

Abstract
Selective derivatization of solvent-exposed cysteine residues in peptides and proteins is achieved by brief irradiation of an aqueous solution containing 3-(hydroxymethyl)-2-naphthol derivatives (NQMPs) with 350 nm fluorescent lamp. NQMP can be conjugated with various moieties, such as PEG, dyes, carbohydrates, or possess a fragment for further selective derivatization, e.g., biotin, azide, alkyne, etc. Attractive features of this labeling approach include an exceptionally fast rate of the reaction and a requirement for low equivalence of the reagent. The NQMP-thioether linkage is stable under ambient conditions, survives protein digestion and MS analysis. Irradiation of NQMP-labeled protein in a dilute solution (<40 μM) or in the presence of a vinyl ether results in a traceless release of the substrate. The reversible biotinylation of bovine serum albumin, as well as capture and release of this protein using NeutrAvidin Agarose resin beads has been demonstrated.
Introduction
Site-specific modification of proteins provides unique opportunities for modifying the properties and expanding the function of these biomolecules.1,2 Among other applications, site-specific modification has been employed for installing tags to study protein trafficking, to alter bioavailability and pharmacokinetics by PEGylation, or for selective drug delivery by attachment of pharmaceuticals. In addition, many proteins undergo co- or post-translational modification thereby altering their biochemical role and introduction of such moieties by synthetic means offers exciting opportunities to access well-defined macromolecules for activity studies.3,4
High nucleophilicity of the thiol moiety of cysteine has long been used for installing a wide variety of functionalities.1,5 Cysteine has a relatively low natural abundance and most of the thiol groups are involved in the formation of intra-protein disulfide bonds. As a result, solvent-exposed free cysteines are rather uncommon in wild-type proteins.6 A unique cysteine residue can, however, be readily introduced by site directed mutagenesis providing opportunities for installing novel functionalities.7,8 Approaches commonly employed for cysteine derivatization include alkylation with bromo- and iodoacetyl derivatives, mixed disulfide formation, or Michael additions to substituted maleimides.7,9 Some of these method allows for the subsequent removal of the installed tag under different conditions.10 Alternatively, cysteine can be converted into dehydroalanine, which then is used for thiol-ene conjugations.11
o-Quinone methides (oQMs) are very susceptible to nucleophilic attack due to the significant positive charge on the methylene carbon atom.12,13 Their reactivity towards DNA is well documented,12,14 while interaction with proteins and peptides is virtually unknown. There are few reports that isomeric p-quinone methides, which are approximately two orders of magnitude less reactive than oQMs,15 alkylate proteins.16 Labeling occurred mostly at cysteines residues, however some nucleophilic amino acid side chains were also tagged.
Here, we report photochemical derivatization of peptides and proteins based on selective reaction of photochemically generated 2-napthoquinone-3-methides (oNQMs, 2) with solvent-exposed cysteine residues (Scheme 1). oNQMs (2) are generated by the efficient photochemical dehydration (Φ = 0.20) of 3-(hydroxymethyl)-2-naphthols (NaphthoQuinone Methide Precursors, NQMP 1, Scheme 1). Attractive features of the labeling approach include an exceptionally fast rate of the reaction and a requirement of low equivalence of the reagent for the quantitative functionalization of available Cys residues. Furthermore, it is shown that photolysis of NQMP-caged substrates in the absence of labeling reagent restores free cysteines and potential utility of this approach is demonstrated by a pull down and subsequent release of a protein.
Scheme 1.
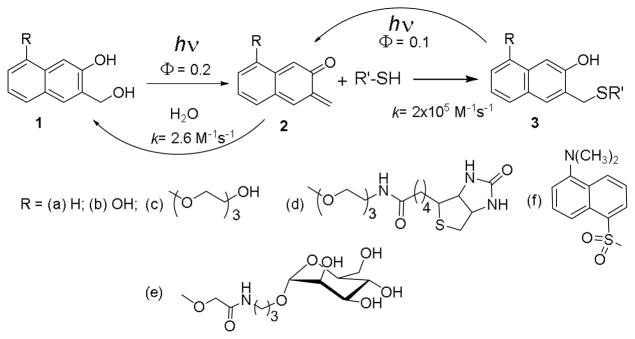
Photochemical alkylation and release of thiol-containing substrates using 300 or 350 nm light.
Results and Discussion
The lifetime of oNQM (2a) in neutral aqueous solutions is only 7 ms as it rapidly adds water (kH2O ~ 2.6 M−1s−1) to regenerate starting NQMP (1a).17 Among endogenous nucleophiles only thiols are reactive enough (kRSH ~ 2.2 × 105 M−1s−1)17 to outcompete water in Michael addition to oNQM. The thioether (3) produced in the reaction of thiols with oNQM is hydrolytically stable but can be quantitatively cleaved under 300 or 350 nm irradiation back to 2 with 10% quantum yield (Scheme 1).18,19
The 3-hydroxy-2-naphthalenemethanol chromophore (1a) has two major absorption bands in UV region above 210 nm: at λmax= 275 nm (log ε = 4.06) and at λmax= 324 nm (log ε = 3.70) Introduction of an alkoxy substituent at the 5-position of the chromophore (e.g., 1c,19 Scheme 1) results in a ca. 10 nm bathochromic shift of both bands. The longer wavelength absorbance band extends past 360 nm making this chromophore suitable for activation using 350 nm fluorescent lamp or 355 nm emission from frequency tripled Nd:YAG laser. o-Naphthoquinone methide precursors 1a–d also show significant fluorescence with a short lifetime (ΦFl = 0.230±0.002 with τFL ~ 7 ns for 1a.17 The emission spectrum of 1a in aqueous solutions contains two major bands at 360 nm and 423 nm. While the parent NQMP 1a and 6-(hydroxymethyl)naphthalene-1,7-diol 1b have rather low aqueous solubility, triethylene glycol (TEG-NQMP, 1c), biotin (Biotin-NQMP, 1d),20 and mannoside (Mann-NQMP, 1e)21 derivatives are freely soluble in wholly aqueous solutions up to at least 5 mM concentrations. 300 or 350 nm irradiation of NQMP 1a–f results in efficient dehydration and the formation of o-naphthoquinone methides (oNQM 2a–e).
The selectivity of oNQM towards nucleophilic amino acid residues commonly present in proteins was tested using three nonapeptide Ac-Cys-Lys-Tyr-Trp-Gly-Arg-Gly-Asp-Ser (4), Met-Lys-Tyr-Trp-Gly-Arg-Gly-His-Ser (4-His), Ac-Met-Lys-Tyr-Trp-Gly-Arg-Gly-Asp-Ser (6), and Met-Lys-Tyr-Trp-Gly-Arg-Gly-His-Ser (6-His, Scheme 2). These nonapeptide contain amino acid residues with nucleophilic side chains: thiol (cysteine), primary amine (lysine and free N-terminus), phenol (tyrosine), indole (tryptophan), guanidine (arginine), carboxylate ion (aspartate), imidazole (histidine), and primary alcohol (serine).
Scheme 2.
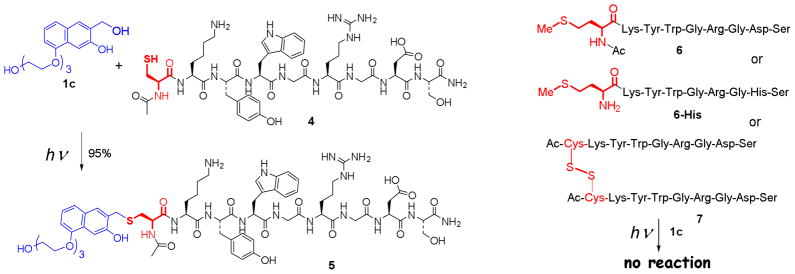
Selective alkylation of cysteine residue in peptides
Irradiation of PBS solution (pH = 7.4) of peptides 4 and 4-His (0.1 mM) containing 4 equivalents of NQMP 1c with fluorescent 300 nm lamp21 results in quantitative conversion of starting peptide into the mono TEG-NQMP-labeled derivatives 5 and 5-His (Scheme 2, Table 1). Peptide 5 was isolated by preparative HPLC in 95% yield and its structure confirmed by ESI-HRMS and MS/MS.20
Table 1.
Efficiency of photo-labeling of peptide 4 with TEG-NQMP (1c) under different conditions.a)
| Irradiation wavelength (nm) | Irradiation time (min) | 4 to 1c ratio | Yield of conjugate 5 (%) | Unreacted 4 (%) |
|---|---|---|---|---|
| 300 | 2 | 1:4 | 94±2 | n.d.b) |
| 350 | 5 | 1:4 | 95±2 | n.d.b) |
| 300 | 2 | 1:3 | 79±3 | 17±2 |
| 300 | 2 | 1:2 | 47±2 | 50±2 |
| 300 | 2 | 1:1 | 20±2 | 76±2 |
| 300 | 12 | 1:1 | 18±2 | 79±2 |
Starting concentration of peptide 4 = 0.1 mM in PBS (pH = 7.4) at r.t.;
Not detected.
When cysteine in the oligopeptide was replaced with methionine (peptides 6 and 6-His), or free thiol was converted into disulfide bond by oxidative homo-dimerization (peptide 7) no labeling was observed upon irradiation in the presence of 4 to 12 equivalents of TEG-NQMP (1c).20 These experiments show the high cysteine specificity of oNQM-tagging procedure. It is important to note that peptides 4 and TEG-NQMP (1c) are photochemically stable under these conditions. No decomposition or losses of materials were detected by HPLC after separate irradiation of 0.1 mM PBS solutions of 1c and peptide 4 for 12 min using both 300 and 350 nm fluorescent lamps. Obviously, NQMP 1c still forms oNQM 2c but in the absence of thiols the latter rapidly undergoes hydration back to starting materials.
Achieving complete conversion of 0.1 mM solution of 4 to 5 requires 2 min of irradiation at 300 nm (Table 1). Photolysis using 350 nm lamp takes somewhat longer, apparently due to the lower extinctions coefficients of NQMP 2c at this wavelength.18 The TEG-NQMP-peptide conjugate (5) is photoreactive itself and produces oNQM 2 upon irradiation. The formation of 5, therefore, represents a photochemically driven equilibrium between peptide 4 and NQMP 1c one side, and 5 on the other (Scheme 3).
Scheme 3.
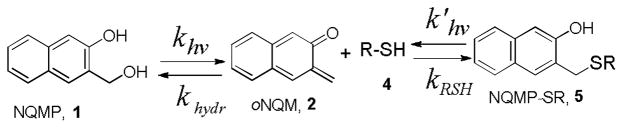
Light-driven equilibrium between NQMP and NQMP-SR
The position of equilibrium is defined by the rate of reaction of the common intermediate 2 with water (khydr = kH2O[H2O]) and cysteine (kRSH), as well as by the efficiency of the photoelimination reaction of NQMP (1, khv) and NQMP-SR (5, k’hv). Since under the established equilibrium conditions concentrations of 1 and 5 remain constant, we can write the following equation (eq. 1), where [NQMP], [RSH], and [NQMP-SR] are equilibrium concentrations of corresponding substrates:
| (1) |
| (2) |
The ratio of caged to free thiol can be further expressed as a function of NQMP concentration (eq. 2). Since kRSH is about five orders of magnitude larger than kH2O, and khv / k’hv ~ 2,17 the equilibrium is shifted in the favor of NQMP–SR (5) formation at relatively low concentrations of thiol and NQMP in aqueous solutions. Using experimental rates of oNQM hydration and reactions with simple thiols,16 as well as photochemical efficiencies of NQMP and NQMP-SR reactions, we can evaluate that 90+% conversion of a substrate is achieved at starting NQMP concentration equal [NQMP] = 3×10−4 M−1 + [Substrate]. In biochemical labeling experiments, where concentration of a substrate is usually in μM range or lower, 400 μM of NQMP derivative should be sufficient to achieve complete functionalization of all available cysteine residues.
Thus, at 0.1 mM concentration of peptide 4 almost quantitative conversion to 5 is achieved with only four equivalents of 1c (Table 1). At lower NQMP to peptide ratios the conversion is reduced, and at the equimolar concentrations of both components yield of the labeled peptide 5 drops to 20%. These results agree well with the conversion predicted by the equation 2. Prolonged irradiation has virtually no effect on peptide 4 to 5 ratio suggesting that system reaches photostationary state after 2 min of irradiation with 300 nm light (Table 1).
Equation 2 also suggests that at low concentration of free NQMP, the process can be reversed. Photolysis of TEG-NQMP-peptide conjugate (5) at a low concentration of free NQMP (1) should result in the traceless release of the peptide 4 (Scheme 4). Indeed, irradiation of 0.1 mM solution of 5 for 2 min with 300 nm lamp produced the mixture containing 75% the free peptide 4 and 21% of caged 5. Longer irradiation does not significantly affect the 4 to 5 ratio (Table 2). Very similar product ratio was obtained in the photolysis of 0.1 mM peptide 4 solution in the presence of equimolar amount of TEG-NQMP (1c) (Table 1). Photo-release of free cysteine residues can be achieved at any concentration of the NQMP-labeled protein when vinyl ethers are employed as oNQM (2) trapping agents. Very facile Diels-Alder addition of ethyl vinyl ether (EVE) to intermediate oNQM converts the latter into a photo-stable benzochroman 8 (Scheme 4).17,18 This reaction efficiently removes oNQM precursors from the reaction mixture and shifts photo-equilibrium towards substrates with free thiol. Due to irreversibility of Diels-Alder addition, even one equivalent of ethyl vinyl ether is sufficient to achieve complete conversion of NQMP-caged peptide 5. The cleavage of NQMP-SR thioethers with or without vinyl ether derivatives can be accomplished using 350 nm light. Complete conversion of the starting material under these conditions requires 2–3 times longer irradiation than with 300 nm lamp (Table 2).
Scheme 4.
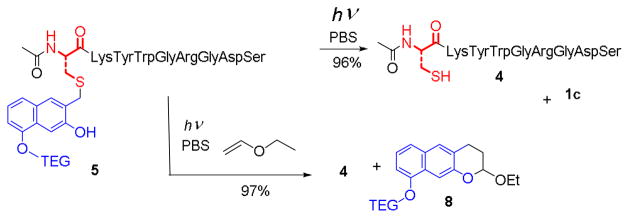
Photochemical Uncaging of Peptide Cysteine Moiety
Table 2.
Uncaging of o-NQMP labeled peptide 5a)
| Irradiation time (min) | Concentration of 5 (μM) | Concentration of EVE (μM) | Unreacted 5 (%) | Yield of peptide 4 (%) |
|---|---|---|---|---|
| 2 | 100 | 0 | 21±2 | 75±3 |
| 12 | 100 | 0 | 19±2 | 77±3 |
| 2 | 50 | 0 | 4±1 | 92±2 |
| 2 | 20 | 0 | n.d.b) | 96±2 |
| 5 | 20 | 0 | n.d.b) | 94±2c) |
| 2 | 100 | 100 | n.d.b) | 97±2 |
| 5 | 100 | 100 | n.d.b) | 96±2c) |
Using 300 nm fluorescent lamps in PBS solution at r.t.;
Not detected.
350 nm fluorescent lamps were used.
These observations indicate that the system photo-equilibrates to same photo-stationary state starting either from NQMP + peptide 4 or from caged peptide 5 side. As expected, further dilution of the solution of 5 results in higher yields of release of the thiol moiety. At 20 μM concentration or below, no caged substrate 5 can be detected after irradiation (Table 2). For practical applications, we can evaluate that 90+% release of NQMP-caged thiol is achieved at of 40 μM or lower substrate concentration.
The successful peptide labeling prompted us to explore the feasibility of the selective photo-caging of free cysteine residues in proteins. Bovine serum albumin (BSA, 9a), which contains a solvent-exposed cysteine residue (Cys-34), was employed as a model protein.22 Solutions of the protein (11 3M) and NQMP-biotin (1d, 100 3M)20 in PBS (pH = 7.4) were incubated in the dark for 15 min or irradiated with 350 nm fluorescent lamp for 2 min (Scheme 5). Low molecular weight compounds were removed by spin filtration or by size-exclusion chromatography (SEC) and resulting protein sample was analyzed by Western blotting using an antibiotin antibody – HRP conjugate (Figure 1A).20 As expected, BSA-NQMP-biotin adduct 10d was formed only in irradiated samples (compare lanes 2 and 3, Fig. 1A; lanes 1 and 4, Fig. 2A). The direct fluorescent labeling of a protein was demonstrated using the conjugate of NQMP with the fluorescent dansyl group (NQMP-DNS, 1f).23 Solutions of protein (12 3M) and 1f (100 3M) were incubated in the dark for 30 or 90 min, or irradiated with 350 nm fluorescent lamps for 6 min (Scheme 5). After removal of unreacted 1f, protein samples were resolved using a 4–20 % Tris-HCl gel. The fluorescent image of the gel clearly demonstrates that dansyl labelling of the protein occurred only under irradiation (Fig. 1B, lane 6).20 The NQMP-DNS derivatization of the protein was confirmed by MALDI-TOF and ESI mass spectra, as well as by digestion analysis.21
Scheme 5.
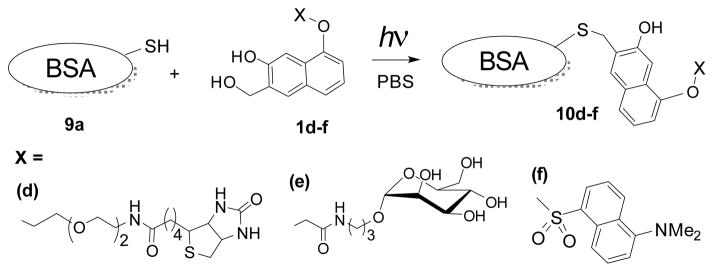
Selective photochemical derivatization of bovine serum albumin.
Fig. 1.

Western blot analysis of photochemical labeling of BSA (9a) with NQMP-biotin (1d) and NQMP-DNS (1f). Membranes were stained by Coomassie Brilliant Blue dye (lower panels) and a-antibiotin antibody-HPR - ECL Plus chemiluminescent substrate (A). Lanes 1A and 1B: molecular weight marker; 2A and 3B: unmodified BSA; 3A: BSA+1d after 2 min of irradiation; 4B & 5B: BSA incubated with 1f in the dark for 30 and 90 min; 6B: BSA + 1f after 6 min of irradiation.
Fig. 2.
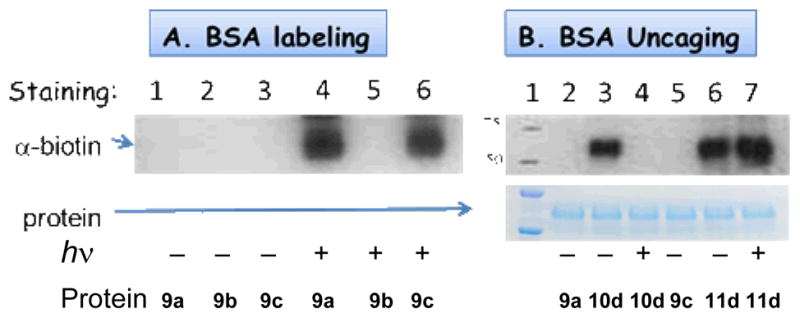
Western blot analysis of photochemical labeling (A) and uncaging (B) of BSA. Membranes were stained by antibiotin antibody-HPR - ECL Plus chemiluminescent substrate (for α-biotin) and Coomassie Brilliant Blue dye (for protein).
It is interesting to note that BSA dimer, which is clearly seen on both gels, apparently possesses at least one free cysteine residue that is photo-labeled with 1d and 1f (Figure 1, Lanes 3A and 6B).
The efficiency of BSA labelling with NQMP-biotin (1d) was highest at pH ~ 7 but was progressively reduced at higher acidities. We believe that this phenomenon is linked to the acidity of Cys-34 in BSA (pKa ~5).24 Under neutral conditions, this residue apparently exists in a very reactive thiolate form. In more acidic medium, equilibrium shifts to less nucleophilic thiol, which loses competition for oNQM intermediate (2, Scheme 3) to the aqueous solvent. The pH-dependence of the NQMP-based cysteine labeling system is currently under investigation.
To further test specificity of NQMP labeling of proteins, we have prepared a BSA analog 9b, in which Cys-34 has been converted into TEG ether (Scheme 6).21 Incubation of 9b with NQMP-biotin (1d) in the dark or under irradiation produces no biotinylated BSA (Fig. 2A, Lanes 2 and 5). This result confirms the cysteine selectivity of the NQMP photo-derivatization technique.
Scheme 6.
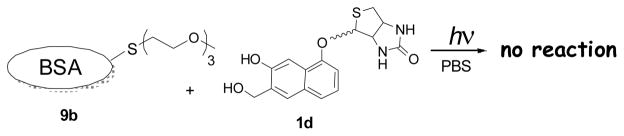
BSA with blocked Cys-34 does not react with NQMP
BSA is notorious for binding lipophilic compounds. However, BSA incubated with NQMP-DNS (1f) in the dark shows virtually no fluorescence, while irradiated sample exhibits strong emission at 490 nm (Fig. 3). Both samples were purified by a triple filtration through a Sephadex column and had the same concentration (9.7 μM). The difference in the absorbance at 335 nm (λmax of dansyl chromophore) between 10f and a control allows us to evaluate concentration of dansyl groups in the sample of 10f at 9.0 μM. This corresponds to almost quantitative labeling of BSA (see discussion below).
Fig. 3.

Emission spectra (λexc= 350 nm) of ~ 10 μM PBS solutions of BSA derivative 10f (solid line) and a control BSA (9a) isolated after incubation with 1f in the dark (dashed line).
The exhaustive NQMP derivatization of BSA (as tested by Ellman’s reagent) requires lower concentrations (100 μM) of NQMP than needed for the peptide 4 (400 μM, vide supra). There are two potential explanations for this phenomenon. First, the hydrophobic interaction between aromatic chromophore and the protein might increase the efficient concentration of NQMP 1d–f at the protein surface thus shifting the position of photo-equilibrium in favor of 10d–f. On the other hand, Cys-34 residue in BSA is apparently more acidic than the cysteine in in peptide 4.24 Therefore, at pH = 7.4 BSA has significantly higher fraction of cysteine in reactive thiolate from than 4. We currently investigate the effect of cysteine S-H acidity in peptides and proteins on the efficiency of NQMP labeling.
To quantify the extent of photo-biotinylation of BSA, total protein concentration in irradiated samples was determined using Bradford and bicinchoninic acid assays. The biotin concentration was measured using biotin quantification kit. Alternatively, BSA to NQMP ratio was determined spectroscopically (Table S1).21 Both methods report near quantitative BSA biotinylation. This was a surprising result because previous reports suggest that only 60 – 80% BSA contains a free thiol group. The rest of Cys-34 moieties form disulfides with cysteine or glutathione. 21,25 It is possible that free thiol contents of BSA is actually higher since Ellman’s test, which uniformly used in these measurements, often underestimates SH contents in proteins.26,27 This is even more plausible for BSA since Cys34 residue is similar or even more acidic than the thiol group in 2-nitro-5-thiobenzoate (NTB).24,27 On the other hand, we cannot exclude photochemically-induced cleavage of disulfide bonds, either directly or through NQMP sensitization.
Cysteine residues are often used for the preparation of glycoproteins.7,10b,28 To test the utility of thiol – oNQM photo-click chemistry for protein glycosylation, a PBS solution of BSA (9a, 11 3M) and mannose-NQMP conjugate (1e, 100 μM)21 was exposed to 350 nm light for 2 min (Scheme 5). The excess of 1e was removed by spin filtration and the resulting BSA-mannose conjugate (10e) was analyzed for total protein. Mannose content in 10e was determined by HPLC after TFA hydrolysis confirming almost quantitative derivatization.21
To explore the reversibility of cysteine photo-derivatization in proteins, a 2 3M PBS solution of biotinylated BSA 10d was irradiated with 350 nm fluorescent lamp for 2 min and protein isolated and purified by spin filtration.20 Western blot analysis of the product showed complete removal of biotin tag (Figure 2B, lane 4), while Coomassie Brilliant Blue staining confirmed the presence of protein in the sample. The product protein has the same mobility as the 9a (Figure 2B, lane 2) and biotinylated 10d, thus confirming clean regeneration of native BSA (Scheme 7).
Scheme 7.
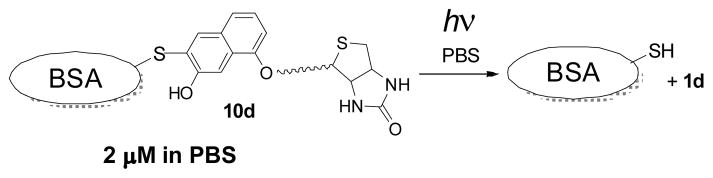
Photo-release of BSA from NQMP caged form 10d.
Photo-stable functionalization of cysteine residues can be achieved via a two-step procedure involving derivatization with a vinyl ether followed by a photo-Diels Alder reaction with NQMP. Thus, BSA (9a) was converted into BSA-vinyl ether conjugate (9c) by the reaction with 2-(2-(vinyloxy)ethoxy)ethyl iodide.21 Two minutes of irradiation at 350 nm of 9c (11 μM) in the presence of NQMP 1d (100 μM) in PBS resulted in the complete conversion of BSA-vinyl ether conjugate (9c) into Diels-Alder adduct 11d (Scheme 8, Figure 2A, lane 6).21 In contrast to 10d, irradiation of BSA-Biotin conjugate 11d, even for extended periods of time, does not result in the removal of a biotin tag (Fig. 2B, lane 7). In fact, electrophoretic mobility of BSA derivative 11d is not affected by irradiation illustrating photochemical stability of this conjugate (Fig. 2B, lane 6 and 7).
Scheme 8.
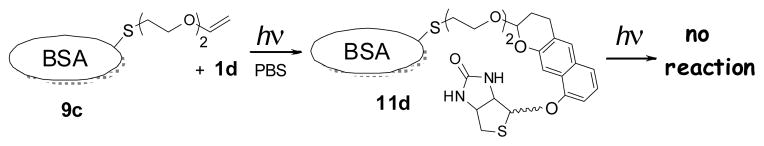
Irreversible photo-biotinylation of BSA
Reversible derivatization of the cysteine residues described in this report can be employed for pulling down and purifying proteins containing or genetically engineered to contain solvent-exposed cysteine residues. To explore the capabilities of the method, we have captured BSA on NeutrAvidin Agarose beads and then photochemically cleaved the protein from the solid support.
Following standard protocol (vide supra) a PBS solution containing BSA 9a (10 μM) and Biotin-NQMP conjugate 1d (100 μM) was irradiated for 5 min at 350 nm. NeutrAvidin Agarose resin (ca. 10% excess) was added to the photolysate and incubated in the dark for 30 min. The resulting resin was separated by centrifugation and washed. Combined supernatant contained no detectable amounts of BSA or 1d according to spectrophotometric analysis and Bradford assay (Scheme 9).
Scheme 9.

Photo-triggered catch and release of BSA using NeutrAvidin Agarose gel
The resulting NeutrAvidin Agarose resin was re-suspended in PBS buffer to achieve ca. 10 μM concentration of bound BSA (and ca. 100 μM of bound 1d). The suspension was exposed to 350 nm light for 5 min under ambient conditions. The analysis of supernatant showed the release of 70 – 80% of immobilized BSA (Scheme 9). When BSA-derivatized NeutrAvidin Agarose resin suspension was diluted to ca. 5 μM concentration of bound protein (~50 μM of 1d), 5 min of 350 nm irradiation resulted in 95% release of BSA from the beads. Similar high yield of the protein photo-cleavage from resin support was obtained at 10 μM of resin-bound BSA when diethylene glycol monovinyl ether (equimolar amount in respect to 1d) was added to a suspension before irradiation. This reagent apparently converts resin-bound NQMP groups into photochemically stable benzochroman moieties (vide supra).
Conclusions
We have described an efficient photochemical method for the selective derivatization of free cysteine residues in peptides and proteins. Irradiation of a neutral aqueous solution containing 3-(hydroxymethyl)-2-naphthol derivatives (NQMPs) and protein or peptide with a hand-held 300 or 350 nm fluorescent lamp results in fast and efficient conversion of free cysteines into NQMP-thioethers, while other amino acid residues remain unchanged. 0.4 mM of NQMP and 2 min of exposure is sufficient for quantitative derivatization of the substrate. NQMP can be equipped with various moieties, such as PEGs, dyes, carbohydrates, or possess a fragment for further selective derivatization, e.g., biotin, azide, alkyne, etc. The utility of this method was demonstrated by derivatizing bovine serum albumin with biotin, fluorescent dye, and mannose in a high yield. Other substrates, which survive short exposure to 350 nm light, can be conjugated with cysteine residues via this technique. Orthogonality of oNQM-based labeling technique to azide click chemistry20,29 allows for the two-step derivatization procedure. First, a target protein is photochemically derivatized with NQMP containing azide or alkyne functionality; the second substrate is then attached using conventional copper-catalyzed or strain-promoted azide-alkyne click chemistry. The NQMP label can be later removed by irradiating the substrate in a dilute solution (40 μM or lower) in the absence of free NQMP. Quantitative release of NQMP-caged substrates at higher concentrations can be achieved in the presence of vinyl ethers. We have demonstrated reversible biotinylation of BSA, as well as capture of biotinylated BSA on NeutrAvidin Agarose resin and then photochemical release of the protein from the beads. The cleavage of protein from the solid support does not require any reagents, simplifying purification. Reversible modification of the cysteine residues in proteins can be employed for the photo-regulation of protein activity either by caging of catalytically active cysteine (e.g., in cysteine proteases) or by attaching activity-modifying fragment to it (e.g., reversibly caging Cys in Acyl carrier proteins). Reversible PEGylation of active peptides and proteins can be employed for light-directed drug delivery. Very fast rate of thiol photo-release from NQMP thioethers17 permits time-resolved activation of caged substrates. Proteins carrying fluorescent NQMP derivative23 can be employed in FRAP-type experiments.
Supplementary Material
Acknowledgments
This research project was supported by the National Science Foundation (Grant No. CHE-0842590, V.V.P.), the National Cancer Institute of the U.S. National Institutes of Health (Grant No. RO1 CA88986, G.-J.B.), and the National Institute of General Medical Sciences of the U.S. National Institutes of Health (Grant No. R01 GM61761, G.-J.B.).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures, preparation and NMR spectra of newly synthesized compounds. See DOI: 10.1039/b000000x/
Contributor Information
Geert-Jan Boons, Email: gjboons@ccrc.uga.edu.
Vladimir V. Popik, Email: vpopik@uga.edu.
References
- 1.Chalker JM, Bernardes GJL, Davis BG. Acc Chem Res. 2011;44:730. doi: 10.1021/ar200056q. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hackenberger CPR, Schwarzer D. Angew Chem Int Ed. 2008;47:10030. doi: 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]; (b) Johnson JA, Lu YY, Van Deventer JA, Tirrell DA. Curr Opin Chem Biol. 2010;14:774. doi: 10.1016/j.cbpa.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lim R, Lin Q. Chem Commun. 2010;46:1589. doi: 10.1039/b925931g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sletten EM, Bertozzi CR. Angew Chem, Int Ed. 2009;48:6974. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Palomo JM. Eur J Org Chem. 2010:6303. [Google Scholar]
- 3.Wang LX, Lomino JV. ACS Chem Biol. 2012;7:110. doi: 10.1021/cb200429n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Díaz-Rodríguez A, Davis BG. Curr Opin Chem Biol. 2011;15:211. doi: 10.1016/j.cbpa.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 5.(a) Chalker JM, Bernardes GJL, Lin YA, Davis BG. Chem - Asian J. 2009;4:630. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]; (b) Godoy CA, de las Rivas B, Filice M, Fernandez-Lorente G, Guisan JM, Palomo JM. Process Biochem. 2010;45:534. [Google Scholar]; (c) Whetstone PA, Butlin NG, Corneillie TM, Meares CF. Bioconj Chem. 2004;15:3. doi: 10.1021/bc034150l. [DOI] [PubMed] [Google Scholar]
- 6.(a) Requejo R, Hurd TR, Costa NJ, Murphy MP. FEBS Journal. 2010;277:1465. doi: 10.1111/j.1742-4658.2010.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Nature. 1997;389:251. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 7.Bernardes GJL, Grayson FJ, Thompson S, Chalker JM, Errey JC, El Oualid F, Claridge DTW, Davis BG. Angew Chem, Int Ed. 2008;47:2244. doi: 10.1002/anie.200704381. [DOI] [PubMed] [Google Scholar]
- 8.Witus LS, Francis MB. Acc Chem Res. 2011;44:774. doi: 10.1021/ar2001292. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wasserberg D, Nicosia C, Tromp EE, Subramaniam V, Huskens J, Jonkheijm P. J Am Chem Soc. 2013;135:3104. doi: 10.1021/ja3102133. [DOI] [PubMed] [Google Scholar]
- 9.Vinzenz X, Grosse W, Linne U, Meissner B, Essen LO. Chem Commun. 2011;47:11071. doi: 10.1039/c1cc12929e. [DOI] [PubMed] [Google Scholar]; Huang R, Holbert MA, Tarrant MK, Curtet S, Colquhoun DR, Dancy BM, Dancy BC, Hwang Y, Tang Y, Meeth K, Marmorstein R, Cole RN, Khochbin S, Cole PA. J Am Chem Soc. 2010;132:9986. doi: 10.1021/ja103954u. [DOI] [PMC free article] [PubMed] [Google Scholar]; Simon MD, Chu F, Racki LR, de La Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. Cell. 2007;128:1003. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; Li QF, Yang Y, Maleckis A, Otting G, Su XC. Chem Commun. 2012;48:2704. doi: 10.1039/c2cc17900h. [DOI] [PubMed] [Google Scholar]; Robin MP, Wilson P, Mabire AB, Kiviaho JK, Raymond JE, Haddleton DM, O’Reilly RK. J Am Chem Soc. 2013;135:2875. doi: 10.1021/ja3105494. [DOI] [PubMed] [Google Scholar]
- 10.(a) Nathani RI, Chudasama V, Ryan CP, Moody PR, Morgan RE, Fitzmaurice RJ, Smith MEB, Baker JR, Caddick S. Org Biomol Chem. 2013;11:2408. doi: 10.1039/c3ob40239h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Macindoe WM, van Oijen AH, Boons GJ. Chem Commun. 1998:847. [Google Scholar]
- 11.Boutureira O, Bernardes GJL, D’Hooge F, Davis BG. Chem Commun. 2011;47:10010. doi: 10.1039/c1cc13524d. [DOI] [PubMed] [Google Scholar]; Chalker JM, Lercher L, Rose NR, Schofield CJ, Davis BG. Angew Chem Int Ed. 2012;51:1835. doi: 10.1002/anie.201106432. [DOI] [PubMed] [Google Scholar]
- 12.Rokita SE, editor. Quinone Methides. Vol. 1. Wiley; Hoboken, NJ: 2009. Wiley Series of Reactive Intermediates in Chemistry and Biology. [Google Scholar]
- 13.Diao L, Yang C, Wan P. J Am Chem Soc. 1995;117:5369. [Google Scholar]; Chiang Y, Kresge AJ, Zhu Y. J Am Chem Soc. 2000;122:9854. [Google Scholar]
- 14.Weinert EE, Dondi R, Colloredo-Melz S, Frankenfield KN, Mitchell CH, Freccero M, Rokita SE. J Am Chem Soc. 2006;128:11940. doi: 10.1021/ja062948k. [DOI] [PMC free article] [PubMed] [Google Scholar]; Di Valentin C, Freccero M, Zanaletti R, Sarzi-Amade M. J Am Chem Soc. 2003;125:8366. doi: 10.1021/ja010433h. [DOI] [PubMed] [Google Scholar]; Wang P, Song Y, Zhang LX, He HP, Zhou X. Curr Med Chem. 2005;12:2893. doi: 10.2174/092986705774454724. [DOI] [PubMed] [Google Scholar]; Richter SN, Maggi S, Mels SC, Palumbo M, Freccero M. J Am Chem Soc. 2004;126:13973. doi: 10.1021/ja047655a. [DOI] [PubMed] [Google Scholar]; Wang P, Liu R, Wu X, Ma H, Cao X, Zhou P, Zhang J, Weng X, Zhang ZL, Qi J, Zhou X, Weng L. J Am Chem Soc. 2003;125:1116. doi: 10.1021/ja029040o. [DOI] [PubMed] [Google Scholar]
- 15.Chiang Y, Kresge AJ, Zhu Y. J Am Chem Soc. 2002;124:6349. doi: 10.1021/ja020020w. [DOI] [PubMed] [Google Scholar]
- 16.Jiang J, Zeng D, Li S. ChemBioChem. 2009;10:635. doi: 10.1002/cbic.200800700. [DOI] [PubMed] [Google Scholar]; Bolton JL, Turnipseed SB, Thompson JA. Chem Biol Interactions. 1997;107:185. doi: 10.1016/s0009-2797(97)00079-3. [DOI] [PubMed] [Google Scholar]; Lo LC, Chiang YL, Kuo CH, Liao HK, Chen YJ, Lin JJ. Biochem Biophys Res Commun. 2004;326:30. doi: 10.1016/j.bbrc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Arumugam S, Popik VV. J Am Chem Soc. 2009;131:11892. doi: 10.1021/ja9031924. [DOI] [PubMed] [Google Scholar]
- 18.Arumugam S, Popik VV. J Am Chem Soc. 2011;133:5573. doi: 10.1021/ja200356f. [DOI] [PubMed] [Google Scholar]
- 19.Arumugam S, Popik VV. J Am Chem Soc. 2012;134:8408. doi: 10.1021/ja302970x. [DOI] [PubMed] [Google Scholar]
- 20.Arumugam S, Popik VV. J Am Chem Soc. 2011;133:15730. doi: 10.1021/ja205652m. [DOI] [PubMed] [Google Scholar]
- 21.Electronic Supplementary Information file
- 22.Rondeau P, Armenta S, Caillens H, Chesne S, Bourdon E. Arch Biochem Biophys. 2007;460:141. doi: 10.1016/j.abb.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Arumugam S, Popik VV. Photochem Photobiol Sci. 2012;11:518. doi: 10.1039/c1pp05317e. [DOI] [PubMed] [Google Scholar]
- 24.Lewis SD, Misra DC, Shafer JA. Biochemistry. 1980;19:6129. doi: 10.1021/bi00567a028. [DOI] [PubMed] [Google Scholar]
- 25.Janatova J, Fuller JK, Hunter MJ. J Biol Chem. 1968;243:3612. [PubMed] [Google Scholar]; Chen SL, Kim KH. Arc Biochem Biophys. 1985;239:163. doi: 10.1016/0003-9861(85)90823-9. [DOI] [PubMed] [Google Scholar]; Yasuhara T, Nokihara K. Anal Chem. 1998;70:3505. doi: 10.1021/ac9802630. [DOI] [PubMed] [Google Scholar]
- 26.Riener CK, Kada G, Gruber HJ. Anal Bioanal Chem. 2002;373:266. doi: 10.1007/s00216-002-1347-2. [DOI] [PubMed] [Google Scholar]; Faulstich H, Tews P, Heintz D. Anal Biochem. 1993;206:357. doi: 10.1006/abio.1993.1061. [DOI] [PubMed] [Google Scholar]; Wright SK, Viola RE. Anal Biochem. 1998;265:8. doi: 10.1006/abio.1998.2858. [DOI] [PubMed] [Google Scholar]
- 27.Brocklehurst K, Little G. Biochem J. 1973;133:67. doi: 10.1042/bj1330067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis NJ, Flitsch SL. Tetrahedron Lett. 1991;46:6793. [Google Scholar]
- 29.Arumugam S, Orski S, Locklin J, Popik VV. J Am Chem Soc. 2012;134:179. doi: 10.1021/ja210350d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.