

Abstract
Cancers originating from digestive system account for 290,000 or ~20% of all new cancer cases annually in the US. We previously developed paclitaxel-loaded tumor-penetrating microparticles (TPM) for intraperitoneal (IP) treatment of peritoneal tumors [1–3]. TPM is undergoing NIH-supported IND-enabling studies for clinical evaluation. The present study evaluated the hypothesis that TPM, via inducing apoptosis and expanding the interstitial space, promotes the delivery and transfection of lipid vectors containing siRNA. The in vivo model was the metastatic human Hs766T pancreatic tumor that, upon IP injection, produced widely distributed solid tumors and ascites in the peritoneal cavity in 100% animals. The target gene was survivin, an anti-apoptotic protein induced by chemotherapy and associated with metastases and poor prognosis of patients with gastric and colorectal cancer. The siRNA carrier was pegylated liposomes comprising cationic and neutral lipids plus a fusogenic lipid (PCat). PCat-loaded with survivin siRNA (PCat-siSurvivin) was active in cultured cells (decreased survivin mRNA and protein levels, reduced cell clonogenicity, enhanced paclitaxel activity), but lost its activity in vivo; this difference is consistent with the well-known problem of inadequate delivery and transfection of siRNA in vivo. In comparison, single agent TPM prolonged animal survival and, as expected, induced survivin expression in tumors. Addition of PCat-siSurvivin reversed the TPM-induced survivin expression and enhanced the antitumor activity of TPM. The finding that in vivo survivin knockdown by PCat-siSurvivin was successful only when it was given in combination with TPM provides the proof-of-concept that tumor priming promotes the delivery and transfection of liposomal siRNA. The data further suggest the TPM/PCat-siSurvivin combination as a potentially useful chemo-gene therapy for peritoneal cancer.
Keywords: Intraperitoneal Chemotherapy, Peritoneal Carcinomatosis, siRNA, Microparticles, Paclitaxel, Pancreatic Cancer
1. Introduction
Cancer originating from the digestive system, including cancers of the esophagus, gallbladder, liver, bill duct, pancreas, stomach, small intestine, colon and rectum, accounts for 290,000 new cases or about 20 percent of all newly diagnosed cancers annually in the United States. Peritoneal carcinomatosis due to loco-regional spread is common and presented with 10 to 30% of cases at the time of initial surgery [4,5].
The survival advantage of intraperitoneal (IP) therapy of peritoneal cancer was first demonstrated 17 years ago and has since been confirmed in multiple clinical trials [6–11]. However, several difficulties, including local toxicity and ineffectiveness against bulky tumors, have prohibited IP therapy from becoming a common standard of care. To address these problems, we developed paclitaxel-loaded tumor penetrating microparticles (TPM). TPM comprises two components, one component releases the drug rapidly to produce tumor priming (i.e., induce apoptosis and thereby expand the interstitial space) to promote the transport of the remaining microparticles and the second component releases the drug slowly to provide sustained drug exposure [12,13]. Studies in mice with metastatic IP pancreatic and ovarian tumors show superior delivery and activity of TPM compared to the intravenous paclitaxel solutions used off-label in previous IP studies [1–3]. Based on its promising preclinical activity, TPM is currently undergoing NIH-supported IND-enabling studies, in preparation for clinical evaluation.
We have since made several observations that suggest it may be possible to further improve the efficacy of IP TPM. We found that tumor priming using either paclitaxel or TPM enhances the delivery and efficacy of doxorubicin-loaded liposomes (85 nm diameter given intravenously), the penetration and dispersion of fluorescent latex beads (2 μm diameter given IP) in peritoneal tumors, and the penetration and dispersion of siRNA-loaded cationic liposomes in 3-dimensional tumor cell spheroids and tumor histocultures [1,13,14]. Based on these observations, we hypothesized that TPM can be used to promote the delivery of siRNA vectors.
siRNA produces post-transcriptional gene silencing and represents a promising approach to reverse the chemotherapy-induced upregulation of chemoresistance genes. Its therapeutic potential is indicated by the demonstration of gene silencing in patients with melanoma [15]. siRNA, typically comprising 20–27 nucleotides, has high negative surface charge that is unfavorable for cellular uptake. A popular siRNA carrier is cationic liposome that forms multilamellar structures with the anionic siRNA upon mixing; the resulting lipoplex protects siRNA from degradation and facilitates cellular uptake. Lipoplex, however, are relatively large in size (e.g., >100 nm diameter) which may impede effective delivery in vivo [16–18].
The present study evaluated whether TPM promotes the delivery of lipid vectors containing siRNA and whether the efficacy of TPM is enhanced by combining it with siRNA targeting the anti-apoptotic survivin. Survivin was selected because (a) it is highly and selectively expressed in a majority of human cancers including gastric and colorectal cancer and peritoneal metastases, compared with most differentiated adult normal tissues including liver [19–25], (b) high survivin expression is correlated with more extensive peritoneal metastases (depth of invasion, lymph node metastasis) and shorter overall survival of patients with gastric and colorectal cancer [26–29], (c) high level survivin expression correlates with chemo/radio-resistance in multiple tumor types and its inhibition enhances cell death induced by chemo/radio-therapy [30,31], and (d) paclitaxel induces survivin expression whereas survivin siRNA (siSurvivin) significantly increases paclitaxel-induced cell death [32].
The present study used pegylated cationic liposomes (PCat) as siRNA carrier and measured the effectiveness of in vivo delivery and survivin knockdown in a metastatic IP pancreatic human H766T xenograft tumor model. This model was selected due to its relative ease; IP injection of Hs766T cells produced carcinomatosis and ascites in the peritoneal cavity, and deaths in 100% animals with median survival time (MST) of 24 days post-implantation [3].
2. Materials and methods
2.1. Chemicals and reagents
Poly-lactide-co-glycolide (PLGA) polymers were purchased from Boehringer Ingelheim (Ingelheim, Germany), paclitaxel from Hande Tech (Houston, TX), lipids (1,2-dioleoyl-3-trimethylammoniumpropane or DOTAP, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine or DOPE, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] or DSPE-PEG, cholesterol) from Avanti Polar Lipids, Inc. (Alabaster, AL, USA), cefotaxime sodium from Hoechst-Roussel (Somerville, NJ), poly(vinyl alcohol) or PVA from Sigma Chemical (St. Louis, MO), chromatography solvents from Fisher Scientific (Fair Lawn, NJ), gentamicin from Solo Pak Laboratories (Franklin Park, IL), and all other cell culture supplies from Life Technologies (Grand Island, NY). Survivin siRNA (siSurvivin, human specific, #6351), survivin monoclonal antibody (71G4B7E), and caspase-3 polyclonal antibody (#9661) were purchased from Cell Signaling Technology (Danvers, MA), survivin polyclonal antibody (C-19, sc-8807) from Santa Cruz Biotechnology (Santa Cruz, CA), and Ki67 (MM1 clone) antigen kit from Novocastra Lab (Newcastle, UK). All chemicals and reagents were used as received.
2.2. Preparation of TPM
TPM comprised two paclitaxel-loaded PLGA microparticles, i.e., Priming and Sustaining TPM. The Priming TPM, which releases paclitaxel rapidly (70% of dose in one day under sink condition [1]), was prepared using 50:50 LA:GA with an inherent viscosity of 0.12 dl/g in chloroform. The Sustaining TPM, which releases paclitaxel slowly (1% of dose in one day under sink condition [1]), was prepared using 75:25 LA:GA with an inherent viscosity of 0.39 dl/g in chloroform. Preparation of paclitaxel-loaded PLGA microparticles was as previously described [1,2]. Briefly, PLGA and paclitaxel were co-dissolved in methylene chloride and emulsified in PVA. After evaporation of methylene chloride, microparticles were collected, washed and lyophilized. Particle size was determined using LS230 laser diffraction particle size analyzer (Beckman Coulter, Brea, CA); the average volume-based diameter was 4.8 μm. Blank PLG microparticles were prepared similarly, except without paclitaxel.
2.3. Preparation of liposomes and lipoplex
PCat liposome comprised two neutral lipids (DOPE, cholesterol), one cationic lipid (DOTAP), and one pegylated lipid (DSPE-PEG2000) at ratio of 50:30:19:1 of DOTAP:Cholesterol:DOPE:DSPE-PEG. DOTAP provides the positive charge. Cholesterol was used to increase the liposome stability. DOPE was used to increase the elasticity of the liposome bilayer and to promote the fusion of liposome with cell membrane and with endosomal membrane [33]. DSPE-PEG was used to provide stealth property and to improve the in vivo stability of liposomes. Briefly, lipids were combined and dissolved in 9:1 v/v mixture of chloroform and methanol (e.g., 10 mg lipids in 5 mL chloroform/methanol). The organic phase was evaporated under nitrogen to yield a thin lipid film, which was dried under vacuum in a desiccator for approximately 12 hr. The lipids were then hydrated with RNase-free buffer (1 ml per 10 mg of lipids) at 60°C for 2 hr, with gentle vortexing every 20 min. The resulting liposomal suspension was passed through a liposome extruder with a 100 nm membrane to obtain liposomes of about 100 nm diameter.
siRNA lipoplex was formed by gently mixing liposomes with siRNA solution (between 2 μM to 10 μM) at room temperature, in a 1:4 siRNA-to-DOTAP charge ratio. Particle size distribution and zeta-potential were measured using Zetasizer Nano ZS90 (Malvern, Westborough, MA). Pilot studies showed that more than 90% of siRNA remained in the PCat lipoplex after 24 hr.
The in vitro and in vivo toxicities of blank PCat carrier (no siRNA) or PCat-siRNA lipoplex were compared to another cationic liposome that had been given to humans, i.e., 50:50 DOTAP:Cholesterol (referred to as DC liposomes [34,35]). DC liposomes were prepared using the procedures described above.
2.4. Effect of paclitaxel on siRNA transfection in vitro
As TPM contains paclitaxel, we evaluated whether paclitaxel affected the siRNA transfection in human pancreatic Hs766T cells (gift from Dr. Byoungwoo Ryu, John Hopkins Medical Institute, Baltimore, MD). Cells were maintained in DMEM medium containing 10% fetal bovine serum, 2 mM L-glutamine, antibiotics (90 μg/ml gentamicin and 90 μg/ml cefotaxime sodium, or 100 units/mL penicillin and 100 ug/mL streptomycin), at 37°C in a humidified atmosphere with 5% CO2. All transfections were carried out in nearly confluent cultures (over 80% confluence); this was to align with the cell density found in vivo.
Cells were treated with vehicle (0.1% ethanol), paclitaxel, PCat loaded with survivin siRNA (PCat-siSurvivin), paclitaxel plus PCat loaded with non-target siRNA (PCat-siNT), or paclitaxel plus PCat-siSurvivin. The study used 10 nM paclitaxel, which was near its IC50 in the clonogenicity assay (see Results). Antitumor activity was measured using the clonogenic assay (cells seeded 24 hr post-siRNA treatment; colonies counted 10 day later). Survivin mRNA and protein levels were measured using RT-PCR and western blot, respectively. Briefly, total RNA was extracted using the Rneasy Plus Mini Kit (Qiagen, Valencia, CA), and reversed transcribed to cDNA using qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD). Real-time RT-PCR (triplicate samples, 5 μL cDNA per reaction) was performed with PerfeCTa® MultiPlex qPCR SuperMix (Quanta Biosciences) in CFX96 Real-Time PCR Detection Systems (Bio-Rad, Hercules, CA). The prime probe sets (Integrated DNA Technologies) used were: survivin, forward: 5’ – CAACCGGACGAATGCTTTT – 3’; reverse: 5’ – AAGAACTGGCCCTTCTTGGA – 3’; probe: 5’ – /5HEX/CCAGATGAC/ZEN/GACCCCATAGAGGAA/3IABkFQ/ - 3’; GAPDH, forward: 5’ – AATCCCATCACCATCTTCCAG – 3’; reverse: 5’ – AAATGAGCCCCAGCCTTC – 3’; probe: 5’ – /5Cy5/CCAGCATCGCCCCACTTGATTTT/3IAbRQSp/ – 3’. The multiplex thermal reaction program was: 3 min at 95°C, 40 cycles of 15 sec at 95°C and 1 min at 61°C. Survivin mRNA expression relative to GAPDH control expression was calculated using the ΔΔCt-method [36]. For protein analysis, cells were collected and lysed, and protein concentrations were measured using BCA kit (Thermo Fisher, Rockford, IL). Cell lysates were run on a Bolt 12% Bis-Tris Plus Gel (Life technologies, Carlsbad, CA). The gel was then transferred to a nitrocellulose membrane using the Trans-blot Turbo Transfer System (Bio-Rad). Blots were probed with survivin and β-actin primary antibodies. The secondary antibody was a HRP-conjugated anti-rabbit antibody. Chemiluminescence was detected and the band intensity analyzed using BioRad Molecular Imager ChemiDoc XRS. Protein levels were normalized to β-actin loading control.
2.5. Toxicity of DC and PCat liposomes in vitro and in vivo
The in vitro cytotoxicity of blank PCat and DC liposomes (i.e., no siRNA) was studied in three human cancer cell lines: ovarian SKOV3, breast MCF7 and prostate PC3, using the microtetrazolium assay [37]. The in vivo toxicity used PCat or DC liposomes containing survivin siRNA, and was studied in immunocompetent CD1 mice (female, 6–8 weeks old; Charles River, Wilmington, MA). Animals were cared in accordance with institutional guidelines. Anesthesia was attained using inhalation Isoflurane® (diluted to 15% in light mineral oil, Abbott Lab, North Chicago, IL), and animals were euthanized using an Isoflurane® overdose. Mice were given intravenous injection of lipoplex via the tail vein; the survivin siRNA dose was 1 nmole and the amount of DOTAP was 12 mg. On day 7 after dosing, liver function tests (i.e., alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels) were performed, and the spleen weight/size was determined.
2.6. In vivo antitumor activity
The antitumor activity study used human xenograft tumors implanted in immunodeficient athymic Nu/Nu mice (female, 6–8 weeks old; Charles River). We used the metastatic IP Hs766T pancreatic tumor model described earlier [1,2]. Briefly, IP implantation of tumor cells (20 millions) resulted in multiple solid tumor nodules throughout the peritoneal cavity and/or aggregates of tumor cells in ascites fluid and death in 100% animals.
Treatments were initiated 10 days after tumor implantation, or at approximately 40% of MST of control animals. All treatments were administered by IP injections, between 10:00 am-3:00 pm. TPM were suspended in 0.01% Tween 80 in physiological saline. Animals were given a single TPM dose (80 mg/kg paclitaxel, 1:1 ratio of Priming and Sustaining TPM; injection volume was 0.033 ml/g body weight). PCat-siSurvivin containing 1 nmole survivin siRNA (0.5 ml/mouse per dose) was administered twice at 72 and 120 hr post-TPM administration. A pilot study in mice (n=3) established that PCat-siNT, given intravenously, had no pharmacological effect on survivin expression, cell proliferation or apoptosis, or animals, and was not further studied. Animals were randomized according to body weight into four treatment groups: (a) blank TPM (without paclitaxel) plus blank PCat liposomes; (b) blank TPM plus PCat-siSurvivin; (c) TPM plus blank PCat liposomes; (d) TPM plus PCat-siSurvivin.
The study used TPM containing 80 mg/kg paclitaxel-equivalent (40 mg/kg each for Priming and Sustaining TPM). This TPM dose was selected because our earlier studies have shown that 40 mg/kg Priming TPM promoted the penetration of fluorescent latex beads (2 μm diameter) into peritoneal tumors [1].
Treatment efficacy was measured as increase in life span (ILS) relative to untreated controls. ILS was calculated as (MSTtreated group-MSTcontrol group) divided by MSTcontrol group × 100%, where MST, median survival time, was expressed in days post-treatment. Tumor-free cures refer to animals that did not show tumor nodules in the peritoneal cavity on the last study day which was 240 days (250 days post-implantation), or 14-times the MST of controls. Moribundity was indicated by slowing or loss of righting reflex and inability to eat or drink. In addition, ascites development led to an increase in body weight with abdominal distension. Moribund-mice or mice with weight gain of 20% over one week or when the weight exceeded 130% of the initial value with visible abdominal swelling, indicative of ascites fluid build-up, were euthanized.
Post-mortem autopsy was performed. Deaths that occurred within 10 days post-treatment and accompanied by significant body weight loss (>15%) were considered toxicity-related deaths. Deaths that occurred at later times and were accompanied by the presence of tumors and ascites in the peritoneal cavity were considered due to disease progression.
2.7. In vivo cellular and molecular pharmacodynamic endpoints
We studied the effects of TPM, alone or in combination with PCat-siSurvivin, on survivin expression, apoptosis and proliferation. Tumor nodules were excised, fixed with 10% formalin, processed, embedded in paraffin, and cut into 6 μm histologic sections. Tumor sections were analyzed for total survivin levels, apoptotic cells (caspase 3 staining), and proliferating cells (Ki67 staining) using previously described immunohistochemical methods [38]. Briefly, paraffin-embedded tumor sections were deparaffinized, rehydrated, and boiled in 10 mM sodium citrate buffer (pH 6) for 15 min for antigen retrieval. After washing, quenching of endogenous peroxidase activity with 3% hydrogen peroxide for 5 min, and blocking with 10 mg/ml bovine serum albumin, tissue samples were incubated with primary antibody (1:500 dilution for Ki67, 1:200 dilution for survivin, and 1:100 dilution for caspase-3) at room temperature for 2 hr, followed by incubation with biotinylated secondary antibody for 30 min and then with streptavidin-peroxidase complex for 30 min. 3,3’-Diaminobenzidine was the chromogen. The histological sections were counterstained with hematoxylin, dehydrated, and mounted using Permount® (Fisher). For negative controls, the primary antibody was replaced with blocking reagent.
Microscopic fields (400× magnification) from the most intensively labeled areas of the tumor were randomly selected. At least 6 microscopic fields per sample were analyzed. On average, about 500 tumor cells were counted per sample for control groups, and about 200 cells for TPM-treated groups. The lower numbers in TPM-treated groups were due to their smaller tumor size and lower cell density. For quantitative image analysis of survivin expression levels in solid tumors, the integrated density or sum of all the pixels of survivin staining was quantified using ImageJ software (NIH, Bethesda, MD), and normalized to the total cell number in the same field.
2.8. Analysis of paclitaxel
Paclitaxel was measured using high performance liquid chromatography, as previously described [1]. For paclitaxel in TPM, TPM were dissolved in methylene chloride and analyzed without extraction. For paclitaxel in tumors, the excised tumors were rinsed free of residual drug-containing peritoneal fluid using distilled water, blot dried, and homogenized with ice-cold PBS; the homogenates were extracted with ethyl acetate using cephalomannine as the internal standard. The stationary phase consisted of a cleanup column (Nova-Pak C8, 4 μm particle, 3.9 mm × 75 mm; Waters, Milford, MA) and an analytical column (Bakerbond C18, 5 μm particle, 4.6 mm × 250 mm; Mallinckrodt Baker, Phillipsburg, NJ). Samples were injected into the cleanup column and eluted with a mobile phase (37.5% acetonitrile in water) at a rate of 1.0 ml/min. The analytical mobile phase (49% acetonitrile in water) was passed through the analytical column at 1.2 ml/min. Paclitaxel was detected by UV absorbance at 229 nm; the detection limit was 1 ng per injection.
2.9. Statistical Analysis
Survival data of different treatment groups were analyzed with the logrank and Wilcoxon test, and Kaplan-Meier plot, using SAS (Cary, NC). Wilcoxon test emphasizes early differences with more weight for early failures, while logrank test has equal weight to all [39]. Differences in antitumor activity among multiple treatment groups were analyzed with the Tukey test after ANOVA. For comparisons of in vitro drug activity and in vivo tumor pharmacokinetics, differences between two treatment groups were analyzed using two-tailed unpaired Student’s t-test. p values of less than 0.05 were considered statistically significant. To our knowledge, previous studies on protein or mRNA knockdown, while typically describing the % decrease on multiple time points, did not provide analysis of the duration of knockdown that is statistically significant compared to, e.g., untreated controls (see examples in [40,41]). We used the following method to establish the duration of knockdown of mRNA and protein expression; this was defined as the duration over which the levels in control and treated groups were separated by at least one 95% confidence interval (CI). First, we obtained the standard-error-of-the-mean (SEM) values of 3 repeated experiments of protein or mRNA levels. Plotting the SEM values against the respective protein or mRNA levels showed increasing SEM with increasing protein or mRNA levels; the best-fitting linear equation was then used to interpolate the representative SEM for each protein or mRNA signal. Next, we constructed bands with width of ½ * 95% CI for individual data points. For example, for n of 3 per time point, the 95% CI was 4.30 * SEM and the band width was 2.15 * SEM. These procedures yielded mRNA or protein levels with their corresponding CI bands, for untreated control and siRNA-treated groups (see Figure 3). The duration over which the bands for the control and treated groups were separated, determined graphically, represented the duration of significant mRNA/protein knockdown.
Figure 3. Effect of paclitaxel on PCat-siRNA transfection.
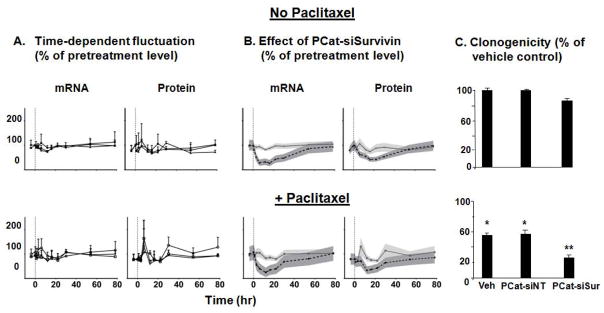
HS766T cells (over 80% confluence) were treated with paclitaxel (10 nM) for 4 hr, followed by PCat-siSurvivin treatment for 6 hr. (A) Fluctuation of mRNA and protein levels in the control groups over time: vehicle or single agent paclitaxel (diamonds), empty PCat vector (squares), PCat-siNT (triangles). 0 hr (indicated by vertical dashed line) refers to initiation of treatments. Baseline samples were taken at 4 hr before initiation of treatment. (B) Effect of PCat-siSurvivin on survivin mRNA and protein levels. The mean values of experimental data of the PCat-siSurvivin groups were compared to the combined results of the three control groups shown in Figure 3A. Symbols are mean values of experimental data (3 experiments). Shaded areas are the 95% confidence intervals of the experimental data. 0 hr (indicated by vertical dashed line) refers to initiation of treatments. (C) In vitro antitumor activity was determined using clonogenic assay. The PCat-siSurvivin concentration was 100 nM. *p<0.01 compared to vehicle control or PCat-siNT control, without paclitaxel treatment (top panel). **p<0.01 compared to single agent PCat-siSurvivin (top panel), and to single agent paclitaxel or paclitaxel+PCat-siNT combination (bottom panel).
3. Results
3.1. Characterization of liposomal siRNA
Figure 1 shows the size and surface charge (zeta potential) of PCat- and DC-liposomes and their lipoplex with siSurvivin. Blank liposomes were stable at 4°C, with no significant changes in particle size and zeta potential after two months. Upon mixing with siRNA, the respective particle size increased from 109±1.3 to 221±1.7 nm and from 111±0.3 to 209±4.8 nm, and the zeta potential reduced from 66±1.7 to 47±3.0 mV and from 75±4.3 to 54±2.4 mV for PCat- and DC-liposomes, respectively (mean±SD, n=3 experiments).
Figure 1. Characterization of liposomes and siRNA lipoplex.

Top panel: DC liposomes and DC-siSurvivin lipoplex. Bottom panel: PCat liposomes and PCat-siSurvivin lipoplex. Solid lines: liposomes. Dotted lines: lipoplex.
Figure 2 compares the in vitro and in vivo toxicity of blank PCat- and DC-liposomes, alone or with siSurvivin. Blank PCat-liposomes were consistently less toxic in three human cancer cells (>10-fold lower). In immunocompetent CD1 mice, PCat-siSurvivin did not alter the liver function and did not cause spleen enlargement (p>0.1), whereas DC-siSurvivin RNA treatment significantly reduced liver function and enlarged the spleen (p<0.05).
Figure 2. In vitro and in vivo toxicity of liposomes and siRNA lipoplex.
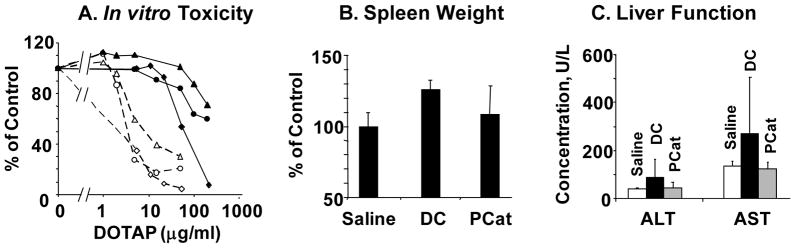
(A) In vitro toxicity. Monolayer cultured ovarian SKOV (diamond), breast MCF7 (circle), and prostate PC3 (triangle) cells were treated with PCat (solid line) or DC (dashed line) liposomes for 6 hr, and cell viability was measured at 72 hr after treatment using MTT assay (n=4). (B) Spleen weight. Immunocompetent CD1 mice were given intravenous injections of physiological saline, PCat-siSurvivin or DC-siSurvivin (n=5 per group). The siSurvivin dose was 1 nmole and the DOTAP dose was 0.12 mg, per injection. Spleen was removed 7 days after treatment and weighed. (C) Liver functions. Animals were treated as above. Blood samples were obtained via cardiac puncture and liver function tests (i.e., alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels) were performed.
3.2. Transfection and antitumor activity of PCat-siRNA in cultured cells
The transfection efficiency of PCat-siSurvivin was measured by monitoring the survivin mRNA and protein levels and the treatment-induced cytotoxicity. The effect of paclitaxel, the active pharmaceutical ingredient in TPM, was also studied. The study comprised three control groups (culture medium, blank PCat vector, PCat-siNT) and the PCat-siSurvivin treated group, with or without paclitaxel treatment (total of 8 groups).
Figure 3 shows the survivin mRNA and protein levels. In general, the control groups (total of six) were indistinguishable; the average mRNA levels showed time-dependent fluctuations over 78 hr of between 70–120% of the baseline level whereas the average protein levels showed greater fluctuation of between 60–170% (Figure 3A).
To facilitate inter-group comparison, the data in the three control groups within each of the two arms of with or without paclitaxel were combined to construct the average mRNA/protein levels and the corresponding 95% confidence intervals (Figure 3B). The averaged control data were compared with the data in the PCat-siSurvivin treated group. The comparisons showed that PCat-siSurvivin significantly suppressed the mRNA level, beginning at 2 hr. The maximum reduction of 60–75% was maintained between 6 to 16 hr, followed by gradual recovery to the pretreatment level at 78 hr. The only difference due to paclitaxel co-treatment was a slightly shorter duration of statistically significant mRNA suppression compared to without paclitaxel (24 vs. 40 hr). Changes in survivin protein levels generally mirrored changes in mRNA levels. The PCat-siSurvivin-treated group showed significantly reduced protein levels, beginning at 5 hr. The maximum reduction of 45–65% was maintained between 12–22 hr, followed by recovery to the pretreatment level at 54 hr. Similar to the observation for the mRNA level, the paclitaxel co-treatment yielded a slight shorter duration of statistically significant protein knockdown compared to without paclitaxel (25 vs. 30 hr).
With respect to antitumor activity, single agent PCat-siNT had no effect on clonogenicity, whereas single agent PCat-siSurvivin yielded a slight but statistically significant inhibition (14±2%) (Figure 3C). In comparison, single agent paclitaxel was more effective (45±3% inhibition), and the combination of paclitaxel and PCat-siSurvivin was the most effective (74±4% inhibition). The above data collectively indicate PCat-SiSurvivin produced survivin knockdown and inhibited the clonogenicity of tumor cells in vitro.
3.3. Effect of TPM on PCat-siSurvivin transfection in vivo
The transfection efficiency of PCat-siSurvivin in vivo was monitored by measuring the treatment efficacy and protein levels in IP tumors.
Figure 4A shows the photographs of animals on day 11 after treatment. The control group (blank TPM plus blank PCat) and the single agent PCat-siSurvivin group showed ascites development (indicated by abdomen distension with 20% weight gain over 1 week). In contrast, the TPM group showed substantially less ascites (i.e., less abdomen distension and less weight gain). Adding PCat-siSurvivin to TPM nearly abolished ascites development.
Figure 4. Antitumor activity in vivo.

Antitumor activity was evaluated in immunodeficient mice bearing IP Hs766T human pancreatic xenograft tumors. All treatments were administered by IP injections. Day 0 represents the day of treatment initiation, which corresponded to 10 days post-tumor implantation. MST is expressed in days post-treatment. The total paclitaxel dose in TPM microparticles was 80 mg/kg (1:1 ratio of Priming and Sustaining TPM, see text). PCat-siSurvivin was administered in two doses (1 nM siRNA per dose), at 72 and 120 hr after TPM. The control group received either blank liposomes (no siRNA) or blank microparticles (no paclitaxel); the results were not different and were combined. (A) Photographs of animals obtained on day 11 after treatment. (B) Kaplan-Meier curves. *p<0.01 (both log rank and Wilcoxon) compare to Control and siSurvivin. **p=0.03 (Wilcoxon) and p=0.06 (logrank) compare to TPM.
Figure 4B shows the Kaplan-Meier analysis of animal survival. Both control and PCat-siSurvivin groups showed MST of 17 days (27 days post-implantation), indicating PCat-siSurvivin had no appreciable activity. In comparison, TPM alone significantly prolonged the MST to 25 days (35 days post-implantation) with 47% of ILS. Adding PCat-siSurvivin to TPM further increased MST to 52 days (62 days post-implantation) with 205% of ILS (Wilcoxon p=0.03 and logrank p=0.06 compared to TPM). The TPM/PCat-siSurvivin combination group yielded higher tumor-free cures (6/24 or 25%), compared to TPM alone (2/25 or 8%); however, the difference is not significant (p=0.11, Chi square test).
Figure 5 shows the micrographs of tumors stained for total survivin, Ki67, and caspase 3, and the corresponding quantitative image analysis results. Single agent PCat-siSurvivin had no effect on these cellular and molecular endpoints. TPM significantly inhibited proliferation, induced apoptosis, and enhanced the survivin protein level. The increased survivin protein level is consistent with the literature reports that chemotherapeutic agents including paclitaxel induce survivin expression [42–44]. These cellular and molecular effects of TPM were maintained for at least 6 days. Addition of PCat-siSurvivin to TPM significantly reduced the survivin protein level and significantly enhanced the TPM-induced antiproliferation and apoptosis (p<0.05 for all three effects).
Figure 5. TPM enhanced functionality of PCat-siSurvivin in animal tumors on molecular and cellular levels.

Immunodeficient mice were implanted with tumors and treated as described in the Figure 4 legend. Tumors were excised and processed for immunohistochemical staining and quantitative image analysis of survivin, Ki67, and caspase-3. *p<0.05 between TPM and TPM+PCat-siSurvivin combination.
With respect to treatment toxicity, single agent PCat-siSurvivin did not cause additional body weight loss compared to untreated controls. TPM treatment resulted in a mild body weight loss (<7%), as would be expected due to the known toxicity of paclitaxel. In comparison, adding PCat-siSurvivin showed similar body weight loss as TPM and did not enhance the toxicity of TPM (p=0.5).
3.4. Improved TPM activity was not due to the changes in tumor pharmacokinetics of paclitaxel
Analysis of paclitaxel concentrations in tumor tissues showed no difference in in animals receiving single agent TPM or the combination of TPM with PCat-siSurvivin (Figure 6). This data indicates the improved efficacy was not due to increases in paclitaxel concentrations in tumors.
Figure 6. PCat-siSurvivin did not alter tumor pharmacokinetics of TPM.
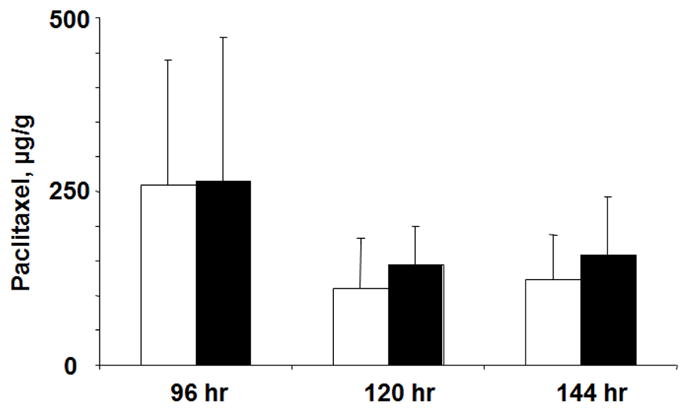
Animals were treated as described in Figure 4. Paclitaxel concentrations in tumor tissues were analyzed; the results showed no difference in paclitaxel concentrations for animals receiving single agent TPM or the TPM+PCat-siSurvivin combination (p=0.97, 0.50 and 0.60 for the 96, 120 and 144 hr samples, respectively).
4. Discussion
Cationic liposomes are popular siRNA carriers. The cationic lipid forms lipoplex with the anionic siRNA and thereby improves siRNA stability and facilitates cellular binding and internalization [18,45–47]. The current study used PCat, a 4-component, positively charged, pegylated lipid siRNA vector. The pegylated DSPE was to provide stealth property in vivo, the cationic DOTAP to achieve cellular internalization, and the fusogenic lipid (DOPE) to destabilize the endosomal membrane and promote siRNA release. The design of PCat was based in part on our previous study on the quantitative relationship between surface charge, pegylation, and endocytosis; the results demonstrate (a) the cell membrane binding and internalization of liposomes show a positive triphasic relationship with surface charge and a negative biphasic relationship with pegylation (breakpoint at 2% pegylation), (b) the negative consequences of pegylation were partially offset by increasing charge, and (c) the equations to optimize liposome stealthness and internalization [48]. The selected PCat contained 1% pegylated DSPE and had a positive surface charge with zeta potential of +66 mV (reduced to +47 mV upon complexing with siRNA). After complexation, PCat-siSurvivin showed broader size distribution. This could be due to the presence of the fusogenic lipid DOPE, as shown in an earlier study [49]. PCat was significantly less toxic compared to another DOTAP-based liposome (50:50 DOTAP:cholesterol) that has been safely administered to humans [34,35], both in cultured cells and in animals. The lower toxicity of PCat is likely due to the inclusion of 1% DSPE-PEG2000, as shown in an earlier study [50], and/or the lower surface cationic charge density.
The in vitro results show that PCat was an efficient siSurvivin carrier in cultured pancreatic Hs766T cells, producing survivin mRNA and protein knockdown and reducing tumor cell clonogenicity. However, under in vivo conditions, PCat-siSurvivin alone did not produce protein knockdown in tumors and had no antitumor activity in animals. The lack of in vivo activity of single agent PCat-siSurvivin in spite of its in vitro activity is consistent with the well-known difficulty in delivering and achieving successful transfection of siRNA in solid tumors in vivo (e.g., [14,18]).
In comparison, paclitaxel showed significant antitumor activity in cultured Hs766T cells, and the paclitaxel-loaded TPM similarly was active in mice bearing IP Hs766T tumors. TPM induced survivin expression in tumors, as previously observed for paclitaxel [32,51]. In tumor-bearing animals, adding PCat-siSurvivin to TPM reversed the drug-induced survivin upregulation, enhanced the apoptosis and antiproliferation drug effects in tumors, and extended the survival time. Because single agent PCat-siSurvivin was inactive in vivo, the enhanced activity for the TPM+PCat-siSurvivin combination indicates synergy, and the ability of TPM to enable the survivin knockdown by PCat-siSurvivin in vivo indicates TPM co-treatment improved the in vivo delivery and transfection of PCat-siSurvivin. The latter is consistent with our previous finding that TPM, by producing tumor priming, promoted the penetration of fluorescent latex beads (2 μm diameter) into peritoneal tumors whereas blank particles had no effect [1].
Paclitaxel, a drug with broad spectrum activity, is used to treat multiple types of solid tumors including ovarian, lung and breast cancer, but has not been used to treat gastrointestinal cancers. On the other hand, results of a recently completed phase III trial established that paclitaxel, in the form of albumin-stabilized paclitaxel nanoparticles (Abraxane®) and given in combination with the standard-of-care gemcitabine therapy, significantly prolonged the survival of chemo-naïve patients with metastatic pancreatic cancer [52].
In summary, the synergy between TPM and PCat-siSurvivin provides the proof-of-concept that TPM-mediated tumor priming improves the delivery and activity of siRNA therapeutics in vivo, and supports the development of the combination of TPM and PCat-siRNA for IP treatment of tumors that reside in the peritoneal cavity and show high survivin expression, e.g., liver, gastric, colorectal, and pancreatic cancer [26–29,53].
Acknowledgments
This work is supported in part by research grants R43CA134047 (J. Wang), RO1CA123159 (D. Cole) and RO1CA158300 (G. Wientjes and J. Au) from National Cancer Institute, and RO1EB015253 (J. Au and G. Wientjes) from National Institute of Biomedical Imaging and Bioengineering, NIH, DHHS.
Abbreviations used
- Blank TPM or blank liposomes
microparticles or liposomes not loaded with drug or siRNA
- DC
liposomes comprising DOTAP:cholesterol in 50:50 ratio
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DOTAP
1,2-dioleoyl-3-trimethylammoniumpropane
- DSPE-PEG
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]
- GA
glycolide
- ILS
increase in life span
- IP
intraperitoneal
- LA
lactide
- MST
median survival time
- PCat
liposomes comprising DOTAP:cholesterol:DOPE:DSPE-PEG in 50:30:19:1 ratio
- PLGA
poly(D,L-lactide-co-glycolide)
- siNT
non-target siRNA
- siSurvivin
survivin siRNA
- TPM
tumor penetrating microparticles loaded with paclitaxel
- CI
confidence interval
- SEM
standard error of the mean
Footnotes
Disclosure:
J.L.-S. Au, M.G. Wientjes, Z. Lu and J. Wang have ownership interests in the siRNA delivery technology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lu Z, Tsai M, Lu D, Wang J, Wientjes MG, Au JL. Tumor-penetrating microparticles for intraperitoneal therapy of ovarian cancer. J Pharmacol Exp Ther. 2008;327:673–682. doi: 10.1124/jpet.108.140095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai M, Lu Z, Wang J, Yeh TK, Wientjes MG, Au JL. Effects of carrier on disposition and antitumor activity of intraperitoneal Paclitaxel. Pharm Res. 2007;24:1691–1701. doi: 10.1007/s11095-007-9298-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai M, Lu Z, Wientjes MG, Au JL. Paclitaxel-loaded polymeric microparticles: Quantitative relationships between in vitro drug release rate and in vivo pharmacodynamics. J Control Release. 2013;172:737–744. doi: 10.1016/j.jconrel.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.del Castillo CF, Warshaw L. Peritoneal metastases in pancreatic carcinoma. Hepatogastroenterology. 1993;40:430–432. [PubMed] [Google Scholar]
- 5.Harmon RL, Sugarbaker PH. Prognostic indicators in peritoneal carcinomatosis from gastrointestinal cancer. Int Semin Surg Oncol. 2005;2:3. doi: 10.1186/1477-7800-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alberts DS, Liu PY, Hannigan EV, O'Toole R, Williams SD, Young JA, Franklin EW, Clarke-Pearson DL, Malviya VK, DuBeshter B. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N Engl J Med. 1996;335:1950–1955. doi: 10.1056/NEJM199612263352603. [DOI] [PubMed] [Google Scholar]
- 7.Carafa M, Santucci E, Lucania G. Lidocaine-loaded non-ionic surfactant vesicles: characterization and in vitro permeation studies. Int J Pharm. 2002;231:21–32. doi: 10.1016/s0378-5173(01)00828-6. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354:34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- 9.Gadducci A, Carnino F, Chiara S, Brunetti I, Tanganelli L, Romanini A, Bruzzone M, Conte PF. Intraperitoneal versus intravenous cisplatin in combination with intravenous cyclophosphamide and epidoxorubicin in optimally cytoreduced advanced epithelial ovarian cancer: a randomized trial of the Gruppo Oncologico Nord-Ovest. Gynecol Oncol. 2000;76:157–162. doi: 10.1006/gyno.1999.5677. [DOI] [PubMed] [Google Scholar]
- 10.Markman M, Bundy BN, Alberts DS, Fowler JM, Clark-Pearson DL, Carson LF, Wadler S, Sickel J. Phase III trial of standard-dose intravenous cisplatin plus paclitaxel versus moderately high-dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small-volume stage III ovarian carcinoma: an intergroup study of the Gynecologic Oncology Group, Southwestern Oncology Group, and Eastern Cooperative Oncology Group. J Clin Oncol. 2001;19:1001–1007. doi: 10.1200/JCO.2001.19.4.1001. [DOI] [PubMed] [Google Scholar]
- 11.Verwaal VJ, van Ruth S, de Bree E, van Sloothen GW, van Tinteren H, Boot H, Zoetmulder FA. Randomized trial of cytoreduction and hyperthermic intraperitoneal chemotherapy versus systemic chemotherapy and palliative surgery in patients with peritoneal carcinomatosis of colorectal cancer. J Clin Oncol. 2003;21:3737–3743. doi: 10.1200/JCO.2003.04.187. [DOI] [PubMed] [Google Scholar]
- 12.Kuh HJ, Jang SH, Wientjes MG, Weaver JR, Au JL. Determinants of paclitaxel penetration and accumulation in human solid tumor. J Pharmacol Exp Ther. 1999;290:871–880. [PubMed] [Google Scholar]
- 13.Lu D, Wientjes MG, Lu Z, Au JL. Tumor priming enhances delivery and efficacy of nanomedicines. J Pharmacol Exp Ther. 2007;322:80–88. doi: 10.1124/jpet.107.121632. [DOI] [PubMed] [Google Scholar]
- 14.Wong HL, Shen Z, Lu Z, Wientjes MG, Au JL. Paclitaxel tumor-priming enhances siRNA delivery and transfection in 3-dimensional tumor cultures. Mol Pharm. 2011;8:833–840. doi: 10.1021/mp1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Au JL, Jang SH, Wientjes MG. Clinical aspects of drug delivery to tumors. J Control Release. 2002;78:81–95. doi: 10.1016/s0168-3659(01)00488-6. [DOI] [PubMed] [Google Scholar]
- 17.Jang SH, Wientjes MG, Lu D, Au JL. Drug delivery and transport to solid tumors. Pharm Res. 2003;20:1337–1350. doi: 10.1023/a:1025785505977. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Lu Z, Wientjes MG, Au JL. Delivery of siRNA Therapeutics: Barriers and Carriers. AAPS J. 2010;12:492–503. doi: 10.1208/s12248-010-9210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li F. Survivin study: what is the next wave? J Cell Physiol. 2003;197:8–29. doi: 10.1002/jcp.10327. [DOI] [PubMed] [Google Scholar]
- 20.O'Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97:13103–13107. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Connor DS, Wall NR, Porter AC, Altieri DC. A p34(cdc2) survival checkpoint in cancer. Cancer Cell. 2002;2:43–54. doi: 10.1016/s1535-6108(02)00084-3. [DOI] [PubMed] [Google Scholar]
- 22.Sarela AI, Verbeke CS, Ramsdale J, Davies CL, Markham AF, Guillou PJ. Expression of survivin, a novel inhibitor of apoptosis and cell cycle regulatory protein, in pancreatic adenocarcinoma. Br J Cancer. 2002;86:886–892. doi: 10.1038/sj.bjc.6600133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sui L, Dong Y, Ohno M, Watanabe Y, Sugimoto K, Tokuda M. Survivin expression and its correlation with cell proliferation and prognosis in epithelial ovarian tumors. Int J Oncol. 2002;21:315–320. [PubMed] [Google Scholar]
- 24.Tonini G, Vincenzi B, Santini D, Scarpa S, Vasaturo T, Malacrino C, Coppola R, Magistrelli P, Borzomati D, Baldi A, Antinori A, Caricato M, Nuzzo G, Picciocchi A. Nuclear and cytoplasmic expression of survivin in 67 surgically resected pancreatic cancer patients. Br J Cancer. 2005;92:2225–2232. doi: 10.1038/sj.bjc.6602632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsuruma T, Hata F, Torigoe T, Furuhata T, Idenoue S, Kurotaki T, Yamamoto M, Yagihashi A, Ohmura T, Yamaguchi K, Katsuramaki T, Yasoshima T, Sasaki K, Mizushima Y, Minamida H, Kimura H, Akiyama M, Hirohashi Y, Asanuma H, Tamura Y, Shimozawa K, Sato N, Hirata K. Phase I clinical study of anti-apoptosis protein, survivin-derived peptide vaccine therapy for patients with advanced or recurrent colorectal cancer. J Transl Med. 2004;2:19–29. doi: 10.1186/1479-5876-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang YJ, Qi WX, He AN, Sun YJ, Shen Z, Yao Y. The Prognostic Value of Survivin Expression in Patients with Colorectal Carcinoma: A Meta-analysis. Jpn J Clin Oncol. 2013;43:988–995. doi: 10.1093/jjco/hyt103. [DOI] [PubMed] [Google Scholar]
- 27.Montorsi M, Maggioni M, Falleni M, Pellegrini C, Donadon M, Torzilli G, Santambrogio R, Spinelli A, Coggi G, Bosari S. Survivin gene expression in chronic liver disease and hepatocellular carcinoma. Hepatogastroenterology. 2007;54:2040–2044. [PubMed] [Google Scholar]
- 28.Tsuburaya A, Noguchi Y, Yoshikawa T, Saito A, Doi C, Okamoto T, Fukuzawa K. An anti-apoptosis gene, survivin and telomerase expression in gastric cancer. Hepatogastroenterology. 2002;49:1150–1152. [PubMed] [Google Scholar]
- 29.Wang ZN, Xu HM, Jiang L, Zhou X, Lu C, Zhang X. Expression of survivin mRNA in peritoneal lavage fluid from patients with gastric carcinoma. Chin Med J (Engl ) 2004;117:1210–1217. [PubMed] [Google Scholar]
- 30.Guan HT, Xue XH, Dai ZJ, Wang XJ, Li A, Qin ZY. Down-regulation of survivin expression by small interfering RNA induces pancreatic cancer cell apoptosis and enhances its radiosensitivity. World J Gastroenterol. 2006;12:2901–2907. doi: 10.3748/wjg.v12.i18.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kami K, Doi R, Koizumi M, Toyoda E, Mori T, Ito D, Kawaguchi Y, Fujimoto K, Wada M, Miyatake S, Imamura M. Downregulation of survivin by siRNA diminishes radioresistance of pancreatic cancer cells. Surgery. 2005;138:299–305. doi: 10.1016/j.surg.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Ling X, Bernacki RJ, Brattain MG, Li F. Induction of survivin expression by taxol (paclitaxel) is an early event, which is independent of taxol-mediated G2/M arrest. J Biol Chem. 2004;279:15196–15203. doi: 10.1074/jbc.M310947200. [DOI] [PubMed] [Google Scholar]
- 33.Simoes S, Moreira JN, Fonseca C, Duzgunes N, de Lima MC. On the formulation of pH-sensitive liposomes with long circulation times. Adv Drug Deliv Rev. 2004;56:947–965. doi: 10.1016/j.addr.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 34.M. D. Anderson Cancer Center, ClinicalTrial. gov. Identifier- NCT00059605, Phase I study of intravenous DOTAP: Cholesterol-Fus1 in patients with advanced non-small cell lung cancer (NSCLC) Previously treated with chemotherapy. ClinicalTrials.Gov. 2007 [Google Scholar]
- 35.Lu C, Stewart DJ, Lee JJ, Ji L, Ramesh R, Jayachandran G, Nunez MI, Wistuba II, Erasmus JJ, Hicks ME, Grimm EA, Reuben JM, Baladandayuthapani V, Templeton NS, McMannis JD, Roth JA. Phase I clinical trial of systemically administered TUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PLoS One. 2012;7:e34833. doi: 10.1371/journal.pone.0034833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 38.Yu CC, Woods AL, Levison DA. The assessment of cellular proliferation by immunohistochemistry: a review of currently available methods and their applications. Histochem J. 1992;24:121–131. doi: 10.1007/BF01047461. [DOI] [PubMed] [Google Scholar]
- 39.Kalbfleisch JD, Prentice RL. The Statistical Analysis of Failure Time Data. New York: John Wiley; 1980. [Google Scholar]
- 40.Bartlett DW, Davis ME. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006;34:322–333. doi: 10.1093/nar/gkj439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strapps WR, Pickering V, Muiru GT, Rice J, Orsborn S, Polisky BA, Sachs A, Bartz SR. The siRNA sequence and guide strand overhangs are determinants of in vivo duration of silencing. Nucleic Acids Res. 2010;38:4788–4797. doi: 10.1093/nar/gkq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lei Y, Geng Z, Guo-Jun W, He W, Jian-Lin Y. Prognostic significance of survivin expression in renal cell cancer and its correlation with radioresistance. Mol Cell Biochem. 2010;344:23–31. doi: 10.1007/s11010-010-0525-3. [DOI] [PubMed] [Google Scholar]
- 43.Rodel F, Sprenger T, Kaina B, Liersch T, Rodel C, Fulda S, Hehlgans S. Survivin as a prognostic/predictive marker and molecular target in cancer therapy. Curr Med Chem. 2012;19:3679–3688. doi: 10.2174/092986712801661040. [DOI] [PubMed] [Google Scholar]
- 44.Zaffaroni N, Pennati M, Colella G, Perego P, Supino R, Gatti L, Pilotti S, Zunino F, Daidone MG. Expression of the anti-apoptotic gene survivin correlates with taxol resistance in human ovarian cancer. Cell Mol Life Sci. 2002;59:1406–1412. doi: 10.1007/s00018-002-8518-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest. 2007;117:3623–3632. doi: 10.1172/JCI33494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oh YK, Park TG. siRNA delivery systems for cancer treatment. Adv Drug Deliv Rev. 2009;61:850–862. doi: 10.1016/j.addr.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 47.Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Adv Drug Deliv Rev. 2009;61:721–731. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, Wang J, Gao Y, Zhu J, Wientjes MG, Au JL. Relationships between liposome properties, cell membrane binding, intracellular processing, and intracellular bioavailability. AAPS J. 2011;13:585–597. doi: 10.1208/s12248-011-9298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muller JP, Aytar BS, Kondo Y, Lynn DM, Abbott NL. Incorporation of DOPE into Lipoplexes formed from a Ferrocenyl Lipid leads to Inverse Hexagonal Nanostructures that allow Redox-Based Control of Transfection in High Serum. Soft Matter. 2012;8:2608–2619. doi: 10.1039/C2SM00047D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santel A, Aleku M, Keil O, Endruschat J, Esche V, Fisch G, Dames S, Loffler K, Fechtner M, Arnold W, Giese K, Klippel A, Kaufmann J. A novel siRNA-lipoplex technology for RNA interference in the mouse vascular endothelium. Gene Ther. 2006;13:1222–1234. doi: 10.1038/sj.gt.3302777. [DOI] [PubMed] [Google Scholar]
- 51.Stauber RH, Mann W, Knauer SK. Nuclear and cytoplasmic survivin: molecular mechanism, prognostic, and therapeutic potential. Cancer Res. 2007;67:5999–6002. doi: 10.1158/0008-5472.CAN-07-0494. [DOI] [PubMed] [Google Scholar]
- 52.patel J. Drug combination extends late-stage pancreatic cancer survival in Phase III clinical trial. Expert Rev Clin Pharmacol. 2013;6:100. [PubMed] [Google Scholar]
- 53.Kami K, Doi R, Koizumi M, Toyoda E, Mori T, Ito D, Fujimoto K, Wada M, Miyatake S, Imamura M. Survivin expression is a prognostic marker in pancreatic cancer patients. Surgery. 2004;136:443–448. doi: 10.1016/j.surg.2004.05.023. [DOI] [PubMed] [Google Scholar]