

Abstract
Background
Newcastle Disease virus (NDV) causes severe and economically significant disease in almost all birds. However, factors that affect NDV replication in host cells are poorly understood. NDV generates long double-stranded RNA (dsRNA) molecules during transcription of single-stranded genomic RNA. Protein kinase R (PKR) is activated by dsRNA. The aim of this study was to elucidate the role of PKR in NDV infection.
Results
NDV infection led to the activation of dsRNA-dependent PKR and phosphorylation of its substrate, translation initiation factor eIF2α, in a dose-dependent manner by either the lentogenic strain LaSota or a velogenic strain Herts/33. PKR activation coincided with the accumulation of dsRNA induced by NDV infection. PKR knockdown remarkably decreased eIF2α phosphorylation as well as IFN-β mRNA levels, leading to the augmentation of extracellular virus titer. Furthermore, siRNA knockdown or phosphorylation of eIF2α or okadaic acid treatment significantly impaired NDV replication, indicating the critical role of the PKR/eIF2α signaling cascade in NDV infection.
Conclusion
PKR is activated by dsRNA generated by NDV infection and inhibits NDV replication by eIF2α phosphorylation. This study provides insight into NDV-host interactions for the development of candidate antiviral strategies.
Keywords: Newcastle disease virus, dsRNA, PKR, eIF2α, HeLa cells
Background
The presence of viral pathogen-associated molecular patterns (PAMPs) is known to trigger innate immune reactions, especially the stimulation of genes downstream type I interferon (IFN) [1]. Among the variety of IFN-stimulated gene products, double-stranded RNA-dependent protein kinase (PKR) is a key executor of this antiviral response [2]. As a pattern recognition receptor (PRR) for viral dsRNA, PKR is considered to be significant for type I IFN production [3]. The ubiquitously expressed PKR is usually present in an inactive form at a low abundance, but is activated by conformational change and autophosphorylation upon binding of dsRNA, generated during viral genome replication [4]. PKR activation leads to the phosphorylation of its natural substrate, the alpha subunit of eukaryotic initiation factor 2 (eIF2) on serine residue 51, which increases the affinity of phosphorylated eIF2α for eIF2β, which is required for protein translation, leading to the cessation of translation and consequent inhibition of viral replication [5,6]. PKR has been demonstrated to serve as a PRR in the mediation of IFN production in West Nile virus (WNV) infection [7]. Consistent with WNV infection, PKR is also required for IFN production in response to other RNA viruses, including encephalomyocarditis virus, Semliki Forest virus (SFV) and Theiler’s murine encephalomyelitis virus (TMEV) [8]. However, HCV infection arrested PKR-activated cap-dependent protein synthesis, thereby suppressing the translation of retinoic acid-inducible gene 1 (RIG-I) preventing the IFN-β induction [9].
Newcastle disease virus (NDV) is a member of the genus Avulavirus within the subfamily Paramyxovirinae of the Paramyxoviridae family [10]. NDV has a negative-sense, non-segmented, single-stranded RNA genome of at least three sizes of 15,186, 15,192, and 15, 198 nucleotides (nt) [11]. Six transcriptional units encode two surface glycoproteins, the fusion protein hemagglutinin-neuraminidase, the matrix protein, and the ribonucleoprotein complex, composed of the nucleocapsid protein (NP), a phosphoprotein, and large polymerase, which is necessary for viral transcription and replication. As is often the case, for its genomic replication, NDV first generates a full-length positive-strand antigenomic RNA and then, in turn, serves as a template for the synthesis of new negative-stranded RNA genome, which forms a dsRNA intermediate [12].
NDV infection reportedly leads to the upregulation of total PKR mRNA in the spleen of specific pathogen-free chickens [13]. NDV infection can also induce PKR autophosphorylation and subsequently limit protein synthesis [14]. Previous reports also showed that PKR is not required for induction of IFNα upon NDV infection [15]. However, there is little information available in the literature regarding PKR inhibitory mechanisms in the context of NDV infection. In the current report, we found that the NDV lentogenic strain LaSota and velogenic strain Herts/33 both triggered PKR activation and eIF2α phosphorylation in HeLa cells. Furthermore, we demonstrated the antiviral effects of PKR and explored the role of eIF2α in response to NDV replication. Our results indicated that the PKR-induced eIF2α phosphorylation is responsible for the antiviral effect against NDV. Notably, NDV-induced IFN-β mRNA levels were decreased in PKR knockdown HeLa cells using specific short interfering RNA (siRNA) for PKR, indicating that PKR was necessary for NDV-induced IFN synthesis.
Results
NDV infection triggers PKR activation through eIF2α phosphorylation
HeLa cells were infected with NDV strain LaSota or Herts/33 at a multiplicity of infection (MOI) of 1. Cell pellets were collected at 6, 12, and 24 h post-infection (hpi), respectively. As shown in Figure 1A and B, infection by NDV strains LaSota or Herts/33 infection strongly induced PKR activation at 12 and 24 hpi. Furthermore, phosphorylation of eIF2α, the substrate of phosphorylated PKR, was observed at 12 and 24 hpi. To further verify this finding, we explored the NDV-induced phosphorylation of PKR and eIF2α at a dose of 0.5, 1, or 2 MOI for 12 h. Notably, western blot analysis results showed that PKR and eIF2α could be phosphorylated by NDV strains LaSota (Figure 1C) and Herts/33 (Figure 1D) in a dose-dependent manner.
Figure 1.

NDV infection induced the phosphorylation of PKR and eIF2α. HeLa cells were infected with NDV strain LaSota (A) or Herts33 (B) at an MOI of 1 and harvested at 6, 12, and 24 hpi, respectively. The cell lysates were collected and analyzed by western blot with anti-PKR, anti-p-PKR (T446), anti-eIF2α, and anti-p-eIF2α (S51) antibodies. β-actin was used as a protein loading control. HeLa cells were infected and harvested with different doses (0.5, 1, and 2 MOI) of NDV strain LaSota (C) or Herts33 (D) for 12 h. Proteins were analyzed by western blot analysis with anti-PKR, anti-p-PKR (T446), anti-eIF2α, and anti-p-eIF2α (S51) antibodies. β-actin was used as a protein loading control. E. Comparison of PKR and eIF2α phosphorylation levels in HeLa cells inoculated with replication-competent or UV-inactivated NDV strain Herts/33.
To investigate whether viral replication is required for NDV-induced PKR and eIF2α activation, ultraviolet (UV) light-inactivated NDV Herts/33 was used for the experiment. The results showed that neither PKR nor eIF2α was phosphorylated at all timepoints post-infection (Figure 1E) and, furthermore, indicated that PKR activation is induced by NDV infection and not by interacting cells with non-infecting virus particles. Interestingly, total PKR protein content was not increased, suggesting that IFN-induced responses were not fully activated in the early phase of NDV infection.
NDV replication generates double-stranded RNA in HeLa cells
Since PKR is considered a PRR for viral dsRNA, we deduced that NDV generates dsRNA during replication. To verify this deduction, monoclonal antibody (mAb) J2 recognizing the dsRNA molecules of more than 40 bp was used to detect intracellular dsRNA using an immunofluorescence assay. As shown in Figure 2, viral dsRNA particles were identified as bright dots distributed around the nuclei of the HeLa cells infected with NDV strain Herts/33 or LaSota, while no positive signals were found in cells treated with UV-inactivated viruses.
Figure 2.

Detection of dsRNA in NDV infected cells. HeLa cells grown on glass coverslips were infected with NDV strain LaSota or Herts/33 at an MOI of 1 or treated with UV-inactivated Herts/33 for 12 h. dsRNAs were detected by immunofluorescenct staining with the specific anti-dsRNA monoclonal antibody J2. The secondary antibody was Cy3-labeled goat anti-mouse IgG (red). Nuclei were stained with 1 μg/mL of DAPI.
Antiviral effect of PKR on NDV replication
To determine the role of PKR in NDV replication, we transfected HeLa cells with PKR-specific siRNA to knockdown PKR and transfected other cells with non-targeting siRNA as a negative control. The results showed that siPKR efficiently impaired endogenous PKR expression (Figure 3A, B). Using this approach, we successfully inhibited PKR expression, which was followed by an apparent reduction of eIF2α phosphorylation. The reduction of endogenous PKR resulted in an obvious increase in viral NP synthesis (Figure 3A). Consistent with the increase of viral NP, the increase of virus titer in the supernatant of PKR knockdown cells was also analyzed and measured. The results showed that the reduction in PKR protein levels resulted in a 1.7-fold increase in extracellular virus yields (Figure 3C).
Figure 3.
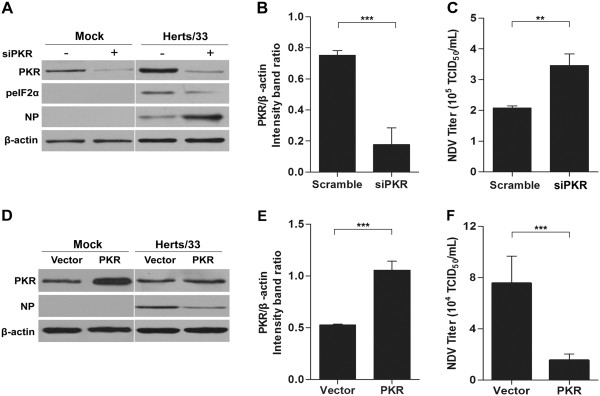
PKR inhibited NDV replication in HeLa cells. A. HeLa cells were transfected with the siRNA specific for PKR (100 pmol/mL) or control siRNA for 48 h and then infected with NDV strain Herts/33 or mock infected. The cells were harvested at 12 hpi and then collected for western blot analysis. The protein levels were determined with anti-PKR, anti-p-eIF2α, and anti-NP antibodies. β-actin was used as a protein loading control. B. The intensity band ratio of PKR to β-actin. Representative results are shown with graphs representing the ratio of PKR to β-actin normalized to the control condition. Data are presented as means from three independent experiments. Significance was analyzed using two-tailed Student’sttest. ***, P ≤ 0.001. C. Determination of the NDV replication in PKR knockdown HeLa cells. The extracellular virus yields were determined at 12 hpi and expressed as TCID50/mL. Data are presented as means from three independent experiments. Significance was analyzed using two-tailed Student’s t-test. **, p ≤ 0.01. D. HeLa cells were transfected with pcDNA3.1 or pcDNA-PKR. At 24 hpi, the cells were infected with NDV strain Herts/33 at an MOI of 1 or mock infected and harvested at 12 hpi. The protein levels were determined with anti-PKR or anti-NP antibodies. β-actin was used as a protein loading control. E. The intensity band ratio of PKR to β-actin. Representative results are shown with graphs representing the ratio of PKR to β-actin normalized to the control condition. Data are presented as means from three independent experiments. Significance was analyzed using two-tailed. Student’sttest. ***, P ≤ 0.001. F. Determination of the NDV replication on PKR overexpression in HeLa cells. The extracellular virus yields were determined at 12 hpi and expressed as TCID50/mL. Data are presented as means from three independent experiments. Significance was analyzed using two-tailed Student’s t-test. ***, p ≤ 0.001.
To further probe the role of PKR, we established HeLa cells overexpressing PKR through transfection of pcDNA-PKR plasmids. PKR protein levels in cells overexpressing PKR were obviously increased compared to that of normal cells (Figure 3D, E). PKR overexpression resulted in a decrease of viral NP synthesis (Figure 3D). Consistently, extracellular viral yields were decreased by 4.9-fold in cells overexpressing PKR infected with NDV strain Herts/33 (Figure 3F). Collectively, these data suggested that PKR plays an antiviral role in NDV-infected cells.
eIF2α plays a critical role in NDV replication
The translation initiation factor eIF2α, which has known involvement in protein translation by composing the ternary complex eIF2α-GTP-MettRNAi, was identified as a PKR substrate [16]. Recently, PKR-induced eIF2α phosphorylation followed by translation inhibition has been shown to play a role in inhibition of viral replication [17]. To investigate whether eIF2α was involved in NDV replication, we established eIF2α knockdown cells by transfecting siRNA specific to eIF2α or non-targeting siRNA into HeLa cells, followed by infection with NDV strain Herts/33. The efficiency of siRNA and the content of eIF2α and NP proteins were analyzed by western blot analysis. Transfection of sieIF2α resulted in a reduction of eIF2α protein level by 4.5 folds in mock-infected cells (Figure 4A, B). As shown in Figure 4A, viral NP protein was decreased by approximately 80% in eIF2α knockdown cells compared to that of control cells. Consistent with this finding, eIF2α knockdown resulted in a 2.6-fold increase in extracellular virus yield, indicating that eIF2α knockdown significantly impaired NDV replication (Figure 4C). Okadaic acid (OA) was defined as a protein phosphatase inhibitor, promoting eIF2α phosphorylation [18]. To further confirm the function of eIF2α in NDV replication, we augmented eIF2α phosphorylation in HeLa cells through OA treatment. PKR and eIF2α phosphorylation were significantly increased in OA-treatment cell, compared with that in mock cells (Figure 4D-E). To intuitively confirm whether eIF2α phosphorylation plays a role in NDV infection, green fluorescent protein (GFP)-expressing NDV ZJ1 strain (ZJ1-GFP) was utilized [19]. As shown in Figure 4G, in contrast to untreated cells, in the presence of OA, the percentage of GFP-positive cells was significantly decreased. Analysis of the spotlights under the fluorescence microscope showed that interfering with eIF2α dephosphorylation pharmacologically led to an approximate 3-fold decrease in intracellular ZJ1-GFP replication. Above all, these results indicated that eIF2α is required for NDV replication and eIF2α phosphorylation by active PKR induces an antiviral effect in NDV-infected cells.
Figure 4.

eIF2α is critical for NDV replication. A. HeLa cells were transfected with the siRNA specific for eIF2α (100 pmol/mL) or control siRNA for 48 h and then processed as described for Figure 3A. B. The intensity band ratio of eIF2α to β-actin. Representative results are shown with graphs representing the ratio of eIF2α to β-actin normalized to the control condition. Data are presented as means from three independent experiments. Significance was analyzed using two-tailed. Student’s t-test. ***, p ≤ 0.001. C. Determination of NDV replication in eIF2α knockdown HeLa cells. The extracellular virus yields were determined at 12 hpi and expressed as TCID50/mL. Data are presented as means from three independent experiments. Significance was analyzed using two-tailed Student’s t-test. *, p ≤ 0.05. D. HeLa cells were infected with ZJ1-GFP at an MOI of 1 or mock infected for 9 h in the presence of OA (10 nM). The cells were harvested and then processed as described for Figure 1A. E and F. The intensity band ratio of peIF2α (E) or pPKR (F) to β-actin. Representative results are shown with graphs representing the ratio of peIF2α (E) or pPKR (F) to β-actin normalized to the control condition. Data are presented as means from three independent experiments. Significance was analyzed using two-tailed Student’s t-test. ***, p ≤ 0.001. G. HeLa cells were infected with ZJ1-GFP at an MOI of 1 for 9 h in the presence of OA (10 nM) and observed by immunofluorescenct microscopy. The number of GFP-positive cells was quantified using Image J software. Data are presented as means from three independent experiments. Significance was analyzed using One-way ANOVA followed by Tukey’s test. **, p ≤ 0.01; ***, p ≤ 0.001, *, p ≤ 0.05.
PKR contributed to IFN-β production during NDV infection
PKR may serve as a PRR that mediates IFN production during WNV, SFV, and TMEV infection. On the other hand, HCV suppressed RIG-I translation thereby inducing IFN-β protein synthesis via PKR activation. To evaluate the role of PKR in type I IFN production after NDV infection, we first infected with HeLa cells with NDV strain Herts/33 and assessed the phosphorylation of PKR and eIF2α. Meanwhile, polyinosinic-polycytidylic acid [poly (I:C)] was used as a positive control. As shown in Figure 5B, IFN-β mRNA levels were increased by 25- and 451-fold in cells treated with poly(I:C) and NDV strain Herts/33, respectively, compared with mock-infected cells. The dramatic elevation of IFN-β mRNA levels in NDV-treated cells coincided with phosphorylation of PKR and eIF2α (Figure 5A). To further identify the role of PKR in IFN-β production, Hela cells were transfected with either siPKR or scrambled siRNA and then infected with NDV strain Herts/33. As expected, PKR knockdown significantly suppressed IFN-β production. These results demonstrated that PKR was involved in type I IFN production during NDV infection.
Figure 5.

PKR contributed to the upregulation of IFN-β after NDV infection. A. HeLa cells were either stimulated with poly (I:C) (5 μg/mL) or infected with N strain Herts33 at an MOI of 1 for 12 h. The cell lysates were collected and analyzed by western blot analysis with anti-PKR, anti-p-PKR (T446), anti-eIF2α, and anti-p-eIF2α (S51) antibodies. β-actin was used as a protein loading control. B. HeLa cells were treated with poly (I:C) (5 μg/mL) or infected with NDV strain Herts33 at an MOI of 1 for 12 h. IFN-β mRNA levels in the cell lysates were quantified by real-time PCR. The folds of mRNA expression were calculated by the formula of comparative CT method. Data are presented as means from three independent experiments. Significance was analyzed using One-way ANOVA followed by Tukey’s test. **, p ≤ 0.01; ***, p ≤ 0.001. C. HeLa cells were transfected with PKR-specific siRNA (100 pmol/mL) or control siRNA for 48 h and then inoculated with NDV strain Herts/33 at an MOI of 1. The cells were harvested and analyzed by western blot analysis to determine the efficiency of siRNAs. D. HeLa cells transfected with siPKRs were infected with NDV strain Herts/33 at an MOI of 1 for 12 h and then harvested for qualification of IFN-β mRNA levels by real-time PCR. The folds of mRNA expression were calculated using the comparative CT method. Data are presented as means from three independent experiments. Significance was analyzed using two-tailed Student’s t-test. ***, p ≤ 0.001.
Discussion
Protein kinase PKR is known to be activated mainly by viral dsRNA molecules as well as by the dsRNA binding protein PKR activating protein (PACT) in human or retina and anterior neural fold homeobox (RAX) in mice [20,21]. Active PKR plays multiple roles in cells by the transmission of signals to eIF2α to control translation and integration of signals from various other factors, such as P53, IкB kinase, P58, and P38 [22]. Previous studies have shown that PKR executes its antiviral effects through control of translation or regulation of apoptosis by phosphorylation of various substrates, including eIF2α, inhibitor of IкB, interferon regulatory factor 1, and P53 [23,24]. Many viruses have evolved various inhibitory mechanisms targeting PKR [25-27]. Viral proteins counteract the PKR pathway at different levels by sequestering dsRNA, inhibiting PKR activation, synthesizing PKR pseudosubstrates, inhibiting PKR dimerization, degrading PKR, or activating antagonist phosphatases [2]. The correlation between PKR and NDV has been suggested by the fact that NDV infection induces PKR phosphorylation and subsequently inhibits host gene translation [14]. A later study showed that the expression of the NDV nonstructural protein V decreased PKR activation [28]. Although these studies primarily indicated the interaction between PKR and NDV infection, detailed evidence for its antiviral mechanism remains limited.
In the current report, we first demonstrated that the activities of phosphorylated PKR and eIF2α were gradually increased during NDV infection in both a time- and dose-dependent manner (Figure 1A–D). The fact that both the lentogenic strain La Sota and velogenic strain Herts/33 could induce the PKR/eIF2α signaling cascade indicated that this response is likely to be a general anti-NDV mechanism. It seems that Herts/33 is able to increase the fraction of phosphorylated eIF2α compared to LaSota. One hypothesis assumed that no trypsin was added in the supernatant of LaSota-infected cells. Therefore LaSota was not able to re-infect after one round of virus replication. The other proposed that the differences of nonstructural protein, such as V, between velogenic Herts/33 and lentogenic LaSota strains contribute to the phosphorylation of eIF2α. PKR and eIF2α were not phosphorylated in the cells treated with UV-inactivated viruses, indicating that NDV replication is required for PKR activation (Figure 1E). PKR is activated by intracellular dsRNA generated by various viruses, such as WNV [8]. On the other hand, Sendai virus C protein restricted PKR activation by limiting dsRNA generation [14]. To verify whether dsRNA is involved in PKR activation during NDV infection, mAb J2 recognizing the dsRNA molecules was used to confirm the generation of dsRNA in the cytoplasm of HeLa cells infected with either lentogenic or velogenic NDV strains, but not UV-inactivated NDV, which was consistent with the activation of PKR after NDV infection. Since UV treatment impairs the ability of NDV to replicate, leading to the inhibition of viral dsRNA generation, subsequent PKR-eIF2α signaling was blocked (Figure 2).
The antiviral effects of PKR have been observed in innate immunity responses to a variety of viral infections. Recently, Zhang et al. [28] reported that PKR inhibition using siRNA PKR, shRNA PKR, or a PKR inhibitor enhanced HCV1α replication and rendered Huh7.5.1 cells more susceptible to HCV1α infection, suggesting that host cells may employ PKR activation to inhibit HCV1α replication. Similarly, PKR functions as a key mediator of IFN-α in opposing HBV replication, and ectopic expression of PKR resulted in the inhibition the expression of HBV capsids with a concomitant increase of phosphorylated eIF2α [29]. In order to determine the role of PKR in NDV infection, viral protein synthesis and extracellular viral yields were evaluated after PKR knockdown or overexpression. As expected, PKR knockdown increased NDV infection, while overexpression decreased NDV replication, thereby demonstrating the antiviral effects of PKR in NDV infection (Figure 3).
PKR was not required to inhibit host translation, but rather sped up this response through eIF2α phosphorylation [30]. A previous study also reported that NDV infection inhibited host protein translation [14], reminding us that translation control by eIF2α phosphorylation might be critical for the antiviral effects of PKR. The γ134.5 protein of the herpes simplex virus 1 binds to protein phosphatase 1a (PP1) and is required to prevent the shut-off of protein synthesis resulting from phosphorylation of the a subunit of eIF-2α [31]. Based on this, we mimic the up-regulation of eIF-2α phosphorylation by using OA, an inhibitor of the serine/theorine protein phosphatases PP1and PP2A [18]. OA reduced intracellular NDV replication, indicating that eIF2α is required for NDV replication (Figure 4A). Furthermore, the western blot and virus titration results of the present study illustrated that NDV replication was significantly inhibited in eIF2α knockdown cells (Figure 4C and D, respectively).These results indicated that PKR inhibits NDV replication by inhibiting cap-mediated eIF2α-dependent protein synthesis [30].
In the present study, dsRNAs were presented as punctuate dots accumulated in NDV-infected HeLa cells, suggesting that dsRNAs might interact with cellular PRRs that recognize dsRNA to form dsRNA-PRR complexes and then mediate downstream signaling, such as the IFN pathway or nuclear translocation of nuclear factor κB [32,33]. As a critical PRR recognizing virus dsRNA, PKR primarily acts as a viral antagonist through activation of the type I IFN signaling pathway. Since the role of PKR in type I IFN production during viral replication varies, we next evaluated the effect of PKR on IFN-β production after NDV infection. Our results showed that IFN-β production coincided with PKR/eIF2α phosphorylation during NDV infection. Moreover, PKR knockdown reduced IFN-β mRNA levels after NDV infection (Figure 5), which was contrary to the findings in previous reports that PKR deficiency did not prevent IFN-β responses to NDV infection in mouse embryonic fibroblasts [34]. In addition, as mentioned, previous report has shown that PKR is not required for induction of IFNα upon NDV infection [15]. Subsequently, the PKR requirement for IFN-α/β production might be virus- and strain-specific [8]. Also, there is a reasonable possibility that these observations might be related to the different cell models used in various experiments. In addition to PKR, other PRRs, such as toll-like receptor 3 (TLR3), RIG-I, and melanoma differentiation-associated protein 5 (MDA5), could also be recognized by dsRNA. Therefore, we could not rule out the possibility that other PRRs evoke IFN-β production through recognition of dsRNA generated by NDV infection [35-37]. To elucidate the mechanism of cross-talk between PKR, TLR3, and RIG/MDA5 during NDV infection, further studies are needed.
Conclusions
In this study, we demonstrated that NDV infection led to the activation of dsRNA-dependent PKR and phosphorylation of its substrate, eIF2α, in both a time- and dose-dependent manner. Inhibition of the PKR/eIF2α signaling cascade significantly impaired NDV infection, indicating the critical role of PKR in restricting NDV infection. The fact that PKR knockdown suppressed IFN-β production in NDV infection indicated that PKR plays an anti-NDV role via IFN-β upregulation. The results of this study should help to improve our understanding of NDV pathogenesis and provides insight for the development of candidate antiviral strategies.
Methods
Viruses and cells
NDV lentogenic strain LaSota was obtained from China Institute of Veterinary Drug Control (Beijing, China). The velogenic strain Herts/33 and recombinant NDV ZJ1 strain expressing GFP (ZJ1-GFP) were kindly provided by Xiufan Liu (Yangzhou University, Jiangsu, China) [19]. To obtain incomplete viral replication, viral suspensions were irradiated with UV-C at 75 mWs/cm2 using a low-pressure mercury vapor discharge lamp [38]. The absence of infective viruses after UV treatment was confirmed by the lack of replication in 9-day-old chicken embryonated eggs and in DF1 monolayer cultures. The human cervical cancer cell line (HeLa) and galline embryonic fibroblast cell line (DF-1) was purchased from ATCC (Manassas, VA, USA). Both cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA), 0.1 mg/mL of streptomycin (Sigma-Aldrich, St. Louis, MO, USA), and 100 U/mL of penicillin (Sigma-Aldrich) at 37°C in an atmosphere of 5% CO2.
Antibodies
Rabbit polyclonal anti-total PKR (Santa Cruz Biotechnology, Inc., Dallas, TX, USA), rabbit polyclonal anti-phospho-PKR threonine 451 (Epitomics, Inc., Burlingame, CA, USA), rabbit polyclonal anti-eIF2α, rabbit polyclonal anti-phospho-eIF2α (Cell Signaling Technology, Inc., Danvers, MA, USA), and mouse monoclonal anti-β-actin antibody (Sigma-Aldrich) were used in the current study. Mouse monoclonal anti-NP mAb was generated using the NP of LaSota strain as an antigen expressed in a prokaryotic system. Briefly, sequences encoding the full length of NP gene was amplified by reverse transcription polymerase chain reaction and cloned into the bacterial expression vector pET32a (Novagen, Madison, WI, USA) in order to express the histidine-tagged protein heterologously in Eshcerichia coli strain BL21. The expression was induced by treatment with 0.5 mM isopropyl-β-D-thiogalactopyranoside at 37°C for 4 h, and the expression product was purified using a His Band kit (Novagen) according to the manufacturer’s instructions. BALB/c mice were immuned by purified proteins for 4 times and sacrificed. The spleen of BALB/c mice was obtained for monoclonal antibodies preparation as previously [39].
Viral infection and drug treatment
HeLa cells were infected with NDV at an MOI of 1 at 37°C. For dose-dependent assay, HeLa cells were infected with different doses (0.5 MOI, 1 MOI, and 2 MOI) of NDV. After a 1-h absorption period, the cells were washed three times with cold phosphate-buffered saline (PBS) to remove unattached viruses and then cultured in DMEM containing 1% FBS. At 6, 12 and 24 hpi, the cell pellets and supernatant were harvested and stored at −80°C for further use. The replication of NDV in the cells was determined as the expression of NDV NP protein with monoclonal antibody against NDV NP protein using western blotting. Extracellular viral yields in cell supernatant were determined by estimating the median tissue culture infective dose (TCID50) in DF1 cells using the Reed and Muench method [40]. In the transfection assay, cells were transfected with eukaryotic expression plasmids or siRNA using lipofectamine 2000 transfection reagent (Invitrogen). 24 h (plasmid transfection) or 48 h (siRNA transfection) post transfection, cells were infected with NDV at an MOI of 1 and harvested at 12 hpi. For pharmacological experiments, HeLa cells were infected with ZJ1-GFP at an MOI of 1 or mock infected for 9 h in the presence of OA (10 nM). The number of GFP-positive cells was quantified in 10 randomly chosen fields (magnification × 100) using Image J software (http://imagej.nih.gov/ij).
Immunoblotting
HeLa cells were lysed in lysis buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 20 mM NaF, 1 mM ethylenediaminetetraacetic acid, 1% Triton, 1 mM phenylmethylsulfonyl fluoride, 1 mM ethylene glycol tetra-acetic acid, 20 mM Na4P2O7, 1 mM Na3VO4) containing the protease inhibitors leupeptin (0.5 μg/mL), aprotinin (0.5 μg/mL), and pepstatin (0.7 μg/mL). Cells were sonicated for 1 second using a Vibra Cell VCX130 sonicator (Sonics Vibra cell; Sonics & Material, Newtown, CT, USA), boiled for 5 min, and then cleared by centrifugation for 10 min at 12,000 g at 4°C. The lysates were further denatured by incubation for 5 min at 95°C in SDS-PAGE sample loading buffer (Beyotime, Nantong, China). The samples were further separated on 10% polyacrylamide gel (Bio-Rad Laboratories, Inc., Hercules, CA, USA), and transferred to a nitrocellulose membrane at 250 mA for 90 min, and then blocked using Tris buffered saline (TBS) solution containing 0.1% Tween 20 and 5% non-fat milk for 2 h. The membrane was incubated overnight at 4°C with the indicated antibodies. After washing three times with TBS/0.1% Tween 20, the membrane was exposed to horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG antibody (Sigma-Aldrich) at a dilution of 1:10000 for 2 h at room temperature. The protein bands were visualized with an enhanced chemiluminescent reagent (Pierce Biotechnology, Rockford, IL, USA) and quantified using Image J software.
Plasmid construction and transfection
Human PKR cDNA was amplified from HeLa cell mRNA and inserted into the HindIII/XhoI sites of pcDNA3.1(+) to construct pcDNA3.1-PKR. The purified plasmid DNA was prepared using the HiSpeed plasmid maxi kit (Qiagen, Hilden, Germany). The transfection procedure was performed essentially as described in the manufacturer’s instructions, but with minor modifications. Briefly, HeLa cells (1 × 106/well) were seeded into each well of a 6-well tissue culture plate and cultured at 37°C overnight. On the following day, 2 μg of plasmid was incubated for 45 min with 5 μL of Lipofectamine 2000 transfection reagent (Invitrogen) and then the transfection was allowed to occur overnight before the media was replaced with fresh DMEM. At 24 h post-transfection (hpt), cells were infected with Herts/33 strain at an MOI of 1 and harvested at 12 hpi.
siRNA treatment
Short interfering RNA oligos against PKR or eIF2α targeted to specific sequences (siPKR: 5′-GCGAGAAACUAGACAAAGU -3′ [41]; eIF2α: 5′-GAGAGGCUUGAAAGAGAAA-3′ [42]; non-targeting siRNA oligos: 5′-UUCUCCGAACGUGUCACGU-3′) were custom synthesized by GenePharma (Shanghai) Co., Ltd. (Shangai, China). HeLa cells were cultured to 60–70% confluency in a 6-well plate at 37°C overnight. PKR-specific siRNA, eIF2α-specific siRNA, and nontargeting siRNA as a negative control (100 pmol/well) were transfected with Lipofectamine 2000 according to the manufacturer’s instructions. At 6 hpt, the culture media was replaced with DMEM supplemented with 1% FBS (v/v). After a 48-h incubation period, the transfected cells were infected with NDV strain Herts/33 at an MOI of 1 and harvested at 12 hpi.
Real-time PCR
Total RNA of HeLa cells infected with NDV strain Hert/33 or PKR-silenced HeLa cells were extracted using the RNeasy kit (Qiagen, Valencia, CA, USA) and then reverse transcribed to cDNA using the SuperScript® III First-Strand Synthesis System kit (Invitrogen) with oligo dT primers (Invitrogen) [43]. To determine mRNA levels of IFN-β and β-actin, real-time PCR was performed using the SYBR Premix Ex Taq kit (TaKaRa Biotechnology (Dalian) Co., Ltd., Dalian, China) according to the instruction manual. The specific primers targeting IFN-β (forward: 5′-ACGACAGCTCTTTCCATGA-3′; reverse: 5′-AGCCAGTGCTCGATGAATCT-3′) and β-actin (forward: 5′-GATCTGGCACCACACCTTCT-3′; reverse: 5′-GGGGTGTTGAAGGTCTCAAA-3′) used in this study were described previously [43,44]. The expression levels of target mRNA were normalized to that of β-actin and calculated by the comparative CT method using the formula: fold change = 2−ΔΔCT, where ΔΔCT = [(CT target mRNAs –CT β-actin) in HeLa cells infected with NDV – (CT target mRNAs – CT β-actin) in mock cells] [45].
Immunofluorescent staining
HeLa cells were grown on coverslips overnight and infected with NDV strain Herts/33 at an MOI of 1 or stimulated with poly (I:C) for 12 h. Cells were fixed in 3% paraformaldehyde, permeabilized with 0.5% Triton X-100 for 10 min, incubated in blocking buffer (3% BSA/PBS), and then stained with mouse mAb J2 (Scicons, Hungary) followed by Cy3-labeled goat anti-mouse IgG (Beyotime). Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific Inc., Rockford, IL, USA) at a dilution of 1:500 for 10 min, and then the coverslips were mounted on slide glasses and visualized using a fluorescence microscope (Nikon Eclipse 80i; Nikon, Tokyo, Japan).
Statistical analysis
Data were expressed as means ± standard deviations. Significance was determined with the two-tailed independent Student’s t test (p < 0.05) between two groups. One-way ANOVA followed by Tukey’s test was used to compare multiple groups (>2).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SZ performed the immunoassays, participated in cell transfection and virus infection, and drafted the manuscript. YS coordinated the experimental design and drafted the manuscript. HC performed the immunofluorescent staining. YD performed the real-time PCR assay. YZ performed the viral titration. LT performed viral propagation in embryonated eggs. SY contributed to editing the manuscript. XQ, CS and CD made substantial contributions to the experimental design. All of the authors have read and approved the final version of this manuscript.
Contributor Information
Shilei Zhang, Email: zsl8600@126.com.
Yingjie Sun, Email: sunyingjie@shvri.ac.cn.
Hongjun Chen, Email: vetchj@163.com.
Yabin Dai, Email: ybdai@163.com.
Yuan Zhan, Email: jekitutu@163.com.
Shengqing Yu, Email: xxyusxx@yahoo.com.
Xusheng Qiu, Email: xsqiu1981@163.com.
Lei Tan, Email: tanlei6664ster@gmail.com.
Cuiping Song, Email: songcp203@163.com.
Chan Ding, Email: shoveldeen@shvri.ac.cn.
Acknowledgments
We wish to thank Professor Xiufan Liu (Yangzhou University) for providing NDV strain Herts/33 and ZJ1-GFP. This work was financially supported by grants from the Chinese National High-tech R&D Program (863 Program) (2011AA10A209), the National Natural Science Foundation of China (31372421), the Chinese Special Fund for Agro-scientific Research in the Public Interest (201003012, 201303033 and 201303038).
References
- Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20:1278–1290. doi: 10.1128/MCB.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey JW. Translational control in mammalian cells. Annu Rev Biochem. 1991;60:717–755. doi: 10.1146/annurev.bi.60.070191.003441. [DOI] [PubMed] [Google Scholar]
- Sudhakar A, Ramachandran A, Ghosh S, Hasnain SE, Kaufman RJ, Ramaiah KV. Phosphorylation of serine 51 in initiation factor 2 alpha (eIF2 alpha) promotes complex formation between eIF2 alpha(P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry. 2000;39:12929–12938. doi: 10.1021/bi0008682. [DOI] [PubMed] [Google Scholar]
- Gilfoy FD, Mason PW. West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J Virol. 2007;81:11148–11158. doi: 10.1128/JVI.00446-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, Takeuchi O, Akira S, Kaufman RJ, Reis e Sousa C. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe. 2010;7:354–361. doi: 10.1016/j.chom.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud N, Dabo S, Maillard P, Budkowska A, Kalliampakou KI, Mavromara P, Garcin D, Hugon J, Gatignol A, Akazawa D, Wakita T, Meurs EF. Hepatitis C virus controls interferon production through PKR activation. PLoS One. 2010;5:e10575. doi: 10.1371/journal.pone.0010575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CB, Pomeroy BS, Schrall K, Park WE, Lindeman RJ. An outbreak of conjunctivitis due to Newcastle disease virus (NDV) occurring in poultry workers. Am J Public Health Nations Health. 1952;42:672–678. doi: 10.2105/AJPH.42.6.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAO Yong-zhong GM, Wen-bo LIU, Xiu-fan LIU. Comparison of 40 complete genome sequences of Newcastle disease virus. J Yangzhou Univ (Argricultural and Life Science Edition) 2008;29:1–5. [Google Scholar]
- Dortmans JC, Rottier PJ, Koch G, Peeters BP. The viral replication complex is associated with the virulence of Newcastle disease virus. J Virol. 2010;84:10113–10120. doi: 10.1128/JVI.00097-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rue CA, Susta L, Cornax I, Brown CC, Kapczynski DR, Suarez DL, King DJ, Miller PJ, Afonso CL. Virulent Newcastle disease virus elicits a strong innate immune response in chickens. J Gen Virol. 2011;92:931–939. doi: 10.1099/vir.0.025486-0. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Komatsu T, Kitagawa Y, Sada K, Gotoh B. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J Virol. 2008;82:10102–10110. doi: 10.1128/JVI.00599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EJ, Marie I, Prakash A, Garcia-Sastre A, Levy DE. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by Vaccinia virus E3L protein. J Biol Chem. 2001;276:8951–8957. doi: 10.1074/jbc.M008717200. [DOI] [PubMed] [Google Scholar]
- Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254–266. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- Toribio R, Ventoso I. Inhibition of host translation by virus infection in vivo. Proc Natl Acad Sci U S A. 2010;107:9837–9842. doi: 10.1073/pnas.1004110107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Liao Y, Yap PL, Png KJ, Tam JP, Liu DX. Inhibition of protein kinase R activation and upregulation of GADD34 expression play a synergistic role in facilitating coronavirus replication by maintaining de novo protein synthesis in virus-infected cells. J Virol. 2009;83:12462–12472. doi: 10.1128/JVI.01546-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YL, Hu SL, Zhang YM, Sun SJ, Romer-Oberdorfer A, Veits J, Wu YT, Wan HQ, Liu XF. Generation of a velogenic Newcastle disease virus from cDNA and expression of the green fluorescent protein. Arch Virol. 2007;152:1241–1249. doi: 10.1007/s00705-007-0961-x. [DOI] [PubMed] [Google Scholar]
- Ito T, Yang M, May WS. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem. 1999;274:15427–15432. doi: 10.1074/jbc.274.22.15427. [DOI] [PubMed] [Google Scholar]
- Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MA, Meurs EF, Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Cuddihy AR, Wong AH, Tam NW, Li S, Koromilas AE. The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene. 1999;18:2690–2702. doi: 10.1038/sj.onc.1202620. [DOI] [PubMed] [Google Scholar]
- Samuel CE. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J Biol Chem. 1993;268:7603–7606. [PubMed] [Google Scholar]
- Myskiw C, Arsenio J, van Bruggen R, Deschambault Y, Cao J. Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-kappaB, and IRF3 pathways. J Virol. 2009;83:6757–6768. doi: 10.1128/JVI.02570-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Tripathi S, Ranjan P, Kumar P, Garten R, Deyde V, Katz JM, Cox NJ, Lal RB, Sambhara S, Lal SK. Influenza A virus nucleoprotein exploits Hsp40 to inhibit PKR activation. PLoS One. 2011;6:e20215. doi: 10.1371/journal.pone.0020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu YC, Yu CY, Liang JJ, Lin E, Liao CL, Lin YL. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J Virol. 2012;86:10347–10358. doi: 10.1128/JVI.00525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang J, Hong SH, Choi D, Choi KS, Kang S, Kim IH. Overexpression of Newcastle disease virus (NDV) V protein enhances NDV production kinetics in chicken embryo fibroblasts. Appl Microbiol Biotechnol. 2010;85:1509–1520. doi: 10.1007/s00253-009-2189-z. [DOI] [PubMed] [Google Scholar]
- Park IH, Baek KW, Cho EY, Ahn BY. PKR-dependent mechanisms of interferon-alpha for inhibiting hepatitis B virus replication. Mol Cells. 2011;32:167–172. doi: 10.1007/s10059-011-1059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry G, Breakwell L, Fragkoudis R, Attarzadeh-Yazdi G, Rodriguez-Andres J, Kohl A, Fazakerley JK. PKR acts early in infection to suppress Semliki Forest virus production and strongly enhances the type I interferon response. J Gen Virol. 2009;90:1382–1391. doi: 10.1099/vir.0.007336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Gross M, Roizman B. The gamma134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J Biol Chem. 1998;273:20737–20743. doi: 10.1074/jbc.273.33.20737. [DOI] [PubMed] [Google Scholar]
- Der SD, Lau AS. Involvement of the double-stranded-RNA-dependent kinase PKR in interferon expression and interferon-mediated antiviral activity. Proc Natl Acad Sci U S A. 1995;92:8841–8845. doi: 10.1073/pnas.92.19.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc Natl Acad Sci U S A. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, Williams BR, Aguet M, Weissmann C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995;14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M Jr. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orinska Z, Bulanova E, Budagian V, Metz M, Maurer M, Bulfone-Paus S. TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood. 2005;106:978–987. doi: 10.1182/blood-2004-07-2656. [DOI] [PubMed] [Google Scholar]
- Wilden H, Fournier P, Zawatzky R, Schirrmacher V. Expression of RIG-I, IRF3, IFN-beta and IRF7 determines resistance or susceptibility of cells to infection by Newcastle Disease Virus. Int J Oncol. 2009;34:971–982. doi: 10.3892/ijo_00000223. [DOI] [PubMed] [Google Scholar]
- Song H, Li J, Shi S, Yan L, Zhuang H, Li K. Thermal stability and inactivation of hepatitis C virus grown in cell culture. Virol J. 2010;7:40. doi: 10.1186/1743-422X-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayner EA, Carter WG. Identification of multiple cell adhesion receptors for collagen and fibronectin in human fibrosarcoma cells possessing unique alpha and common beta subunits. J Cell Biol. 1987;105:1873–1884. doi: 10.1083/jcb.105.4.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer-Oberdorfer A, Werner O, Veits J, Mebatsion T, Mettenleiter TC. Contribution of the length of the HN protein and the sequence of the F protein cleavage site to Newcastle disease virus pathogenicity. J Gen Virol. 2003;84:3121–3129. doi: 10.1099/vir.0.19416-0. [DOI] [PubMed] [Google Scholar]
- Rahman MM, Liu J, Chan WM, Rothenburg S, McFadden G. Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog. 2013;9:e1003465. doi: 10.1371/journal.ppat.1003465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM, Pae HO, Zheng M, Park R, Kim YM, Chung HT. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ Res. 2007;101:919–927. doi: 10.1161/CIRCRESAHA.107.154781. [DOI] [PubMed] [Google Scholar]
- Sun Y, Ding N, Ding SS, Yu S, Meng C, Chen H, Qiu X, Zhang S, Yu Y, Zhan Y, Ding C. Goose RIG-I functions in innate immunity against Newcastle disease virus infections. Mol Immunol. 2013;53:321–327. doi: 10.1016/j.molimm.2012.08.022. [DOI] [PubMed] [Google Scholar]
- Taura M, Eguma A, Suico MA, Shuto T, Koga T, Komatsu K, Komune T, Sato T, Saya H, Li JD, Kai H. p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol Cell Biol. 2008;28:6557–6567. doi: 10.1128/MCB.01202-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]