammonium lauroyl sarcosine (AmLS) can replace SDS in lysis buffers. Figure S2. A standard protein mixture was reduced and alkylated with combinations of DTT, TCEP, IAN, and 4-VP, prior to trypsin digestion in the presence or absence of 0.1% DCA. Figure S3. Quantitative comparison of peptide yield from eFASP processing of E. coli lysate with 0.2% and 0.1% DCA in the digestion buffer. Figure S4. Quantitative comparison of peptide yield from eFASP-0.2DCA processing of E. coli lysate that was either reduced with DTT and alkylated in-filter with IAN or reduced with TCEP and alkylated in-solution with 4-VP. Table S1. E. coli lysates were prepared with solubilization buffer containing either DTT or TCEP. Supplementary Protocols for eFASP standard and express. Analysis and documentation of peptide and protein identifications. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Manza L. L.; Stamer S. L.; Ham A. L.; Codreanu S. G.; Liebler D. C. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 2005, 5, 1742–1745. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Mann M. Spin filter–based sample preparation for shotgun proteomics. Nat. Methods 2009, 6, 785–786. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zielinska D. F.; Mann M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Anal. Biochem. 2011, 410, 307–309. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zougman A.; Mann M. Combination of FASP and StageTip-Based Fractionation Allows In-Depth Analysis of the Hippocampal Membrane Proteome. J. Proteome Res. 2009, 8, 5674–5678. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Nagaraj N.; Zougman A.; Gnad F.; Mann M. Brain Phosphoproteome Obtained by a FASP-Based Method Reveals Plasma Membrane Protein Topology. J. Proteome Res. 2010, 9, 3280–3289. [DOI] [PubMed] [Google Scholar]
- Zielinska D. F.; Gnad F.; Jedrusik-Bode M.; Wisniewski J. R.; Mann M. Caenorhabditis elegans Has a Phosphoproteome Atypical for Metazoans That Is Enriched in Developmental and Sex Determination Proteins. J. Proteome Res. 2012, 8, 4039–4049. [DOI] [PubMed] [Google Scholar]
- Zielinska D. F.; Gnad F.; Wisniewski J. R.; Mann M. Precision Mapping of an In Vivo N-Glycoproteome Reveals Rigid Topological and Sequence Constraints. Cell 2010, 141, 897–907. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Mann M. Consecutive Proteolytic Digestion in an Enzyme Reactor Increases Depth of Proteomic and Phosphoproteomic Analysis. Anal. Chem. 2012, 84, 2631–2637. [DOI] [PubMed] [Google Scholar]
- Tran B. Q.; Hernandez C.; Waridel P.; Potts A.; Barblan J.; Lisacek F.; Quadroni M. Addressing trypsin bias in large scale (phospho)proteome analysis by size exclusion chromatography and secondary digestion of large post-trypsin peptides. J. Proteome Res. 2011, 10, 800–811. [DOI] [PubMed] [Google Scholar]
- Silva J. C.; Gorenstein M. V.; Li G.; Vissers J. P.; Geromanos S. J. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 2006, 5, 144–156. [DOI] [PubMed] [Google Scholar]
- Geromanos S. J.; Vissers J. P. C.; Silva J. C.; Dorschel C. A.; Li G.; Gorenstein M. V.; Bateman R. H.; Langridge J. I. The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics 2009, 9, 1683–1695. [DOI] [PubMed] [Google Scholar]
- Silva J. C.; Denny R. R.; Dorschel C. C.; Gorenstein M. V.; Li G.; Richardson K. K.; Wall D. D.; Geromanos S. J. Simultaneous qualitative and quantitative analysis of the Escherichia coli proteome: a sweet tale. Mol. Cell. Proteomics 2006, 5, 589–607. [DOI] [PubMed] [Google Scholar]
- Masuda T.; Tomita M.; Ishihama Y. Phase Transfer Surfactant-Aided Trypsin Digestion for Membrane Proteome Analysis. J. Proteome Res. 2008, 7, 731–740. [DOI] [PubMed] [Google Scholar]
- Masuda T.; Sugiyama N.; Tomita M.; Ishihama Y. Microscale Phosphoproteome Analysis of 10,000 Cells from Human Cancer Cell Lines. Anal. Chem. 2011, 83, 7698–7703. [DOI] [PubMed] [Google Scholar]
- Masuda T.; Saito N.; Tomita M.; Ishihama Y. Unbiased Quantitation of Escherichia coli Membrane Proteome Using Phase Transfer Surfactants. Mol. Cell. Proteomics 2009, 8, 2770–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Zhou T.; Cao R.; Liu Z.; Shen J.; Chen P.; Wang X.; Liang S. Evaluation of the Application of Sodium Deoxycholate to Proteomic Analysis of Rat Hippocampal Plasma Membrane. J. Proteome Res. 2006, 5, 2547–2553. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Zhou J.; Bi D.; Chen P.; Wang X.; Liang S. Sodium-deoxycholate-assisted tryptic digestion and identification of proteolytically resistant proteins. Anal. Biochem. 2008, 377, 259–266. [DOI] [PubMed] [Google Scholar]
- Proc J. L.; Kuzyk M. A.; Hardie D. B.; Yang J.; Smith D. S.; Jackson A. M.; Parker C. E.; Borchers C. H. A Quantitative Study of the Effects of Chaotropic Agents, Surfactants, and Solvents on the Digestion Efficiency of Human Plasma Proteins by Trypsin. J. Proteome Res. 2010, 9, 5422–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Liu Y.; Li J.; Zhao Y.; He Q.; Han W.; Chen P.; Wang X.; Liang S. Evaluation and optimization of removal of an acid-insoluble surfactant for shotgun analysis of membrane proteome. Electrophoresis 2010, 31, 2705–2713. [DOI] [PubMed] [Google Scholar]
- Yeung Y.; Nieves E.; Angeletti R. H.; Stanley E. R. Removal of detergents from protein digests for mass spectrometry analysis. Anal. Biochem. 2008, 382, 135–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Vissers J. P. C.; Silva J. C.; Golick D.; Gorenstein M. V.; Geromanos S. J. Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 2009, 9, 1696–1719. [DOI] [PubMed] [Google Scholar]
- Silva J. C.; Denny R.; Dorschel C. A.; Gorenstein M.; Kass I. J.; Li G.; McKenna T.; Nold M. J.; Richardson K.; Young P.; Geromanos S. Quantitative Proteomic Analysis by Accurate Mass Retention Time Pairs. Anal. Chem. 2005, 77, 2187–2200. [DOI] [PubMed] [Google Scholar]
- Kyte J.; Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [DOI] [PubMed] [Google Scholar]
- Katayama H.; Nagasu T.; Oda Y. Improvement of in-gel digestion protocol for peptide mass fingerprinting by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2001, 15, 1416–1421. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Ostasiewicz P.; Mann M. High Recovery FASP Applied to the Proteomic Analysis of Microdissected Formalin Fixed Paraffin Embedded Cancer Tissues Retrieves Known Colon Cancer Markers. J. Proteome Res. 2011, 10, 3040–3049. [DOI] [PubMed] [Google Scholar]
- Passivation of Amicon Centricon Concentrators for Improved Recovery; Millipore: Bedford, MA, 1999; pp 1–2.
- Sebastiano R.; Citterio A.; Lapadula M.; Righetti P. G. A new deuterated alkylating agent for quantitative proteomics. Rapid Commun. Mass Spectrom. 2003, 17, 2380–2386. [DOI] [PubMed] [Google Scholar]
- Righetti P. G. Real and imaginary artefacts in proteome analysis via two-dimensional maps. J. Chromatogr., B 2006, 841, 14–22. [DOI] [PubMed] [Google Scholar]
- Bai F.; Liu S.; Witzmann F. A. A “de-streaking” method for two-dimensional electrophoresis using the reducing agent tris(2-carboxyethyl)-phosphine hydrochloride and alkylating agent vinylpyridine. Proteomics 2005, 5, 2043–2047. [DOI] [PubMed] [Google Scholar]
- Liu S.; Bai F.; Witzmann F.. Destreaking Strategies for Two-Dimensional Electrophoresis. In Separation Methods in Proteomics; CRC: Boca Raton, FL, 2006; pp 207–217. [Google Scholar]
- Boja E. S.; Fales H. M. Overalkylation of a protein digest with iodoacetamide. Anal. Chem. 2001, 73, 3576–3582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.